
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer brushes in solid-state nanopores form an impenetrable entropic barrier for proteins†
Gustav
Emilsson
a,
Kunli
Xiong
a,
Yusuke
Sakiyama
b,
Bita
Malekian
a,
Viktor
Ahlberg Gagnér
a,
Rafael L.
Schoch
b,
Roderick Y. H.
Lim
 b and
Andreas B.
Dahlin
*a
b and
Andreas B.
Dahlin
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Göteborg, Sweden. E-mail: adahlin@chalmers.se
bBiozentrum and the Swiss Nanoscience Institute, University of Basel, 4056 Basel, Switzerland
First published on 6th February 2018
Abstract
Polymer brushes are widely used to prevent the adsorption of proteins, but the mechanisms by which they operate have remained heavily debated for many decades. We show conclusive evidence that a polymer brush can be a remarkably strong kinetic barrier towards proteins by using poly(ethylene glycol) grafted to the sidewalls of pores in 30 nm thin gold films. Despite consisting of about 90% water, the free coils seal apertures up to 100 nm entirely with respect to serum protein translocation, as monitored label-free through the plasmonic activity of the nanopores. The conclusions are further supported by atomic force microscopy and fluorescence microscopy. A theoretical model indicates that the brush undergoes a morphology transition to a sealing state when the ratio between the extension and the radius of curvature is approximately 0.8. The brush-sealed pores represent a new type of ultrathin filter with potential applications in bioanalytical systems.
Polymer brushes consist of end-grafted macromolecules at a surface coverage high enough to promote stretching of the chains perpendicular to the surface.1 Brushes have long been attracting attention because of their ability to modify surface properties such as charge and wettability,2 even in a responsive manner.3 One particularly important application that can be realized by hydrophilic polymer brushes is resistance to protein adsorption. Reducing the adsorption of proteins is vital for medical applications such as implants and drug delivery vehicles as well as for analytical devices or any surface that needs “non-fouling” properties. Numerous studies have shown that solvated polymer brushes strongly reduce protein adsorption under physiological conditions. This in turn prevents cell attachment, making brushes essential for a broad range of applications in vitro and also in vivo. In particular, poly(ethylene glycol) (PEG) brushes are widely used to create inert surfaces and grafting PEG to nanoparticles is known to mediate a “stealth effect” bypassing the immune system.4
However, the physical mechanisms by which brushes regulate protein adsorption have remained unclear and heavily debated for 40 years. The problem has occupied theoreticians already since the initial considerations by de Gennes.5 Important work by the group of Szleifer predicted that when the polymer does not interact favorably with the surface, a brush with a reasonably high thickness can form a kinetic barrier, rather than an equilibrium prevention against protein adsorption.6 In this view, protein binding is energetically favorable on the solid surface beneath the brush, but not inside the brush or on top of it. Still, the theory of brushes as kinetic barriers is hard to verify experimentally. This is mainly because regardless of whether protein adsorption is detected or not for a given system, the role of potential polymer–surface interactions outcompeting the protein adsorption7 (equilibrium prevention) cannot be distinguished from the kinetic barrier effect. For instance, although surface plasmon resonance (SPR) can confirm qualitatively that brushes have protein excluding properties,8–10 it does not provide the concentration profile perpendicular to the surface. Furthermore, neutron reflectometry can analyze where in the brush proteins are located,11,12 but if adsorption occurs on the solid surface it remains unknown if the kinetic barrier was insufficient or if the polymer did not successfully outcompete the protein–surface attraction. In addition, all techniques based on averaging over a planar interface will have contributions from impurities and various types of defects. The literature is particularly inconsistent regarding potentially favorable interactions between proteins and PEG,12 which severely complicates reaching an understanding of, for instance, the fate of PEG-modified particles in the body.4
In this work, we provide the first conclusive evidence that a hydrophilic polymer brush can be a strong kinetic barrier for proteins by preparing PEG brushes inside different types of plasmonic nanopores.13 In this configuration proteins can either pass through or be repelled by the brush barrier without interacting with the solid surface. Protein translocation through the nanopores (or the lack thereof) is analyzed label-free in real-time by spectroscopy of surface plasmons in a thin gold film containing the pores. By systematically varying the polymer molecular weight and nanopore diameter we establish conditions for sealing the apertures. The brushes inside the pores are further analyzed by atomic force microscopy (AFM) in both indentation and high-speed operations. Our study provides new insights into the physics of macromolecules inside nanopores in general14 and illustrates the repulsive nature of PEG–protein interactions in particular. We also expect an impact on other research fields ranging from the nuclear pore complex (NPC) in molecular biology15 to biomolecular filters16 and single-molecule sensors based on nanopores.17
We prepared four different types of plasmonic nanopores as previously described18–21 (Fig. 1a). In all cases, colloidal self-assembly was used to produce a short-range ordered monolayer on silica, where each colloid in the end gives rise to one pore. Oxygen plasma was then used to tune the diameter,21 followed by deposition of 30 nm gold and 20 nm alumina, after which colloids were removed by rubbing the surface in liquid. Simple “nanoholes” on glass are then achieved by only removing the alumina film.21 Undercut-etched “nanocaves” are obtained by dipping the sample in a wet etchant for ∼40 s in order to remove ∼100 nm of the underlying glass in a more isotropic manner20 (more images in the ESI†), thereby creating a suspended gold film. The so-called “nanowells” are obtained by instead using reactive ion etching19 of the silica underneath the gold. Finally, “membrane pores” are obtained just like nanowells but by etching through a 50 nm thin support of silicon nitride,18 thereby connecting two reservoirs by an array of parallel pores. Regardless of the structure prepared, the nanoscale apertures in gold appeared identical when imaged by electron microscopy from above, except that for nanowells and membrane pores some of the underlying material was visible.18,19Fig. 1b shows an example image (nanocaves) with an average diameter of 70 nm and a standard deviation of a few nm. Note that the diameter distribution is more narrow and that the apertures are more cylindrical than for conventional pore arrays,22 even in ultrathin membranes.23
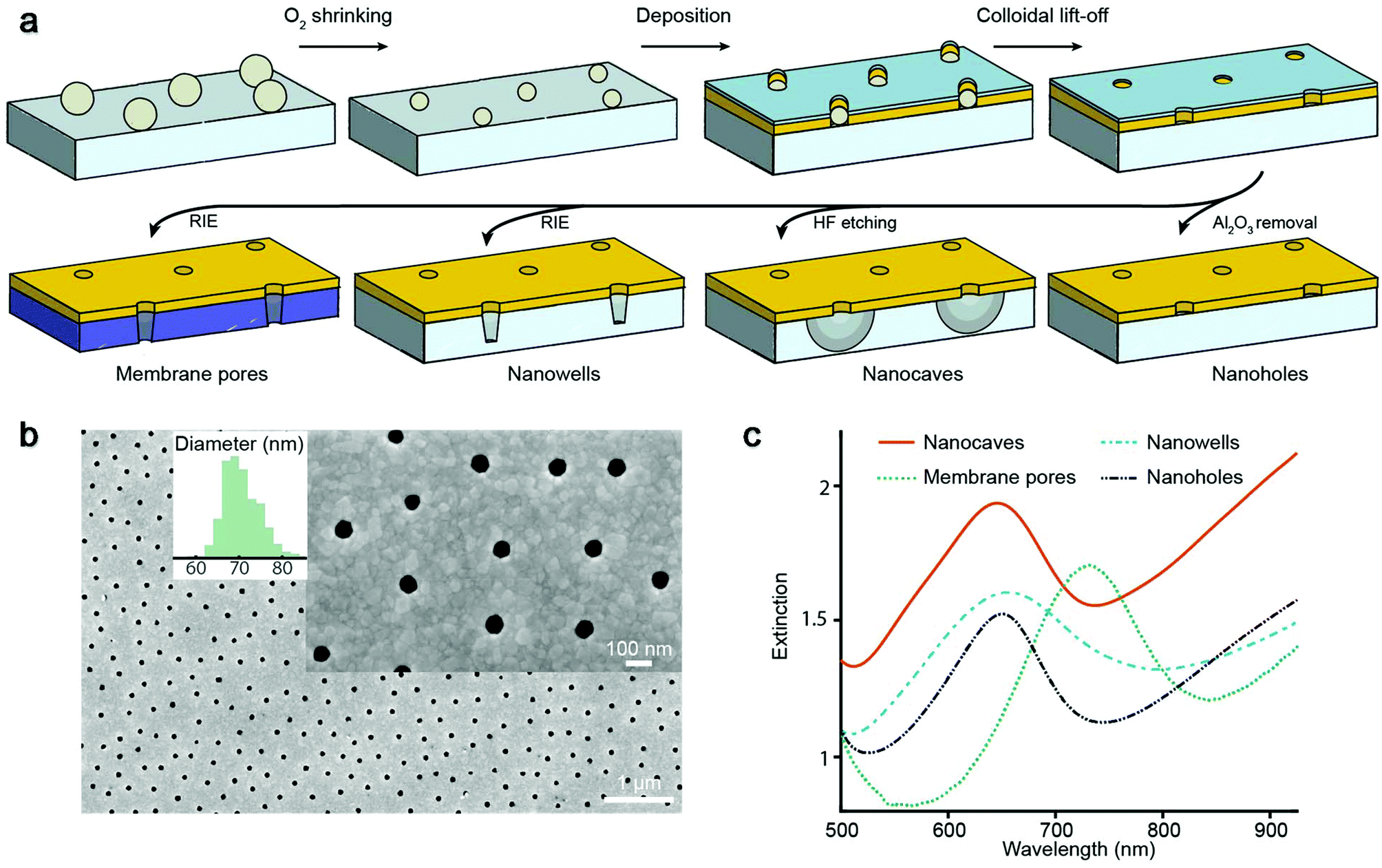 | ||
| Fig. 1 Nanostructure fabrication and characterization. (a) Preparation of different plasmonic nanopores using colloidal lithography on various supports followed by different etching steps. (b) Electron microscopy imaging of nanopores from above with diameter distribution. (c) Extinction spectra of the different nanopores in water showing plasmon resonance peaks. | ||
The extinction spectra of the different nanopores (Fig. 1c) showed the characteristic asymmetric resonance feature associated with the plasmonic activity.13 The extinction peak corresponds to the grating-type excitation of bonding mode surface plasmons induced by the short-range ordering of the apertures18–21 and offers a way to detect refractive index changes if they occur no longer than ∼100 nm from the metal surface.24 In the data below we plot the shift in the resonance peak wavelength as is conventional in refractometric sensing.13 For all samples in this study the characteristic center to center distance is ∼300 nm, which gives a resonance in the red region for a gold thickness of 30 nm and a water environment.18–21 For membrane pores the peak is shifted towards longer wavelengths (Fig. 1c) due to the higher refractive index of the silicon nitride membrane.18 Note that the number of pores analyzed is defined by the area from which transmitted light is collected, which is always much larger (∼100 μm or more) than the average distance between neighboring pores. Although single pores can be addressed both electrically16 and optically,17 averaging over many pores greatly improves the resolution in the plasmonic detection.
Polymer brushes were prepared inside the nanopores by introducing monodisperse (PDI 1.03 ± 0.02) thiol-terminated PEG with various molecular weights at room temperature using a theta solvent. This grafting-to strategy produces strongly stretched brushes on gold8,10 without influencing silica.19,20 Using conventional SPR we determined the brush height in air and in PBS buffer as previously reported8,9 (ESI†). The volume fractions of water were 57%, 69% and 84% for 2, 10 and 20 kg mol−1, respectively (the degree of polymerization N is equal to 45, 227 and 456, respectively). The contour lengths are 13–15 nm, 64–76 nm and 128–150 nm for 2, 10 and 20 kg mol−1, respectively. Fig. 2a shows the plasmon resonance peak shift during the binding of PEG with different molecular weights to nanocaves with a diameter of 70 nm. The plasmonic signal makes it possible to verify that the chemisorption of PEG reaches a saturation and that the signal magnitude is reasonable for a hydrated organic layer tens of nm in thickness.19–21
 | ||
| Fig. 2 Sealing nanopores with polymer brushes. (a) Grafting of PEG with different molecular weights (introduced at 0 min) in 0.9 M Na2SO4 monitored by the plasmonic signal from nanocaves. (b) Plasmonic signal (or the absence thereof) from the adsorption of avidin: Graphs I–III: 70 nm nanocaves modified with 2, 10 and 20 kg mol−1 PEG. Graph IV: 20 kg mol−1 PEG on 90 nm nanowells. Graph V: the same as IV but selective grafting to pore walls. Graph VI: the same as IV but theta solvent conditions. (c) Fluorescence intensity next to the “exit” side of a membrane with pores modified with PEG when labelled molecules are added on the opposite side. (d) Plasmonic time trace when complete serum is added to nanocaves (70 nm) and nanowells (90 nm) modified with 20 kg mol−1 PEG. | ||
To investigate the sealing ability of the PEG-modified nanopores we introduced the average sized protein avidin (NeutrAvidin, 60 kg mol−1) in PBS at a relatively high concentration (50 μg mL−1) and monitored the irreversible adsorption onto the silica surface19,20 exposed underneath the apertures using nanocaves and nanowells (Fig. 2b). There is no protein adsorption on the PEG-modified gold8 and the plasmonic signals come solely from the refractive index changes inside the cavities under the gold. (All material-selective chemistry was double checked as described in the ESI.†) Fig. 2b shows the response from avidin added to 70 nm nanocaves for different molecular weights of PEG (Graphs I–III). The smallest PEG (2 kg mol−1) does not create a significant barrier because it cannot possibly seal the apertures. Indeed, the time until saturation (∼1 h) is what is expected for open pores,19–21 showing that there is no significant hindrance for the diffusing proteins in this case. The situation changes when the nanocaves are modified with 10 kg mol−1 PEG, for which the signal starts to saturate at a lower value. For 20 kg mol−1 PEG brushes we consistently observed complete pore sealing, i.e. no detectable protein binding during the timescale of the experiment (Fig. 2b), as long as the average aperture diameter was 90 nm or less. (The detection limit of the nanocaves and nanowells is ∼8 proteins per cavity with our experimental setup, which is slightly more than for nanoholes which can detect a single protein per aperture.) For 20 kg mol−1 PEG on apertures >100 nm the response corresponding to complete adsorption inside the cavities was always observed, just like for 2 kg mol−1 PEG on 70 nm apertures. Thus, even the longest PEG does not block the entire cavity underneath the metal, i.e. the silica surface is indeed exposed. For 20 kg mol−1 PEG on apertures with an average diameter around 100 nm we observed signals that were a fraction of the response for open pores, just like that for the 10 kg mol−1 PEG on 70 nm apertures.
Fig. 2b further shows that the sealing behavior was the same on nanocaves and nanowells (Graph IV), which illustrates that the structure of the void below the metal is irrelevant. In fact, the sealing ability remained even when PEG was selectively grafted only to the walls of the apertures (Graph V in Fig. 2b). This was achieved by keeping the alumina layer during polymer binding and removing it afterwards, followed by grafting a shorter PEG to the planar Au. Thus, the conditions for sealing are solely determined by the radius of negative curvature for a given grafting density and molecular weight of PEG. Furthermore, the protein binding rate was always similar, even in the transition range of pore sizes where a fraction of the maximum response was observed. Our interpretation is that the pores in the upper region of the diameter distribution (Fig. 1b) are open and allow fast protein translocation resulting in a plasmonic signal. In other words, we could not identify any “semi-sealing regime” where the protein translocation still occurred, but at a considerably slower rate, given the resolution in the experiments. We could also confirm that when introducing proteins in the grafting solvent (0.9 M Na2SO4) the pores were open (Graph VI in Fig. 2b), i.e. reversible solvent-mediated gating was possible.25 This demonstrates the importance of the solvent quality for sufficient coil stretching which may be of interest for the active control of permeability.22
Nanowells and nanocaves were normally used to measure protein translocation because of the ease of fabrication and good stability in the plasmonic measurements. However, we also verified the sealing of membrane pores by using fluorescently labelled bovine serum albumin (BSA). The protein was introduced on one side and the intensity was measured next to the membrane on the opposite side (Fig. 2c). The membranes contained over 105 pores connecting two compartments in parallel. Still there was barely any signal from BSA (50 μg mL−1) when using 20 kg mol−1 PEG (in contrast to 2 kg mol−1 PEG) even though a single defect pore should result in high leakage (∼1000 molecules per second) for membranes this thin.23 We also verified that a sufficiently small molecule (biocytin Alexa Fluor 488, 975 g mol−1) could translocate the PEG-sealed pores. Furthermore, no significant increase in liquid flow resistance was detected when modifying the pores with PEG and wetting became simple.
To further characterize the blocking ability of sealed pores we also introduced complete serum at a concentration of 0.5% to pores with diameters in the range of 70–90 nm (Fig. 2d). The dilution in PBS was simply to avoid too large changes in the bulk refractive index and the protein concentration remains very high26 (∼350 μg mL−1). Still we could not detect any significant shift in the plasmon resonance. (The decrease of ∼0.04 nm in the signal upon injection of serum is due to a weak light scattering effect of the bulk liquid and the uncertainty is ±0.02 nm over 1 h.) Although one cannot expect every protein species present in serum to adsorb onto silica under these conditions, complementary experiments showed high adsorption from serum on silica as expected (ESI†).
Conventional liquid-phase AFM was used to characterize the polymer brush morphology over the pores. Fig. 3a shows the scans on nanoholes with 20 kg mol−1 PEG obtained in indentation mode so that for each point on the surface the brush height is obtained from the force–distance data together with the substrate topography.8,27 The brush height (1 pN threshold) was evaluated by comparing with the solid surface topography at lateral positions either on planar gold (set to 0 nm) or over the apertures (surface topography <−5 nm). Clearly, the coils stretch out to occupy the free volume above the holes due to the entropy loss in confined space, in agreement with simulations.28 For diameters below 100 nm, the brush becomes equal in height or even higher above the apertures compared to the planar regions. Hence, there must be a considerable number of polymers grafted to the walls because otherwise the free volume at the holes would relax the stretching perpendicular to the surface. At the same time, the grafting density Γ on the walls cannot be higher than on the planar surface, since only a positive curvature promotes higher Γ when grafting the polymer.29 Based on the available wall area and the measured brush heights, it was confirmed that the volume fraction of monomers at the nanoholes is very similar (within a few percent) or slightly lower than that on the planar surface (if Γ is lower).
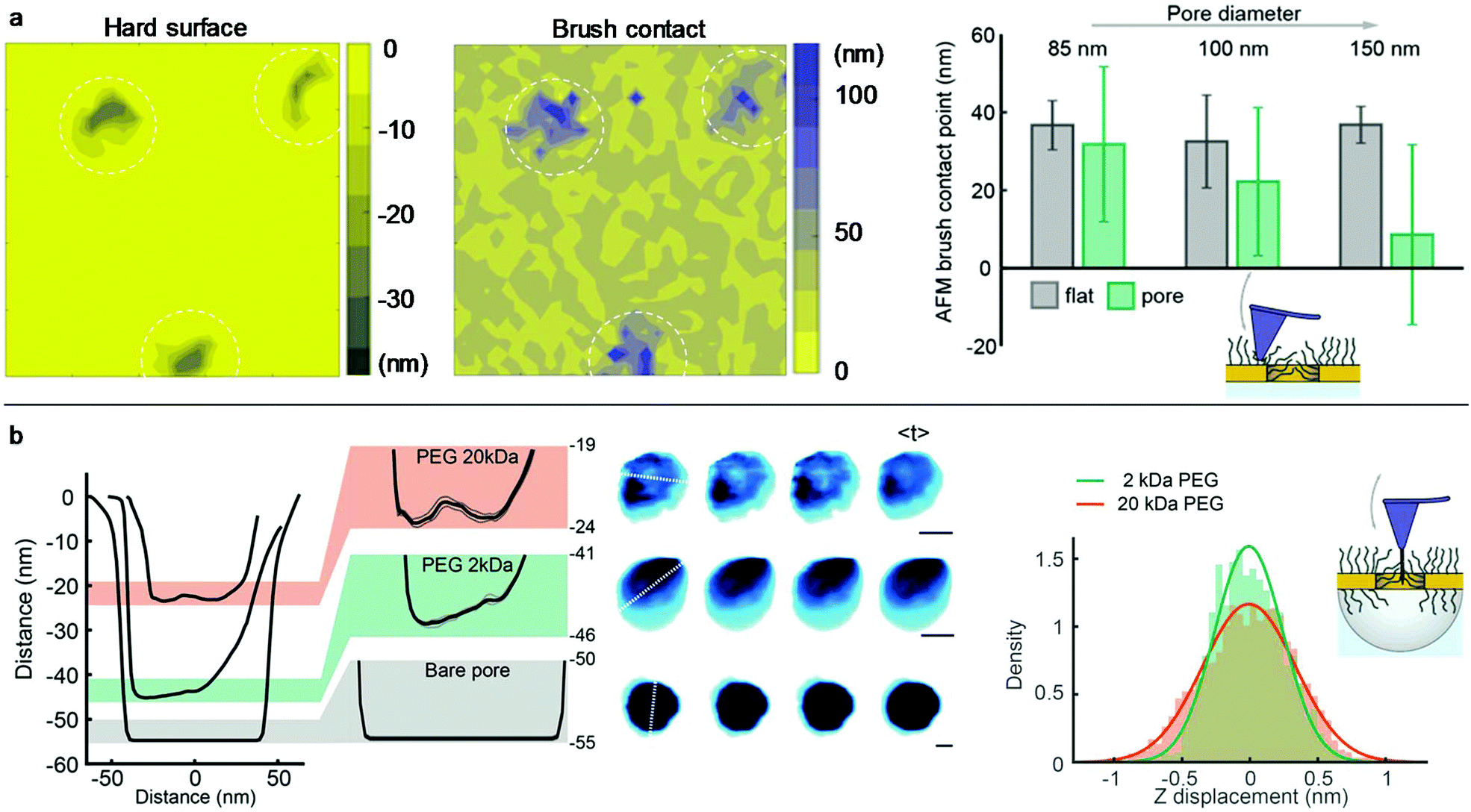 | ||
| Fig. 3 Characterization of brush morphology by AFM. (a) Examples of indentation maps on nanoholes with 20 kg mol−1 PEG. The right bar plots summarize the brush contact height over the pore regions compared to the planar gold (z = 0). (b) High-speed AFM data of nanocaves with 20 kg mol−1 PEG, 2 kg mol−1 PEG or no PEG. Typical line profiles are shown together with snapshot and time-averaged images. Scale bars are 30 nm. The right histogram shows the average z-displacement over time for pixels in the pore area. | ||
The polymer-modified pores were further investigated by high-speed AFM on 70 nm nanocaves (Fig. 3b) using an electron beam deposited high aspect ratio amorphous carbon tip.30 This oscillating (frequency ∼400 kHz) probe was sufficiently sharp (radius down to 5 nm) to penetrate into the brush but it could not go through the pores containing 20 kg mol−1 PEG as shown by the line profiles (Fig. 3b). This confirms the strong repulsion of the brush since a tip comparable in size to proteins and oscillating with a mechanical energy of tens of kBT31 cannot penetrate the barrier. In contrast, when the same nanocaves were functionalized with 2 kg mol−1 PEG the probe easily penetrated the pores (depth >40 nm) and reached even deeper for bare nanocaves. Furthermore, the line profiles for 20 kg mol−1 PEG did not exhibit a minimum at the pore center. Instead, the monomer density appeared quite homogeneous in the radial direction. This is expected since the parabolic monomer density profile for planar surfaces1 is compensated by the geometrical constraints set by the pore when the brush extension is comparable to the radius, in excellent agreement with the simulations by Dimitrov et al.32 We could also detect fluctuations in the brush morphology inside the cylindrical void. Although the scan rate (5 frames per s) essentially only provides snapshots, the data still show that the PEG barrier is dynamic and similar to the intrinsically disordered proteins in the NPC.30 The mean fluctuations inside the nanopore over time were higher for 20 than for 2 kg mol−1 PEG (Fig. 3b). Examples of real-time AFM movies are provided online as the ESI.†
In order to analyze our results from a theoretical point of view we present a simple “de Gennes style” model33 of the polymer brush in a negative cylindrical surface curvature of radius R. The free energy G as a function of extension h (derivation in the ESI†) can be written as:
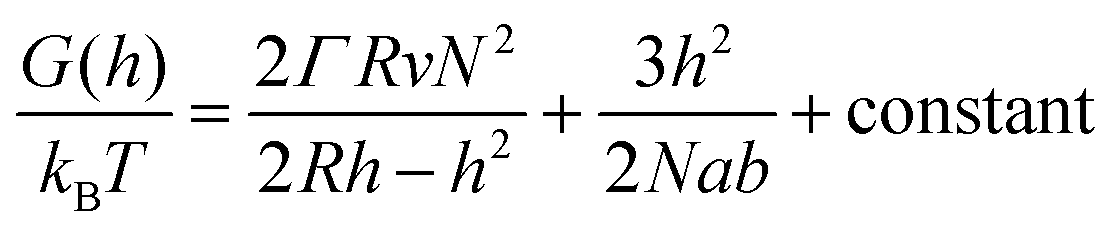 | (1) |
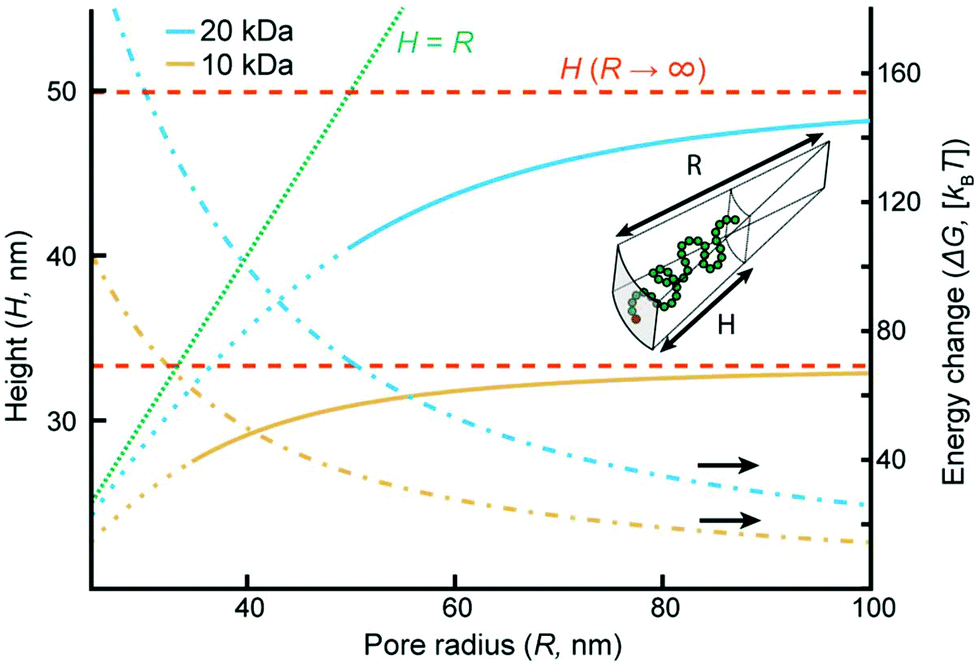 | ||
| Fig. 4 Theory of polymer brushes inside the pores for 10 and 20 kg mol−1 PEG at Γ of 0.68 and 0.28 nm−2, respectively. The equilibrium extension H and free-energy change per coil ΔG as functions of the radius R of the negative cylindrical curvature. The dashed lines show the heights on a planar surface (R → ∞ and ΔG = 0). The curves become dotted at the R values below which sealed pores were observed experimentally because then the model is not applicable. | ||
Although more sophisticated theoretical approaches certainly exist,28,32,38 our simple model still captures several essential features. Assuming that all variables except for R remain constant, the pore geometry leads to a decrease in H compared to a planar surface. The reason is that stretching towards the center of a channel is not associated with the same excluded volume entropy gain as on a planar surface, while the chain stretching (entropic spring) term is the same. For the same reason there is a considerable increase in the free energy per coil (tens of kBT), in agreement with the tendency of coils to stretch out from the pore (Fig. 3a). The main limitation with the model is that it assumes a sharp decrease in monomer density at H,33 while in reality some monomers extend beyond zero (up to 1.3H for a planar surface).8 Still, by comparing our model with experimental data we can estimate when sealing occurs. The values of R below which sealing was observed experimentally were 35 and 50 nm for 10 and 20 kg mol−1 PEG, respectively. As indicated in Fig. 4, the transition to the sealed state occurs when H/R ≈ 0.8 (0.77 and 0.82 for 10 and 20 kg mol−1, respectively). Our interpretation is that at this ratio the pores become so crowded that an entropy gain is obtained by allowing the free ends to spread out beyond R and deviate from the axis defined by the grafting point and the channel center, even though this means mixing with other coils.28 This, as well as our other results, is in excellent agreement with the simulations by Nasrabad et al. on brushes in pores with similar segment densities.38
How can an ultrathin layer of PEG coil containing almost 90% water withstand so high protein concentrations? As PEG is not charged, electrostatic/ionic contributions can be ignored. Dehydration effects should also be irrelevant since any protein easily fits physically into the sparse brush. Osmotic pressure needs to be considered,10,34 but it is weak at low polymer volume fractions. The intrinsic entropy loss of the chains upon protein insertion may be more important. Gu et al. calculated the conformational entropy penalty for inserting inert particles into brushes at similar grafting densities.39 The free-energy penalty was ∼10kBT when the ratio between the particle diameter and the Kuhn segment (b) reached ∼5. Since even relatively small proteins are at least 3 nm in size, this suggests that blocking is feasible because of the high flexibility of PEG (b = 0.7 nm). However, the dynamics of the polymer may also come into play and that is not easily accounted for in simulations. In this view, larger objects which diffuse slower will experience more collisions from the free chains while trying to cross the barrier. Such a blocking mechanism was recently suggested based on AFM data on the NPC.30
In conclusion, we have shown conclusive experimental evidence that hydrophilic polymer brushes can be extremely strong entropic barriers which prevent the passage of serum proteins, even when the thickness is only tens of nm. Despite the claims of attractive interactions between PEG and proteins,12 the repulsive nature of the brush clearly dominates. Considering that the smallest serum proteins in reasonable abundance are below 10 kg mol−1 (ref. 26) the blocking ability is more efficient than that of the NPC.16 We emphasize that besides having one terminal anchored to the surface, the coils are “free” and repel each other,34i.e. they do not interact to form a gel-like network. Furthermore, the brushes consist mainly of water and allow a high diffusive flux of small molecules as well as a high volumetric liquid flow. Our results form a foundation for developing new types of ultrathin membrane filters23 where the selectivity is solely based on the properties of the polymer brush, in contrast to merely using surface chemistry to tune the effective pore diameter or the local chemical environment. The PEG has the additional benefit of preventing fouling of the surface, enabling long-term operation. Other applications include single molecule analysis17 and improved detection limits in sensors by pre-filtration.40 Future work will investigate brushes with more sophisticated chemistry such as copolymers and functional end-groups. With inspiration from the NPC,16 this might enable highly efficient and selective transport of predetermined targets.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded by the Swedish Research Council, the Swedish Foundation for Strategic Research, the EU FP7 Marie Curie Actions and the Knut & Alice Wallenberg Foundation. YS, RLS and RYHL are supported by the Swiss Nanoscience Institute and the Biozentrum at the University of Basel.References
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS PubMed.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- S. Schottler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. de Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- J. Satulovsky, M. A. Carignano and I. Szleifer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9037–9041 CrossRef CAS PubMed.
- U. R. Dahal, Z. Wang and E. E. Dormidontova, Macromolecules, 2017, 50, 6722–6732 CrossRef CAS.
- G. Emilsson, R. L. Schoch, L. Feuz, F. Hook, R. Y. H. Lim and A. B. Dahlin, ACS Appl. Mater. Interfaces, 2015, 7, 7505–7515 CAS.
- G. Emilsson, R. L. Schoch, P. Oertle, K. Xiong, R. Y. H. Lim and A. B. Dahlin, Appl. Surf. Sci., 2017, 396, 384–392 CrossRef CAS.
- R. L. Schoch, G. Emilsson, A. B. Dahlin and R. Y. H. Lim, Polymer, 2017, 132, 362–367 CrossRef CAS.
- E. Schneck, A. Schollier, A. Halperin, M. Moulin, M. Haertlein, M. Sferrazza and G. Fragneto, Langmuir, 2013, 29, 14178–14187 CrossRef CAS PubMed.
- V. M. Latza, I. Rodriguez-Loureiro, I. Kiesel, A. Halperin, G. Fragneto and E. Schneck, Langmuir, 2017, 33, 12708–12718 CrossRef CAS PubMed.
- A. B. Dahlin, Analyst, 2015, 140, 4748–4759 RSC.
- M. Tagliazucchi and I. Szleifer, Soft Matter, 2012, 8, 7292–7305 RSC.
- T. Jovanovic-Talisman and A. Zilman, Biophys. J., 2017, 113, 6–14 CrossRef CAS PubMed.
- S. W. Kowalczyk, T. R. Blosser and C. Dekker, Trends Biotechnol., 2011, 29, 607–614 CrossRef CAS PubMed.
- T. Gilboa and A. Meller, Analyst, 2015, 140, 4733–4747 RSC.
- A. B. Dahlin, M. Mapar, K. L. Xiong, F. Mazzotta, F. Hook and T. Sannomiya, Adv. Opt. Mater., 2014, 2, 556–564 CrossRef CAS.
- J. Junesch, T. Sannomiya and A. B. Dahlin, ACS Nano, 2012, 6, 10405–10415 CrossRef CAS PubMed.
- B. Malekian, K. Xiong, G. Emilsson, J. Andersson, C. Fager, E. Olsson, E. M. Larsson-Langhammer and A. B. Dahlin, Sensors, 2017, 17, 1444 CrossRef PubMed.
- K. Xiong, G. Emilsson and A. B. Dahlin, Analyst, 2016, 141, 3803–3810 RSC.
- X. Hou, Adv. Mater., 2016, 28, 7049–7064 CrossRef CAS PubMed.
- C. C. Striemer, T. R. Gaborski, J. L. McGrath and P. M. Fauchet, Nature, 2007, 445, 749–753 CrossRef CAS PubMed.
- F. Mazzotta, T. W. Johnson, A. B. Dahlin, J. Shaver, S.-H. Oh and F. Hook, ACS Photonics, 2015, 2, 256–262 CrossRef CAS.
- R. Y. H. Lim and J. Deng, ACS Nano, 2009, 3, 2911–2918 CrossRef CAS PubMed.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CAS.
- J. T. Hyotyla, J. Deng and R. Y. H. Lim, ACS Nano, 2011, 5, 5180–5187 CrossRef CAS PubMed.
- O. Peleg, M. Tagliazucchi, M. Kroger, Y. Rabin and I. Szleifer, ACS Nano, 2011, 5, 4737–4747 CrossRef CAS PubMed.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS PubMed.
- Y. Sakiyama, A. Mazur, L. E. Kapinos and R. Y. H. Lim, Nat. Nanotechnol., 2016, 11, 719–723 CrossRef CAS PubMed.
- T. Ando, Microscopy, 2013, 62, 81–93 CrossRef CAS PubMed.
- D. I. Dimitrov, A. Milchev and K. Binder, J. Chem. Phys., 2006, 125, 34905 CrossRef CAS PubMed.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- P. L. Hansen, J. A. Cohen, R. Podgornik and V. A. Parsegian, Biophys. J., 2003, 84, 350–355 CrossRef CAS PubMed.
- F. Kienberger, V. P. Pastushenko, G. Kada, H. J. Gruber, C. Riener, H. Schindler and P. Hinterdorfer, Single Mol., 2000, 1, 123–128 CrossRef CAS.
- S. Liese, M. Gensler, S. Krysiak, R. Schwarzl, A. Achazi, B. Paulus, T. Hugel, J. P. Rabe and R. R. Netz, ACS Nano, 2016, 11, 702–712 CrossRef PubMed.
- F. Oesterhelt, M. Rief and H. E. Gaub, New J. Phys., 1999, 1, 6 CrossRef.
- A. E. Nasrabad, D. Jasnow, A. Zilman and R. D. Coalson, J. Chem. Phys., 2016, 145, 064901 CrossRef.
- C. Gu, R. D. Coalson, D. Jasnow and A. Zilman, J. Phys. Chem. B, 2017, 121, 6425–6435 CrossRef CAS PubMed.
- J. Breault-Turcot and J.-F. Masson, Chem. Sci., 2015, 6, 4247–4254 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods. Additional nanostructure images. Quartz crystal microbalance verification of material-specific surface chemistry. Measurements of the planar brush height with SPR. High-speed AFM tip images and videos. Theory of polymer brushes in pores. Selective grafting to pore walls. See DOI: 10.1039/c7nr09432a |
| This journal is © The Royal Society of Chemistry 2018 |