
Composition-driven shape evolution to Cu-rich PtCu octahedral alloy nanocrystals as superior bifunctional catalysts for methanol oxidation and oxygen reduction reaction†

Abstract
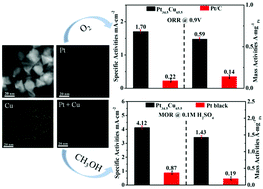
Synergetic effects between Pt and a cheap metal, downshift of the d-band center of Pt and the shape can boost the catalytic performance of Pt-based nanocrystals. Therefore, tailoring the shape and composition within the nanoscale is the key to designing a robust electrocatalyst in electrochemical energy conversion. Here, Cu-rich PtCu octahedral alloys achieved by a composition-driven shape evolution route have been used as outstanding bifunctional electrocatalysts for both methanol oxidation (MOR) and oxygen reduction reaction (ORR) in an acid medium. When benchmarked against commercial Pt black or Pt/C, for MOR, the specific activity/mass activity on Pt34.5Cu65.5 octahedra is 4.74/7.53 times higher than that on commercial Pt black; for ORR, the specific activity/mass activity on Pt34.5Cu65.5 octahedra is 7.7/4.2 times higher than that on commercial Pt/C. After a current–time test for 3600 s, the remaining mass activity on Pt34.5Cu65.5 octahedra is 35.5 times higher than that on commercial Pt black for MOR. After undergoing 5000 cycles for ORR, the remaining mass activity on Pt34.5Cu65.5 octahedra is 4.2 times higher than that on commercial Pt/C.
- This article is part of the themed collection: International Year of the Periodic Table : Single Atoms as Active Catalysts
 Please wait while we load your content...
Please wait while we load your content...

