
Sensing in-plane nanomechanical surface and sub-surface properties of polymers: local shear stress as function of the indentation depth
Christian
Dietz

Physics of Surfaces, Institute of Materials Science, Technische Universität Darmstadt, Alarich-Weiss-Str. 2, 64287 Darmstadt, Germany. E-mail: dietz@pos.tu-darmstadt.de
First published on 6th December 2017
Abstract
Dynamic force microscopy (DFM) is an excellent tool for the study of the compositional and nanomechanical properties of polymers that exploits the flexural eigenmodes of a cantilever comprising a sharp tip vibrating perpendicular to the sample surface. However, the in-plane nanomechanical properties of a specimen cannot be detected by this technique. Here, a bimodal approach was developed where flexural and torsional eigenmodes are driven simultaneously. The corresponding vibrational amplitude and phase shift of the vertical tip motion were utilized for topographical feedback and out-of-plane dissipative interaction acquisition, respectively, whereas the frequency shift and the drive amplitude of the lateral tip motion mapped the in-plane conservative and dissipative interactions of two heterogeneous polymers: an elastomeric polypropylene (ePP) and a polystyrene-block-polybutadiene diblock copolymer (SB). The shear stress at different sub-surface levels revealed an amorphous cover layer as well as a “slipping” plane on the ePP crystallites. The imaging of SB supported by dynamic force spectroscopy experiments showed that SB exhibits considerably different in- and out-of-plane nanomechanical properties at certain areas due to the complex polymer conformation of this diblock copolymer accompanied by inter- and intramolecular interactions that give rise to its viscoelastic behavior.
Introduction
The macroscopic mechanical properties of materials, in particular those of polymers, strongly depend on their composition and, consequently, on their nanomechanical heterogeneities due to chain configurations, conformations, lengths and interactions. Atomic force microscopy (AFM) operated in the amplitude modulation mode, also known as an intermittent contact mode or tapping mode, is a popular and efficient surface characterization method for the study of the mechanical properties at the nanoscale.1 This mode is based on a resonantly driven cantilever, the tip of which oscillates perpendicular to the sample surface. Dissipative tip–sample interactions can be analyzed from the phase lag of the tip oscillation with respect to the cantilever excitation2,3 and mapped in two-dimensional images when the surface is scanned over a certain area. However, this type of nanomechanical characterization is incomplete because in-plane surface properties, i.e., physical properties, directed along the sample surface are inaccessible and cannot be visualized. Moreover, the phase lag does not provide direct information about the elastic sample properties.Many insightful and thoroughly conducted studies have been published in the field of morphological, compositional and nanomechanical characterization of polymers and soft matter using phase imaging.4 The reader is referred to ref. 5 for a comprehensive review. In contrast, a comparatively small number of studies have addressed the nanomechanical properties directed along the sample surface, including frictional characterization using DFM techniques.6–19 In recent years, multifrequency approaches have grown increasingly significant because of the additionally available observables and their potential to elucidate important qualitative as well as quantitative physical sample properties.20 For this purpose, two21 or more cantilever eigenmodes22 or frequencies in their vicinity23,24 were simultaneously excited, and their corresponding amplitude, phase or frequency changes were detected when the tip interacted with the sample surface. Various techniques have been developed with great success in order to investigate polymers,25 biomolecules,26,27 living cells28 and crystal structures19 that provided quantitative sample information29 with unprecedented lateral and depth resolution.30–32
To simultaneously measure the in-plane and out-of-plane nanomechanical surface properties of materials with the AFM, the cantilever has to be resonantly driven in a combined flexural and torsional vibration.33,34 In this study, a bimdoal approach is introduced to quantify and image the nanomechanical surface and sub-surface properties of polymers as a function of the tip-indentation. Fig. 1 shows the experimental setup of the bimodal flexural and torsional force microscope. In addition to the detection laser, a second laser (wavelength λ = 405 nm) is placed at the fixed end of the cantilever and at a considerable distance off the length symmetry axis to enable the simultaneous photothermal excitation of both the flexural and torsional eigenmodes.33 The excitation laser is power-modulated using frequencies matching the resonant frequencies ωflex and ωtor of the flexural and the torsional cantilever vibrations, respectively. A lock-in amplifier is used to analyze the vertical deflection signal of the photosensitive diode to obtain the amplitude and phase shift of the flexural cantilever oscillation. For imaging, this amplitude is kept constant to track the topography of the sample (as in conventional amplitude modulation mode), while the phase shift provides useful information about the energy dissipated between the tip and the sample in the vertical tip motion.2,3 Additionally, the lateral deflection signal is fed into a phase-locked loop to track the instantaneous torsional resonant frequency when the tip interacts with the sample surface and to keep the cantilever oscillating at torsional resonance. The power (amplitude) of the excitation laser is varied during scanning to hold the lateral oscillation amplitude Ator at a constant value (constant amplitude configuration) using a proportional-integral controller. Then, the excitation amplitude ator is a measure of the energy dissipated between the tip and the sample in the lateral motion direction. Hereinafter, this excitation and detection scheme is abbreviated as the bimodal AMFlex-FMTor mode. The strength of this method lies in the fact that the lateral tip motion (unaffected by the feedback loop) can sense the local in-plane nanomechanical properties of the sample, while the indentation of the tip into the sample surface can be accurately controlled and adjusted by the amplitude setpoint of the vertical vibration. The frequency shift Δωtor(d) between the torsional resonant frequency ωtor when the tip interacts with the sample and that of the “freely” vibrating cantilever ωtor,0 as a function of the tip–sample distance d can be related to the local shear force Ftor(d), provided that lateral and torsional tip motions can be considered independently. Then, the shear force can be approximated as a half-fractional derivative of the frequency shift by the Sader–Jarvis method35,36 for the large amplitude case, when the oscillation amplitude is larger than the characteristic length scale of the interaction force given by:
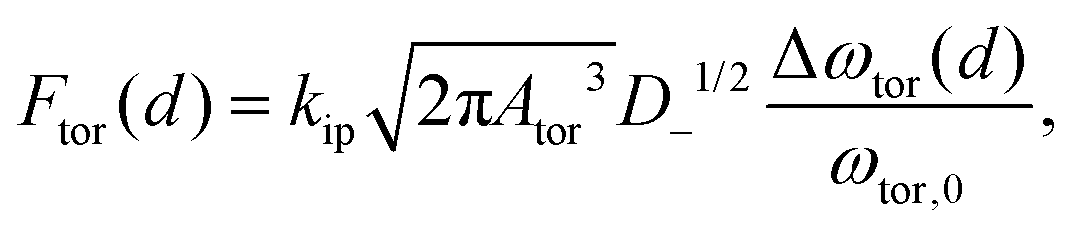 | (1) |
| Ac = π[R2 − (R − δ)2] for δ ≤ R |
Ac = π[R + (δ − R)tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) φ]2, for δ > R φ]2, for δ > R | (2) |
| τ(δ) = Ftor/Ac. | (3) |
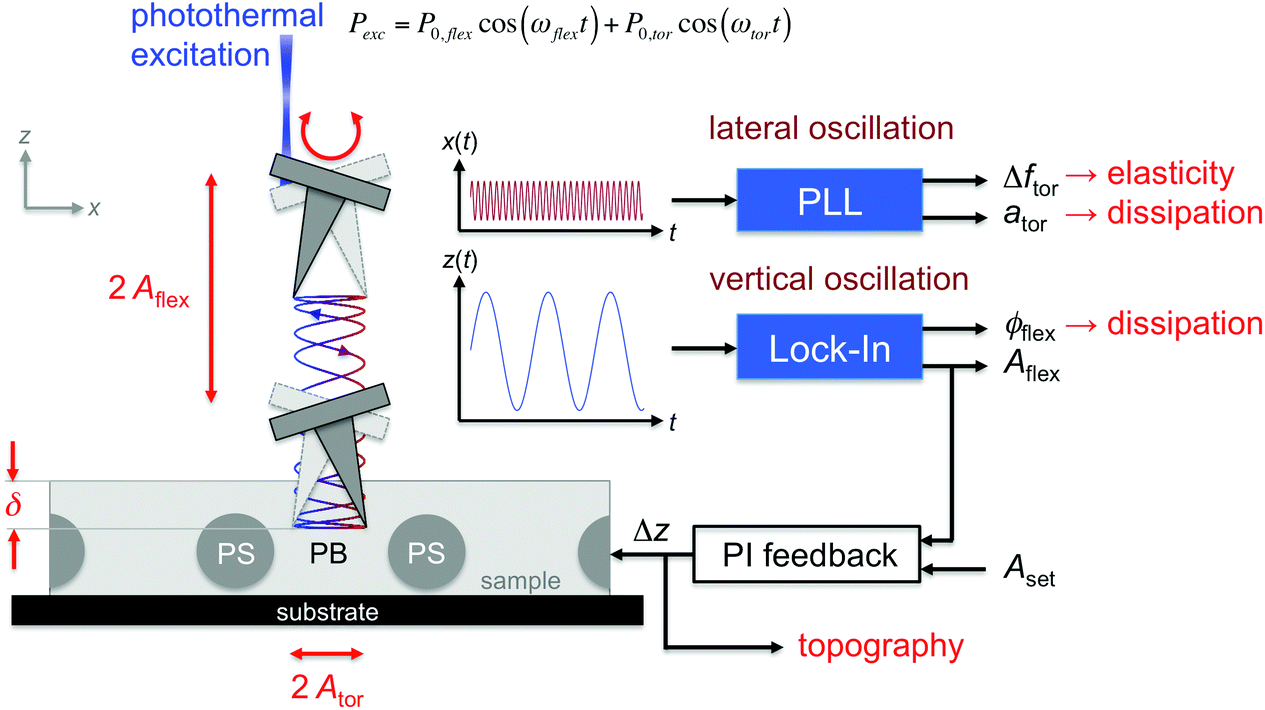 | ||
| Fig. 1 Schematic illustration of the functional principle of a bimodal AMFlex-FMTor force microscope. The vertical tip oscillation senses the out-of-plane nanomechanical sample properties, whereas the lateral tip oscillation is sensitive to in-plane forces. | ||
In the amplitude modulation mode, the indentation depth at a given amplitude setpoint can be deduced from the dependence of the amplitude on the z-piezo position by subtracting the z-piezo position of an infinitely stiff material (no indentation, i.e., moving the cantilever toward the sample surface by a certain distance reduces the amplitude by the same amount) from the z-piezo position of the “soft” material.37,38 Because the contribution of the tip motion caused by the torsional vibration of the cantilever to the vertical oscillation of the tip is negligible for amplitudes considerably smaller than the tip height, the indentation depth for the bimodal AMFlex-FMTor mode can be determined in the same manner. It is important to mention that the mechanical properties of viscoelastic materials, e.g., (shear) storage and (shear) loss moduli, depend strongly on the drive frequency of the sensor, and the results presented in this study were determined at a specific frequency, providing only a small window of the full frequency spectra of the nanomechanical sample properties. However, this issue can be overcome by using cantilevers with the specific dimensions that lead to the desired resonant frequencies.
Results and discussion
Imaging
To test the imaging capabilities of the bimodal AMFlex-FMTor mode for simultaneous mapping of the nanomechanical in- and out-of-plane properties of polymers, two test samples were studied: the semi-crystalline polymer elastomeric polypropylene (ePP) and the diblock copolymer polystyrene(PS)-block-polybutadiene(PB) (SB). Fig. 2 shows the topography images (Fig. 2a and e), the phase images of the vertical cantilever deflection (Fig. 2b and f), the images of the excitation amplitude (Fig. 2c and g) and the images of the frequency shift of the lateral cantilever deflection (Fig. 2d and h) obtained for the SB (Fig. 2a–d) and for the ePP samples (Fig. 2e–h). The alignment of the cantilever with respect to the fast scan axis (left to right) is indicated by the large blue arrow in Fig. 2a. The topography and the phase images of the SB sample clearly reflect the self-organized cylindrical structure of polystyrene (bright) distributed in a polybutadiene matrix (dark). Note that dark areas are regions with higher phase values, i.e., closer to 90° compared to those of the bright areas, corresponding to regions of greater energy dissipated between the tip and sample per oscillation cycle. This is also evident in the dissipation channel of the lateral tip oscillation represented by the drive amplitude ator (note the color scale: dark corresponds to high values for better comparison) that maintained the oscillation amplitude at a constant value (Fig. 2c), even though the cylinders appear to be slightly more broad. The image of the frequency shift reveals higher values for the PS cylinders due to their higher stiffness compared to PB, in agreement with the relationship between the frequency shift and the shear force (cf.Eqn (1)). The bimodal AMFlex-FMTor mode can also unambiguously resolve the crystalline structure within an amorphous matrix of an elastomeric polypropylene specimen in the topography (Fig. 2e), vertical phase (Fig. 2f), torsional drive amplitude (Fig. 2g) and torsional frequency shift images (Fig. 2h). Crystallites appear in bright (low dissipation between tip and sample, high stiffness) whereas the amorphous matrix appears in dark (high dissipation, low stiffness). | ||
| Fig. 2 Structural and nanomechanical mapping of a polystyrene-block-polybutadiene diblock copolymer (SB) (a–d) and elastomeric polypropylene (ePP) (e–h) by bimodal AMFlex-FMTor force microscopy. (a, e) Topography, (b, f) vertical (out-of-plane) phase (c, g) torsional (in-plane) drive amplitude and (d, h) torsional frequency shift images. Arrows indicate regions with differences between the in-plane and out-of-plane nanomechanical properties. The areas framed by the yellow dashed boxes are shown enlarged in Fig. 3. The alignment of the cantilever with respect to the fast scan axis (left to right) is indicated by the large blue arrow in (a). The free flexural amplitudes were A0,flex = 73 nm for SB and 32 nm for ePP, the torsional amplitude for ePP and SB was A0,tor = 7 nm (kept constant during imaging by a PID controller), and the setpoint amplitude was adjusted to ≈60% of A0,flex. Scale bars: (a) 200 nm, (e) 400 nm. | ||
At first glance, the distribution of PS and PB in Fig. 2d looks very similar to those of Fig. 2a–c. However, in certain regions, the PS structure shows a slight deviation in the frequency images that are noted by red arrows. The subtle differences become clearly apparent when comparing the areas framed by the yellow boxes (Fig. 2b and d) enlarged in Fig. 3 showing a direct comparison between the out-of-plane (vertical phase, Fig. 3a) and in-plane (torsional frequency shift, Fig. 3b) nanomechanical properties. At the position highlighted by the yellow arrow, two cylinders appear to be disconnected in the vertical phase (Fig. 3a), whereas they appear connected in the torsional frequency shift image (Fig. 3b). The same phenomenon is visible in the lower right corner of the figures (turquoise arrow). In contrast, at the position indicated by the green arrow, a horizontally aligned PS cylinder connected to another cylinder (Fig. 3a) appears as single dot in Fig. 3b. A ring-like structure of PS can be seen in the vertical phase image (Fig. 3a) whereas two disjoined vertically aligned cylinders appear in the torsional frequency shift image (Fig. 3b) at the same position (red arrow). Considering the lateral motion of the tip parallel to the surface and oriented along the fast scan axis (horizontal scan lines), the sample exhibits a high shear storage modulus at the positions of the connections of the two cylinders that are only apparent in the torsional frequency shift image (Fig. 3b, yellow and turquoise arrows) and a high shear loss modulus (cf. torsional drive amplitude, Fig. 2c) with respect to this particular direction. Consistently, at the positions of the broken cylinders (red and green arrows), the sample has a comparably low shear storage modulus and a low shear loss modulus. It is reasonable to assume that the differences between in-plane and out-of-plane nanomechanical properties are originated from the chain configuration and conformation of PS and PB in the distinct spatial orientations and the structure beneath the visible top layer. Upon closer examination of ePP, subtle differences in the crystalline structure between the torsional frequency shift image and the others can be observed here as well. To demonstrate the differences, the indicated region in Fig. 2f and h (yellow boxes) is shown enlarged in Fig. 3c and d, respectively. At the position highlighted by the yellow arrow, the vertical phase image clearly exhibits two individual crystallites separated by an amorphous region with distinct out-of-plane dissipation properties between crystallites and the amorphous interspace, whereas the frequency shift images give rise to the assumption that both crystallites and the amorphous joint are physically connected to each other in the respective in-plane direction. The red and green arrows point out further positions, where in-plane and out-of-plane nanomechanical properties vary as can be determined by different color scale values in Fig. 3c (bright) and 3d (dark), most likely caused by the local distribution of isotactic and atactic polymer chains and their interaction. Hence, the additional channels based on the torsional cantilever vibration provide important information that is complementary to the information provided by the standard amplitude modulation mode.
 | ||
| Fig. 3 High-resolution bimodal AMFlex-FMTor force microscopy images revealing differences between out-of-plane and in-plane nanomechanical properties of a polystyrene-block-polybutadiene diblock copolymer (SB) (a–b) and elastomeric polypropylene (ePP) (c–d). (a, c) Vertical (out-of-plane) phase and (b, d) torsional (in-plane) frequency shift images. Arrows indicate regions with differences in the in-plane and out-of-plane nanomechanical properties. The free flexural amplitudes were A0,flex = 73 nm for SB and 32 nm for ePP, the torsional amplitude for ePP and SB was A0,tor = 7 nm (kept constant during imaging by a PID controller), and the setpoint amplitude was adjusted to ≈60% of A0,flex. Scale bars: (a) 100 nm, (c) 200 nm. | ||
Spectroscopy
To gain further insight into the mechanism underlying the interaction of superimposed vertical and lateral tip motions with the sample surface, spectroscopy experiments were performed on both polymers at individual locations. For this purpose, the topographical feedback adjusted on the vertical oscillation amplitude was switched off, and the tip was placed on pure PS, PB, regions of crystalline (isotactic polypropylene, iPP) or amorphous polypropylene (atactic polypropylene aPP) or on silicon (Si) as a stiff reference material after imaging of the respective structure. Subsequently, the vibrating cantilever/tip was moved toward the sample surface and beneath by means of the z-piezo until a chosen trigger value for the vertical amplitude was reached. Simultaneously, the vertical amplitude Aflex and phase shift ϕflex, torsional drive amplitude ator and frequency shift Δftor were recorded. Fig. 4 shows the distribution of vertical amplitudes (Fig. 4a and d), phase shifts (Fig. 4b and e), and torsional frequency shifts (Fig. 4c and f) versus the z-piezo position detected on the silicon sample (Fig. 4a–c) for three different free oscillation amplitudes (A0,flex = 8, 16, 32 nm) and on the individual locations of the polymer samples (Fig. 4d–f) for a given free amplitude A0,flex = 32 nm. During the approach, the flexural amplitude linearly decreased with the extension of the z-piezo (from left to right) on silicon upon tip–sample interaction, regardless of the choice of the free oscillation amplitude. The vertical phase shift adopted values above 90° (net attractive interaction regime) after the onset of tip–sample interactions and before it switched to values below 90° (net repulsive interaction regime), except for the measurement performed with the lowest free amplitude (A0,flex = 8 nm). The transition occurs at the lowest z-piezo position for the largest free amplitude. This is in accordance with experiments conducted in standard amplitude modulation mode, where the free oscillation amplitude and the choice of the setpoint are the decisive parameters for the selection of the operation regime.39 The torsional frequency shift (Fig. 4c) increased substantially approximately at the z-piezo position where the vertical phase shift reached its maximum (cf. dashed vertical lines). Additionally, kinks are apparent in these curves, denoting a change in slope at the position where the transition from net attractive to repulsive regime was completed during the approach (see arrows). For the polymers (Fig. 4d–f), similar observations can be conducted, with the main differences being that the transition from net attractive to repulsive interaction is accompanied by a more pronounced change of the amplitude and that it is unlikely to access the net repulsive interaction regime on the soft polymers (PB, aPP) with reasonable scan parameters.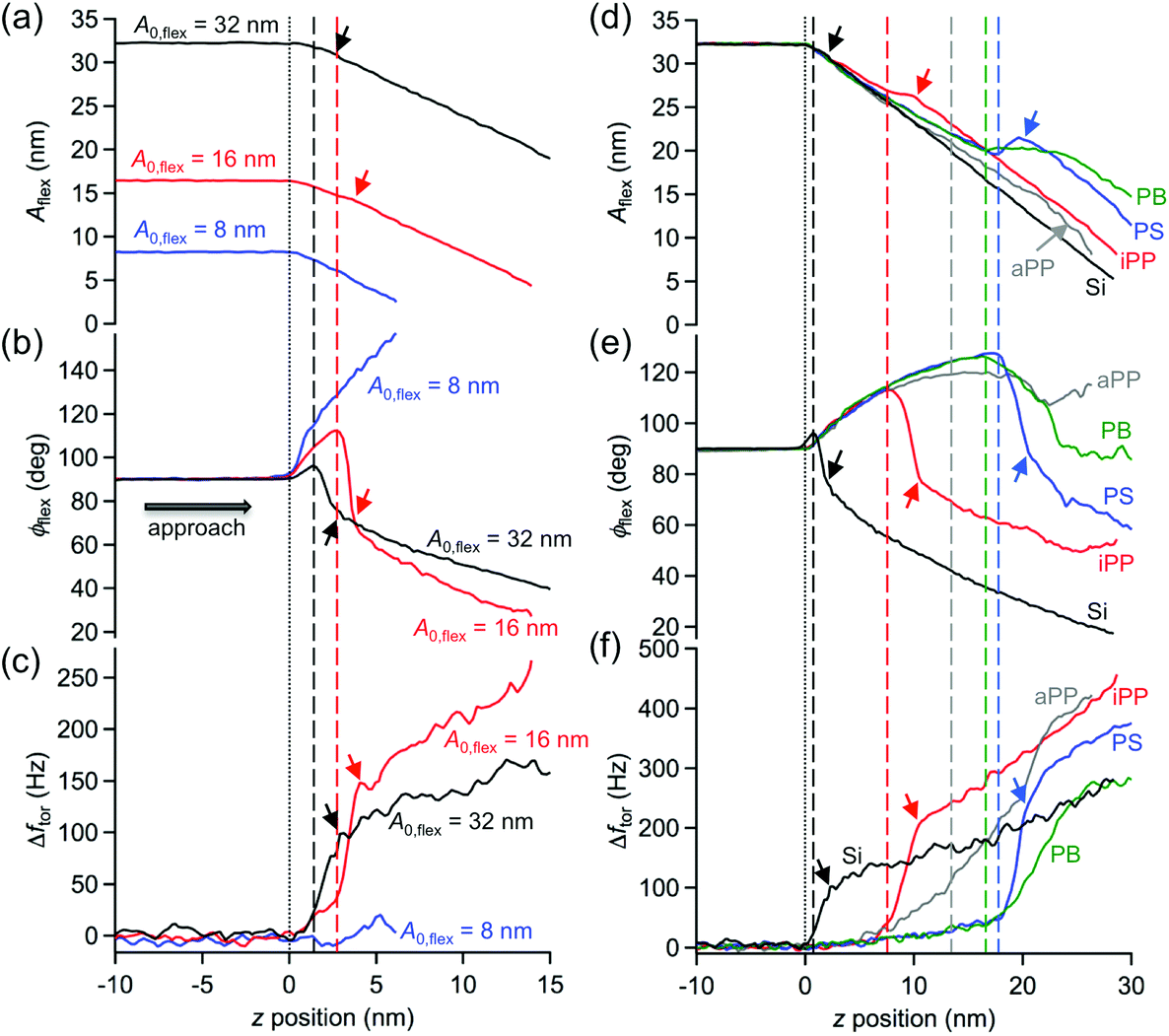 | ||
| Fig. 4 Spectroscopy experiments, exhibiting the flexural amplitude (a, d), phase shift (b, e), and torsional frequency shift (c, f) vs. z-piezo position performed on Si(100) (a–c) with different free amplitudes (A0,flex = 8, 16, 32 nm) and on individual regions of an elastomeric polypropylene and polystyrene-block-polybutadiene diblock copolymer (SB) samples (d–f), including the measurement on Si(100) for comparison. The vertical dotted lines indicate the z-piezo position where the tip starts to interact with the sample during the approach (left to right). The vertical dashed lines show the z-piezo position where the transition from net attractive to net repulsive tip–sample interaction starts. Upon further approach kinks are apparent, denoting a change in slope at the position where the transition from net attractive to repulsive regime was completed (see arrows). Abbreviations (d–f): Si = Silicon(100), aPP = atactic polypropylene (amorphous), iPP = isotactic polypropylene (crystalline), PS = polystyrene (one block of SB), PB = polybutadiene (the other block of SB). | ||
The considerable deviations of the amplitude curves Aflex(z) acquired for the polymers from the one measured on Si (Fig. 4d) are caused by the increasing indentation depth δ of the tip into the sample surfaces for increasing the z-piezo position. The indentation depth of the tip into a particular polymer can be directly deduced from that deviation (see Experimental section). Accordingly, as shown in Fig. 5, all physical observables accessible by the bimodal AMFlex-FMTor mode, i.e., the flexural amplitude Aflex(δ) (Fig. 5a) and phase shift ϕflex(δ) (Fig. 5b) as well as the torsional frequency shift Δftor(δ) (Fig. 5c) and drive amplitude (inset, Fig. 5a), can be plotted as a function of the indentation depth, providing valuable information about the in-plane and out-of-plane mechanical properties in different surface layers of the polymer. Additionally, from eqn (1) the shear force, and thus the shear stress τ(δ) (eqn (3)) dependence on the tip indentation, can be derived (Fig. 5d). For aPP, a continuous decrease in the amplitude with increasing indentation was observed until the maximum depth of δ = 4 nm. Upon further approach, δ decreased again, while the amplitude further decreased. However, for iPP, the amplitude first slightly decreased for δ ≤ 4 nm and then abruptly dropped to low values. The amplitude values measured on PS and PB almost coincide for δ ≤ 10 nm, for which the trend is characterized by a decrease of approximately 12 nm over the first 5 nm of depth, followed by a constant value plateau and a subsequent more rapid decrease for δ ≥ 10 nm on the PS cylinder. The setpoint amplitude corresponding to the amplitude ratio Asp/A0 ≈ 0.6 chosen for imaging is indicated as a horizontal dotted line in Fig. 5a. The normalized flexural drive amplitude (Fig. 5a, inset) reveals an increase in in-plane tip–sample dissipation with increasing indentation depth for all polymers under study. The indentation depths necessary for a transition from net attractive to the net repulsive interaction with respect to the flexural tip motion (δtrans) can be read out from the phase shift diagram (golden horizontal dashed line, Fig. 5b) and are approximately 4 nm, 9 nm, and 12 nm for iPP, PS, and PB, respectively. However, for aPP, the net repulsive interaction regime could not be achieved. For all polymers, the frequency shift continuously increased during the approach, including a change in the slope at approximately the position where the net flexural interaction changed its sign. It appears that there exists a maximum possible tip indentation for each polymer that is slightly larger than δtrans. It is anticipated, although not systematically studied in this work, that this tip indentation is characteristic for each polymer and strongly depends on the intrinsic cantilever and measurement parameters such as flexural and torsional stiffness, vertical and lateral vibration frequencies and amplitudes. Fig. 5d shows the local shear stress as a function of the indentation depth calculated using eqn (1)–(3). When the oscillating tip approached the aPP regions, the shear stress was initially low (≈1 MPa) for δ ≤ 1 nm and then strongly increased to the constant level of ≈11 MPa (note the split axes) upon further indentation. For iPP, two different levels of almost constant shear stress with a sudden jump to high values could be observed: τ(δ ≤ 2 nm) ≈ 2 MPa, τ(2 nm < δ ≤ 4 nm) ≈ 3 MPa, and τ(δ > 4 nm) up to 150 MPa. The areas containing PS and PB exhibited a different shear stress behavior and allowed considerably higher indentation depths than those for iPP or aPP. After an increase of the shear stress from the initial 2 MPa to 4 MPa at δ = 1 nm on PS, the shear stress decreased again to τ = 2 MPa. For δ > 4 nm, the shear stress plateaus at slightly above 2 MPa before increasing to 6 MPa at maximum indentation (δ = 10 nm). The curve deduced for PB shows two maxima in the shear stress, one at δ = 1 nm with τmax = 14 MPa and the other at the maximum indentation depth (δ = 12 nm) with τmax = 8 MPa. In the interval 1 nm < δ ≤ 4 nm, the shear stress decreased from 14 MPa to 1 MPa, exhibiting a local maximum close to δ = 4 nm. Upon further tip indentation, the shear stress remained constant at approximately 1 MPa.
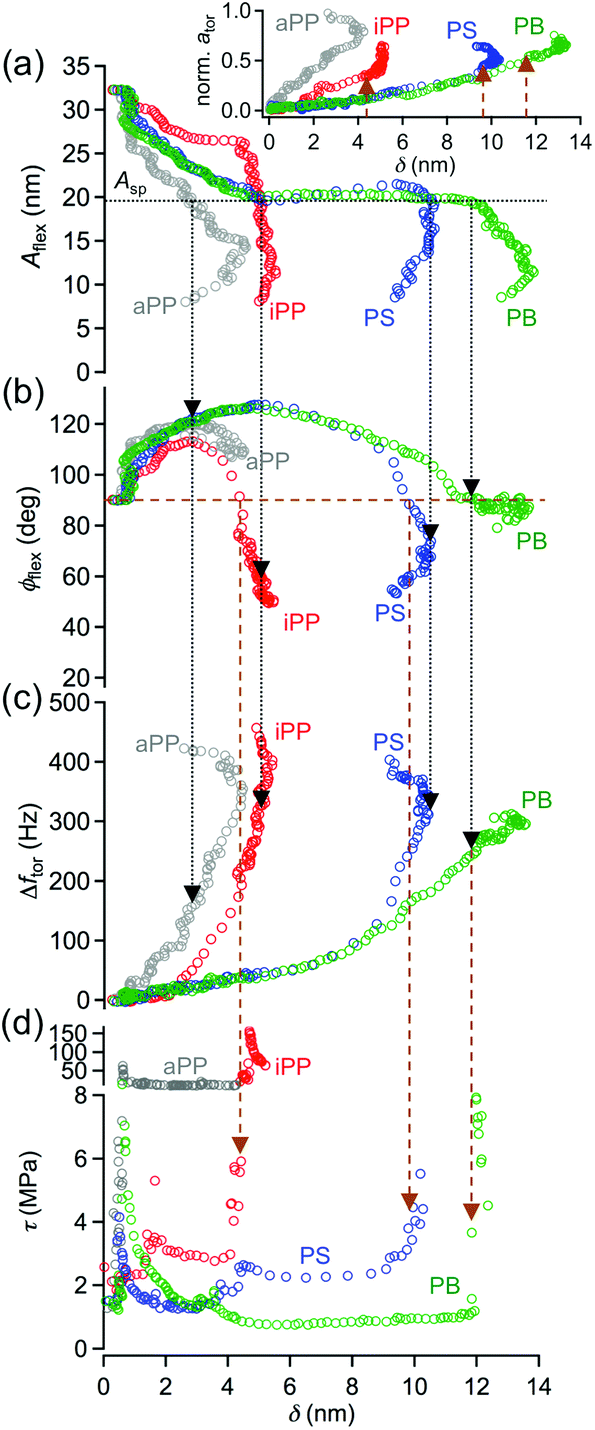 | ||
| Fig. 5 In- and out-of-plane nanomechanical properties of the different components of an elastomeric polypropylene and a polystyrene-block-polybutadiene diblock copolymer (SB) sample, represented by the flexural amplitude (a), phase shift (b), torsional frequency shift (c) and shear stress (d) as a function of the indentation depth. The horizontal dotted line (black) in (a) indicates the setpoint amplitude Asp ≈ 60% of the free (flexural) oscillation amplitude A0,flex chosen for imaging – vertical arrows guide the eye to the corresponding phase (b) and frequency shift values (c). The transition from the net attractive to net repulsive tip–sample interaction regime can be read out from (b) indicated by the horizontal (golden) dashed line. The corresponding values for the shear stress (d) and normalized torsional drive amplitude (inset in (a)) are pointed out by the vertical arrows. Abbreviations: aPP = atactic polypropylene (amorphous), iPP = isotactic polypropylene (crystalline), PS = polystyrene (one block of SB), PB = polybutadiene (the other block of SB). Inset in (a) shows the torsional drive amplitude vs. indentation depth as a measure for the in-plane tip-sample dissipation. | ||
The initially low shear stress (1–2 MPa) detected on iPP and aPP can be explained by an insufficient mechanical coupling between the tip motion and the sample surface that gives rise to friction effects at the interface. Due to the low indentation depth, it is assumed that only the polymer chains located near the surface are involved in this friction process. The corresponding friction force deduced from eqn (1) was below 500 pN. When the tip was further indented into the aPP region, the applied vertical tip load given by the flexural cantilever vibration was sufficiently high to provoke shear stress in deeper regions of that polymer, counteracted by the entanglements and intrinsic elasticity of the atactic polypropylene chains which was then independent of the applied load and hence, the indentation depth in the range investigated in this study. The crystalline regions sensed by the tip at the sample surface of this polymer are covered by a thin amorphous layer.40–43 Prior to the indentation of the tip through that surface layer and being able to detect the shear stress directly on crystallites (δ > 4 nm, τ > 100 MPa), the tip sensed the shear force arising from an interfacial layer between the atactic chains accumulated at the surface and the isotactic chains forming the crystallites or the atactic chains connected to the isotactic regions. It is reasonable to assume that this interface acts as a slipping plane or “loose” connection, giving rise to a shear stress value between those of molecules located at the surface and the crystallites found at a sample depth of 4 nm. The shear stress measured on crystallites themselves was lower than expected due to the amorphous embedding that leads to a lower effective shear modulus in these regions.
The surface arrangement of PS and PB in thin films of a polystyrene-block-polybutadiene-block-polystyrene triblock copolymer has been well studied and widely understood.38 It is assumed that the SB diblock top layer studied here also consists of polybutadiene because of its lower surface energy compared to polystyrene. When the tip approaches the surface, it first senses the polybutadiene top layer. After the initial friction force for δ < 1 nm, the sudden increase in the shear force or stress observed on PS as well as on PB can be explained by the penetration of the tip through an accumulation of homopolymers from the original solution7 and/or a thin oxidized PB layer comprising cross-linking,44 for which the polymer chains interact more strongly in the in-plane direction than those of pure polybutadiene. However, it should be noted that at small indentation depths and in particular for soft polymers such as polybutadiene, the obtained shear stress values have to be interpreted with care. A simple geometrical contact behavior was considered in this study to estimate the contact area between the tip and the sample surface. There are more sophisticated contact mechanics models, such as Johnson–Kendall–Roberts (JKR) model45 which provide a more reliable contact area in that case. The effect of the contact model is most significant at small indentation depths because the polymer immediately forms a much larger contact during the approach than expected from the geometrical model used here. Hence, the contact area is underestimated leading to an overestimation of the shear stress at small indentation depth, which contributes to the high shear stress values obtained at δ = 1 nm for PS and PB. Since the shear stress vs. indentation measured on both components, PS and PB, exhibited a local maximum at δ ≈ 4 nm, and the thickness of the PB top layer is larger than 4 nm, it is assumed that for δ > 4 nm, the tip mainly senses the intramolecular interaction between the PS and PB chains, which is stronger in regions directly above the polystyrene cylinders than in the regions between the cylinders. At δ ≈ 10 nm, the shear stress of the actual PS cylinder embedded in the PB matrix was determined. Thus, the thickness of the PB surface layer can be estimated as 10 nm, which concurs with the study of Knoll et al.38 Upon further indentation on the PB regions, the tip is directly located between two cylinders, measuring a rapid increase in the shear stress due to the strong intermolecular interactions at the sample depth corresponding to the level of the first cylinders (δ ≈ 14 nm).
Conclusion
A bimodal dynamic force microscopy technique that combines the vertical and lateral tip motion by simultaneously driving the cantilever at flexural and torsional resonance can provide valuable insights into the in-plane and out-of-plane nanomechanical surface properties of heterogeneous polymers, which are not accessible by standard atomic force microscopy methods. Because of the amplitude-modulated topographical feedback, the variation of the setpoint allows for the controlled indentation of the tip into the sample surface, facilitating the creation of nanomechanical maps at different sample depths for distinct spatial directions. Based on the phase shift of the flexural cantilever vibration with respect to the excitation, the net tip–sample interaction regime can be deduced. Two different heterogeneous polymers with elastic and dissipative properties spanning a broad range were examined: elastomeric polypropylene and polystyrene-block-polybutadiene diblock copolymer. Imaging of these polymers revealed heterogeneities in the in- and out-of-plane nanomechanical properties at certain areas caused by the spatial chain conformation, intermolecular interactions, and intramolecular elasticity of the polymer chains. Spectroscopy experiments performed on specific polymer regions allowed the calculation of the shear stress sensed by the tip at different sample depths where peculiarities in the stress-indentation relationship revealed the existence of a thin amorphous layer covering the elastomeric polypropylene sample and an interfacial region between the crystallites and amorphous embedding that acted as a slipping plane. On the polystyrene-block-polybutadiene diblock copolymer, two surface layers, a modified polybutadiene or homopolymer top layer and the polybutadiene phase from the diblock, showing slightly different shear stress, were revealed by this technique. The targeted location of the tip between two polystyrene cylinders allowed the evaluation of the shear stress originating from the intermolecular interaction between the polymer blocks involved in this cylinder conformation. In future work, a simultaneous excitation of the fundamental flexural and torsional cantilever vibration in combination with the second flexural eigenmode will provide additional observables for the quantification of the in-plane and out-of-plane elastic and dissipative interactions between the tip and the sample. The use of two scans of the same sample spot in lateral directions perpendicular to each other through a 90°-rotation of the sample will pave the way for a complete three-dimensional nanomechanical surface and sub-surface quantitative characterization.Experimental section
Polymers and sample preparation
To prepare elastomeric polypropylene (ePP) samples, a 100 μL droplet from a 5 mg mL−1 ePP solution in decaline (weight-average molecular weight Mw = 160 kg mol−1 of ePP and an [mmmm]-pentade content of 36% (m = meso-conformation) synthesized by Dietrich et al.46) was drop-casted on a Si(100) surface. Subsequently, the solution dried for approximately 24 h in a fume hood at the temperature of 22 ± 1 °C, resulting in a polymer film with the thickness greater than 1 μm. The polystyrene(PS)-block-polybutadiene(PB) diblock copolymer (SB), with molecular weights of the blocks Mw,PS = 13.6 kg mol−1 and Mw,PB = 33.7 kg mol−1 was purchased from Polymer Source Inc. (Dorval, Canada). Thin films of SB were prepared by spin coating a 100 μL droplet from a 20 mg mL−1 SB solution in toluene onto a silicon(100) with 2000 rpm for 60 s. The thickness of the film was ≈125 nm as determined by AFM measuring a cross-sectional topography profile of a scratch made by a sharp needle to expose the silicon substrate.Imaging and spectroscopy
Imaging and spectroscopy were performed in the AMFlex-FMTor mode using a Cypher atomic force microscope (Asylum Research, Oxford Instruments, Santa Barbara, CA) and PPP-FM-Au Cantilevers (NanoWorld, Neuchâtel, Switzerland). The fundamental flexural and torsional resonant frequencies of these cantilevers in air were f0,flex ≈ 60 kHz and f0,tor ≈ 820 kHz, respectively, and the flexural and torsional spring constant as determined by the Sader method47,48 were kflex = 2.6 N m−1 and ktor = 2.0 × 10−8 N m rad−1. Taking the nominal tip height htip = 15 μm provided by the manufacturer, an in-plane force constant of kip = 88.0 N m−1 could be deduced. The quality factor of the cantilever first flexural eigenmode measured in air was Q ≈ 200, and the value of the tip radius R = 10 nm and half cone angle φ = 10° were estimated based on the cantilever data provided by the manufacturer. The vertical deflection sensitivity was calibrated by pushing a Si(100) sample against the tip and measuring the respective laser deflection on the photosensitive diode. To minimize optical interference between the laser light reflected at the cantilever backside and the light reflection from the sample, a superluminescence photo diode from Asylum Research was used. The lateral deflection sensitivity was determined by scanning a 4 × 4 nm2 area on a Si(100) surface in contact mode while applying the vertical force of 220 nN to provoke static friction between the tip and surface and reading out the linear dependence of the lateral laser deflection on the photosensitive diode in volts versus the distance that the tip was moved by the sample motion in nanometers (averaged over 128 scan lines). The “freely” vibrating flexural (A0,flex = 32 nm for ePP, 73 nm for SB) and torsional amplitudes (A0,tor = 7 nm for imaging both, ePP and SB) were adjusted 2.5 μm above the sample surface, and setpoint amplitudes ≈60% of A0,flex and tip velocities of ≈8 μm s−1 were chosen for scanning. Spectroscopy on the particular polymer regions was accomplished by moving the sample towards the vibrating tip with the z-velocity of 2 μm s−1 while recording the vertical amplitude and phase as well as the torsional drive amplitude and frequency shift. The frequency shift was tracked with a phase locked loop (HF2PLL, Zurich Instruments, Zurich, Switzerland), and a PID controller of the same instrument was used to adjust the drive amplitude for constant amplitude of the lateral tip vibration. All measurements (imaging and spectroscopy experiments) were performed in the constant amplitude mode for the torsional cantilever vibration. Proper gains for the PID controller were found by toggling the lateral setpoint amplitude between two values and assuring that the lateral oscillation amplitude accurately follows the setpoints. The indentation depth δ for each data point was calculated as δ = z − (A0,flex − Aflex), where z is the z-piezo position, A0,flex is the initial flexural amplitude of the oscillating tip, and Aflex is the amplitude corresponding to the z-piezo position.Data processing
Topography images were first-order leveled to remove sample tilt and to correct for thermal drift during the measurement, whereas phase shift, drive amplitude, and frequency shift images remained untreated. The raw data of the spectroscopy experiments were processed with MATLAB R2013a (MathWorks Inc., Natick, MA) to reconstruct the lateral tip–sample forces using a script provided by Welker et al.49 and Igor Pro v6.36 software (WaveMetrics Inc., Lake Oswego, OR) for graphic representation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The author thanks the Center of Smart Interfaces and the Deutsche Forschungsgemeinschaft (Sachbeihilfe DI 2176/2-1) for financial support.References
- Q. Zhong, D. Inniss, K. Kjoller and V. B. Elings, Surf. Sci., 1993, 290, L688–L692 CrossRef CAS.
- J. P. Cleveland, B. Anczykowski, A. E. Schmid and V. B. Elings, Appl. Phys. Lett., 1998, 72, 2613–2615 CrossRef CAS.
- J. Tamayo and R. Garcia, Appl. Phys. Lett., 1998, 73, 2926–2928 CrossRef CAS.
- R. Garcia, R. Magerle and R. Perez, Nat. Mater., 2007, 6, 405–411 CrossRef CAS PubMed.
- H. Schönherr, in Polymer Morphology, John Wiley & Sons, Inc., 2016, pp. 100–117, DOI:10.1002/9781118892756.ch6.
- T. Kawagishi, A. Kato, Y. Hoshi and H. Kawakatsu, Ultramicroscopy, 2002, 91, 37–48 CrossRef CAS PubMed.
- A. Yurtsever, A. M. Gigler, C. Dietz and R. W. Stark, Appl. Phys. Lett., 2008, 92, 143103 CrossRef.
- M. Reinstadtler, T. Kasai, U. Rabe, B. Bhushan and W. Arnold, J. Phys. D: Appl. Phys., 2005, 38, R269–R282 CrossRef.
- P. A. Thoren, A. S. de Wijn, R. Borgani, D. Forchheimer and D. B. Haviland, Nat. Commun., 2016, 7, 13836 CrossRef CAS PubMed.
- Y. X. Song and B. Bhushan, J. Appl. Phys., 2005, 97, 083533 CrossRef.
- N. Mullin and J. Hobbs, Appl. Phys. Lett., 2008, 92, 053103 CrossRef.
- G. Weissmuller, A. Yurtsever, L. T. Costa, A. B. F. Pacheco, P. M. Bisch, W. M. Heckl and R. W. Stark, Nano, 2008, 3, 443–448 CrossRef.
- A. Yurtsever, A. M. Gigler and R. W. Stark, Ultramicroscopy, 2009, 109, 275–279 CrossRef CAS PubMed.
- A. Sikora and L. Bednarz, Cent. Eur. J. Phys., 2011, 9, 372–379 Search PubMed.
- I. S. Hwang, C. W. Yang, P. H. Su, E. T. Hwu and H. S. Liao, Ultramicroscopy, 2013, 135, 121–125 CrossRef CAS PubMed.
- C. W. Yang, R. F. Ding, S. H. Lai, H. S. Liao, W. C. Lai, K. Y. Huang, C. S. Chang and I. S. Hwang, Nanotechnology, 2013, 24, 305702 CrossRef PubMed.
- J. D. Adams, B. W. Erickson, J. Grossenbacher, J. Brugger, A. Nievergelt and G. E. Fantner, Nat. Nanotechnol., 2016, 11, 147–151 CrossRef CAS PubMed.
- W. Cai and N. Yao, Sci. Rep., 2016, 6, 27874 CrossRef CAS PubMed.
- S. Kawai, C. M. Pina, A. Bubendorf, G. Fessler, T. Glatzel, E. Gnecco and E. Meyer, Nanotechnology, 2013, 24, 055702 CrossRef PubMed.
- R. Garcia and E. T. Herruzo, Nat. Nanotechnol., 2012, 7, 217–226 CrossRef CAS PubMed.
- T. R. Rodriguez and R. Garcia, Appl. Phys. Lett., 2004, 84, 449–451 CrossRef CAS.
- S. D. Solares and G. Chawla, J. Appl. Phys., 2010, 108, 054901 CrossRef.
- S. Jesse, S. V. Kalinin, R. Proksch, A. P. Baddorf and B. J. Rodriguez, Nanotechnology, 2007, 18, 435503 CrossRef.
- D. Platz, E. A. Tholen, D. Pesen and D. B. Haviland, Appl. Phys. Lett., 2008, 92, 153106 CrossRef.
- D. Forchheimer, R. Forchheimer and D. B. Haviland, Nat. Commun., 2015, 6, 6270 CrossRef CAS PubMed.
- A. Cartagena, M. Hernando-Perez, J. L. Carrascosa, P. J. de Pablo and A. Raman, Nanoscale, 2013, 5, 4729–4736 RSC.
- X. Xu, J. Melcher, S. Basak, R. Reifenberger and A. Raman, Phys. Rev. Lett., 2009, 102, 060801 CrossRef PubMed.
- A. Raman, S. Trigueros, A. Cartagena, A. P. Z. Stevenson, M. Susilo, E. Nauman and S. A. Contera, Nat. Nanotechnol., 2011, 6, 809–814 CrossRef CAS PubMed.
- E. T. Herruzo, A. P. Perrino and R. Garcia, Nat. Commun., 2014, 5, 3126 Search PubMed.
- H. T. Thompson, F. Barroso-Bujans, J. G. Herrero, R. Reifenberger and A. Raman, Nanotechnology, 2013, 24, 135701 CrossRef PubMed.
- L. Tetard, A. Passian and T. Thundat, Nat. Nanotechnol., 2010, 5, 105–109 CrossRef CAS PubMed.
- D. Ebeling, B. Eslami and S. D. Solares, ACS Nano, 2013, 7, 10387–10396 CrossRef CAS PubMed.
- T. Meier, B. Eslami and S. D. Solares, Nanotechnology, 2016, 27, 085702 CrossRef PubMed.
- Y. Naitoh, R. Turansky, J. Brndiar, Y. J. Li, I. Stich and Y. Sugawara, Nat. Phys., 2017, 13, 663 CrossRef CAS.
- J. E. Sader and S. P. Jarvis, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 012303 CrossRef.
- J. E. Sader and S. P. Jarvis, Appl. Phys. Lett., 2004, 84, 1801–1803 CrossRef CAS.
- C. Dietz, M. Zerson, C. Riesch, M. Franke and R. Magerle, Macromolecules, 2008, 41, 9259–9266 CrossRef CAS.
- A. Knoll, R. Magerle and G. Krausch, Macromolecules, 2001, 34, 4159–4165 CrossRef CAS.
- R. Garcia and R. Perez, Surf. Sci. Rep., 2002, 47, 197–301 CrossRef CAS.
- A. Sakai, K. Tanaka, Y. Fujii, T. Nagamura and T. Kajiyama, Polymer, 2005, 46, 429–437 CrossRef CAS.
- C. Dietz, M. Zerson, C. Riesch, A. M. Gigler, R. W. Stark, N. Rehse and R. Magerle, Appl. Phys. Lett., 2008, 92, 143107 CrossRef.
- C. Dietz, M. Schulze, A. Voss, C. Riesch and R. W. Stark, Nanoscale, 2015, 7, 1849–1856 RSC.
- S. Liparoti, A. Sorrentino and V. Speranza, Int. J. Polym. Sci., 2017, 127, DOI:10.1155/2017/9037127.
- R. G. Bauman and S. H. Maron, J. Polym. Sci., 1956, 22, 203–212 CrossRef CAS.
- K. L. Johnson, K. Kendall and A. D. Roberts, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CrossRef CAS.
- U. Dietrich, M. Hackmann, B. Rieger, M. Klinga and M. Leskelä, J. Am. Chem. Soc., 1999, 121, 4348–4355 CrossRef CAS.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS.
- C. P. Green, H. Lioe, J. P. Cleveland, R. Proksch, P. Mulvaney and J. E. Sader, Rev. Sci. Instrum., 2004, 75, 1988–1996 CrossRef CAS.
- J. Welker, E. Illek and F. J. Giessibl, Beilstein J. Nanotechnol., 2012, 3, 238–248 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |