
Enhanced topical delivery of dexamethasone by β-cyclodextrin decorated thermoresponsive nanogels†
M.
Giulbudagian
a,
S.
Hönzke
b,
J.
Bergueiro
a,
D.
Işık
a,
F.
Schumacher
cf,
S.
Saeidpour
d,
S. B.
Lohan
e,
M. C.
Meinke
e,
C.
Teutloff
d,
M.
Schäfer-Korting
b,
G.
Yealland
b,
B.
Kleuser
c,
S.
Hedtrich
b and
M.
Calderón
 *a
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Takustrasse 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de
bFreie Universität Berlin, Institute for Pharmacy (Pharmacology and Toxicology), Königin-Luise-Str. 2+4, 14195 Berlin, Germany
cUniversity of Potsdam, Department of Nutritional Toxicology, Institute of Nutritional Science, Nuthetal, Germany
dFreie Universität Berlin, Berlin Joint EPR Laboratory, Fachbereich Physik, Berlin, Germany
eCharité - Universitätsmedizin Berlin, Department of Dermatology, Venerology and Allergology, Center of Experimental and Applied Cutaneous Physiology, Berlin, Germany
fUniversity of Duisburg-Essen, Department of Molecular Biology, Essen, Germany
First published on 29th November 2017
Abstract
Highly hydrophilic, responsive nanogels are attractive as potential systems for the topical delivery of bioactives encapsulated in their three-dimensional polymeric scaffold. Yet, these drug carrier systems suffer from drawbacks for efficient delivery of hydrophobic drugs. Addressing this, β-cyclodextrin (βCD) could be successfully introduced into the drug carrier systems by exploiting its unique affinity toward dexamethasone (DXM) as well as its role as topical penetration enhancer. The properties of βCD could be combined with those of thermoresponsive nanogels (tNGs) based on dendritic polyglycerol (dPG) as a crosslinker and linear thermoresponsive polyglycerol (tPG) inducing responsiveness to temperature changes. Electron paramagnetic resonance (EPR) studies localized the drug within the hydrophobic cavity of βCD by differences in its mobility and environmental polarity. In fact, the fabricated carriers combining a particulate delivery system with a conventional penetration enhancer, resulted in an efficient delivery of DXM to the epidermis and the dermis of human skin ex vivo (enhancement compared to commercial DXM cream: ∼2.5 fold in epidermis, ∼30 fold in dermis). Furthermore, DXM encapsulated in βCD tNGs applied to skin equivalents downregulated the expression of proinflammatory thymic stromal lymphopoietin (TSLP) and outperformed a commercially available DXM cream.
Introduction
The topical administration of drugs is the route of choice for the treatment of many inflammatory skin diseases. Likewise, it serves as a powerful tool for the non-invasive systemic application of drugs subjected to strong hepatic first-pass effect and/or short duration of action following oral intake.1 Yet, the challenge to overcome the physical and biological barriers of the skin demands the use of advanced formulations, e.g. by the use of drug carriers or penetration enhancers.2 To overcome that, a variety of carrier systems in the nanometric size have been developed in the recent years.3 The encapsulation capabilities of both hydrophilic and hydrophobic drugs in their three dimensional scaffold have been investigated to meet the pharmacokinetics needs in specific pathological condition.4–6 Polymeric carrier systems can be highly effective for topical drug delivery, increasing the effective drug concentration in the viable tissue.2,3,7 Most studies report on the accumulation of the carriers in the outermost physical skin barrier, the stratum corneum (SC), while enabling the penetration of the encapsulated moiety into viable skin layers, the site of action.8Enhanced penetration of drugs into the skin can also be achieved by using various chemical penetration enhancers. For instance, fatty acids, alcohols, and sulfoxides enhance the percutaneous absorption of drugs.9 Nevertheless, out of a tested library of existing penetration enhancers, 99.50% were eliminated due to caused irritancy or low potency.10 Cyclodextrins (CDs) are a group of cutaneous penetration enhancers, however not comparable with other chemical enhancers in their mode of action, i.e. penetration into the barrier.11,12 CDs are interesting building blocks for different drug delivery systems because of their biocompatibility and a unique ability to build host–guest complexes with a variety of especially hydrophobic biologically active molecules.13,14 Rather than direct interaction with the SC components and the perturbation of their organization, CDs enhance topical delivery of drugs by increasing the solubilization and the partition of the drug in the skin.15,16 Other studies showed the ability of βCDs to extract SC lipids, resulting in enhanced absorption of hydrophilic as well as hydrophobic drugs, independent of inclusion complex formation.17,18 Importantly, excellent biocompatibility was demonstrated on various studies for CDs after topical application.19–22 Dexamethasone (DXM) is one of the most frequently used corticosteroid for topical application. Its inclusion complex with βCD has been applied for the ocular delivery of the drug as well as a topical ointment formulation known as Glymesason.23,24 While such complexation could substantially improve DXM absorption, βCD itself was found to penetrate into the viable tissue in particular into barrier disrupted models.25 Hence, the integration of βCD properties with polymeric particulate systems could provide an advantageous platform for cutaneous drug delivery.
Nanogels are three-dimensional scaffolds of polymeric building blocks which form nanosized particles due to chemical bonds and/or physical interaction.26 Their unique behavior to absorb large amounts of water in their polymeric network in aqueous media enables also the ability to encapsulate biologically active molecules, providing a protective environment from degradation or rapid clearance.27–30 Thermoresponsive nanogels (tNGs) in particular are attractive carriers for the topical delivery of drugs due to their unique ability to undergo a transition from highly hydrophilic to hydrophobic state upon a temperature trigger using the thermal gradient of the skin (32 °C–37 °C).31–33 Their ability to hydrate the SC by repealing the inner water molecules, resulted in the perturbation of the lipid and protein organization.34 While different model molecules and therapeutic biomacromolecules could be delivered into the viable tissue with the aid of tNGs, the carriers remained mainly accumulated in the SC.34,35 Yet, tNGs, as other hydrophilic carrier systems suffer from low encapsulation capacities and inefficient delivery of highly hydrophobic drugs.
Herein we describe a modular approach for the surface modification of tNGs, simultaneously inducing skin penetration enhancement and affinity towards hydrophobic drugs. The multifunctional surface of dendritic polyglycerol (dPG) enabled its function as a macro-crosslinker between linear thermoresponsive polyglycerol (tPG) units. These carriers were previously proven to be highly tolerated by primary normal human keratinocytes in terms of cell viability, genotoxicity, reactive oxygen species formation, as well as eye irritancy.36,37 The developed synthetic pathway served as a convenient platform for tNG surface decoration, enabling the accessibility towards βCD grafting or fluorescent labeling of the carriers. Electron paramagnetic resonance (EPR) studies were employed to investigate the localization and possible encapsulation of DXM within the βCD hydrophobic cavity. The βCD decorated tNGs were examined for their ability to deliver DXM into the viable epidermis and dermis of human skin ex vivo, compared to a commercially available DXM cream. Human-based skin equivalents emulating clinical characteristics of atopic dermatitis (AD) due to a knockdown of the filaggrin gene (FLG−) and exposure to Th2 cytokines IL-4 and IL-13,38 served for the investigation of DXM effect. The suppression of thymic stromal lymphopoietin (TSLP), a key factor in AD treatment, was followed upon the application of the developed carriers.
Materials and methods
The chemicals were purchased from Acros Organics, Alfa Aesar, Roth, Sigma-Aldrich, Deutero GmbH, and used as received. DXM-d4 was purchased from C/D/N Isotopes (Quebec, Canada). For the column chromatography silica gel 60 from Merck-Schuchardt® (40–60 nm, 230–400 mesh) was used. The thin layer chromatography (TLC) plates were purchased from Merck® (silica gel 60, fluorescence indicator F254, thickness 0.25 mm). Water used for the synthesis was obtained from a Millipore water purification system. Dendritic polyglycerol (dPG) with average Mw of 10 kDa (PDI = 1.27) was purchased from Nanopartica GmbH (Berlin, Germany). The amine functionalization of dPG was performed by a previously reported protocol39 while the degrees of functionalization for dPG-BCN15% are given as the percentage of the total hydroxyl groups of the dPG (∼ 135 hydroxyl groups for 10 kDa dPG). Glycidyl methyl ether (GME) (85%) and ethyl glycidyl ether (EGE) (98%; both TCI Europe, Eschborn, Germany) were dried over CaH2, distilled, and stored over molecular sieves (5 Å). The crosslinking reagent (1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethyl (4-nitrophenyl) carbonate (BCN) was purchased from Synaffix (AE Oss, Netherlands).40 The synthesis and functionalization of linear thermoresponsive polymers was performed according to a slightly modified procedure as already reported.41,42All NMR spectra were recorded on a JEOL ECX400 spectrometer operating at 400 MHz and JEOL ECP500 spectrometer operating at 500 MHz at concentrations of 5–20 mg mL−1. All data were analyzed using MestReNova software. DLS measurements were performed using a Malvern Zetasizer Nano ZS ZEN3600. IR spectra were recorded with a Nicolet iS10 FT-IR spectrometer.
Cloud point temperature (Tcp) determination by turbidimetry measurement
The Tcp were measured on a Lambda 950 UV/Vis/NIR spectrometer, PerkinElmer Life and Analytical Sciences (Connecticut, USA) equipped with a temperature-controlled, six-position sample holder. Buffer phosphate pH 7.4 nanogel solutions were heated at 0.2 °C min−1 while monitoring both the transmission at 500 nm (1 cm path length) and the solution temperature (from 18 to 55 °C), as determined by the internal temperature probe. The Tcp of each nanogel was defined as the temperature at the inflection point of the normalised transmission curves.Atomic force microscopy (AFM)
AFM measurements were recorded by a tapping mode with a MultiMode 8 AFM equipped with a Nanoscope V controller from Veeco Instruments, Santa Barbara, California. The data was analyzed using NanoScope Analysis 1.3 software and statistical analysis were performed in a 10 μm × 10 μm image. The tNGs aqueous solutions (2.5 mg mL−1) were air dried on a Mica sheet. Samples were analyzed by Nano World tips, Soft Tapping Mode (SNK-10), with a resonance frequency of 56–75 kHz and a force constant of 0.24 N m−1.Transmission electron microscopy (TEM)
TEM samples were prepared by applying a droplet (5 μL) of the sample solution (10 mg mL−1 in Milli-Q water) on a hydrophilised (60 s low discharging at 8 W using a BALTEC MED 020 device) carbon-coated copper grid (400 meshes, Quantifoil Micro Tools GmbH, Großlöbichau, Germany) for 60 s. The supernatant fluid was removed by blotting with filter paper. Then a droplet (5 μL) of 1% (w/v) uranyl acetate solution was applied and kept for another 60 s. The excess contrasting material was removed by means of filter paper and the sample was allowed to dry in air. The measurements were carried out using the TEM mode of a Hitachi Scanning Electron Microscope (SU8030, Hitachi High-Technologies Corporation, Tokyo, Japan) (20 kV) at different magnifications.mono-6-(p-Toluenesulfonyl)-6-oxy-βCD (βCD-Ts)
In a 1 L three-necked round bottom flask 25 g of βCD (22 mmol, 1 eq.) were dried under vacuum for 1 h while 8.4 g of p-toluenesulfonyl chloride (44 mmol, 2 eq.) were dissolved in 30 mL of freshly opened dry pyridine. Then 400 mL of dry pyridine was added to the βCD under dry conditions. The p-toluenesulfonyl chloride was added dropwise to the βCD at 0 °C under vigorous stirring. After the addition the reaction mixture was stirred at 0 °C for 1 h, temperature allowed to raise to room temperature (r.t.), and kept overnight under continuously stirring. Intermittently the progress of the reaction was monitored with TLC (1-butanol/ethanol/water = 5/4/3; stained by p-anisaldehyde). The solution was concentrated to half of the volume and precipitated using 600 mL of acetone while vigorously stirring. The crude product was then recrystallized in water three times and lyophilized to obtain 12.6 g (44.4%) of βCD-Ts as a white solid. 1H-NMR (400 MHz, dimethyl sulfoxide (DMSO)-d6): δ = 7.75 (d, J = 8.3 Hz, 2H, Ar–![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.43 (d, J = 8.3 Hz, 2H, Ar–), 5.98–5.55 (m, 14H, C2,3–O), 4.97–4.67 (m, 7H, C1–), 4.55–4.16 (m, 6H, C6–O), 3.87–3.43 (m, 28H, C2,3,4,5–), 3.47–3.15 (m, 14H, –C6–2; overlap with HDO), 2.43 ppm (s, 3H, –C3).43,44
), 7.43 (d, J = 8.3 Hz, 2H, Ar–), 5.98–5.55 (m, 14H, C2,3–O), 4.97–4.67 (m, 7H, C1–), 4.55–4.16 (m, 6H, C6–O), 3.87–3.43 (m, 28H, C2,3,4,5–), 3.47–3.15 (m, 14H, –C6–2; overlap with HDO), 2.43 ppm (s, 3H, –C3).43,44
mono-6-Azido-6-deoxy-βCD (βCD-N3)
In a 100 mL round flask 650 mg of mono-6-(p-toluenesulfonyl)-6-oxy-βCD (0.503 mmol, 1 eq.) and 653.3 mg of sodium azide (10.1 mmol, 20 eq.) were dissolved in 50 mL of deionized water and refluxed at 90 °C overnight. The crude product was then precipitated and washed using 100 mL of acetone to result in 574.3 mg (98%) of βCD-N3 as a white solid. 1H-NMR (400 MHz, DMSO-d6): δ = 5.75 (s, 14H, C2,3–O), 4.94–4.76 (m, 7H, C1–), 4.63–4.37 (m, 6H, C6–O), 3.81–3.50 (m, 28H, C2,3,4,5–), 3.47–3.15 ppm (m, 14H, –C6–2; overlap with HDO); IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3301 (O–H str & brd), 2923 (C–H med), 2102 (–N3 med & shp), 1152 (C–O–C med) 1021 (C–O str & shp), 996 cm−1 (C–O str).44,45
= 3301 (O–H str & brd), 2923 (C–H med), 2102 (–N3 med & shp), 1152 (C–O–C med) 1021 (C–O str & shp), 996 cm−1 (C–O str).44,45
Functionalization of dPG with βCD (dPG-BCN-βCD)
The functionalization of dPG with BCN units was performed as previously reported.42 For βCD conjugation, 20 mg of dPG-BCN(15%) (54 μmol BCN-groups, 1 eq.) were dissolved in 3 mL of dimethylformamide (DMF). To this solution a: 3.14 mg and b: 9.4 mg of βCD-N3 (a: 0.54 μmol, 0.01 eq. for 1% functionalization; b: 1.6 μmol, 0.03 eq. for 3% of functionalization) were added dissolved in 1 mL of DMF. The reaction mixture was stirred overnight at r.t. The crude product was then dialyzed against methanol for 1 d. 1H-NMR (500 MHz, CH3OD) δ = 5.72 (s, 14H, C2,3–O), 4.92–4.38 (m, 13H, C1–; C6–O), 4.13–3.98 (m, 2H, C7–2), 3.78–3.21 (m, 300H, dPG/CD backbone), 2.30–2.00 (m, 4H, C12,15–2), 1.36–1.20 (m, 4H, C11,16–2), 1.15–0.78 ppm (m, 3H, cyclopropyl ring).
Synthesis of thermoresponsive nanogels (tNG_tPG)
For tPG based nanogels (tNG_tPG) 20 mL of Milli-Q water in a glass vial were heated in an oil bath to 45 °C. In parallel azide functionalized tPG (tPG-(N3)2) (15 mg) and dPG-(BCN)10% (5 mg) were dissolved separately in DMF (0.5 mL). The solutions were cooled down in an ice bath to 0 °C. Both solutions were mixed together under cooling and injected via a syringe into the water at 45 °C. After keeping the reaction at 45 °C for 6 h the unreacted alkynes were quenched with azidopropanol and let to react for another 2 h. For βCD grafting, the reaction was quenched with β-CD-N3. For fluorescent labelling, the quenching was performed with indodicarbocyanine azide (IDCC-N3). Following, the nanogel solution was dialyzed (regenerated cellulose, molecular weight cut off (MWCO) 50 kDa) against water for 1 d, concentrated and purified by a Sephacryl S-100 column.βCD grafted thermo-responsive nanogels (tNG_tPG_βCD)
In a 5 mL glass vial 5.6 mg of dPG-BCN(15%) (11.35 μmol BCN-groups, 3 eq.) (or alternatively dPG-BCN-βCD) were dissolved in 0.5 mL of DMF. Separately, a solution of 9.5 mg of tPG-(N3)2 (1.9 μmol) in 0.5 mL of DMF was prepared. After cooling both solutions to 0 °C, they were mixed for half a minute and injected into 20 mL of MilliQ water at 45 °C. The reaction mixture was stirred overnight, quenched with 35 mg of βCD-N3 (34 μmol, 3 eq.) while stirring for an additional hour, and purified by dialysis against water for 3 d: 1H-NMR (700 MHz, DMF-d7) δ = 3.70–3.49 (m, 412H, dPG, tPG, and βCD backbone), 3.35 (s, 3H, –OC3), 1.20–1.14 ppm (m, 3H, –OCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3).
H3).
DXM loading and release
For loading of dexamethasone (DXM), 5 mg of DXM were added to tNG_tPG or tNG_tPG_βCD (10 mg) in 2 mL of MilliQ water at 37 °C. The nanogels were let to swell by cooling the dispersions in an ice bath, followed by sonication in a water bath for 30 min. The dispersions were stirred overnight at 25 °C and the insolubilized fraction was filtered off with a 0.45 μm regenerated cellulose syringe filter. Following, 100 μL of the encapsulated fraction were lyophilized for determination of the total concentration. The loading capacity was determined by LC-MS/MS, as described previously.36 The nanogels with the encapsulated drugs were stored at 4 °C. For the in vitro release studies, the tNGs with the encapsulated drug were placed in a centrifugal filtering device Vivaspin 300 kDa and incubated at 25 °C or 37 °C. At certain time intervals, the incubated solutions were centrifuged (5 min, 6000 rpm) and replaced with a fresh PBS buffer. The filtrate was lyophilized, dissolved in acetonitrile and analyzed by UV-Vis (absorbance read at 241 nm).Analysis of the association of DXM with the cavities of βCD by electron paramagnetic resonance (EPR) spectroscopy
To study the localization of DXM to the cavities of βCD, dual frequency EPR spectroscopy was performed. For this purpose, the various tNGs (tNG_tPG and tNG_tPG_βCD) were loaded with DXM which was labelled by the spin marker 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxy (PCA).46–48 DXM-PCA in water (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 1.89), in DMSO and an aqueous solution of βCD served as references. A X-band EPR spectrometer (ν = 9.5 GHz, Magnettech MiniScope MS5000, Berlin, Germany) with the following parameter settings was used: microwave power 10 mW, sweep width 20 mT, sweep time 940 s, modulation frequency 100 kHz, modulation amplitude 0.1 mT. The temperature was adjusted by a TC H014 temperature controller (Magnettech GmbH). The particle solutions were measured in quartz capillaries (QSIL GmbH, Langewiesen, Germany) sized 2 mm/1.0 mm outer/inner diameters (OD/ID).
P = 1.89), in DMSO and an aqueous solution of βCD served as references. A X-band EPR spectrometer (ν = 9.5 GHz, Magnettech MiniScope MS5000, Berlin, Germany) with the following parameter settings was used: microwave power 10 mW, sweep width 20 mT, sweep time 940 s, modulation frequency 100 kHz, modulation amplitude 0.1 mT. The temperature was adjusted by a TC H014 temperature controller (Magnettech GmbH). The particle solutions were measured in quartz capillaries (QSIL GmbH, Langewiesen, Germany) sized 2 mm/1.0 mm outer/inner diameters (OD/ID).
The nitrogen hyperfine coupling and the Zeeman interaction (g- and A-matrix) of the unpaired electron on a nitroxide are highly sensitive parameters for the surrounding microenvironment properties.49,50 To investigate these magnetic parameters for DXM-PCA loaded to different tNGs and in various reference solvents, high field (W-band, 94 GHz) EPR spectroscopy with a modulation amplitude of 0.5 mT and a power of 0.5 μW at cryogenic temperature (−193 °C) were performed on a Elexsys E680 EPR spectrometer equipped with a Teraflex EN600-1021H probe head (both Bruker Biospin, Karlsruhe, Germany). The temperature was adjusted by an ITC503 temperature controller (Oxford Instruments, Oxfordshire, United Kingdom). Samples were filled into quartz glass capillaries with 0.87 mm/0.7 mm (OD/ID) (VitroCom Inc. Mountain Lakes, NJ, USA). The magnetic field was calibrated by using N@C60 for each measurement.51 All X- and W-band EPR spectra were analyzed with Easyspin,52 a toolbox for Matlab (The MathWorks Inc., Natick, MA, USA).
Penetration of DXM into excised human skin
Cutaneous absorption of DXM into human skin was studied using abdominal human skin obtained from plastic surgeries (with informed consent; ethics vote from the Charité Mitte, Berlin, EA1/081/13) according validated protocols.53 After surgical removal, the subcutaneous layer of the skin was removed, the skin was washed with PBS and kept in a freezer (−20 °C, <6 months). During the experiment, the defrosted skin was placed in cell culture well inserts with a growth area of 4.2 cm2 (BD Biosciences, Heidelberg, Germany). Commercially available DXM LAW® cream and DXM loaded tNGs (3 wt%) were applied on the skin surface (5 μg cm−2 DXM). To mimic temperature gradient within the skin, temperature was increased from 32 to 37 °C in the first 3 h and held there for the remaining incubation. The donor compartment under cell culture inserts was filled with PBS. After incubation, surplus material was carefully removed and the skin was stripped twice with adhesive tape. In order to quantify the DXM amount in the skin layers, epidermis and dermis were heat separated (1 min in 60 °C hot water) and dermis was horizontally cut into 8 sections á 50 μm at 24 °C using a freeze microtome (Frigocut 230 2800N, Leica Microsystems Holding GmbH, Bensheim, Germany). Epidermis and dermis skin slices were subjected to 5 freeze–thaw cycles. Following the addition of 40 pmol DXM-d4 as internal standard, samples were extracted 3 times with 500 μL ethyl acetate. Combined extracts were exsiccated by vacuum rotation and the dried residues were dissolved in 200 μL acetonitrile. DXM concentrations were quantified by LC-MS/MS as described previously.54Generation of skin equivalents and treatment with DXM loaded cream and nanogels
Normal (FLG+) and filaggrin-deficient (FLG−) skin equivalents were generated according to previously published procedures.55 Primary human keratinocytes and fibroblasts were isolated from juvenile foreskin, acquired from circumcision surgeries (ethics vote from the Charité Mitte, Berlin, EA1/081/13). To induce gene knockdown, keratinocytes were transfected (HiPerFect®; Qiagen, Hilden, Germany) with FLG specific siRNA (Sequence: CAGCUCCAGACAAUCAGGCACUCAU; NM_002016, Invitrogen, Darmstadt, Germany). Primary human fibroblasts, FCS Superior (Merck Millipore, Darmstadt, Germany) and bovine collagen I (PureCol; Advanced BioMatrix, San Diego, USA) were brought to neutral pH and poured into 3D cell culture well inserts with a growth area of 4.2 cm2 (BD Biosciences, Heidelberg, Germany). After 2 h at 37 °C, defined cell culture medium was added and the system transferred to an incubator with 5% CO2 and 95% humidity. After 2 h, primary human keratinocytes (with or without filaggrin knockdown) were added on top of the collagen matrix. After 24 h, the skin equivalents were lifted to the air-liquid interface and a differentiation medium was added. The equivalents were cultivated for 14 d with media change every second day. Starting at day 10, the culture media was supplemented with 15 ng mL−1 IL-4 and IL-13. At day 11 and day 13 skin models were treated with DXM LAW® cream or loaded in tNGs (3 wt%) in a dose of 2 μg cm−2 (4 μg cm−2 in total) and their respective drug free vehicles. To mimic temperature gradient within the skin, temperature was increased from 32 to 37 °C in the first 3 h and held there for the remaining incubation. Carrier concentration within the aqueous solution was set at 5 mg mL−1.To investigate the penetration of the tNGs, they were labeled with the fluorescent dye IDCC and applied on the skin models at the same carrier amount used above. For histological analysis, skin models were immediately frozen, and cut into 8 μm vertical slices using a Leica CM 1510 S cryotome (Leica Biosystems, Nussloch, Germany). Skin sections were fixed with 4% formaldehyde, embedded in 4′,6-diamidin-2-phenylindol (DAPI) antifading mounting medium afterwards and analyzed under a fluorescence microscope (BZ-8000, objectives 20×/0.75, zoom 10×, Plan-Apo, DIC N2, Keyence, Neu-Isenburg, Germany).
Western blot
The epidermis was gently removed from the models and proteins were extracted with radioimmunoprecipitation assay (RIPA) buffer. Total protein concentrations were determined using the Pierce® BCA Protein Assay Kit (Thermo Fisher Scientific, Schwerte, Germany). Subsequently, samples (30 μg protein) were boiled in standard SDS-PAGE sample buffer and separated by 10% SDS polyacrylamide gel electrophoresis (Bio-Rad, Munich, Germany). Gels were blotted onto nitrocellulose membranes (Bio-Rad, Munich, Germany). After blocking with 5% skimmed-milk powder for 1 h at 37 °C, membranes were incubated with primary antibodies at 4 °C overnight (rabbit polyclonal to filaggrin 1:1000, rabbit polyclonal to involucrin 1:1000, rabbit polyclonal to TSLP 1:1000, mouse monoclonal to β-actin 1:10000). Blots were washed and incubated with anti-rabbit horseradish-peroxidase-conjugated secondary antibody (Cell Signaling, Frankfurt/Main, Germany) for 1 h in a dilution of 1:1000. Afterwards, blots were developed with SignalFire™ ECL reagent (Cell Signaling, Frankfurt/Main, Germany) and visualized by a PXi/PXi Touch gel imaging system (Syngene, Cambridge, UK). Protein expression was semi quantified by densitometry and normalized to β-actin levels using ImageJ version 1.46r (National Institutes of Health, Bethesda, USA). Potential off-target effects on respective protein expression levels due to siRNA transfection were excluded previously by using skin models generated with siRNA negative control.38
Results and discussion
Synthesis of thermoresponsive nanogels
The chemical ligation of βCD to the polymeric building blocks requires its modification to allow efficient coupling reactions. The controlled and selective modification of βCD was performed according to a protocol of Ng et al.56 by initial tosylation of one primary alcohol followed by the displacement with sodium azide (Fig. 1a). The products were characterized by means of 1H-NMR and IR spectroscopy to assure the modification on a single hydroxide (C6) of the βCD. This functionalization was selected to enable the decoration of polymers with βCD-N3, since more than one functional group would possibly cause to undesired crosslinking reactions. Following, the dPG-(NH2)15% was reacted with BCN to enable the strain-promoted alkyne–azide cycloaddition (SPAAC) reaction57 allowing the facile and mild grafting of βCD-N3 on to the dPG surface (Fig. 1b). Three different conjugates were synthesized differing in the percentage of βCD units per dPG: 1%, 3%, and 5% of the total hydroxyl groups of the dPG. The remaining alkynes were left to react with the thermoresponsive polymer, during nanogels synthesis. Interestingly, already at the lowest percentage of βCD grafting, the water solubility of the dPG-(βCD)1% dropped, appearing as a slightly turbid solution. Obviously, the solubility decreased with increasing amount of βCD, resulting in a non-soluble conjugate for the dPG-(βCD)5%.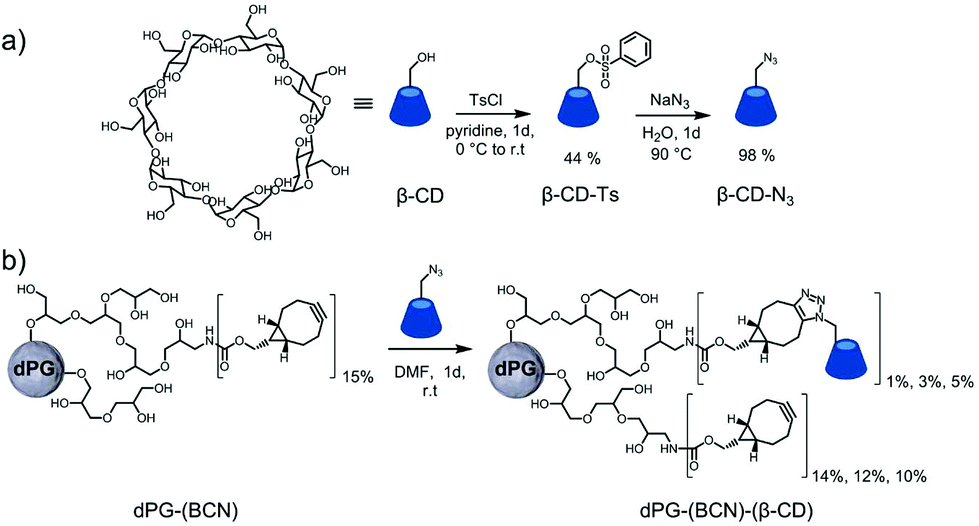 | ||
| Fig. 1 Functionalization of the macromolecular precursors. | ||
The nanogels were prepared by the thermonanoprecipitation approach where the presynthesized polymeric precursors, i.e. dPG-BCN and tPG-(N3)2, were dissolved in DMF and injected into water at 45 °C, above the Tcp of the thermoresponsive polymers. The crosslinking occurred via the strain-promoted alkyne–azide cycloaddition (SPAAC) between the orthogonally functionalized polymers. Two routes were exploited for the introduction of βCD onto the nanogels (Fig. 2).
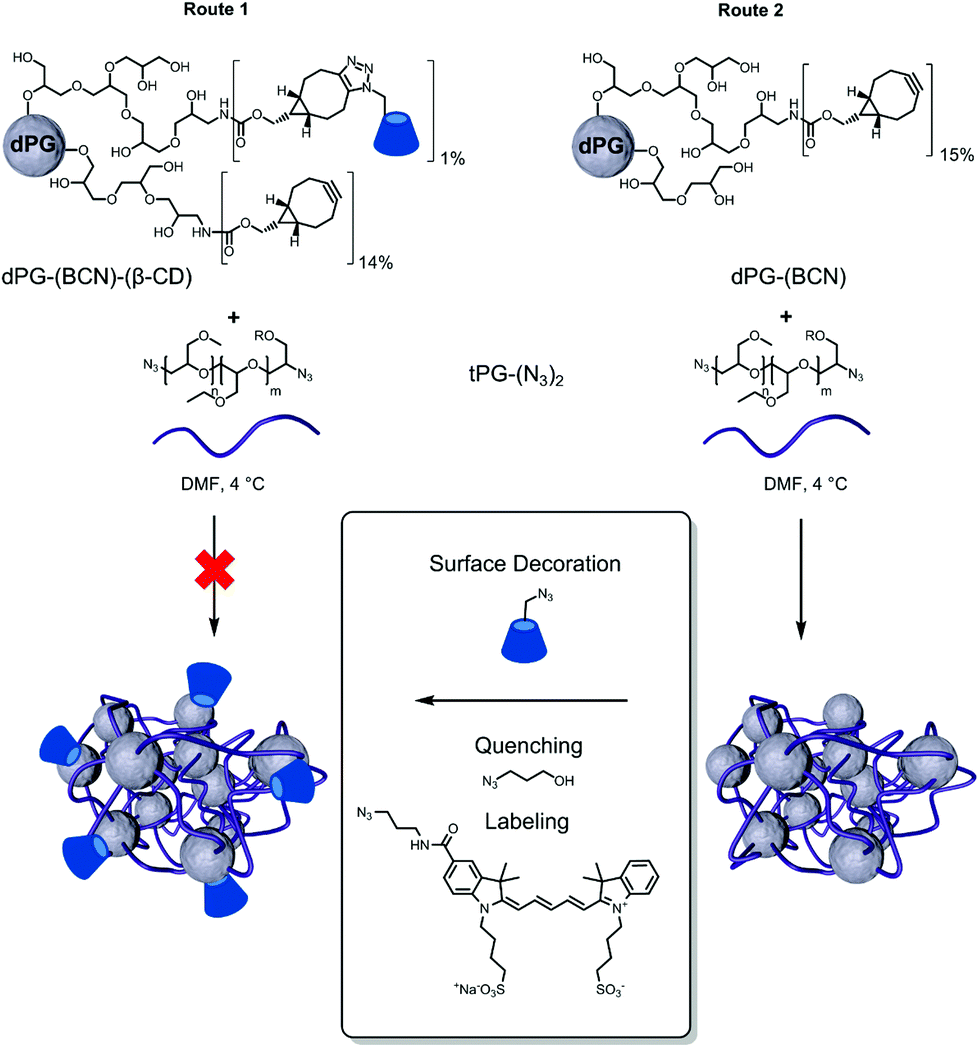 | ||
| Fig. 2 Synthesis of β-CD grafted thermoresponsive nanogels. | ||
Route 1 follows the functionalization of the macro-crosslinker (dPG) with the βCD prior to the nanogels synthesis. Such approach would enable more controlled and quantitatively precise introduction of the βCD on the formed nanogels. Using this route however, no stable nanoparticles could be obtained, most probably due to uncontrolled aggregation of the dPG-(BCN)-(βCD) adduct resulting from its low water solubility.
Nevertheless, stable βCD grafted thermoresponsive nanogels could be obtained using route 2 (Fig. 2). In this case dPG-(BCN) was crosslinked with tPG-(N3)2via thermonanoprecipitation to form stable polymeric nanoparticles. In a second step the excess of cyclooctyne moieties were quenched with βCD-N3 as both quenching and post-functionalization agent to form the surface decorated nanogels. Depending on the functionalization of dPG with -(BCN)5% or -(BCN)15% (Table 1) it could be shown that the thermoresponsive behavior of the resulting nanogel is influenced by the amount of tPG used for the crosslinkage. As a result of the low functionalization percentage on dPG-(BCN)5%, the weight ratio between the two macromolecular precursors had to be adjusted accordingly to 47–62 wt% of dPG-(BCN)5%, to ensure constant ratios between the functional groups. However, in that case the synthesis resulted in nanoparticles with broad PDIs and no thermoresponsive behavior. Therefore, a minimum amount of tPG is necessary for the thermonanoprecipitation to occur, which should not fall below 53 wt%.
| FG-ratioa | Wt% | Sizeb [nm] | PDIb | |||
|---|---|---|---|---|---|---|
| BCN | –N3 | (dPG) | 25 °C | 45 °C | ||
| a Functional group (FG) feed ratio. b Size and polydispersity index (PDI) by DLS in water at 25 °C. Measurements were performed in triplicates; intensity average mean value presented. | ||||||
| dPG-BCN5% | 3 | 1 | 62 | 200 | — | 0.405 |
| 2 | 1 | 53 | 140 | — | 0.472 | |
| 1.5 | 1 | 47 | 120 | — | 0.500 | |
| dPG-BCN15% | 3 | 1 | 37 | 70 | 60 | 0.170 |
| 2 | 1 | 28 | 80 | 62 | 0.200 | |
| 1.5 | 1 | 23 | 75 | 63 | 0.108 | |
Nevertheless, the increasing weight percentage (wt%) of tPG in the nanogels formed with dPG-(BCN)15% result in narrow PDI values and thermally triggered size reduction of the nanoparticles, as analyzed by DLS. The narrow PDI values for increasing wt% of tPG are most probably resulted by the fact that the thermonanoprecipitation process is governed by the phase transition of the tPG (from soluble below its transition temperature, and precipitated when the transition temperature is crossed). Hence, its domination created the suitable conditions for the formation of the uniform precursor particles. Interestingly, the introduction of βCD to the tNGs had only a minor influence on their Tcp (Fig. 3a). The Tcp of tNG_tPG was measured to occur at 33.9 °C while decreasing to 32.3 °C for tNG_tPG_βCD. This tendency can be attributed to the rather hydrophobic nature of βCD, while the minor influence arises from the fact the surface decoration of the tNGs was achieved by utilizing the multifunctionality of the crosslinker dPG, without modifying the thermoresponsive polymer itself. Moreover, this approach enables the decoration of the nanogels surface with the desired functionality: i.e. with IDCC-N3 for fluorescent labelling or azidopropanol for quenching of the unreacted alkynes. TEM and AFM measurments of tNG_tPG_βCD confirmed the presence of spherical particles in the sizes of 230 ± 70 nm (Fig. 3b and c). The enlarged surface visible in the microscopy images is caused by the flattening of the dehydrated particles expressed by a height profile of 20.6 nm by AFM. Furthermore, the ability of βCD to form an inclusion complex with 8-anilinonaphthalene-1-sulfonic acid (ANS) was employed to qualitatively characterize the tNG surface decoration.58–61 ANS is a known guest molecule for the cyclic sugar, which complexation leads to gain in its fluoresce intensity. Thus, tNG_tPG_βCD were added to an aqueous solution of ANS which resulted in increase of the fluorescent signal until a saturation point (Fig. S1†), suggesting successful grafting of βCD.
 | ||
| Fig. 3 Characterization of tNG_tPG_βCD. (a) Transmission vs. temperature measured by UV-Vis at 500 nm in PBS (1 mg mL−1), compared to tNG_tPG (b) TEM, and (c) AFM. | ||
The encapsulation of the DXM in the tNGs could be achieved by sonication of the tNG/DXM suspension, followed by filtration of the excess of drug. The maximum encapsulation capacity reached for tNG_tPG was 5.8 ± 0.2 wt% as reported elsewhere.36 The introduction of βCD to the tNG scaffold did not improve the encapsulation capacity of DXM, but rather decreased it to 4.1 ± 0.2 wt%. This can be attributed to aggregation patterns that the βCD can induce to the tNGs, while the entrapment of DXM in the hydrophobic pocket of βCD is not expected to have a large impact on the encapsulation efficiencies. Both tNGs with the encapsulated drug were incubated below (25 °C) and above (37 °C) their Tcp to study the in vitro release kinetics of DXM (Fig. 4). While a slight increase in the released DXM amount was observed for both tNGs incubated at 37 °C, this could not be attributed to temperature triggered release. In line with previous reports, the release of a low Mw drug could not be triggered by the conformational change of the tNGs but occurred by slow diffusion of the drug from the polymer matrix. Moreover, the presence of βCD on the tNG surface, did not influence the release kinetics of the drug incubated below and above the Tcp.
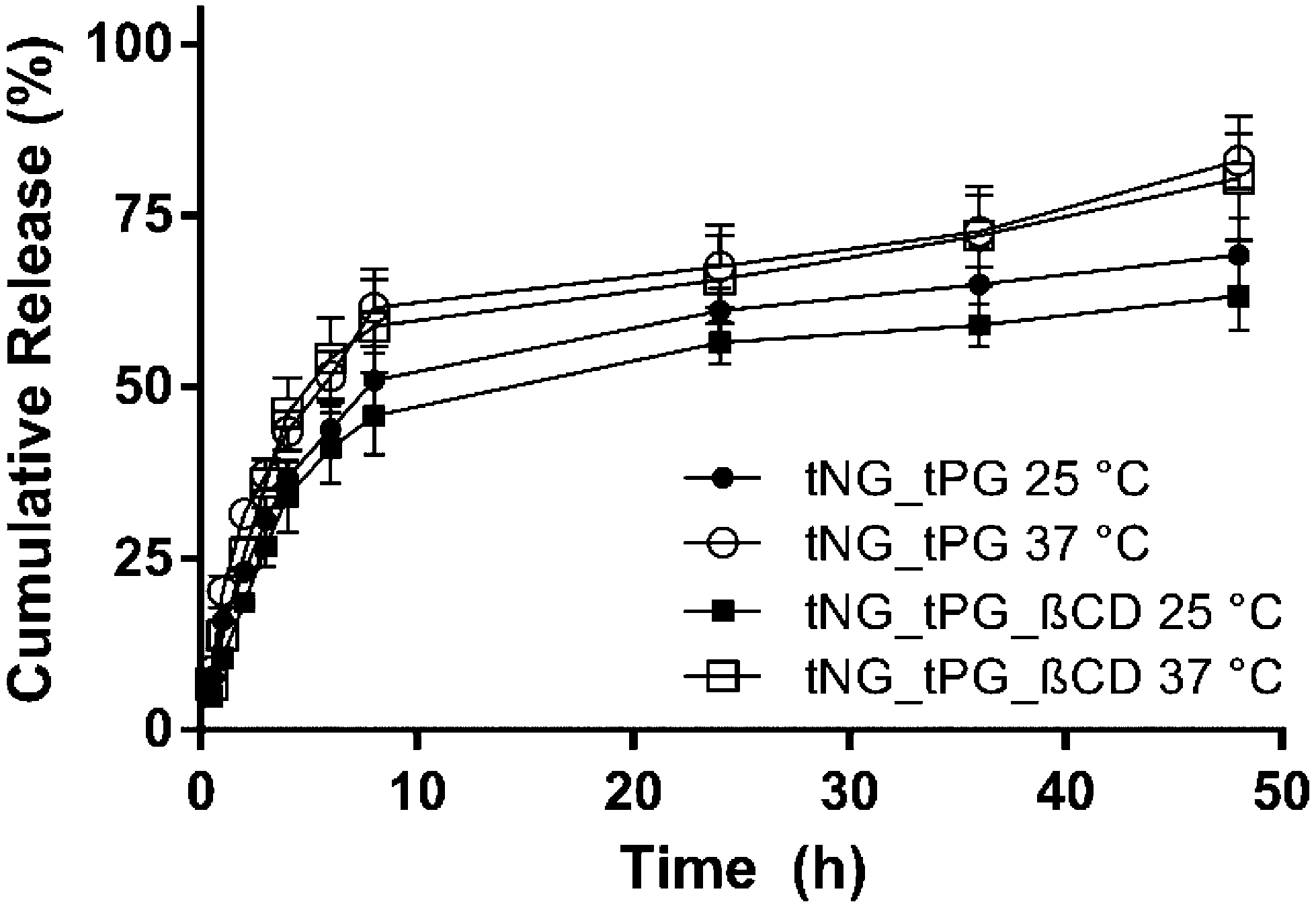 | ||
| Fig. 4 Cumulative release of DXM from tNG_tPG at 25 °C (black circles) and 37 °C (hollow circles) and from tNG_tPG_βCD at 25 °C (black squares) and 37 °C (hollow squares). | ||
EPR study on the association of DXM to βCD
EPR investigations pointed out different environments for DXM-PCA loaded to the tNGs with and without βCD, taking into account DXM-PCA dissolved in various reference solvents. The EPR spectrum of DXM-PCA in water is characterized by three sharp lines of almost identical height (Fig. 5). For the aqueous dispersion of the tNG_tPG loaded with the spin labeled drug the spectral shape is comparable, representing a freely tumbling molecule in solution. For DXM-PCA in DMSO already a slight reduction of the line intensity at 336 mT can be noticed owing to the higher viscosity of this solvent, thus leading to lower mobility.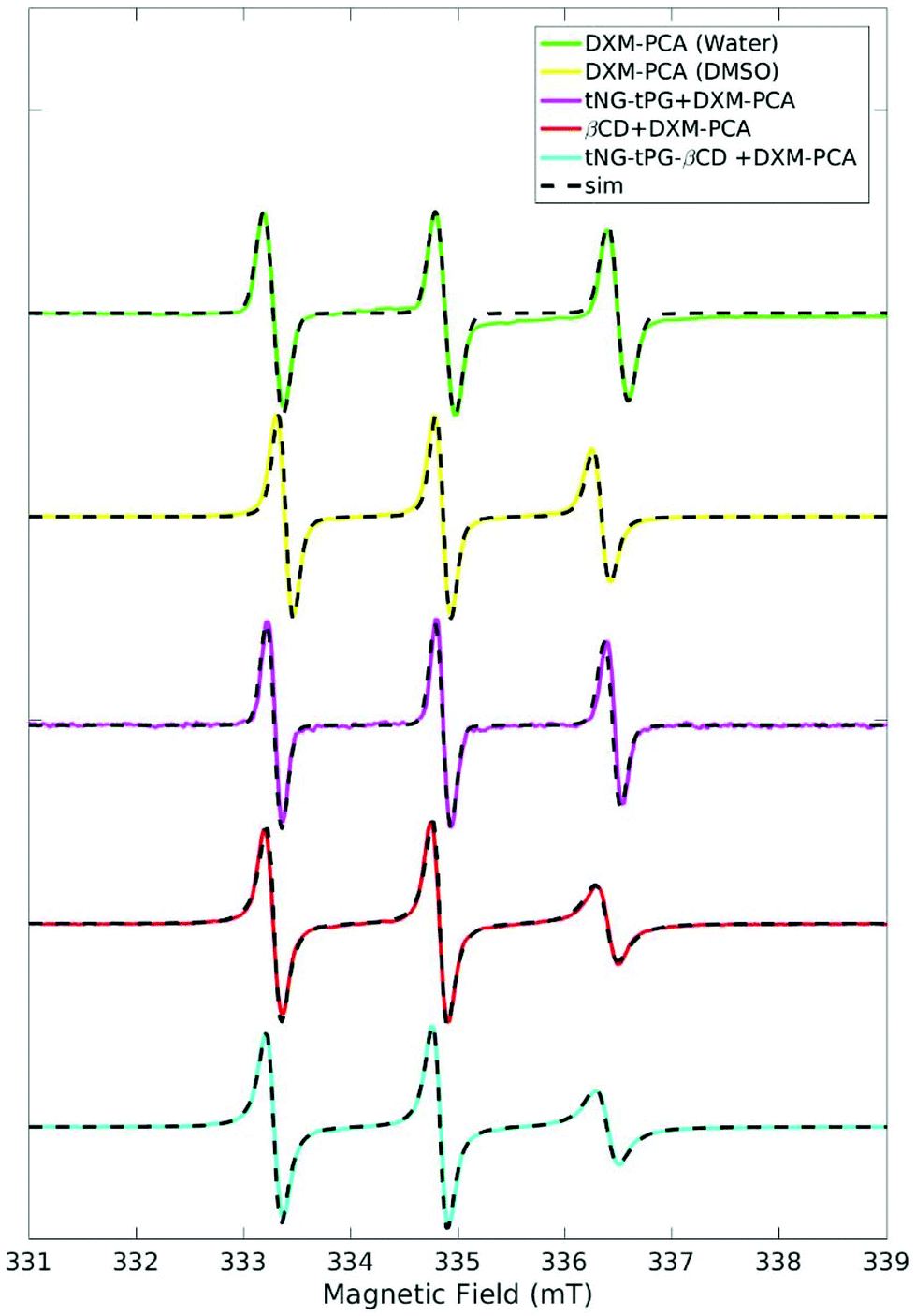 | ||
| Fig. 5 X-band EPR spectra of DXM-PCA subjected to tNG_tPG, tNG_tPG_βCD, DMSO, and water at 25 °C, normalized to the maximum height of the middle peak. Simulations are shown in dashed lines. | ||
The EPR spectrum of DXM-PCA loaded to tNG_tPG_βCD differs remarkably recognizable by spectral broadening of the high-field line (at 336 mT) in comparison to tNG_tPG (DMSO and water) (Fig. 5). By comparison, the EPR spectra of DXM-PCA loaded to tNG_tPG_βCD and in an aqueous solution of βCD (Fig. 5) illustrate that the cyclic oligosaccharide seems to be responsible for the restricted mobility of DXM-PCA.
In general, the mobility of a spin label is encoded into the spectral shape of an EPR spectrum as incomplete time-averaging over the anisotropic magnetic interactions by the tumbling motion of the molecule.62 A measure for the mobility is the rotational correlation time tcorr, which can be deduced by simulation of the EPR spectrum. For a consistent simulation of the EPR spectra at r.t., the magnetic parameters (g- and 14N hyperfine matrices) should be measured independently. Therefore EPR spectra at 94 GHz were acquired, from which by simulation the g-matrix and the 14N hyperfine coupling could be estimated (see Table 2, Fig. S2, and Table S1†).
The obtained magnetic parameters (gxx, and Azz, see Table 2) illustrate the polarity of DXM-PCA's environment loaded to the various tNG dispersions and dissolved in different reference solutions. The gxx value and the Azz component are known to be sensitive to the polarity of the environment.49,50 DXM-PCA loaded to tNG_tPG shows a similar EPR spectrum as in water (Table 2), indicating a high mobility of DXM-PCA in this dispersion. A little tendency towards a more apolar environment for tNG_tPG is given, recognizable by the slight upshift of gxx and correspondingly downshift of Azz (Table 2). For DXM-PCA loaded to tNG_tPG_βCD and dissolved in the aqueous βCD solution the change is noticeably higher reflecting a more apolar environment. Interestingly, for both βCD containing solutions the same magnetic as well as mobility parameters were obtained indicating a similar environment for DXM-PCA.
Comparing the parameters obtained from EPR the following conclusions can be drawn: (a) for the tNG_tPG_βCD dispersion as well as the βCD solution DXM-PCA is located in a more apolar environment compared to the tNG_tPG dispersion, (b) the rotational correlation time reports for the same partial immobilization of the drug in these βCD containing solutions. Taking these two facts together, the most probable location of DXM-PCA is within the hydrophobic pocket of βCD, leading to a more apolar environment and hindered rotation compared to the tNG_tPG dispersion and aqueous solution.
Dexamethasone delivery into human skin ex vivo
Topical corticosteroid administration, such as DXM cream, is a benchmark therapy for inflammatory skin diseases such as AD.64 To investigate the delivery efficacy by the tNGs, 3 wt% DXM was incorporated into the nanogels. In line with previous studies,65 distinct penetration of 0.2 μg cm−2 DXM into the epidermis was found by topical application of the LAW® cream for 6 h (Fig. 6a). Loading DXM in tNGs_tPG had no further influence on the delivery into the epidermis, but led to a distinct increase in drug amounts in the dermis. These results may be explained by recent findings, where the tNG_tPG act as penetration enhancers by increasing the hydration of the SC with consequent perturbation of the lipids and proteins organization.34 Interestingly, βCD functionalized tNGs significantly increased the amounts of DXM in both skin layers, when compared to the standard cream treatment (∼2.5 fold in epidermis; ∼30 fold in dermis). These results demonstrated the ability of βCD to act as a topical penetration enhancer when used as an integral part of a polymeric carrier system. The direct interaction between DXM and βCD shown by the EPR data, suggests enhanced penetration of DXM due to its increased solubilization, directly influencing the availability of the drug. Along with that, the coordination complex did not retain the drug in the carrier, but allowed its diffusion into the deeper skin layers faster than the control groups (Fig. 6a). Notably, the temperature ramp from 32 °C–37 °C was chosen to simulate the natural thermal gradient of human skin: the outermost layer having a surface temperature of 32 °C which gradually increases with SC depth ultimately reaching 37 °C in the viable epidermis.35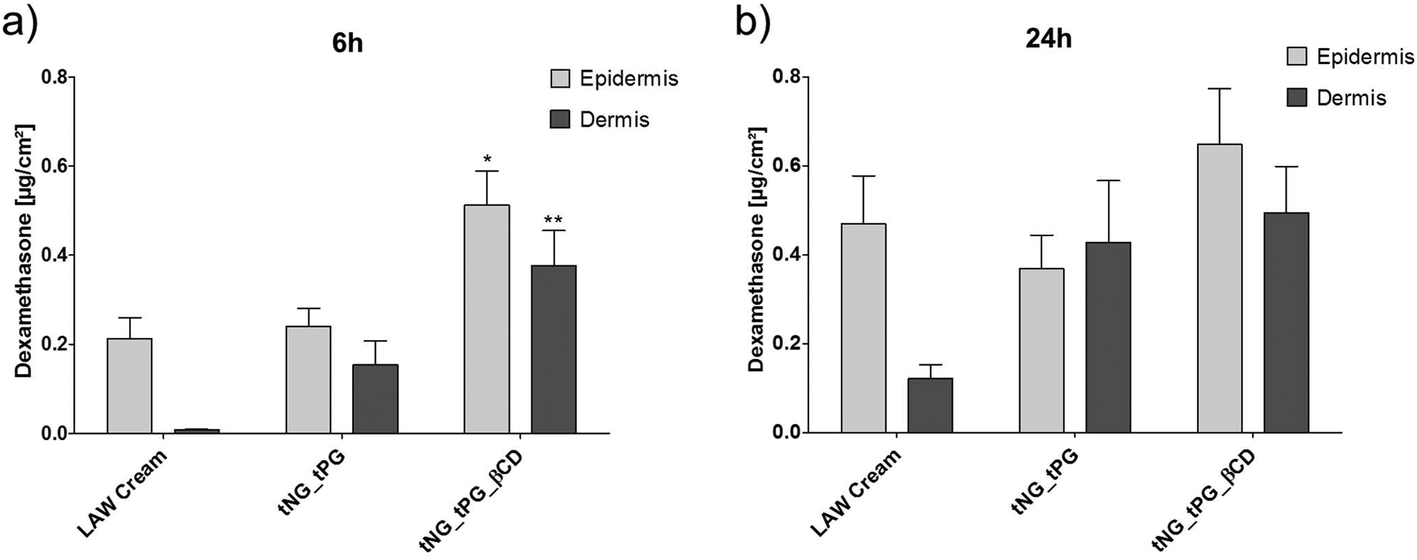 | ||
| Fig. 6 DXM penetration into human skin assessed by LC-MS/MS. DXM amount [μg cm−2] in human epidermis and dermis after topical exposure for 6 h (a) and 24 h (b) incorporated in a commercially available LAW® cream or tNGs. Temperature was increased from 32 to 37 °C in the first 3 h and held there for the remaining incubation. Statistical differences were assessed by one-way ANOVA and Dunett post analyses compared to the LAW® cream treatment (mean ± SEM; n = 3). | ||
After 24 h, no distinct variation of DXM amounts were found in skin samples treated with tNG_tPG_βCD compared to the short treatment, however, cream treatment and tNG_tPG almost doubled the DXM amount (Fig. 6b). Nevertheless, the βCD decorated tNGs retained DXM concentration in both skin layers, dominating over the control groups after 24 h of incubation, with a recovered drug concentration of ∼1.2 μg cm−2, while the rest removed from skin surface by tape stripping or discarded from the acceptor compartment. This might suggest the ability of tNG_tPG_βCD to act as drug reservoirs, maintaining DXM concentration over longer periods of time. Data-based modeling of DXM penetration depth based on its diffusivity and free energy profiles, has previously shown the considerably moderate drug diffusion through the SC while reaching a distinct free energy barrier (high solubility) in the viable epidermis.66 Thus, the penetration profile of DXM with the aid of tNG_tPG_βCD, might alternatively suggest an increased diffusivity through the SC reaching a dynamic equilibrium state of DXM concentration in the viable epidermis and the dermis layers.
Therapeutic efficacy of DXM loaded βCD functionalized thermoresponsive nanogels
To evaluate the pharmacological efficacy of delivered DXM we treated previously established skin equivalent exhibiting characteristics of AD in vitro.38 These characteristics were described by altered skin histology, changes in the expression of epidermal barrier proteins and defects in the cutaneous innate immune response. Significant changes in the expression of the cytokine TSLP, which is considered a key player in AD pathogenesis,67 were confirmed in the present study and used as a pharmacological read out parameter for the anti-inflammatory treatment. Compared to untreated, normal (FLG+) skin equivalents, a distinct upregulation of TSLP was observed in diseased skin equivalents, which exhibiting a deficiency of filaggrin (FLG−) and a simultaneous Th2 triggered inflammation (IL-4/IL-13) (Fig. 7a and Hönzke et al.).38 TSLP primes differentiation of naïve proliferative T cells into Th2 cells by either direct activation68 or indirect activation trough dendritic cells.69 These activation of immune cells is followed by secretion of high levels of other pro-inflammatory cytokines such as TNFα.69 Glucocorticoids such as DXM have the anti-inflammatory potency to suppress the activation of the immune response by modulating the activation of various transcription factors within the inflamed tissue.70 Interestingly for TSLP it is known, that the expression within keratinocytes is hampered by glucocorticoids trough binding to a negative glucocorticoid response element.71 In line with these findings we found decreased TSLP levels in the DXM treated skin equivalents compared to the untreated FLG−IL-4/IL-13 equivalent (Fig. 7a). In accordance with the ex vivo penetration data (Fig. 6), we found a significant downregulation of the inflammation, exclusively in the skin equivalents, which were treated with the DXM loaded tNG_tPG_βCD. These findings further demonstrated the downregulating effect of DXM on the expression of TSLP, as previously shown in a mouse model of AD.72 Furthermore, it must be noted that both tNGs did not overcome the SC of the skin equivalents indicating that the observed effects result from enhanced DXM delivery (Fig. 7b and c). Notably, human-based skin disease models were specifically chosen, since inter-species related effects, which occur for example with mice, are avoided.73 Moreover, their applicability for assessing drug effects has been repeatedly demonstrated qualifying them as valuable alternatives to animal studies.74–77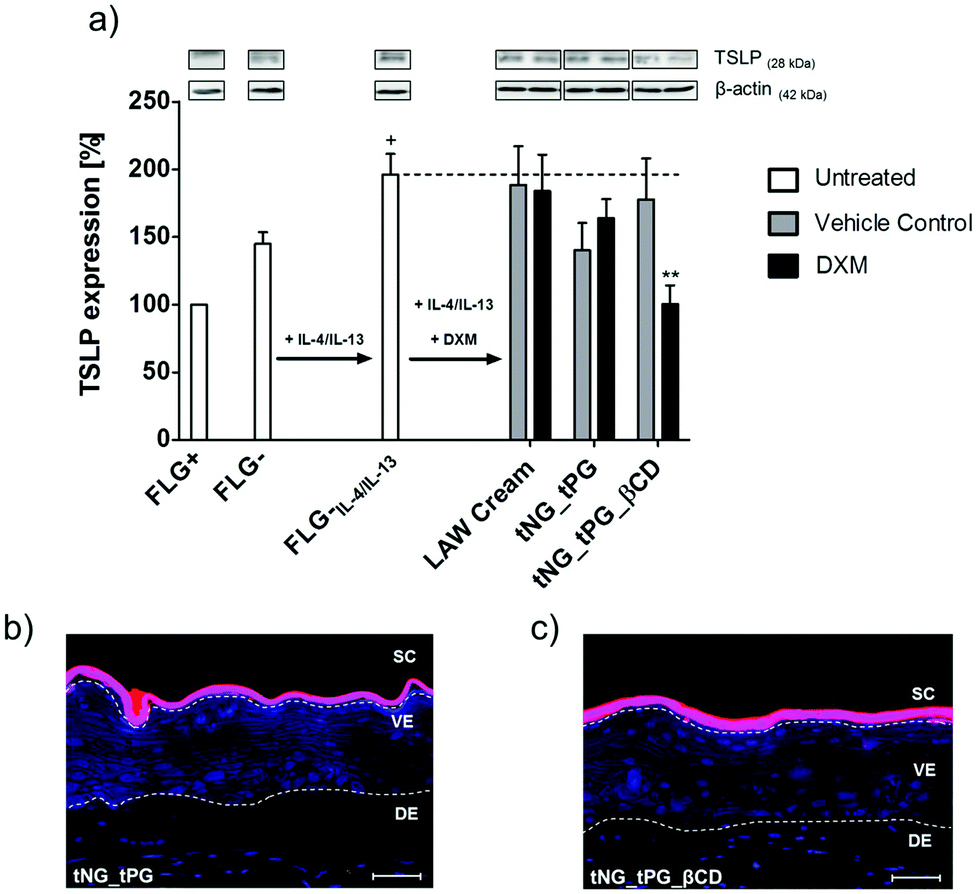 | ||
| Fig. 7 (a) Western blots and relative protein expression of TSLP semi-quantified via densitometry of untreated normal (FLG+) and filaggrin knockdown (FLG−) (untreated and IL-4/IL-13 supplemented) skin equivalents. Skin equivalents were topically treated at day 11 and day 13 with DXM loaded onto LAW® cream, tNG_tPG or tNG_tPG_βCD. Temperature was increased from 32 to 37 °C in the first 3 h and held there for the remaining incubation. Statistical differences were calculated by Student‘s t-test compared to FLG−IL-4/IL-13 (** = p ≤ 0.01) or compared to FLG+ (+ = p ≤ 0.05, Mean ± SEM; n = 5). (b, c) Representative fluorescence imaging of fluorescent labeled tPG (red) (b) and tPG_βCD (c) nanogels in FLG−IL-4/IL-13 skin equivalents. The skin equivalent sections were counterstained with DAPI (blue). Dashed lines indicate the SC, viable epidermis (VE), dermis (DE). Scale bar = 50 μm. | ||
Conclusions
To conclude, we present here a joint approach for the topical delivery of hydrophobic drugs by utilizing polymeric nanoparticulate delivery systems combined with conventional penetration enhancers. The tNGs were decorated with βCD units by strain promoted click chemistry of the orthogonally functionalized precursors. The chosen synthetic pathway of post functionalization allowed to overcome aggregations and low solubility of the polymeric precursors caused by the rather hydrophobic βCD. Similar approach was used for the decoration of the tNGs with a fluorescent dye or quenching of the unreacted moieties. DXM could be encapsulated within the tNGs while interestingly, βCD did not change the properties of the carrier nor the in vitro release kinetics of DXM. EPR studied could explicitly locate the encapsulated drug within the hydrophobic cavity of βCD, while retaining its mobility in the bare tNGs. Nevertheless, when applied on excised human skin, tNG_tPG_βCD were superior in their ability to deliver DXM to the viable epidermis and the dermis compared to the unfunctionalized tNGs, but more importantly, to a commercial DXM formulation. This effect was further pronounced when the βCD decorated tNGs were the only ones able to down regulate the TSLP expression in the treatment of inflammatory skin equivalents. Exploiting nanocarriers coupled to topical penetration enhancers presents a promising strategy for the topical delivery of various hydrophobic drugs while ensuring the retention of the delivery system in the SC, avoiding potential irritation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Sonderforschungsbereich 1112, projects A04, B01, C02 and Z01, the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (13N12561, Thermonanogele), and the Focus Area NanoScale of the Freie Universität Berlin (http://www.nanoscale.fu-berlin.de).References
- K. S. Paudel, M. Milewski, C. L. Swadley, N. K. Brogden, P. Ghosh and A. L. Stinchcomb, Ther. Delivery, 2010, 1, 109–131 CrossRef CAS.
- A. Vogt, C. Wischke, A. T. Neffe, N. Ma, U. Alexiev and A. Lendlein, J. Controlled Release, 2016, 242, 3–15 CrossRef CAS PubMed.
- M. S. Roberts, Y. Mohammed, M. N. Pastore, S. Namjoshi, S. Yousef, A. Alinaghi, I. N. Haridass, E. Abd, V. R. Leite-Silva, H. A. E. Benson and J. E. Grice, J. Controlled Release, 2017, 247, 86–105 CrossRef CAS PubMed.
- J. Kreuter, Nanoparticles, Marcel Dekker Inc, 1994 Search PubMed.
- Y.-S. Rhee and H. M. Mansour, Int. J. Nanotechnol., 2010, 8, 84–114 CrossRef.
- L. Fernandez, M. Calderón, M. Martinelli, M. Strumia, H. Cerecetto, M. González, J. J. Silber and M. Santo, J. Phys. Org. Chem., 2008, 21, 1079–1085 CrossRef CAS.
- F. Rancan, U. Blume-Peytavi and A. Vogt, Clin., Cosmet. Invest. Dermatol., 2014, 7, 23–34 CrossRef PubMed.
- T. W. Prow, J. E. Grice, L. L. Lin, R. Faye, M. Butler, W. Becker, E. M. T. Wurm, C. Yoong, T. A. Robertson, H. P. Soyer and M. S. Roberts, Adv. Drug Delivery Rev., 2011, 63, 470–491 CrossRef CAS PubMed.
- H. Trommer and R. H. H. Neubert, Skin Pharmacol. Physiol., 2006, 19, 106–121 CrossRef CAS PubMed.
- P. Karande, A. Jain and S. Mitragotri, Nat. Biotechnol., 2004, 22, 192–197 CrossRef CAS PubMed.
- K. Wang, Y. Yan, G. Zhao, W. Xu, K. Dong, C. You, L. Zhang and J. Xing, Polym. Chem., 2014, 5, 4658–4669 RSC.
- T. Loftsson and M. Masson, Int. J. Pharm., 2001, 225, 15–30 CrossRef CAS PubMed.
- T. Irie and K. Uekama, J. Pharm. Sci., 1997, 86, 147–162 CrossRef CAS PubMed.
- M. D. Moya-Ortega, C. Alvarez-Lorenzo, H. H. Sigurdsson, A. Concheiro and T. Loftsson, Carbohydr. Polym., 2012, 87, 2344–2351 CrossRef CAS.
- R. J. Babu and J. K. Pandit, Int. J. Pharm., 2004, 271, 155–165 CrossRef CAS PubMed.
- A. C. Williams, S. R. S. Shatri and B. W. Barry, Pharm. Dev. Technol., 1998, 3, 283–296 CrossRef CAS PubMed.
- M. V. Bentley, R. F. Vianna, S. Wilson and J. H. Collett, J. Pharm. Pharmacol., 1997, 49, 397–402 CrossRef CAS PubMed.
- A. M. Totterman, N. G. Schipper, D. O. Thompson and J. P. Mannermaa, J. Pharm. Pharmacol., 1997, 49, 43–48 CrossRef CAS PubMed.
- Y. Yan, J. Xing, W. Xu, G. Zhao, K. Dong, L. Zhang and K. Wang, Int. J. Pharm., 2014, 474, 182–192 CrossRef CAS PubMed.
- N. Kaur, R. Puri and S. K. Jain, AAPS PharmSciTech, 2010, 11, 528–537 CrossRef CAS PubMed.
- G. Piel, S. Moutard, E. Uhoda, F. Pilard, G. E. Piérard, B. Perly, L. Delattre and B. Evrard, Eur. J. Pharm. Biopharm., 2004, 57, 479–482 CrossRef CAS PubMed.
- L. H. Peng, W. Wei, Y. H. Shan, T. Y. Zhang, C. Z. Zhang, J. H. Wu, L. Yu, J. Lin, W. Q. Liang, G. Khang and J. Q. Gao, J. Biomed. Nanotechnol., 2015, 11, 680–690 CrossRef CAS PubMed.
- M. Tanito, K. Hara, Y. Takai, Y. Matsuoka, N. Nishimura, P. Jansook, T. Loftsson, E. Stefánsson and A. Ohira, Invest. Ophthalmol. Visual Sci., 2011, 52, 7944–7948 Search PubMed.
- T. Loftsson and D. Duchene, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS PubMed.
- M. Tanaka, Y. Iwata, Y. Kouzuki, K. Taniguchi, H. Matsuda, H. Arima and S. Tsuchiya, J. Pharm. Pharmacol., 1995, 47, 897–900 CrossRef CAS PubMed.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- N. Smeets and T. Hoare, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3027–3043 CrossRef CAS.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed.
- M. Molina, M. Giulbudagian and M. Calderón, Macromol. Chem. Phys., 2014, 215, 2414–2419 CrossRef CAS.
- J. Bergueiro and M. Calderón, Macromol. Biosci., 2015, 15, 183–199 CrossRef CAS PubMed.
- M. Asadian-Birjand, A. Sousa-Herves, D. Steinhilber, J. C. Cuggino and M. Calderon, Curr. Med. Chem., 2012, 19, 5029–5043 CrossRef CAS PubMed.
- F. F. Sahle, M. Giulbudagian, J. Bergueiro, J. Lademann and M. Calderon, Nanoscale, 2017, 9, 172–182 RSC.
- M. Giulbudagian, F. Rancan, A. Klossek, K. Yamamoto, J. Jurisch, V. C. Neto, P. Schrade, S. Bachmann, E. Rühl, U. Blume-Peytavi, A. Vogt and M. Calderón, J. Controlled Release, 2016, 243, 323–332 CrossRef CAS PubMed.
- M. Witting, M. Molina, K. Obst, R. Plank, K. M. Eckl, H. C. Hennies, M. Calderón, W. Frieß and S. Hedtrich, Nanomedicine, 2015, 11, 1179–1187 CrossRef CAS PubMed.
- C. Gerecke, A. Edlich, M. Giulbudagian, F. Schumacher, N. Zhang, A. Said, G. Yealland, S. B. Lohan, F. Neumann, M. C. Meinke, N. Ma, M. Calderón, S. Hedtrich, M. Schäfer-Korting and B. Kleuser, Nanotoxicology, 2017, 11, 267–277 CrossRef CAS PubMed.
- A. Edlich, C. Gerecke, M. Giulbudagian, F. Neumann, S. Hedtrich, M. Schafer-Korting, N. Ma, M. Calderon and B. Kleuser, Eur. J. Pharm. Biopharm., 2017, 116, 155–163 CrossRef CAS PubMed.
- S. Hönzke, L. Wallmeyer, A. Ostrowski, M. Radbruch, L. Mundhenk, M. Schäfer-Korting and S. Hedtrich, J. Invest. Dermatol., 2016, 136, 631–639 CrossRef PubMed.
- W. Fischer, M. Calderón, A. Schulz, I. Andreou, M. Weber and R. Haag, Bioconjugate Chem., 2010, 21, 1744–1752 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- M. Gervais, A.-L. Brocas, G. Cendejas, A. Deffieux and S. Carlotti, Macromolecules, 2010, 43, 1778–1784 CrossRef CAS.
- M. Giulbudagian, M. Asadian-Birjand, D. Steinhilber, K. Achazi, M. Molina and M. Calderon, Polym. Chem., 2014, 5, 6909–6913 RSC.
- A. Kuzuya, T. Ohnishi, T. Wasano, S. Nagaoka, J. Sumaoka, T. Ihara, A. Jyo and M. Komiyama, Bioconjugate Chem., 2009, 20, 1643–1649 CrossRef CAS PubMed.
- Y. Wang, H. Chen, Y. Xiao, C. H. Ng, T. S. Oh, T. T. Y. Tan and S. C. Ng, Nat. Protocols, 2011, 6, 935–942 CAS.
- T. Osawa, K. Shirasaka, T. Matsui, S. Yoshihara, T. Akiyama, T. Hishiya, H. Asanuma and M. Komiyama, Macromolecules, 2006, 39, 2460–2466 CrossRef CAS.
- S. Saeidpour, S. B. Lohan, A. Solik, V. Paul, R. Bodmeier, G. Zoubari, M. Unbehauen, R. Haag, R. Bittl, M. C. Meinke and C. Teutloff, Eur. J. Pharm. Biopharm., 2017, 110, 19–23 CrossRef CAS PubMed.
- S. B. Lohan, S. Saeidpour, A. Solik, S. Schanzer, H. Richter, P. Dong, M. E. Darvin, R. Bodmeier, A. Patzelt, G. Zoubari, M. Unbehauen, R. Haag, J. Lademann, C. Teutloff, R. Bittl and M. C. Meinke, Eur. J. Pharm. Biopharm., 2017, 116, 102–110 CrossRef CAS PubMed.
- S. Saeidpour, S. B. Lohan, M. Anske, M. Unbehauen, E. Fleige, R. Haag, M. C. Meinke, R. Bittl and C. Teutloff, Eur. J. Pharm. Biopharm., 2017, 116, 94–101 CrossRef CAS PubMed.
- T. Kamawamura, S. Matsunami and T. Yonezawa, Bull. Chem. Soc. Jpn., 1967, 40, 1111–1115 CrossRef.
- O. H. Griffith, P. J. Dehlinger and S. P. Van, J. Membr. Biol., 1974, 15, 159–192 CrossRef CAS PubMed.
- A. Weidinger, M. Waiblinger, B. Pietzak and T. Almeida Murphy, Appl. Phys. A, 1998, 66, 287–292 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. Schafer-Korting, A. Mahmoud, S. Lombardi Borgia, B. Bruggener, B. Kleuser, S. Schreiber and W. Mehnert, ATLA, Altern. Lab. Anim., 2008, 36, 441–452 Search PubMed.
- N. Döge, S. Hönzke, F. Schumacher, B. Balzus, M. Colombo, S. Hadam, F. Rancan, U. Blume-Peytavi, M. Schäfer-Korting, A. Schindler, E. Rühl, P. S. Skov, M. K. Church, S. Hedtrich, B. Kleuser, R. Bodmeier and A. Vogt, J. Controlled Release, 2016, 242, 25–34 CrossRef PubMed.
- L. Wallmeyer, K. Dietert, M. Sochorová, A. D. Gruber, B. Kleuser, K. Vávrová and S. Hedtrich, Sci. Rep., 2017, 7, 774 CrossRef PubMed.
- W. Tang and S.-C. Ng, Nat. Protocols, 2008, 3, 691–697 CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. J. Michels, M. W. P. L. Baars, E. W. Meijer, J. Huskens and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2, 2000, 1914–1918, 10.1039/B002689L.
- P. A. L. Jacobsen, J. L. Nielsen, M. V. Juhl, N. Theilgaard and K. L. Larsen, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 173–181 CrossRef CAS.
- H. Wei and S. F. Y. Li, J. Chromatogr., A, 1998, 800, 333–338 CrossRef CAS.
- J. Nishijo and M. Nagai, J. Pharm. Sci., 1991, 80, 58–62 CrossRef CAS PubMed.
- J. H. Freed, in Spin Labeling, Academic Press, Amsterdam, 1976, pp. 53–132, DOI:10.1016/B978-0-12-092350-2.50008-4.
- S. Saeidpour, S. B. Lohan, M. Anske, M. Unbehauen, E. Fleige, R. Haag, M. C. Meinke, R. Bittl and C. Teutloff, Eur. J. Pharm. Biopharm., 2017, 116, 94–101 CrossRef CAS PubMed.
- M. G. Lebwohl, W. R. Heymann, J. Berth-Jones and I. Coulson, Treatment of skin disease: comprehensive therapeutic strategies, Elsevier Health Sciences, 2013 Search PubMed.
- B. Balzus, F. F. Sahle, S. Hönzke, C. Gerecke, F. Schumacher, S. Hedtrich, B. Kleuser and R. Bodmeier, Eur. J. Pharm. Biopharm., 2017, 115, 122–130 CrossRef CAS PubMed.
- R. Schulz, K. Yamamoto, A. Klossek, R. Flesch, S. Hönzke, F. Rancan, A. Vogt, U. Blume-Peytavi, S. Hedtrich, M. Schäfer-Korting, E. Rühl and R. R. Netz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3631–3636 CrossRef CAS PubMed.
- S. F. Ziegler, Curr. Opin. Immunol., 2010, 22, 795–799 CrossRef CAS PubMed.
- L. Wallmeyer, K. Dietert, M. Sochorova, A. D. Gruber, B. Kleuser, K. Vavrova and S. Hedtrich, Sci. Rep., 2017, 7, 774 CrossRef PubMed.
- V. Soumelis, P. A. Reche, H. Kanzler, W. Yuan, G. Edward, B. Homey, M. Gilliet, S. Ho, S. Antonenko, A. Lauerma, K. Smith, D. Gorman, S. Zurawski, J. Abrams, S. Menon, T. McClanahan, R. de Waal-Malefyt Rd, F. Bazan, R. A. Kastelein and Y. J. Liu, Nat. Immunol., 2002, 3, 673–680 CrossRef CAS PubMed.
- D. Ratman, W. Vanden Berghe, L. Dejager, C. Libert, J. Tavernier, I. M. Beck and K. De Bosscher, Mol. Cell. Endocrinol., 2013, 380, 41–54 CrossRef CAS PubMed.
- M. Surjit, K. P. Ganti, A. Mukherji, T. Ye, G. Hua, D. Metzger, M. Li and P. Chambon, Cell, 2011, 145, 224–241 CrossRef CAS PubMed.
- K. Mizuno, S. Morizane, T. Takiguchi and K. Iwatsuki, J. Dermatol. Sci., 2015, 80, 45–53 CrossRef CAS PubMed.
- A. Gerber Peter, A. Buhren Bettina, H. Schrumpf, B. Homey, A. Zlotnik and P. Hevezi, Biol. Chem., 2014, 395, 577 Search PubMed.
- L. Wallmeyer, D. Lehnen, N. Eger, M. Sochorová, L. Opálka, A. Kováčik, K. Vávrová and S. Hedtrich, J. Dermatol. Sci., 2015, 80, 102–110 CrossRef CAS PubMed.
- E. Desmet, A. Ramadhas, J. Lambert and M. V. Gele, Exp. Biol. Med., 2017, 242, 1158–1169 CrossRef CAS PubMed.
- C. Zoschke, M. Ulrich, M. Sochorová, C. Wolff, K. Vávrová, N. Ma, C. Ulrich, J. M. Brandner and M. Schäfer-Korting, J. Controlled Release, 2016, 233, 10–18 CrossRef CAS PubMed.
- S. Hönzke, C. Gerecke, A. Elpelt, N. Zhang, M. Unbehauen, V. Kral, E. Fleige, F. Paulus, R. Haag, M. Schäfer-Korting, B. Kleuser and S. Hedtrich, J. Controlled Release, 2016, 242, 50–63 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr04480a |
| This journal is © The Royal Society of Chemistry 2018 |