
Facile synthesis of 1,4-benzodiazepine-2,5-diones and quinazolinones from amino acids as anti-tubercular agents†
Seegehalli M.
Anil
ab,
Rangappa
Shobith
c,
Kuppalli. R.
Kiran
a,
Toreshettahally R.
Swaroop
d,
Ningegowda
Mallesha
b and
Maralinganadoddi P.
Sadashiva
 *a
*a
aDepartment of Studies in Chemistry, University of Mysore, Manasagangothri, Mysuru, Karnataka 570 006, India. E-mail: mpsadashiva@gmail.com
bSri Ram Chem, Plot No.: 31, JCK industrial park, Mysuru, Karnataka 570 016, India
cAdichunchanagiri Institute for Molecular Medicine, Nagamangala, Karnataka 571448, India
dDepartment of Studies in Organic chemistry, University of Mysore, Manasagangothri, Mysuru, Karnataka 570 006, India
First published on 14th November 2018
Abstract
A family of 1,4-benzodiazepine-2,5-diones and quinazolinones with diverse substituents at the C-3 position were synthesized via a novel, simple and convenient methodology using H2PtCl6 as the catalyst. The substitution at the C-3 position was varied using pre-defined amino acids as precursors. The synthesized benzodiazepines were screened for anti-mycobacterial tuberculosis (anti-TB) activity. The results revealed that the 1,4-benzodiazepine-2,5-diones displayed promising activity in comparison with their open chain precursors, which indicates that the diazepine frame is vital for their activity. The compounds 4h and 4f were found to be the lead nominees in the series with MIC values of 1.55 and 2.87 μg mL−1, respectively. A docking study was carried out on the enoyl acyl carrier protein to provide a better understanding of the mechanism of action of these compounds. Based on this study, the 1,4-benzodiazepine-2,5-dione framework is a good starting point for the development of new lead drug candidates in the treatment of multi-drug resistant tuberculosis.
Introduction
The 1,4-benzodiazepine-2,5-dione scaffold, a subset of benzodiazepinone, and its derivatives possess diverse therapeutic properties and are ubiquitous in medicinal chemistry. Functionalized benzodiazepinone structures exhibit a diverse array of biological activities and have stimulated substantial interest in the scientific community.1 Moreover, due to its physicochemical properties, this scaffold is considered a novel non-peptide peptidomimetic and exists as active core structure in many therapeutics.2 As an illustration of its significance, it is involved in the development of drugs against some of the most deadly diseases in the word, such as HIV, diabetes and obesity, by inhibiting metabolic pathways such as capsid assembly3 and monoacylglycerol acyltransferase-2.4 Also, it is present in several natural products and their derivatives are active anticholinesterase agents,5 histone deacetylase inhibitors,6 melanocortin agonists,7 glycoprotein IIb–IIIa antagonism agents,8 endothelin receptor antagonists,9 HDM2-p53 interaction antagonists,10 anxiolytic agents,11 antileishmanial agents12 and herbicidal agents.13On the other hand, quinazolinones are fused heterocycles, which are present in many naturally occurring alkaloids.14 They are extensively explored in both the synthetic and biological fields due to their diverse range of biological properties such as antimicrobial,15 antimalarial,16 anticancer,17 anti-inflammatory,18 anticonvulsants,19 antitubercular,20 antidepressant21 and anti-diabetic.22 Some important biologically active 1,4-benzodiazepine-2,5-diones and quinazolinones are given in Fig. 1. Besides, amino acid/peptide conjugated heterocyclic compounds are reported to have diverse biological activities.23 This prompted us to develop a new method involving amino acids for the construction of heterocycles.
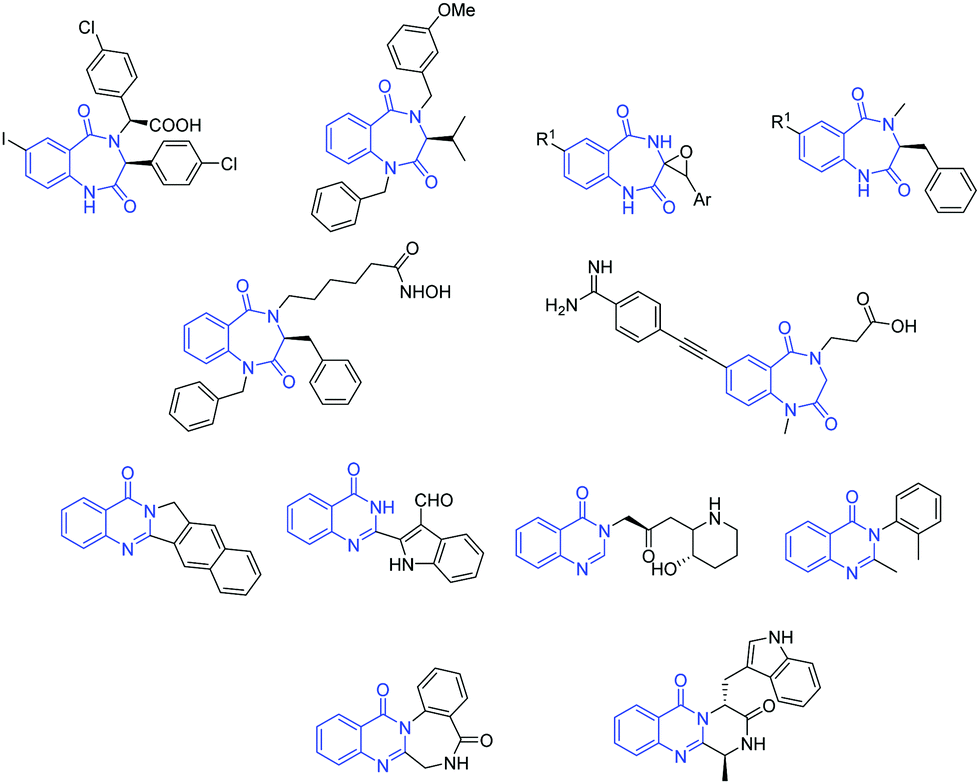 | ||
| Fig. 1 Selected structures of some biologically active compounds containing 1,4-benzodiazepine-2,5-dione and quinazolinone motifs. | ||
The development of efficient and environmentally benign synthetic methodologies for biologically important heterocycles from the same intermediate is one of the major challenges in modern organic synthesis. A literature survey revealed that three major strategies are used for the synthesis of 1,4-benzodiazepine-2,5-dione. The first is the condensation of an anthranilic acid or its derivative with α-amino acid followed by seven-membered ring closure mediated by either acid or base.24 The second is a versatile Ugi four-component condensation employing a substituted N-Boc protected anthranilic acid, aldehyde, isonitrile and amine.25 The third approach is the base-induced rearrangement of 3-aminoquinoline-2,4-diones to yield 1,4-benzodiazepine-2,5-dione.26 The first is less advantageous than the other two methods, which generally uses harsh reaction conditions, long reaction time and results in a low yield of products.
Similarly, there are several methodologies available for the synthesis of quinazolinone. Among them, the most common approach involves amidation followed by base-mediated oxidative cyclization of 2-aminobenzonitrile, 2-aminobenzoic acid or 2-aminobenzamide.27 Several related approaches are employed through the condensation of 2-aminobenzoic acid derivatives with imidate,28 polymer-bound isothioureas,29 guanidine,30 alkyl or aromatic diamines31 or aldehydes32 followed by oxidative ring closure under basic conditions or intramolecular cyclodehydration under acidic conditions. Other methods involve microwave-promoted cyclocondensation33 or the use of catalysts/reagents such as ionic liquids,34 T3P®,35 PEG-4000,36 H3PW12O40·13H2O,37 silica-supported Preyssler nanoparticles38 I2/KI,39 InCl3,40 iron,41 Pd2(dba)3/Xantphos42 and Pd(OAc)2/PPh3.43
Due to the considerable synthetic and biological importance of 1,4-benzodiazepine-2,5-dione and quinazolinone derivatives, it is necessary to discover new strategies for their synthesis. As a part of our interest in exploring the applicability of chloroplatinic acid as a catalyst in organic synthesis,44 herein, we report a novel approach for the synthesis of C-3 substituted 1,4-benzodiazepine-2,5-diones and quinazolinones from isatoic anhydride and amino acid methyl esters as starting materials in a simple two-step protocol. The anti-TB activity of the synthesized compounds with a docking study correlation is also presented.
Results and discussion
Chemistry
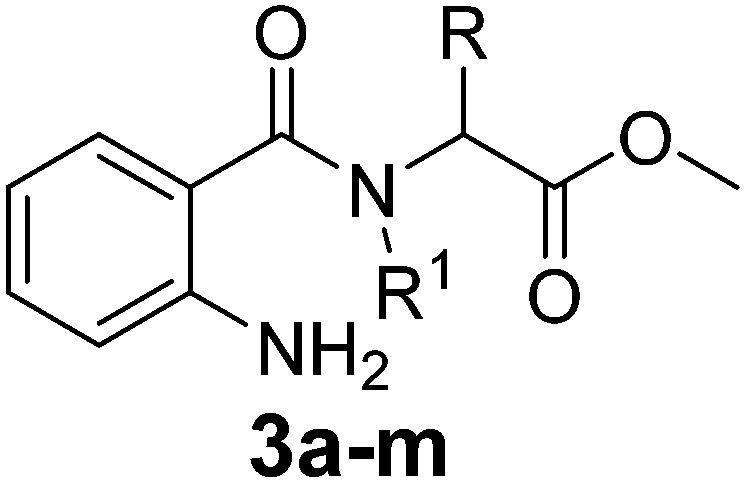
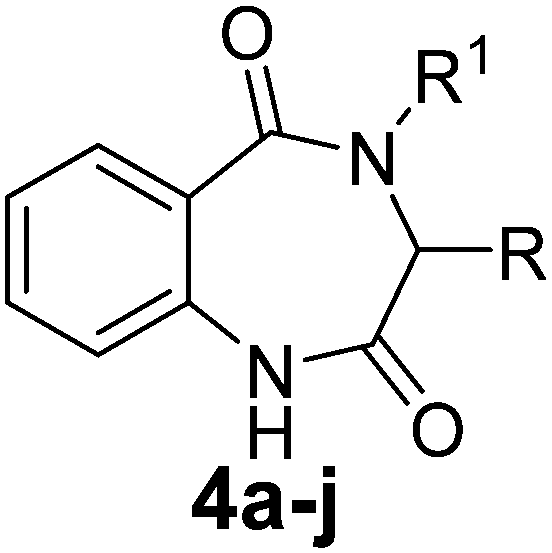
Various functionalized precursors, 2-aminobenzamido methyl ester (3a–m), required for this study were synthesized from commercially available isatoic anhydride 1 and L-amino acid methyl ester hydrochlorides (2a–m) in 61–68% yield (Scheme 1). To optimize the reaction conditions for the synthesis of 3-methyl-3,4-dihydro-1H-benzo[e][1,4]diazepine-2,5-dione (4b), initially we conducted several screening experiments using different solvents with compound 3b as a model substrate with 10 mol% H2PtCl6 as the catalyst. Amongst the solvents used, THF was found to be the best solvent at its reflux temperature with respect to reaction time and yield (30 min, 71%, Table 1a, entry 6). However, other solvents including methanol, acetonitrile, acetone, toluene and diethyl ether failed to give the product in satisfactory yields (Table 1a, entries 1–5). It is reasonable to assume that the chloroplatinic acid catalyst assisted the ring closure. Further, reaction in THF at rt gave 4b in a lower yield (Table 1a, entry 7). Thus, we selected THF as the reaction medium to explore the effect of catalyst concentration on the product yield and reaction time. It was found that an increase in catalyst concentration from 5 mol% to 20 mol% resulted in an increase in product yield and decrease in reaction time (Table 1b). Thus, yield (86%) and reaction time (30 min, Table 1) were saturated at 15 mol% of H2PtCl6 (Table 1b, entry 3). A further increase in the amount of catalyst had no effect on the reaction yield and time.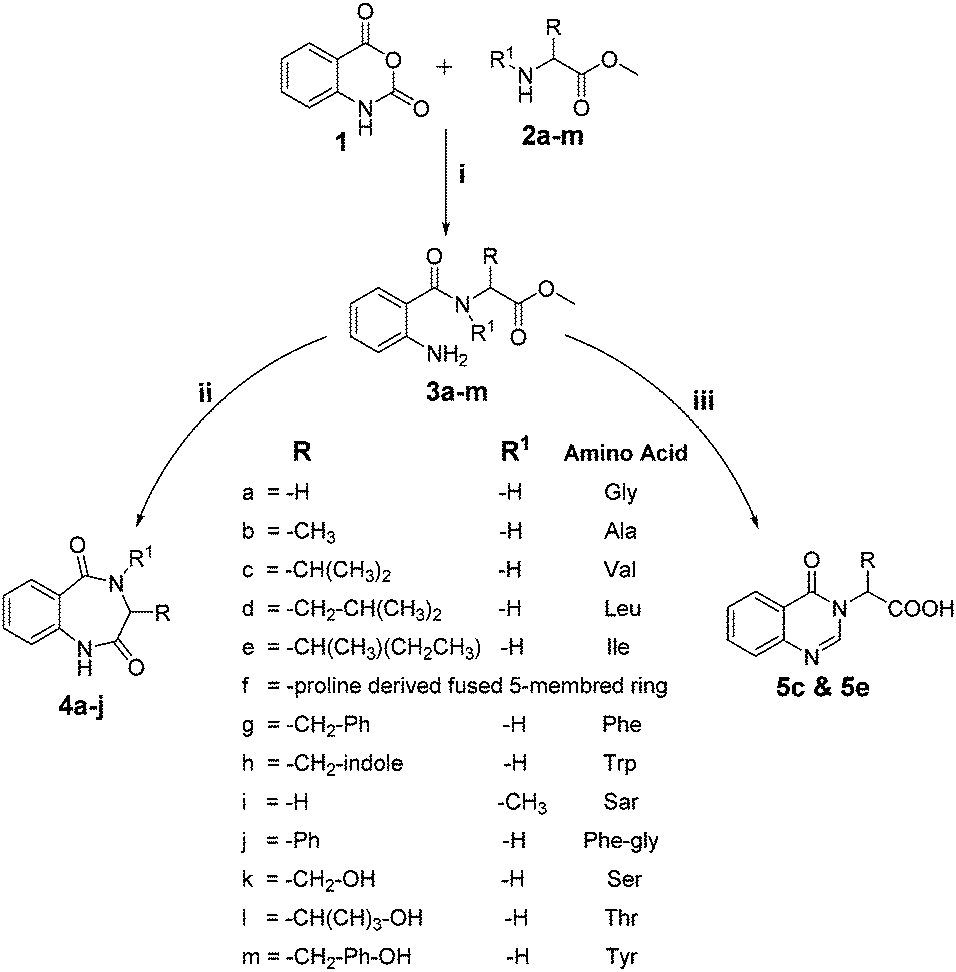 | ||
| Scheme 1 Reagents and reaction conditions: (i) Na2CO3, ACN:H2O, 4 h, and rt; (ii) 15 mol% H2PtCl6, THF, reflux, and 30 min; and (iii) (a) hydrated or anhydrous H2PtCl6, DMF, reflux, and 30 min and (b) 1 N NaOH, rt, and 30 min. | ||
| (a) Solvent optimization with 10 mol% of H2PtCl6 catalyst and 3b as the model substrate | |||
|---|---|---|---|
| S. no. | Solvent | Temperature | Yield in % |
| 1 | Methanol | Reflux | 62 |
| 2 | Acetonitrile | Reflux | 34 |
| 3 | Acetone | Reflux | 48 |
| 4 | Toluene | Reflux | 53 |
| 5 | Diethyl ether | Reflux | 29 |
| 6 | THF | Reflux | 71 |
| 7 | THF | rt | 18 |
| (b) Optimization of catalyst loading with 3b as the model substrate in THF at reflux temperature | ||
|---|---|---|
| S. no. | Catalyst load in mol% | Yield in % |
| 1 | 5 | 68 |
| 2 | 10 | 71 |
| 3 | 15 | 86 |
| 4 | 20 | 83 |
| (c) Catalyst screening with 3b as the model substrate in THF at reflux temperature | |||
|---|---|---|---|
| S. no. | Catalyst | Catalyst load in mol% | Yield in % |
| 1 | Pt metal powder (Pt) | 15 | 13 |
| 2 | PtO2 | 15 | 39 |
Later, we screened the above reaction using 15 mol% of PtO2 and platinum metal powder (Pt) with compound 3b as a model substrate in refluxing THF, which gave the product in 56% and 36% yield, respectively (Table 1c). It appeared that the efficiency of this cyclization depends on the acidic character of the catalyst; thus, H2PtCl6 was the best.
The optimized reaction conditions (15 mol% H2PtCl6 in refluxing THF) were used for the synthesis of 4a–j in varying yields (59–86%). The N-unsubstituted substrates (R1 = H), required 15 mol% of catalyst for efficient cyclization; whereas, the substituted precursor 4i (R1 = Me) required only 5 mol% for efficient cyclization. The intermediates 3k–m failed to give the respective products, which may be due to suppression of the catalytic activity of chloroplatinic acid by the –OH group of the substrates. In contrast, the reactions with moist H2PtCl6 were too slow and/or resulted in unacceptable low conversions for practical application for preparative purposes.
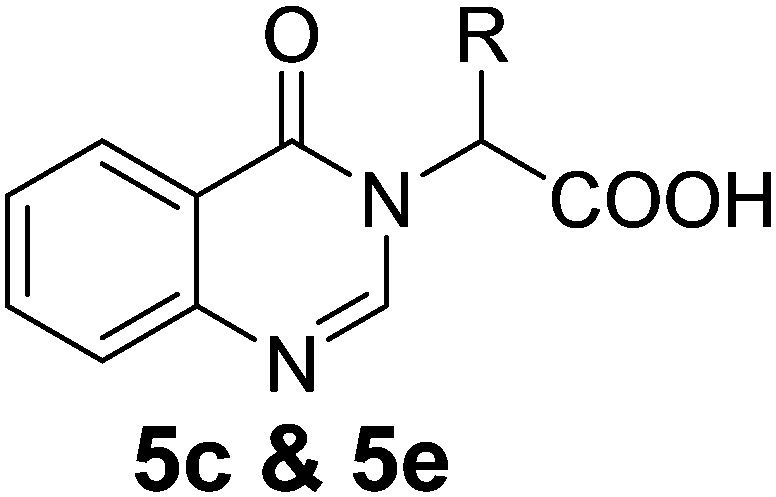
We thought an increase in reaction temperature using a high boiling point solvent would enhance the product yield. Consequently, when DMF was used as the solvent, unexpectedly, quinazolinones 5c and 5e were obtained as products in 72% and 78% yield, respectively (Scheme 1). The chemical compositions of all the compounds under investigation were well-characterized and confirmed using standard spectroscopic and analytical methods. The detailed experimental procedures and spectroscopic data of the synthesized compounds are provided in the supplementary material, which is in accordance with the assigned structures of the compounds.
Pharmacology
The preliminary anti-TB screening of all compounds (3, 4 and 5) was performed against Mycobacterium tuberculosis (Vaccine strain, MtbH37Rv (ATCC-27294)) using the microplate alamarBlue assay (MABA), as reported by Maria et al.45 The minimum inhibitory concentration (MIC) of the compounds against the MtbH37Rv strain and its correlation with the docking studies are presented in Table 2. The in vitro studies indicated that six compounds (3g, 3h, 4c, 4f, 4g and 4h) have moderate to good activity, while the rest showed lower inhibition against Mycobacterium tuberculosis (Mtb) in comparison with the standards. The MIC values of the non-peptide peptidomimetics 1,4-benzodiazepine-2,5-diones (4) were found to be better than their acyclic precursors (3), signifying that the diazepine ring is vital for the Mtb inhibition. The molecules 3g and 3h with phenyl and indole side chains were the most active among the acyclic intermediates (3), showing inhibitory concentrations of 6.08 and 5.75 μg mL−1 respectively. It was observed from structural activity relationship studies that substitution at the C-3 position enhances activity due to the increase in hydrophobicity in the molecule. The 1,4-benzodiazepine-2,5-dione 4c with an isopropyl group at the C-3 position had a high docking score of −7.631 kcal mol−1 with an MIC value of 5.75 μg mL−1, which is comparable with that of the standards. The 1,4-benzodiazepine-2,5-dione 4f derived from proline having three fused rings, structurally resembles the naturally occurring aszonalenin. This compound showed a high docking score of −7.908 kcal mol−1 and in vitro activity at 2.87 μg mL−1 against Mtb, which is comparable with that of the standards. The molecule 4g with phenyl substitution at C-3, with much more hydrophobicity, showed equal activity against Mtb (6.08 μg mL−1) as its acyclic precursor 3g, leaving no explanation for this deviation. The molecule of 4h tethered with an indole side chain derived from tryptophan was found to be the most active among the series examined, exhibiting a superior docking score of −9.221 kcal mol−1 and lowest MIC value of 1.55 μg mL−1, which is much lower than that of the standards. Notably, the 4-N-methylated benzo[1,4]diazepine-2,5-dione 4i derived from the amino acid sarcosine showed decreased biological activity, which may be due the presence of N-substitution, indicating the need for an unconstrained –CO–NH- bond in the 1,4-benzodiazepine-2,5-dione ring for better receptor interaction. The 1,4-benzodiazepine-2,5-diones derived from tryptophan (4h) and proline (4f) were found to be the most efficient in the series. Thus, the presence of active indole or fused pyrrolidine moieties on the 1,4-benzodiazepine-2,5-dione may have added a key-boost for their activity. These molecules can open a new door for the development of active anti-TB drug candidates in the future.|
|
|
|
||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | Anti-Mtb activity | |||
| MICa (μg mL−1) | Docking score (kcal mol−1) | H-Bond interactions with AAb residue | π–π stacking interaction | |||
| a MIC is defined as the minimum concentration of compound required to inhibit bacterial growth completely (0% growth). b Amino acid. c The values are the means of three determinations; and NF: not formed. | ||||||
| 3a | –H | –H | 50 | −6.508 | Tyr-158 | NF |
| 3b | –CH3 | –H | 100 | −6.696 | Tyr-158 | NF |
| 3c | –CH(CH3)2 | –H | 25 | –7.631 | Tyr-158 | NF |
| 3d | –CH2–CH(CH3)2 | –H | 25 | –8.279 | Tyr-158 | NF |
| 3e | –CH(CH3)–CH2–CH3 | –H | 25 | −8.437 | Tyr-158 | NF |
| 3f | Proline derived | 12.5 | −8.53 | Tyr-158 | NF | |
| 3g | –CH 2 –Ph | –H | 6.08 ± 0.81 | −10.51 | Tyr-158 | Phe-149 |
| 3h | –CH 2 –Indole | –H | 5.75 ± 0.43 | −11.1 | Tyr-158 | Phe-149 |
| 3i | –H | –CH3 | 50 | −7.636 | Tyr-158 | Tyr-158 |
| 3j | –Ph | –H | 25 | −9.0206 | NF | NF |
| 3k | –CH2–OH | –H | 25 | −8.16 | Tyr-158 | Phe-149 |
| 3l | –CH(OH)–CH3 | –H | 25 | −8.105 | Tyr-158 | NF |
| 3m | –CH2–Ph–OH | –H | 25 | −10.08 | Tyr-158 | Phe-149 |
| 4a | –H | –H | 100 | −6.003 | NF | Tyr-158 |
| 4b | –CH3 | –H | 25 | −6.522 | NF | Tyr-158 |
| 4c | –CH(CH 3 ) 2 | –H | 5.75 ± 0.43 | −7.972 | Tyr-158 | NF |
| 4d | –CH2–CH(CH3)2 | –H | 12.5 | −8.706 | Tyr-158 | Tyr-158 |
| 4e | –CH(CH3)–CH2–CH3 | –H | 12.5 | −8.667 | Tyr-158 | NF |
| 4f | Proline derived | 2.87 ± 0.21 | −7.908 | NF | Tyr-158 | |
| 4g | –CH 2 –Ph | –H | 6.08 ± 0.81 | −8.855 | NF | NF |
| 4h | –CH 2 –Indole | –H | 1.55 ± 0.05 | −9.221 | NF | Tyr-158 |
| 4i | –H | –CH3 | 50 | −7.248 | Tyr-158 | NF |
| 4j | –Ph | –H | 50 | −8.26 | Tyr-158 | NF |
| 5c | –CH(CH3)2 | –H | 12.5 | −9.317 | Tyr-158 | NF |
| 5e | –CH(CH3)–CH2–CH3 | –H | 12.5 | −8.863 | Tyr-158 | NF |
| Pyrazinamide | — | — | 3.12 | −4.233 | Tyr-158 | NF |
| Ciprofloxacin | — | — | 3.12 | −7.279 | NF | Phe-149 |
| Streptomycin | — | — | 6.25 | −9.63 | Tyr-158 | NF |
Docking study
A molecular docking study was performed to support the mode of action, interaction and preferred binding sites of the targeted compounds with the active sites of the bacterial protein. The enoyl acyl carrier reductase (InhA, PDB ID-2NSD) of Mtb was chosen as the bacterial protein-bearing active site. InhA is a key enzyme involving the type II fatty acid biosynthesis pathway of Mtb and has been identified as a high-confidence drug target.46 Thus, we focused and docked the target compounds using the GLIDE module of Schrodinger. The docking scores of all the targeted compounds are listed in Table 2. The docking studies showed that docking scores over the range of −6.003 to −11.1, which are comparable to the standard drug pyrazinamide and ciprofloxacin with docking scores of −4.233 and −7.279, respectively. The higher negative docking value of the targeted candidates is marked as efficient binding for the ligand. The result showed that the compounds 3g, 3h, 4c, 4f, 4g and 4h exhibited better receptor interaction with superior docking-score values of −10.51, −11.1, −7.972, −7.908, −8.855 and −9.221, respectively, which is in agreement with our observed in vitro anti-TB result. The lead compounds (4c, 4f, 4g and 4h) representing the 2D and 3D dock pictures are shown in Fig. 2. Among them, compound 4h had the highest docking score due to having one π–π staking interaction with the amino acid residues of the protein.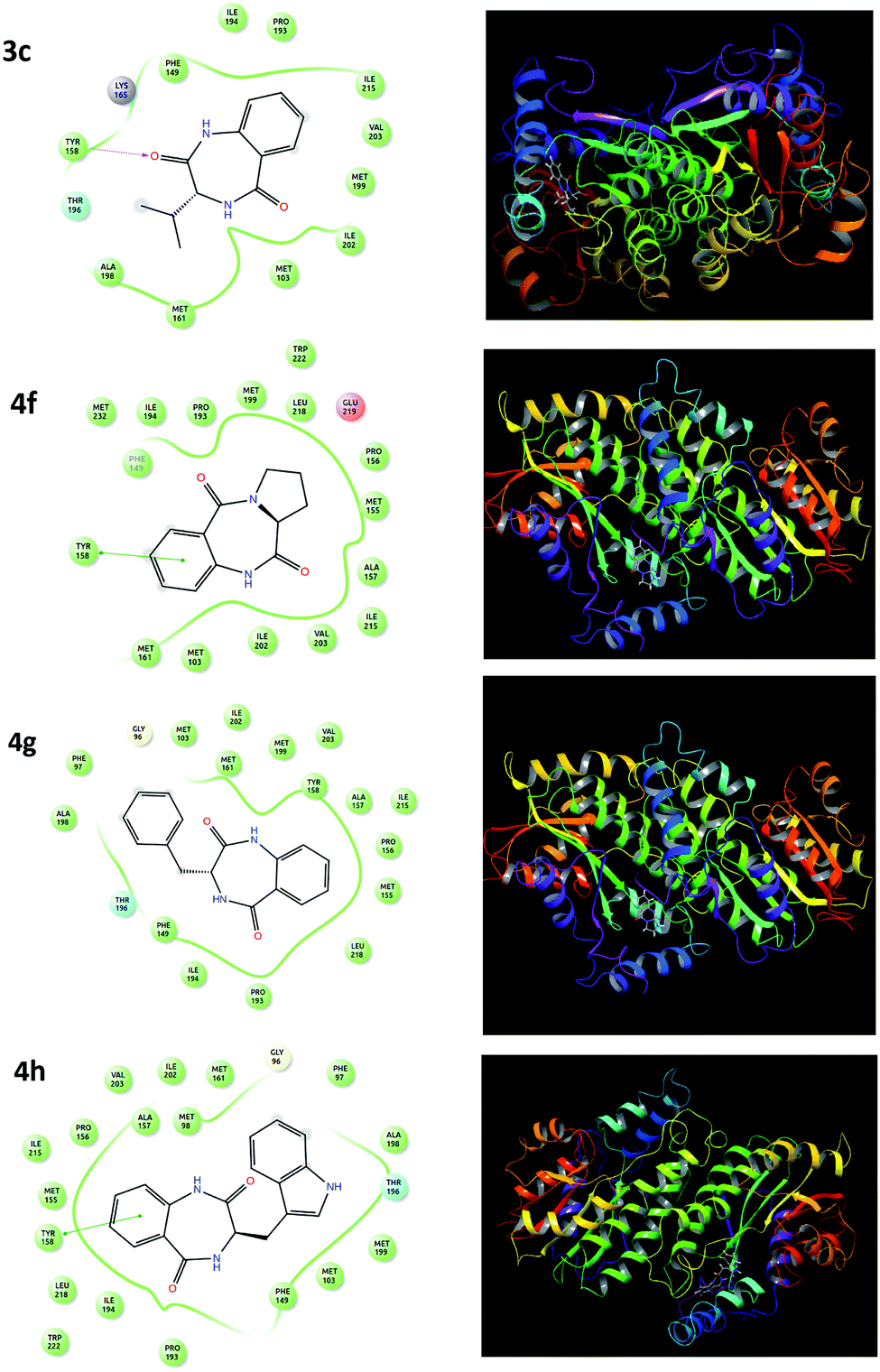 | ||
| Fig. 2 2D and 3D representations of the 4c, 4f, 4g and 4h molecular docking studies with the InhA protein. | ||
This π–π staking interaction occurred from the aromatic ring of the diazepine-2,5-dione (4h) to the Tyr-158 of the bacterial protein (InhA). Similarly, the benzodiazepinediones derived from valine (4c), proline (4f) and phenylalanine (4g) also exhibited hydrogen bonding interaction/π–π staking interaction with the amino acid residues of the protein, which are depicted in Fig. 2.
Conclusions
In conclusion, a novel approach to 1,4-benzodiazepine-2,5-dione and quinazolinone scaffolds from the same precursor, 2-aminobenzamido methyl ester (3), using chloroplatinic acid under mild reaction conditions was reported. The structure of 1,4-diazepine-2,5-dione was varied using various L-amino acid methyl ester hydrochlorides. Furthermore, we studied the anti-mycobacterial activity and docking correlations of the synthesized compounds. All the compounds were found to be active against Mtb, and among them 4f and 4h derived from proline and tryptophan, respectively, showed better activity compared to traditional anti-TB drugs. The 1,4-diazepine-2,5-dione derived from the tryptophan 4h was found to be the most active in terms of in vitro screening, which had an MIC value of 1.55 μg mL−1 with a docking score of −10.58 kcal mol−1. These amino acid-derived, biocompatible, non-peptide peptidomimetics open a new door in anti-TB drug design perspective by providing an entire range of highly specific and non-toxic boost candidates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MPS thank UGC-SAP DRS III (Grant No. UGC F-540/10/DRS-III (SAP-I) Dated 14–09-2016). Authors are grateful to Institution of Excellence, University of Mysore, Mysuru for spectral analysis and HPC Lab, University potential for Excellence, University of Mysore, Mysuru for providing docking software facility. N. Rajeev thanks UGC, New-Delhi for providing RGNF Fellowship.Notes and references
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103(3), 893–930 CrossRef CAS; (b) S. G. Smith, R. Sanchez and M. M. Zhou, Chem. Biol., 2014, 21(5), 573–583 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57(24), 10257–10274 CrossRef CAS.
- (a) P. Verdié, G. Subra, L. Feliu, P. Sanchez, G. Bergé, G. Garcin and J. Martinez, J. Comb. Chem., 2007, 9(2), 254–262 CrossRef; (b) N. Cabedo, X. Pannecoucke and J. C. Quirion, Eur. J. Org. Chem., 2005, 1590–1596 CrossRef CAS.
- (a) L. D. Fader, R. Bethell, P. Bonneau, M. Bös, Y. Bousquet, M. G. Cordingley and N. Goudreau, Bioorg. Med. Chem. Lett., 2011, 21(1), 398–404 CrossRef CAS; (b) L. D. Fader, S. Landry, S. Goulet, S. Morin, S. H. Kawai, Y. Bousquet and J. Rancourt, Bioorg. Med. Chem. Lett., 2013, 23(11), 3401–3405 CrossRef CAS.
- J. G. Barlind, L. K. Buckett, S. G. Crosby, Ö. Davidsson, H. Emtenäs, A. Ertan and G. O’Mahony, Bioorg. Med. Chem. Lett., 2013, 23(9), 2721–2726 CrossRef CAS.
- L. W. Mohamed and M. F. El-yamany, Arch. Pharmacal Res., 2012, 35(8), 1369–1377 CrossRef CAS.
- L. Loudni, J. Roche, V. Potiron, J. Clarhaut, C. Bachmann, J. P. Gesson and I. Tranoy-Opalinski, Bioorg. Med. Chem. Lett., 2007, 17(17), 4819–4823 CrossRef CAS PubMed.
- C. G. Joseph, K. R. Wilson, M. S. Wood, N. B. Sorenson, D. V. Phan, Z. Xiang and C. Haskell-Luevano, J. Med. Chem., 2008, 51(5), 1423–1431 CrossRef CAS PubMed.
- (a) R. S. McDowell, B. K. Blackburn, T. R. Gadek, L. R. McGee, T. Rawson, M. E. Reynolds and E. D. Thorsett, J. Am. Chem. Soc., 1994, 116(12), 5077–5083 CrossRef CAS; (b) B. K. Blackburn, A. Lee, M. Baier, B. Kohl, A. G. Olivero, R. Matamoros and R. S. McDowell, J. Med. Chem., 1997, 40(5), 717–729 CrossRef CAS.
- M. F. Cheng, H. M. Yu, B. W. Ko, Y. Chang, M. Y. Chen, T. I. Ho and J. M. Fang, Org. Biomol. Chem., 2006, 4(3), 510–518 RSC.
- (a) D. J. Parks, L. V. LaFrance, R. R. Calvo, K. L. Milkiewicz, V. Gupta, J. Lattanze and D. Maguire, Bioorg. Med. Chem. Lett., 2005, 15(3), 765–770 CrossRef CAS; (b) M. D. Cummings, C. Schubert, D. J. Parks, R. R. Calvo, L. V. LaFrance, J. Lattanze and T. Lu, Chem. Biol. Drug Des., 2006, 67(3), 201–205 CrossRef CAS.
- W. B. Wright Jr, H. J. Brabander, E. N. Greenblatt, I. P. Day and R. A. Hardy Jr, J. Med. Chem., 1978, 21(10), 1087–1089 CrossRef CAS.
- R. L. Clark, K. C. Carter, A. B. Mullen, G. D. Coxon, G. Owusu-Dapaah, E. McFarlane and S. P. Mackay, Bioorg. Med. Chem. Lett., 2007, 17(3), 624–627 CrossRef CAS.
- G. M. Karp, J. Org. Chem., 1995, 60(18), 5814–5819 CrossRef CAS.
- (a) C. Wattanapiromsakul, P. I. Forster and P. G. Waterman, Phytochemistry, 2003, 64(2), 609–615 CrossRef CAS; (b) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62(42), 9787–9826 CrossRef CAS.
- G. Grover and S. G. Kini, Eur. J. Med. Chem., 2006, 41(2), 256–262 CrossRef CAS.
- P. Verhaeghe, N. Azas, M. Gasquet, S. Hutter, C. Ducros, M. Laget and P. Vanelle, Bioorg. Med. Chem. Lett., 2008, 18(1), 396–401 CrossRef CAS PubMed.
- (a) P. M. Chandrika, T. Yakaiah, A. R. R. Rao, B. Narsaiah, N. C. Reddy, V. Sridhar and J. V. Rao, Eur. J. Med. Chem., 2008, 43(4), 846–852 CrossRef CAS PubMed; (b) B. Marvania, P. C. Lee, R. Chaniyara, H. Dong, S. Suman, R. Kakadiya and T. L. Su, Bioorg. Med. Chem., 2011, 19(6), 1987–1998 CrossRef CAS PubMed.
- (a) V. Alagarsamy, V. Raja Solomon, R. V. Sheorey and R. Jayakumar, Chem. Biol. Drug Des., 2009, 73(4), 471–479 CrossRef CAS; (b) V. Alagarsamy, V. Raja Solomon, M. Murugan, K. Dhanabal, P. Parthiban and G. V. Anjana, J. Enzyme Inhib. Med. Chem., 2008, 23(6), 839–847 CrossRef CAS.
- A. S. El-Azab and K. E. ElTahir, Bioorg. Med. Chem. Lett., 2012, 22(5), 1879–1885 CrossRef CAS.
- (a) U. Pandit and A. Dodiya, Med. Chem. Res., 2013, 22(7), 3364–3371 CrossRef CAS; (b) H. K. Maurya, R. Verma, S. Alam, S. Pandey, V. Pathak, S. Sharma and A. Gupta, Bioorg. Med. Chem. Lett., 2013, 23(21), 5844–5849 CrossRef CAS.
- M. Dukat, K. Alix, J. Worsham, S. Khatri and M. K. Schulte, Bioorg. Med. Chem. Lett., 2013, 23(21), 5945–5948 CrossRef CAS.
- M. S. Malamas and J. Millen, J. Med. Chem., 1991, 34(4), 1492–1503 CrossRef CAS.
- (a) M. Wang, K. P. Rakesh, J. Leng, W. Y. Fang, L. Ravindar, D. C. Gowda and H. L. Qin, Bioorg. Chem., 2018, 76, 113–129 CrossRef CAS PubMed; (b) X. Chen, J. Leng, K. P. Rakesh, N. Darshini, T. Shubhavathi, H. K. Vivek and H. L. Qin, MedChemComm, 2017, 8(8), 1706–1719 RSC; (c) X. Zhang, K. P. Rakesh, S. N. A. Bukhari, M. Balakrishna, H. M. Manukumar and H. L. Qin, Bioorg. Chem., 2018, 80, 86–93 CrossRef CAS; (d) W. Y. Fang, R. Dahiya, H. L. Qin, R. Mourya and S. Maharaj, Mar. Drugs, 2016, 14(11), 194 CrossRef PubMed.
- (a) N. Kaur and D. Kishore, Synth. Commun., 2014, 44(10), 1375–1413 CrossRef CAS; (b) D. H. Kim, J. Heterocycl. Chem., 1975, 12(6), 1323–1324 CrossRef CAS.
- (a) I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1(1), 8–21 CrossRef; (b) T. A. Keating and R. W. Armstrong, J. Org. Chem., 1996, 61(25), 8935–8939 CrossRef CAS.
- F. Křemen, M. Gazvoda, S. Kafka, K. Proisl, A. Srholcová, A. Klásek and J. Košmrlj, J. Org. Chem., 2016, 82(1), 715–722 CrossRef.
- D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61(43), 10153–10202 CrossRef CAS.
- D. J. Connolly and P. J. Guiry, Synlett, 2001, 1707–1710 CrossRef CAS.
- R. Y. Yang and A. Kaplan, Tetrahedron Lett., 2000, 41(36), 7005–7008 CrossRef CAS.
- H. J. Hess, T. H. Cronin and A. Scriabine, J. Med. Chem., 1968, 11(1), 130–136 CrossRef CAS PubMed.
- M. de Fatima Pereira, V. Thiéry and T. Besson, Tetrahedron, 2007, 63(4), 847–854 CrossRef.
- R. J. Abdel-Jalil, W. Voelter and M. Saeed, Tetrahedron Lett., 2004, 45(17), 3475–3476 CrossRef CAS.
- T. Besson and E. Chosson, Comb. Chem. High Throughput Screening, 2007, 10(10), 903–917 CrossRef CAS.
- (a) M. K. Kathiravan, R. R. Jalnapurkar, T. S. Chitre, R. S. Tamboli and K. V. Srinivasan, Green Sustainable Chem., 2011, 1(01), 12 CrossRef CAS; (b) H. R. Safaei, M. Shekouhy, V. Shafiee and M. Davoodi, J. Mol. Liq., 2013, 180, 139–144 CrossRef CAS.
- M. Desroses, M. Scobie and T. Helleday, New J. Chem., 2013, 37(11), 3595–3597 RSC.
- C. Xie, H. X. Li, M. G. Liu and M. W. Ding, Chin. Chem. Lett., 2008, 19(5), 505–508 CrossRef CAS.
- K. Ighilahriz, B. Boutemeur, F. Chami, C. Rabia, M. Hamdi and S. M. Hamdi, Molecules, 2008, 13(4), 779–789 CrossRef CAS.
- M. M. Heravi, S. Sadjadi, S. Sadjadi, H. A. Oskooie and F. F. Bamoharram, Ultrason. Sonochem., 2009, 16(6), 708–710 CrossRef CAS.
- M. Bakavoli, A. Shiri, Z. Ebrahimpour and M. Rahimizadeh, Chin. Chem. Lett., 2008, 19(12), 1403–1406 CrossRef CAS.
- N. Mulakayala, B. Kandagatla, R. K. Rapolu, P. Rao, C. Mulakayala, C. S. Kumar and S. Oruganti, Bioorg. Med. Chem. Lett., 2012, 22(15), 5063–5066 CrossRef CAS.
- H. Wang, X. Cao, F. Xiao, S. Liu and G. J. Deng, Org. Lett., 2013, 15(18), 4900–4903 CrossRef CAS.
- B. Li, L. Samp, J. Sagal, C. M. Hayward, C. Yang and Z. Zhang, J. Org. Chem., 2013, 78(3), 1273–1277 CrossRef CAS.
- Z. Zheng and H. Alper, Org. Lett., 2008, 10(5), 829–832 CrossRef CAS.
- S. M. Anil, A. C. Vinayaka, N. Rajeev, T. R. Swaroop, N. Mallesha, K. S. Rangappa and M. P. Sadashiva, ChemistrySelect, 2018, 3(7), 1999–2003 CrossRef CAS.
- M. C. Lourenço, M. V. de Souza, A. C. Pinheiro, M. D. L. Ferreira, R. S. Gonçalves, T. C. M. Nogueira and M. A. Peralta, ARKIVOC, 2007, 15, 181–191 Search PubMed.
- K. Raman, K. Yeturu and N. Chandra, BMC Syst. Biol., 2008, 2(1), 109 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed experimental procedures and characterization data for all compounds. See DOI: 10.1039/c8nj04936j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |