
Structure and catalytic properties of novel copper isatin Schiff base complexes†
Andrey S.
Smirnov
ab,
Luísa M. D. R. S.
Martins
 *c,
Dmitriy N.
Nikolaev
d,
Roman A.
Manzhos
e,
Vlad V.
Gurzhiy
b,
Alexander G.
Krivenko
e,
Kirill O.
Nikolaenko
a,
Alexander V.
Belyakov
a,
Alexander V.
Garabadzhiu
a and
Pavel B.
Davidovich‡
*a
*c,
Dmitriy N.
Nikolaev
d,
Roman A.
Manzhos
e,
Vlad V.
Gurzhiy
b,
Alexander G.
Krivenko
e,
Kirill O.
Nikolaenko
a,
Alexander V.
Belyakov
a,
Alexander V.
Garabadzhiu
a and
Pavel B.
Davidovich‡
*a
aSaint-Petersburg Technological Institute, Moskovskii av. 26, St. Petersburg, 190013, Russia
bSaint-Petersburg State University, University emb. 7/9, St. Petersburg, 199034, Russia
cCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: luisammartins@tecnico.ulisboa.pt
dResearch Institute of Experimental Medicine, Akad. Pavlova str. 12, St. Petersburg, 197376, Russia
eInstitute of Problems of Chemical Physics, Russian Academy of Sciences, 1 Akad. Semenov Av., Chernogolovka, 142432, Russia
First published on 2nd November 2018
Abstract
Isatin Schiff base ligands in combination with redox active metal ions have the potential to behave as non-innocent ligands facilitating industrially important chemical reactions. The ligand structure is easily modified by introducing substituents in three different positions, affecting the electron density distribution that was evaluated by Mulliken charge analysis and cyclic voltammetry for a range of isatin derivatives. It was noticed that coordination of these ligands to copper(II) bromide in alcohol resulted in copper reduction to Cu(I) species and alcohol oxidation. Compared to organic chemistry, the inorganic chemistry of these ligands remains poorly examined. Here, we present the structural study of sixteen novel copper complexes with mononuclear [Cu(L)2]Hal2/1 and halo-bridged binuclear [Cu2(μ-Hal)2(L)2] structures (L = isatin Schiff base ligand; Hal = Cl, Br and I). Finally, application of the above complexes for alcohol oxidation was evaluated: the complexes selectively catalyse benzyl alcohol oxidation to the corresponding aldehyde with almost quantitative yields and high selectivity.
Introduction
Copper complexes with organic ligands are widely used in organic synthesis as catalysts, e.g., in the click azide alkene/nitrile synthesis of tri- and tetrazoles; in arene coupling, selective oxidation reactions1,2etc.3 Naturally, copper is present as a cofactor in the active sites of different metal dependent enzymes, such as catechol and galactose oxidase, lysine oxidase, superoxide dismutase, N2O reductase etc.4 Researchers are constantly trying to mimic enzyme metal binding sites and improve catalytic properties by varying the surrounding ligands. A special place in the ligand space is allotted to the so-called non-innocent ligands that can, along with metal centers, participate in RedOx reactions. Ligand structure design allows the properties of the formed complexes to be finely tuned. Many studies have focused on Schiff base derivatives of a naturally existing non-innocent ligand pair – catechol/quinone.5 Jacquet et al. reported iminobenzoquinone copper complexes that actively promoted the oxidation of alcohols,6 thus mimicking alcohol oxidase activity. Another natural compound resembling the quinone structure – isatin – remained innocent in the sense of studies by inorganic chemists. Despite its popularity caused by a plethora of biological activities,7–9 reports on isatin imine (Schiff base) coordination chemistry remain scarce.10 Earlier studies evaluated copper(II) isatin Schiff base complexes [Cu(isapn)](ClO4)2 as active catalysts towards carbohydrate oxidation via intermediate reactive oxygen species formation.11 Taking together the data mentioned above, we decided to prepare copper complexes with different isatin Schiff base derivatives (ISBDs) and to explore their structural features and their potential to catalyze the oxidation of alcohols.Results and discussion
Synthesis and structure
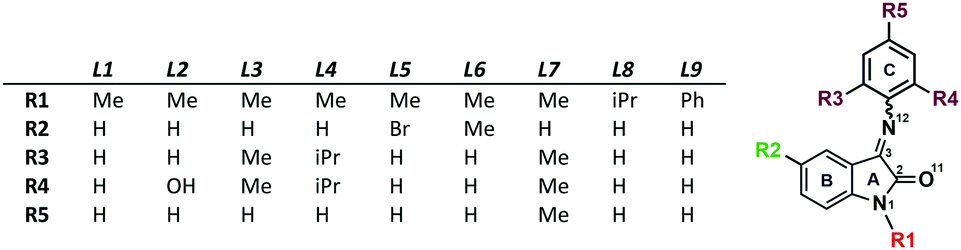 | ||
| Fig. 1 Isatin Schiff bases used in the current study. | ||
On the other hand, in accordance with the anisotropy of current induced density (ACID),13,14 the ISBD system is highly conjugated (Fig. 2 and Fig. S32, ESI†) and different derivatization positions could be used to tune the electronic properties of these ligands. This assumption finds proof in the analysis of the Mulliken charges on the heavy atoms of the ligands (Table S5, ESI†). Derivatization of ring “C” has no feasible effect on the carbonyl oxygen charge, while alkylation of the amide nitrogen with iPr decreases the charge on oxygen and arylation with a Ph substituent increases the negative charge; in turn, the charge on the imine nitrogen is significantly changed upon substitution of ring “C”; ring “B” modifications do not really effect the electron density distribution on the donor atoms.
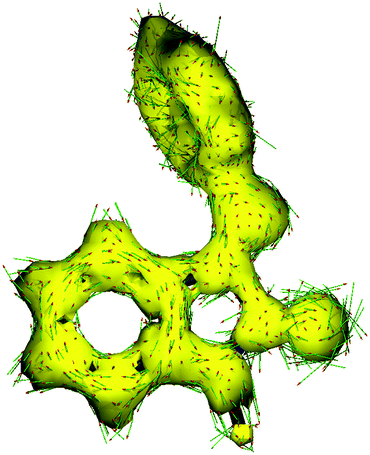 | ||
| Fig. 2 ACID isosurface for L1 at an isosurface value of 0.05 a.u. Current density vectors are plotted onto the ACID isosurface to indicate dia- and paratropic ring currents. The magnetic field vector is orthogonal relative to the ring plane and points upward. | ||
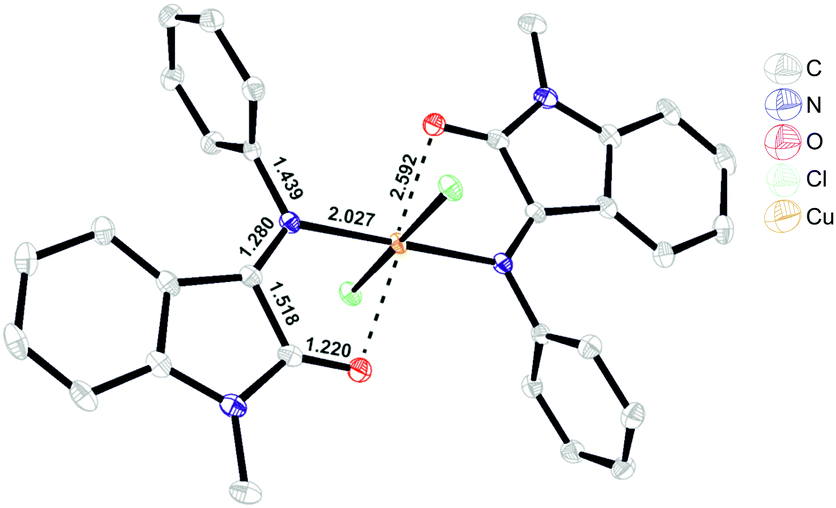 | ||
| Fig. 3 ORTEP representation of the X-ray resolved molecular structure of [Cu(L1)2]Cl2 (C1).§ | ||
Despite the fact that coordination to the copper(II) center proceeds via carbonyl and imine groups of L1, Cu–N and Cu–O bond formation had almost no effect on the ligand C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO bond lengths, but it did affect the carbonyl group vibration frequency that shifted from 1734 cm−1 (L1) to 1728 cm−1 (C1) and the imine frequency that shifted from 1653(L1) to 1658(C1) cm−1.
N and CO bond lengths, but it did affect the carbonyl group vibration frequency that shifted from 1734 cm−1 (L1) to 1728 cm−1 (C1) and the imine frequency that shifted from 1653(L1) to 1658(C1) cm−1.
As potential alcohol oxidation by a complex would probably include the reduction of copper(II), an attempt to obtain a Cu(I) complex with L1 was performed. Previous studies on the homologues acenaphthoquinone and camphor hydrazone ligands reported the formation of two structurally distinct Cu(I) complexes:18,19 mono [Cu(2,6-iPrC6H3-BIAO)]Cl and binuclear [Cu2(μ-Hal)2(iPrNNC10H14O)2] species. Thus, four types of copper complexes could potentially be formed (Scheme 1).
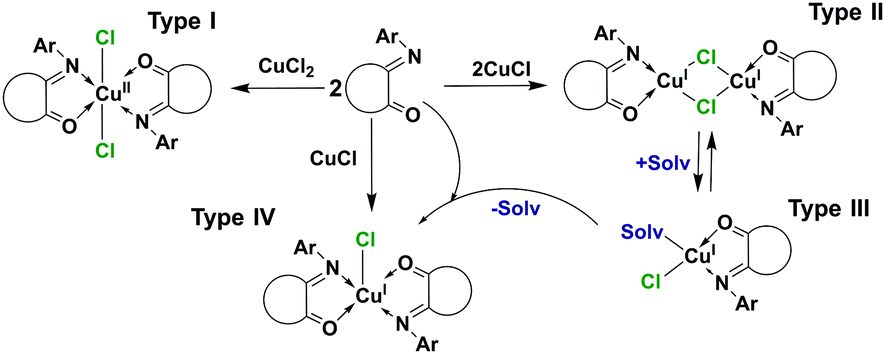 | ||
| Scheme 1 Possible types of copper complexes with ISBDs. | ||
A heterogeneous reaction between solid CuCl and a methanolic solution of ligand L1 resulted in the formation of two products: a brown MeOH soluble sample and a green sample soluble in MeCN. ESI solution analysis showed that the green crystals correspond to [Cu(L1)]+ particles with an m/z ratio of 299.02, while the red crystals correspond to bischelate {[Cu(L1)2]Cl}+ with chloride unbound m/z = 535.12 and bound m/z = 570.09 species. To avoid heterogeneous reactions, Cu(I) ion was generated in situ by the reduction of Cu(II) with isopropyl thiol (i-PrSH) (C1 reacts stoichiometrically with thiols) (Fig. S1, ESI†). The ESI analysis of the [Cu(L1)2]Cl2 + RSH reaction solution confirmed the presence of the same peaks as in the case of the heterogeneous reaction. Attempts to crystallize the individual products in a way that would allow X-ray suitable crystals to be obtained failed. Substitution at the 2nd and 6th positions of the ring “C” of the ISBD structure could result in steric hindrance between the halogen and ligand atoms and shift the reaction to monochelate products. To validate this assumption, complexes with 2,6-dimethyl (L3), 2,6-diisopropyl (L4) and 2,4,6-trimethyl derivatives (L7) of L1 were prepared according to Scheme 2.
 | ||
| Scheme 2 Complex metal center reduction with thiol. | ||
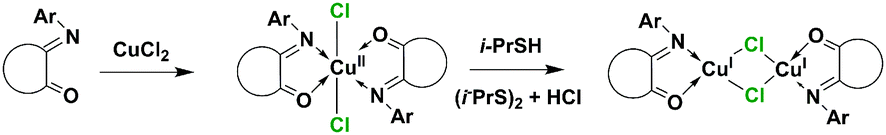
Only with the L7 ligand X-ray quality crystals of complex C2 were obtained and characterized. The XRD analysis confirmed the binuclear monochelate structure of the C2 complex (Fig. 4) with a Cu′⋯Cu distance of 2.66 Å, which is 0.2 Å shorter than in an analogous [Cu2(μ-Hal)2(PhenoxBQ)2] complex described by Speier et al.20 In the binuclear species [Cu2(μ-Hal)2(L7)2], the carbonyl vibration frequency shifts from 1729 cm−1 to 1719 cm−1 and the imine vibration at ∼1650 cm−1 almost totally disappears. To compare the effect of a bridging halogen on the complex structure, complexes C3 and C14 were prepared from CuBr and CuI salts and L7. The formed compounds resembled the binuclear structure of [Cu2(μ-Hal)2(L7)2] described above with no significant geometry differences (Fig. 4).¶
 | ||
| Fig. 4 ORTEP representation of type II binuclear complex [Cu2(μ-Hal)2(L7)2], Hal = Cl (C2), Br (C7) and I (C14) structures. | ||
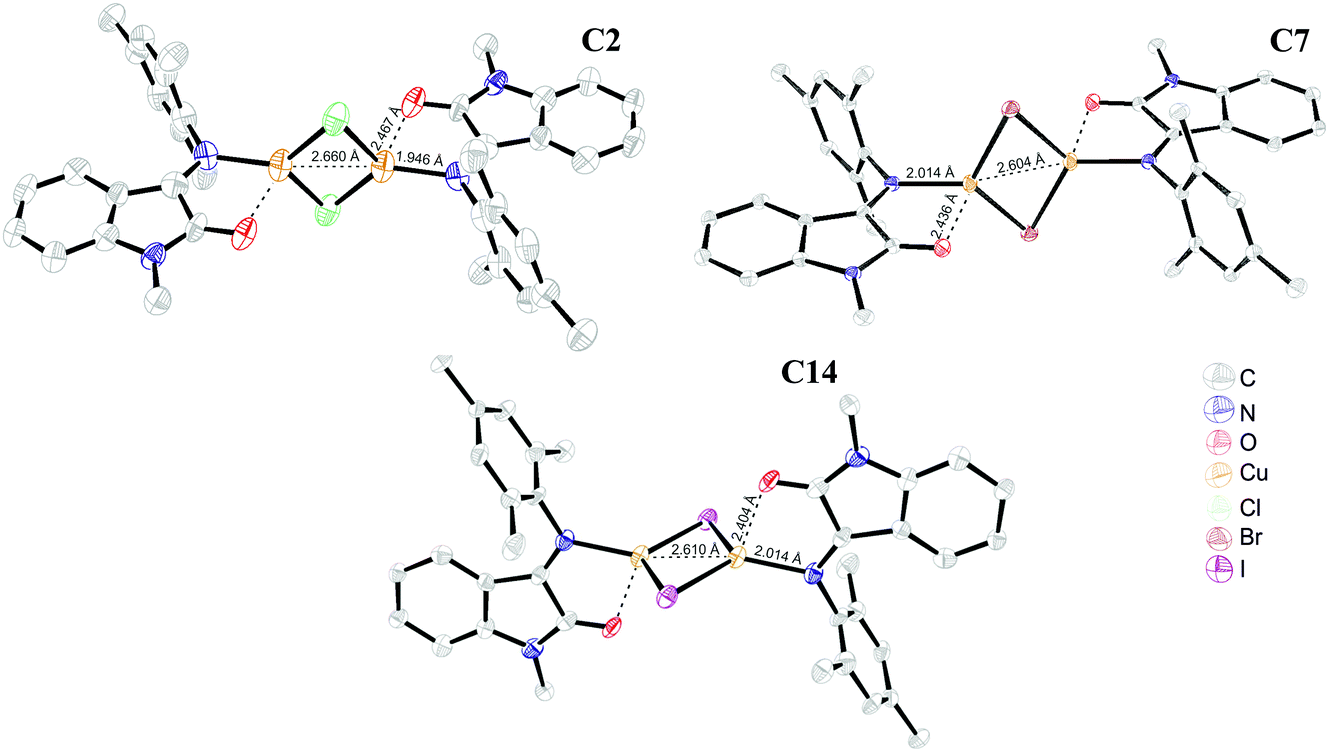
Copper(I) bromide reaction with the more electronically saturated and sterically demanding ligand L4 resulted in the isolation of a green crystalline product, [Cu2(μ-Br)2(L4)2]. Interestingly, the crystal consists of molecules in two separate conformations differing in Cu⋯Cu distance: first, a C5 molecule with a metal–metal distance of 2.875 Å and a second one, C5′, with a much longer 3.176 Å Cu⋯Cu distance (Fig. 5). In all the other complexes, the Cu⋯Cu distances (Table S1, ESI,†) are shorter than the sum of copper van der Waals radii (= 2.8 Å), which might be evidence of closed shell metallophilic d10–d10 interactions;22 in the case of the elongated metal–metal distance, these interactions are most probably lost because of the strong influence of neighboring molecules that form short contacts with bridging bromide ions. The bond lengths in the coordinated ligand are almost identical for both conformations and do not differ from the ones in unbound ligand (Table S1, ESI†), suggesting that no change in the ligand or metal redox state occurred.
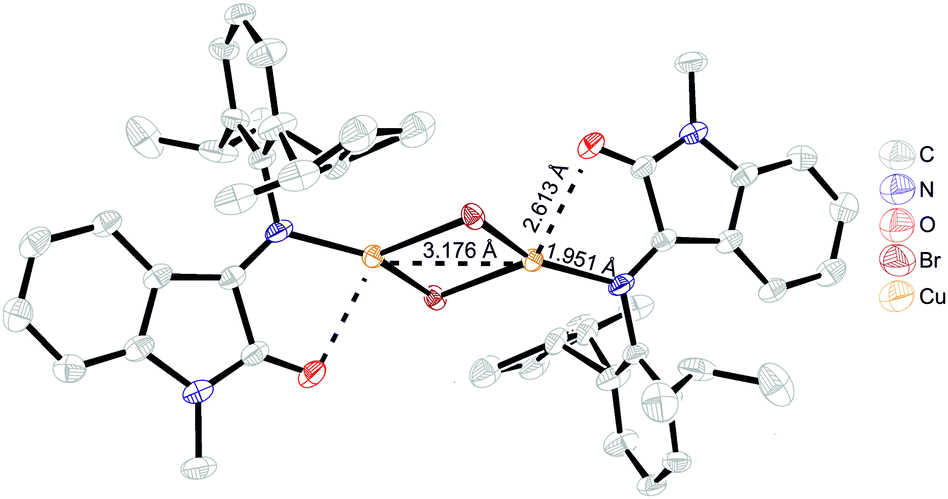 | ||
| Fig. 5 ORTEP representation of type II binuclear complex (C5′). | ||
The reaction of CuBr with the dimethylated derivative L3 resulted in the formation of the acetonitrile soluble complex C4. Slow MeCN evaporation resulted in XRD quality crystals that confirmed the binuclear structure for product, [Cu2(μ-Br)2(L3)2], with a Cu⋯Cu distance of 2.739 Å. The reaction of ligand L3 with CuI leads to the formation of binuclear complex species C16 with a Cu⋯Cu distance of 2.754 Å that is close to the bromide analogue.
Interestingly, PhenoxBQ ligand reacted with CuCl2 to form the binuclear halo-bridged paramagnetic Cu(II) complex [Cu2(μ-Cl)2(PhenoxBQ)]Cl2.20 Here, reactions of methanol dissolved CuBr2 with L3 resulted in the reduction of copper(II) and formation of the binuclear type II copper(I) complex C4 [Cu2(μ-Br)2(L3)2]. Bond distances (N–CAr 1.445 Å, CN 1.294 Å, C–C 1.512 Å and CO 1.218 Å) in the coordinated ligand show almost no difference with the free non-coordinated form (N–CAr 1.422 Å, CN 1.274 Å, C–C 1.527 Å and CO 1.215 Å), which points to the fact that the ligand “redox” state is not changed upon copper center reduction (Fig. S24, ESI†). The same behavior and complex [Cu2(μ-Br)2(Ln)2] formation was observed when ligands L4 and L7 were used (C5 and C3, accordingly). As these redox reactions proceeded without any external reducing agent, two possible reduction processes could occur: copper(II) catalyzed oxygen mediated alcohol transformation to aldehyde,23,24 like in the case of the homologue bis(o-iminosemiquinonato) Cu(II),25 or bromide ion could reduce the ligand molecule to form anion L− that could transfer an electron to the Cu(II) ions, as reported by Mukherjee.26
Having observed the spontaneous reduction of CuBr2 (complexes C3, C4 and C5), a similar reaction of CuBr2 with L1 in methanol solution was performed. As expected, the reaction also proceeded with metal center reduction and the formation of a [CuI(L1)2]Br trigonal-monopyramidal C9 type IV complex.
Unexpectedly, the reaction between CuBr and L1 resulted in the isolation of a bischelate type IV mononuclear [Cu(L1)2]Br complex (C9), but not a halo-bridged complex. Compared to C1, Cu(I) complex formation resulted in the elongation of the Cu–O distance from 2.592 to 2.754 Å and shortening of the Cu–N bond from 2.027 to 1.954 Å (Fig. 6). Upon reduction, bonds between the imine nitrogen and copper strengthen, but the bonds with carbonyl oxygen weaken compared to the Cu(II) species.
 | ||
| Fig. 6 ORTEP structural representation of type IV complexes C9–C12. | ||
Different behavior was observed in the case of L5 and L6 ligands that have substituents in the 5th position of ring “B”. Complexes with these ligands were prepared to evaluate the possible effects of donor (Me) and acceptor (Br) substituents in ring “B” on the structural and electronic features of the obtained compounds. Compared to L1, the bromide-bridged complexes C6 and C7 were obtained from CuBr and ligands, but not [Cu(Ln)2]Br. Preferential formation of the binuclear species [Cu2(μ-Br)2(Ln)2] could be the reason for the steric effects caused by the bromine and methyl substituents. No substantial difference between C6 and C7 complex core geometry was observed (Table 1). These results generally correspond to previous observations that substitutions in an isatin benzene ring have no serious impact on the electronic parameters of the isatin dione fragment,27,28 but could sterically affect reactivity.16
| μ-X | Cu⋯Cu | Cu–O | Cu–N | CN |
CO |
C2–C3 | |
|---|---|---|---|---|---|---|---|
| C2 (L7) | Cl | 2.660 | 2.467 | 1.946 | 1.261 | 1.237 | 1.514 |
| C3 (L7) | Br | 2.604 | 2.436 | 1.979 | 1.289 | 1.210 | 1.517 |
| C4 (L3) | Br | 2.739 | 2.418 | 1.992 | 1.294 | 1.218 | 1.520 |
| C5 (L4) | Br | 2.835 | 2.689 | 1.948 | 1.282 | 1.223 | 1.518 |
| C5′ (L4) | Br | 3.176 | 2.613 | 1.951 | 1.295 | 1.215 | 1.527 |
| C6 (L5) | Br | 2.548 | 2.429 | 1.974 | 1.287 | 1.215 | 1.519 |
| C7 (L6) | Br | 2.561 | 2.434 | 1.979 | 1.288 | 1.222 | 1.525 |
| C14 (L7) | I | 2.610 | 2.404 | 2.014 | 1.284 | 1.226 | 1.526 |
| C15 (L1) | I | 2.527 | 2.513 | 1.995 | 1.289 | 1.219 | 1.524 |
| C16 (L3) | I | 2.724 | 2.377 | 2.041 | 1.283 | 1.224 | 1.515 |
The copper iodide complex C15 was obtained from CuI and L1 in acetonitrile. XRD analysis of magenta colored crystals confirmed the formation of a binuclear type II structure, [Cu2(μ-I)2(L1)2].|| Switching to the less electronically saturated L1 ligand resulted in shortening of the metal-to-metal distance compared to other μ-I complexes, such as [Cu2(μ-I)2(L7)2] C14 and [Cu2(μ-I)2(L7)2] C16 (Table 1), but it was close to the bromide bridged C6 and C7 ones. Thus, generally, the Cu⋯Cu distance is not significantly dependent on the halogen bridge.
Similar to the described reduction of Cu(II) by thiols, the reaction between the sulfate complex of L1 [CuII(L1)2(H2O)2]SO4 and KI was performed according to Scheme 3.
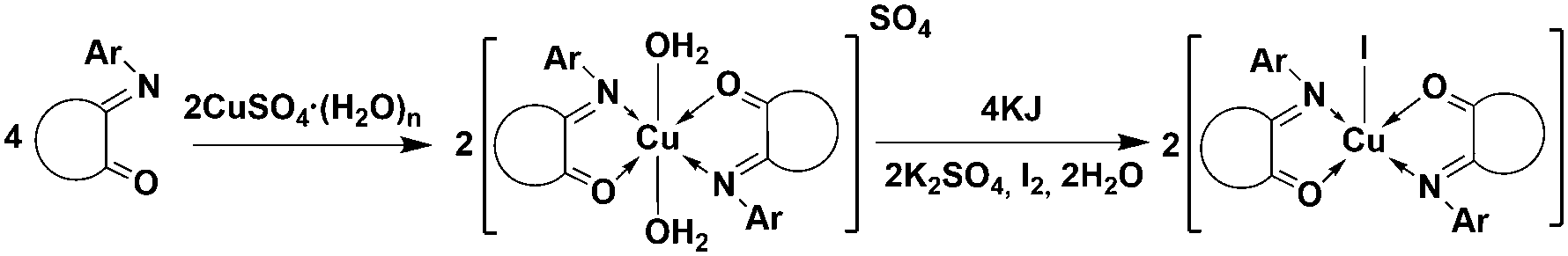 | ||
| Scheme 3 Cu(I) bischelate iodide complex formation. | ||
Mass-spectrum analysis of the reaction solution showed a major peak with m/z = 535.13 corresponding to the dissociated [Cu(L1)2]+ form of a bischelate type IV complex, but no fragments of binuclear species were detected. Solvent evaporation resulted in crude powder product isolation, and the consequent crystallization proceeded with the formation of good quality crystals of complex C10 (Fig. 6).
As in solution, the complex is most probably present as a dissociated salt, the halogen binding effect on the Cu(I) complex geometry was evaluated by obtaining a complex with a non-coordinating counter BF4− ion. The reaction of [Cu(MeCN)4](BF4) with L7 resulted in heteroleptic complex [Cu(L7)2(MeCN)](BF4) (C11) formation. As confirmed by the crystal structure data, in the absence of a halogen atom, the coordination site is occupied by the solvent MeCN molecule (Fig. 6), and this “substitution” results in Cu–O bond elongation to 2.584 Å with no feasible effect on the Cu–N bond (see Table 2).
| X | Cu–O | Cu–N | CN |
CO |
C2–C3 | |
|---|---|---|---|---|---|---|
| a Cu–Br distance of 2.387 Å is shorter than the ionic radius of the ions. | ||||||
| C1 (L1) | Cl | 2.592 | 2.027 | 1.280 | 1.220 | 1.518 |
| C8 (L6) | Cl | 2.518 | 2.036 | 1.275 | 1.220 | 1.522 |
| C9 (L1) | Br | 2.754 | 1.954 | 1.278 | 1.206 | 1.519 |
| C10 (L1) | I | 2.728a | 1.953 | 1.289 | 1.221 | 1.512 |
| C11 (L7) | BF4 | 2.584 | 1.980 | 1.284 | 1.214 | 1.512 |
| C12 (L2) | Br | 2.675 | 1.953 | 2.277 | 2.215 | 1.519 |
A copper–oxygen bond length of 2.58 Å in complex C11 is close to the one in the type I Cu(II) species. Possibly, halogen coordination to Cu(I) stabilizes a lower oxidation state, while the non-coordinating BF4− anion is less likely to do that. According to the theoretically optimized geometry of the reduced ligand L− (Fig. S24, ESI†), electron acceptance leads to carbon–carbon (C2–C3) bond shortening and that is reflected in the C11 structure, where the ligand carbon–carbon (C2–C3) distance is shortened by 0.02 Å. Electron relocation to the ISBD ligand can occur from Cu(I), pointing to electron density conjugation between metal and ligand. A similar tendency was observed in the case of copper(I) complex C12 with ligand L2 (hydroxyl in ring “C”), where the Cu–O bond distance of 2.675 Å is significantly shorter than in the analogous C9 complex (Table 2 and Fig. 6). The bromide–copper(I) bond of 2.435 Å is attenuated compared to complex C9 (2.387 Å) because of two hydrogen bonds between the hydroxyl groups of the ligands and Br− anion. Lower charge compensation on Cu(I) leads to electron density redistribution and shortening of the carbon–carbon (C2–C3) bond by 0.013 Å.
Alcohol oxidation catalysis
Benzyl alcohol (PhCH2OH), an important industrial raw-material, was used as a model substrate to investigate the potential application of copper complexes C1, C7, C8 and C9 as heterogeneous catalysts for the oxidation of primary alcohols. Under ambient conditions, the complexes promoted aerobic alcohol oxidation with moderate conversion rates of ∼22% and thus were further evaluated using microwave (MW) assisted alcohol oxidation assay. The oxidation conditions were optimized relative to several parameters such as reaction time and temperature, copper catalyst and oxidant amount, and the presence of additives: TEMPO radical, a well-known promoter in the catalytic oxidation of alcohols,29 or 0.1 M aqueous potassium carbonate solution, recognised to facilitate the deprotonation of alcohol.30 The experiments showed that the complexes selectively catalyse the MW assisted oxidation of benzyl alcohol to benzaldehyde with tert-butyl hydroperoxide (tBuOOH, TBHP),31 under low irradiation power and in the absence of any solvent or promotor. Under the adopted conditions, the presence of TEMPO led to a strong inhibiting effect of the catalytic activity of all compounds, whereas K2CO3 had almost no effect (Fig. 7).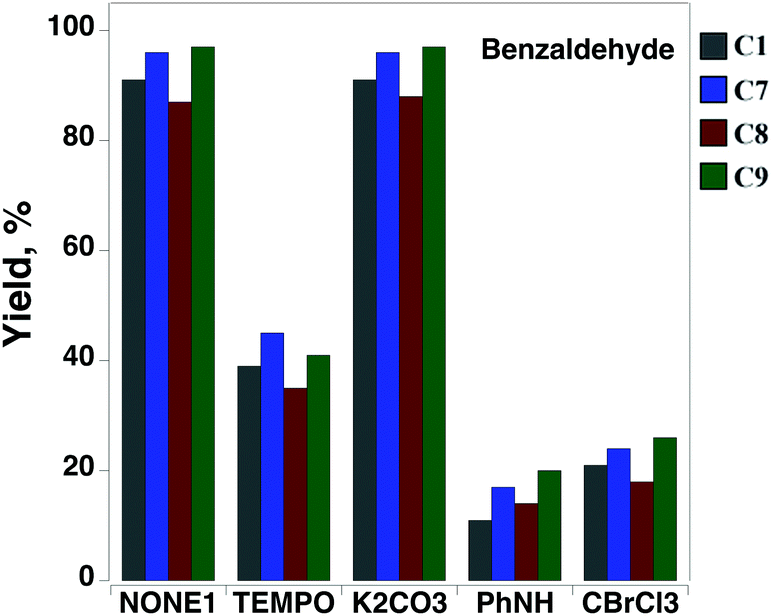 | ||
| Fig. 7 Effect of additives on the oxidation of PhCH2OH catalysed by C1 and C7–C9. | ||
The results obtained for benzyl alcohol oxidation are presented in Table 3. Most probably, the mononuclear complex structure is maintained in the solution and during the catalytic reaction, while binuclear type II complex C7 can undergo dissociation to the mononuclear type III species [Cu(L)]Hal (Scheme 1), similar to those obtained by Anga, [Cu(2,6-iPrC6H3-BIAO)]Cl.18
| Complex | Yield, % | TOF, h−1 | Selectivity, % |
|---|---|---|---|
| — | 0.9 | — | — |
| CuBr | 16 | 1.6 × 102 | 55 |
| CuCl | 21 | 2.1 × 102 | 73 |
| CuCl2 | 13 | 1.3 × 102 | 64 |
| C1 | 91 | 9.1 × 102 | 97 |
| C7 | 96 | 9.6 × 102 | 98 |
| C8 | 87 | 8.7 × 102 | 97 |
| C9 | 97 | 9.7 × 102 | 97 |
All complexes were found to be much more active than their precursor salts (Table 3), suggesting the favourable involvement of ligands in the metal-assisted steps of this catalytic oxidation reaction. The ligands themselves are not able to catalyse the benzyl alcohol to benzaldehyde oxidation under the same reaction conditions (yields in the 0–2% range). Accordingly, the addition to the reaction mixture of Ph2NH or CBrCl3, well known oxygen- and carbon-radical traps, respectively,32,33 led to a significant yield drop of over 80% compared to the reaction carried out under the same conditions but in the absence of a radical trap (Fig. 7). Thus, the oxidation of benzyl alcohol by the complexes proceeds via a radical mechanism involving carbon and oxygen-centred radicals.
One of the important parameters for any catalyst is recycling capacity. Complex C9 was tested for up to 5 consecutive cycles to evaluate its catalyst recycling capacity (Fig. 8). A slight (6%) decrease in product yield was observed after the second cycle, but on the following consecutive cycles, much more pronounced values were obtained, as depicted in Fig. 8, suggesting the deactivation of the active species by the reaction medium (or during the wash steps). However, the recovery of the catalyst might not be an issue due to its low cost. As the low recycling capacity of a novel catalyst may arise from the irreversibility of oxidation/reduction cycles of the complex or ligands, their redox behavior was studied.
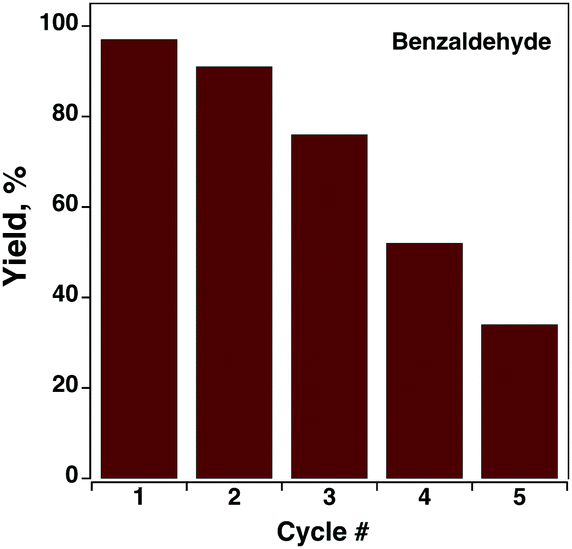 | ||
| Fig. 8 Benzaldehyde yield as a function of C9 catalytic cycle. | ||
Ligand and complex redox behavior
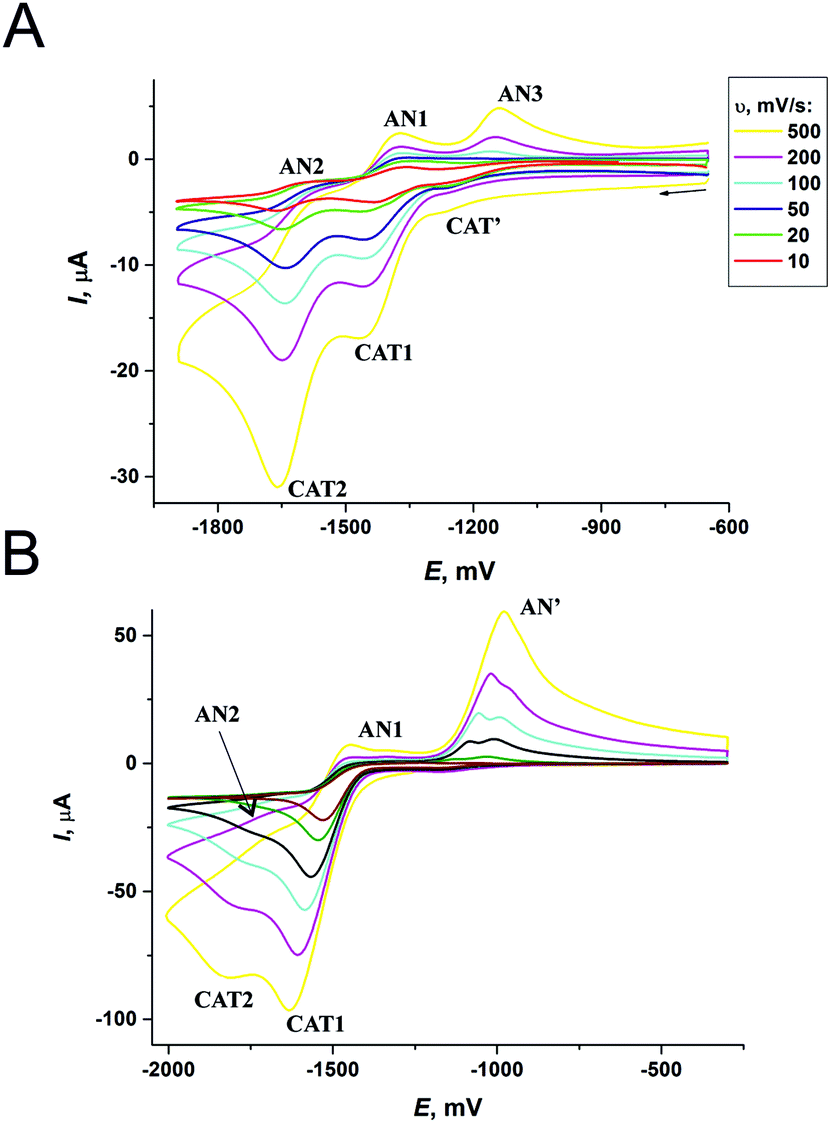 | ||
| Fig. 9 CVs measured for C1 complex (A) and L1 ligand (B); potential values vs. Fc+/Fc. | ||
| e transfer | First | Second |
|---|---|---|
| E C, V | −1.44 | −1.64 |
| E A, V | −1.37 | −1.57 |
| ΔE, V | 0.07 | 0.07 |
| E 0, V | −1.41 | −1.61 |
The difference between the cathodic and anodic peak potentials is about 70 mV and attests that the 1st and 2nd electron transfers occur electrochemically reversibly (quasi-reversibly). The values of anodic peak current are lower than the cathodic ones, which is especially evident in the case of the second electron transfer. That may be the reason for the unstable product formation that partially decomposes during the experiment and results in the reduction of peak AN2. Peak AN3 resembles, most probably, the oxidation of intermediates formed after the second electron addition.** Thus, the general scheme for the complex RedOx behavior could be represented as follows:
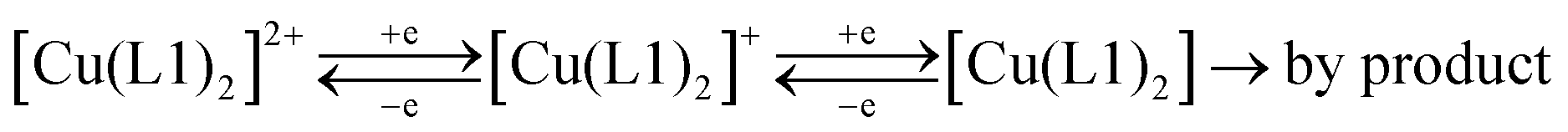
| L1 | L3 | L4 | L6 | L7 | L8 | L9 | |
|---|---|---|---|---|---|---|---|
| a Reaction of Cu(I) with the bulkier phenyl substituent in the 1st position of L9 resulted in polymeric I-bridged (Cu⋯Cu 4.468 Å) complex C13 (Fig. S25, ESI) formation close to the one obtained in the reaction between CdBr2 and L1.15 | |||||||
| E C, V | −1.62 | −1.70 | −1.73 | −1.60 | −1.76 | −1.70 | −1.57 |
| E 0, V | −1.57 | −1.66 | −1.69 | −1.56 | −1.72 | −1.66 | −1.52 |
In the potential region of −1500 to −2000 mV and at mid scan rates of 10–100 mV s−1, the only cathodic peak CAT1 is observed (Fig. 9B). ΔE increases from 80 to 120 mV with the increase of scan rate from 200 to 500 mV s−1.
These data suggest that the first electron transfer occurs by the EC-mechanism with reverse electrochemical and irreversible chemical steps.36 Below scan rates of ν < 200 mV s−1, the product of electron addition to the ligand (L1−) virtually completely manages to degrade with the formation of byproducts (P). At the higher rates of ν, the reasonable part of L− can be further oxidized and the shape of the CV curve transforms to that characteristic of a quasi-reversible process with a formal potential of −1570 mV for L1. At E >−1200 mV, the anodic peak AN′ is present, but the corresponding cathodic peak is not observed and this indicates that the process is non-reversible. Thus, the peak AN′ corresponds to the oxidation of the products formed during the decomposition of L1−. At the more negative potentials, the peak CAT2†† and the corresponding anodic peak AN2 appear on the CV curve. CAT2 grows with the increase in potential scan rate and thus, most probably, corresponds to the second step of L1 reduction. During a slow potential scan, L1− manages to majorly transform into product P, and peaks CAT2 and AN2 do not appear. Finally, we can suggest that the ligand RedOx processes proceed via the ECE-mechanism.
As can be seen from Table 5, the introduction of a substituent in ring “B” (L6) has a very modest effect on reduction potential. Propylation in ring “A” increases the reduction potential and the opposite effect is observed for N-arylated modification. The introduction of ring “C” donor substituents in all cases (L3, L4, and L7) increases the reduction potential. These tendencies are in agreement with the Mulliken charge analysis distribution discussed above. Modification of ligand structures can be used as a strategy to affect complex redox properties, and furthermore, the introduction of more strongly chelating ISBDs with extra coordinating centers could augment the redox stability of a novel catalyst.
Conclusions
As the ISBD system is conjugated, these ligands and can potentially described as non-innocent (analogous to the widely studied iminobenzoquinones). Isatin Schiff base derivatives are redox sensitve probes and undergo two quasi-reversible electron reduction steps. The redox potentials can be varied by the introduction of different substituents. The combination of these molecules and redox active d-metals could lead to the discovery of structurally interesting and catalytically active complexes. Here, we obtained and structurally characterized sixteen novel copper(II) and (I) complexes with ISBDs. Depending on the ligand structure and copper oxidation state, two different types of products with ISBDs were formed: mononuclear para- or diamagnetic [Cu(L)2]Halx and neutral binuclear halo-bridged [Cu2(μ-Hal)2(L)2] diamagnetic species. The obtained complexes showed the ability to catalyze the oxidation of a model substrate, benzyl alcohol (BnOH), to the corresponding aldehyde, suggesting that ISBD ligands can behave as non-innocent ligands in combination with copper ions.Optimization of ligand structures by introducing substituents and heteroatoms into the ligand core fragments or increasing the chelation ability could potentialy solve the issue of low redox stability of the complexes and increase their catalytic efficiency.
Experimental section
Instrumental methods
Elemental analysis was performed on a Perkin Elmer 2400 Series II CHNS/O analyzer. Single crystals were selected for data collection (Tables S2–S4, ESI†) under an optical microscope, encased in an oil-based cryoprotectant and mounted on cryoloops. Data for L2–L6, L9, C1, C2, C4–C8, C10, C12–C14, C16 and C18 were collected using a Rigaku Oxford Diffraction SuperNova diffractometer equipped with an Atlas CCD area detector operated with monochromated microfocused CuKα radiation (λ[CuKα] = 1.54184 Å) and data for C3 and C11 were collected using the same diffractometer operated with monochromated microfocused MoKα radiation (λ[MoKα] = 0.71073 Å). Data for L7, L8, C15 and C17 were collected using a Rigaku Oxford Diffraction Excalibur diffractometer equipped with an Eos CCD area detector operated with monochromated MoKα radiation. Data for C9 were collected using a Bruker Smart diffractometer equipped with an Apex II CCD area detector operated with monochromated MoKα radiation. Data were integrated and corrected for background, Lorentz, and polarization effects by means of the Bruker programs APEX2 and XPREP (for C9) and the CrysAlisPro program complex (for the other datasets). Absorption correction was applied using the empirical spherical model within the SADABS program (for C9) and in the CrysAlisPro program complex using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm (for the other datasets). The unit-cell parameters (see Synthesis and Tables S2–S4, ESI†) were refined by the least-squares techniques. The structures were solved by direct methods and refined using the SHELX programs incorporated in the OLEX2 program package. The final models included coordinates and anisotropic displacement parameters for all atoms. The carbon-bound H atoms were placed in calculated positions and were included in the refinement in the ‘riding’ model approximation, with Uiso(H) set to 1.5Ueq(C) and a C–H bond length of 0.96 Å for the CH3 groups, with Uiso(H) set to 1.2Ueq(C) and a C–H bond length of 0.98 Å for the tertiary CH groups, and with Uiso(H) set to 1.2Ueq(C) and a N–H bond length of 0.93 Å for the CH groups of the cyclic fragments. The positions of H atoms of H2O molecules and OH− groups were localized from difference Fourier maps and kept fixed during refinement. The FT-IR spectra were recorded using pellets with KBr in the range of 4000–400 cm−1 on a Shimadzu IR-Affinity-1 spectrometer at rt. All 1H and 13C NMR spectra were recorded on a Bruker Avance III spectrometer at 400.13 and 100.61 MHz at room temperature in d3-MeCN. MS-spectra were recorded in positive mode in the range of 100–1000 m/z with ESI ionization on a Shimadzu Maxima-Resonance spectrometer. CV measurements were carried out in a three-electrode cell. The glassy carbon disk, 1.6 mm in diameter, served as a working electrode. Platinum mesh was used as a counter electrode. For the control potential estimation and correct usage of CV data, preliminary experiments were carried out in a 0.25 M Bu4NPF6 solution of CH3CN, containing 5.4 mM ferrocene. The formal potential of the reference redox system Fc/Fc+ in CH3CN measured relative to the reference electrode (silver wire in 0.01 M AgNO3, 0.1 M Bu4NPF6 in CH3CN) was 60 mV. The resistance of the solution was estimated to be ∼0.1 kOhm in CH3CN. All the potential values were corrected accounting for current resistance and referred to the Fc/Fc+ redox system formal potential. DFT calculations were performed using the Gaussian 09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 software package; structures were optimized with the B3LYP cc-PVTZ level of theory. The calculation of vibrational frequencies confirmed that all structures are in local minima on the potential energy surface. Solvent-free microwave-assisted oxidation of BnOH. Benzyl alcohol (2.5 mM), complexes C1, C7, C8 and C9 (5 μM, 0.2 mol% vs. substrate) and an aqueous solution of t-BuOOH (5.0 mM, aq. 70%) were introduced into a cylindrical Pyrex tube that was subsequently sealed. The tube was placed in the MW reactor and the system was stirred and irradiated (5–20 W) at 60 to 100 °C for 0.5 to 1.5 h (conditions for Table 3: 70 °C, 10 W, 0.5 h). In the experiments with additives, TEMPO (2.5 mol% vs. substrate), K2CO3 (2.5 mol% vs. substrate) or radical traps (2.5 mol% vs. substrate) were added to the reaction mixture. After cooling to room temperature, 100 μL of cyclopentanone (as an internal standard) and 2.0 mL of MeCN (to extract the organics from the reaction mixture) were added. This mixture was stirred for ca. 5 min and a sample of 1 μL was subsequently taken from the organic phase and analysed using GC on a FISONS Instruments GC 8000 series gas chromatograph with a DB-624 (J&W) capillary column (FID detector) and the Jasco-Borwin v.1.50 software. The temperature of injection was 240 °C. The initial temperature was maintained at 120 °C for 1 min, then increased by 10 °C min−1 to 200 °C and held at this temperature for 1 min. Helium was used as the carrier gas. Reaction products were identified by comparison of their retention times with known reference compounds. Control experiments (no catalyst) were performed under the studied reaction conditions and no conversion was detected. Molar yield (%) based on substrate, i.e. moles of benzaldehyde per 100 moles of benzyl alcohol, was determined by GC. Turnover frequency (TOF) = number of moles of benzaldehyde per mole of catalyst per hour. Catalyst re-usability in consecutive runs was tested by separating the used catalyst from the reaction mixture by centrifugation followed by filtration of the supernatant solution, washing with methanol and drying in air; the new run was initiated by addition of fresh amounts of reagents besides the catalyst. After completion of each run, the products were analyzed by GC.
37 software package; structures were optimized with the B3LYP cc-PVTZ level of theory. The calculation of vibrational frequencies confirmed that all structures are in local minima on the potential energy surface. Solvent-free microwave-assisted oxidation of BnOH. Benzyl alcohol (2.5 mM), complexes C1, C7, C8 and C9 (5 μM, 0.2 mol% vs. substrate) and an aqueous solution of t-BuOOH (5.0 mM, aq. 70%) were introduced into a cylindrical Pyrex tube that was subsequently sealed. The tube was placed in the MW reactor and the system was stirred and irradiated (5–20 W) at 60 to 100 °C for 0.5 to 1.5 h (conditions for Table 3: 70 °C, 10 W, 0.5 h). In the experiments with additives, TEMPO (2.5 mol% vs. substrate), K2CO3 (2.5 mol% vs. substrate) or radical traps (2.5 mol% vs. substrate) were added to the reaction mixture. After cooling to room temperature, 100 μL of cyclopentanone (as an internal standard) and 2.0 mL of MeCN (to extract the organics from the reaction mixture) were added. This mixture was stirred for ca. 5 min and a sample of 1 μL was subsequently taken from the organic phase and analysed using GC on a FISONS Instruments GC 8000 series gas chromatograph with a DB-624 (J&W) capillary column (FID detector) and the Jasco-Borwin v.1.50 software. The temperature of injection was 240 °C. The initial temperature was maintained at 120 °C for 1 min, then increased by 10 °C min−1 to 200 °C and held at this temperature for 1 min. Helium was used as the carrier gas. Reaction products were identified by comparison of their retention times with known reference compounds. Control experiments (no catalyst) were performed under the studied reaction conditions and no conversion was detected. Molar yield (%) based on substrate, i.e. moles of benzaldehyde per 100 moles of benzyl alcohol, was determined by GC. Turnover frequency (TOF) = number of moles of benzaldehyde per mole of catalyst per hour. Catalyst re-usability in consecutive runs was tested by separating the used catalyst from the reaction mixture by centrifugation followed by filtration of the supernatant solution, washing with methanol and drying in air; the new run was initiated by addition of fresh amounts of reagents besides the catalyst. After completion of each run, the products were analyzed by GC.
Synthesis
All necessary chemicals were of analytical grade and were used without additional purification, unless otherwise stated.1-Methyl-3(phenylimino)indolinone-2-one (L1). The ligand was prepared in an analogous way to a previously described method.11,15 Briefly, in a round bottom flask equipped with a Dean–Stark receiver, a solution of 3.0 g (18.5 mmol) of N-methyl-isatin, 2.4 g (25 mM) of aniline and a catalytic amount of PTSA in 60 mL of toluene was refluxed for 6 h. The reaction mixture was cooled, washed with water, dried by mixing with MgSO4 and solvent evaporated in a vacuum. The crude precipitate was purified by crystallization from ethanol. Yield 80%. IR (KBr) ν, cm−1: 1734 (νC
O), 1653 (νCN). 1H NMR (CD3CN), δ, ppm: 7.62 (dd, J = 7.4, 0.6 Hz, 0.18H), 7.54–7.43 (m, 1.75H), 7.42–7.31 (m, 1.13H), 7.30–7.24 (m, 0.79H), 7.18–7.09 (m, 0.36H), 7.02–6.91 (m, 2.8H), 6.73 (td, J = 7.7, 0.9 Hz, 0.82 H), 6.47 (dd, J = 7.7, 0.6 Hz, 0.79H), 3.21 (s, 2.48H), 3.09 (s, 0.47H). 13C NMR (CD3CN), δ, ppm: 163.8, 155.7, 151.9, 149.4, 135.2, 135.0, 130.6, 129.5, 126.5, 126.0, 125.6, 123.9, 123.2, 120.0, 118.3, 116.6, 110.6, 110.1, 26.7, 26.3.
1-Methyl-3(2-hydroxy-phenylimino)indolinone-2-one (L2). IR (KBr) ν, cm−1: 1701 (νC
O), 1608 (νCN). MS (ESI), m/z (%): [M + H]+ 253.09, [M + Na]+ 254.09. Prismatic orange crystals suitable for X-ray diffraction analysis were obtained after crystallization from a toluene:hexane 1:1 mixture at 4 °C. C15H12N2O2, P21/c, a = 10.1494(6), b = 7.7078(5), c = 15.2882(7) Å, β = 101.259(6)°, V = 1172.97 Å3, Z = 4, R1 = 0.037, CCDC 1412864.†
1-Methyl-3(2,6-dimethyl-phenylimino)indolinone-2-one (L3). IR (KBr) ν, cm−1: 1733 (νC
O), 1653 (νCN). ESI (MeOH): 265.13 [M + H], 267.12 [M + Na]. 1H NMR (CDCl3), δ, ppm: 7.40 (td, J = 7.8, 1.2 Hz, 1H), 7.14 (d, J = 7.5 Hz, 2H), 7.07 (m, 1H), 6.96 (d, J = 7.9 Hz, 1H), 6.74 (td, J = 7.6, 0.7 Hz, 1H), 6.24 (dd, J = 7.6, 0.5 Hz, 1H), 6.24 (dd, J = 7.6, 0.5 Hz, 1H), 3.23 (s, 3H), 3.09 (s, 3H), 1.96 (s, 6H). 13C NMR (CD3CN), δ, ppm: 163.6, 156.4, 149.4, 148.9, 135.5, 129.3, 125.3, 125.0, 123.7, 117.4, 110.5, 26.7, 17.9. C17H16N2O, C2/c, a = 13.6078(3), b = 19.5284(6), c = 11.3494(3) Å, β = 108.884(3)°, V = 2853.64 Å3, Z = 8, R1 = 0.038, CCDC #1538859.†
1-Methyl-3(2,6-diisopropyl-phenylimino)indolinone-2-one (L4). IR (KBr) ν, cm−1: 1725 (νC
O), 1663 (νCN). ESI (MeOH): 321.20 [M + H], 343.18 [M + Na]. 1H NMR (CD3CN), δ, ppm: 7.39 (td, J = 7.8, 1.1 Hz, 1H), 7.31–7.15 (m, 3H), 6.97 (d, J = 7.9 Hz, 1H), 6.73–6.69 (m, 1H), 6.20 (d, J = 7.2 Hz, 1H), 3.24 (s, 3H), 3.09 (s, 3H), 2.73 (sep, J = 13.7, 6.9 Hz, 2H), 1.11 (d, J = 6.8 Hz, 6H), 0.95 (d, J = 6.9 Hz, 6H) (E/Z ratio – 14:1). 13C NMR (CD3CN), δ, ppm: 163.5, 156.8, 149.1, 135.5, 126.2, 126.0, 124.6, 123.4, 117.3, 110.6, 29.1, 26.7, 23.6, 23.3. C21H24N2O, P21/c, a = 8.47970(10), b = 15.0831(2), c = 28.0078(4) Å, β = 93.3600(10)°, V = 3576.04 Å3, Z = 8, R1 = 0.038, CCDC #1538860.†
1-Methyl-3(phenylimino)-5-bromo-indolinone-2-one (L5). IR (KBr) ν, cm−1: 1729 (νC
O), 1653 (νCN). MS (ESI), m/z (%): 339.12.09, 254.09. 1H NMR (CD3CN), δ, ppm: 7.79–7.75, 7.68–7.64, 7.53–7.48, 7.37–7.30, 7.17, 7.01–6.96, 6.95–6.89 (m, 6.69H), 6.5 (d, J = 6.8 Hz), 3.21 (s, 2.19 H), 3.09 (s, 0.56 H). 13C NMR (CD3CN), δ, ppm: 151.5, 148.6, 137.5, 130.8, 129.6, 129.0, 126.5, 120.1, 118.3, 114.9, 112.6, 26.9. C15H11BrN2O, C2/c, a = 8.1114(2), b = 16.2246(4), c = 19.1312(4) Å, β = 95.696(2)°, V = 2505.32 Å3, Z = 8, R1 = 0.035, CCDC #1538858.†
1-Methyl-3(phenylimino)-5-methyl-indolinone-2-one (L6). IR (KBr) ν, cm−1: 1737 (νC
O), 1646 (νCN). MS (ESI), m/z (%): 271.118, 273.100. 1H NMR (CD3CN), δ, ppm: 7.53–7.41, 7.39–7.06, 6.99–6.93, 6.66–6.63 (m), 6.28 (s, br), 3.19 (s, J = 6.2 Hz, 12H), 3.14 (s, 2H), 3.07 (s, 3H), 2.00 (s, 13H). 13C NMR (CD3CN), δ, ppm: 207.5, 163.9, 155.9, 151.9, 147.3, 139.7, 135.5, 132.7, 130.6, 130.1, 129.9, 127.1, 126.0, 125.5, 123.7, 119.9, 118.3, 115.6, 112.1, 110.9, 110.5, 110.0, 89.9, 89.6, 30.9, 26.7, 20.8. C16 H14 N2 O1, P21/m, a = 9.0075(8), b = 6.8883(8), c = 10.3867(11) Å, β = 98.932(8)°, V = 636.642 Å3, Z = 2, R1 = 0.070, CCDC 1538861.†
1-Methyl-3(2,4,6-trimethyl-phenylimino)-indolinone-2-one (L7). IR (KBr) ν, cm−1: 1729 (νC
O), 1656 (νCN). MS (ESI), m/z (%): 279.150. 1H NMR (CD3CN), δ, ppm: 7.39 (td, J = 7.8, 1.2 Hz, 1H), 6.96 (s, 2.5H), 6.94 (s, 0.5H), 6.74 (td, J = 7.6, 8.0 Hz, 1H), 6.31 (dd, J = 7.6, 0.6 Hz, 1H), 6.31 (dd, J = 7.6, 0.6 Hz, 1H), 3.22 (s, 3H), 2.30 (s, 3H), 1.92 (s, 6H). 13C NMR (CD3CN), δ, ppm: 163.6, 156.6, 148.9, 147.0, 135.4, 134.8, 130.0, 125.4, 124.9, 123.7, 117.5, 110.5, 26.7, 20.9, 17.8.
C18H18N2O2, P21/c, a = 6.9194(4), b = 8.6089(5), c = 24.9101(16) Å, β = 96.141(6)°, V = 1475.34 Å3, Z = 4, R1 = 0.044, CCDC 1538862.†
1-Isopropyl-3(phenylimino)indolinone-2-one (L8). IR (KBr) ν, cm−1: 1724 (νC
O), 1662 (νCN). MS (ESI), m/z (%): 265.134, 287.116. 1H NMR (CD3CN), δ, ppm: 7.64 (dd, J = 7.4, 0.6 Hz, 0.18H), 7.52–7.42 (m, 1.72H), 7.39–7.30 (m, 1.13H), 7.30–7.22 (m, 0.77H), 7.18–7.06 (m, 1.37H), 6.99–6.91 (m, 1.87H), 6.70 (td, J = 7.7, 0.8 Hz, 0.8H), 6.48 (dd, J = 7.7, 0.9 Hz, 0.8H), 4.57 (hept, J = 7.0 Hz, 0.8H), 4.49–4.37 (m, 0.2H), 1.50 (d, J = 7.0 Hz, 4.75H), 1.42 (d, J = 7.0 Hz, 1H). 13C NMR (101 MHz, CD3CN), δ, ppm: 163.4, 155.8, 152.0, 148.3, 134.9, 130.6, 129.4, 126.8, 125.9, 123.5, 122.7, 119.7, 118.1, 117.1, 111.9, 111.5, 45.3, 45.0, 19.5. C17H16N2O, Pna21, a = 9.6713(4), b = 9.0619(4), c = 15.8838(7) Å, V = 1392.06 Å3, Z = 4, R1 = 0.030, CCDC 1538863.†
1-Phenyl-3(phenylimino)indolinone-2-one (L9). IR (KBr) ν, cm−1
:1737 (νCO), 1653 (νCN). MS (ESI), m/z (%): 299.119, 321.101. 1H NMR (CD3CN), δ, ppm: 7.73 (dd, J = 7.5, 0.7 Hz, 0.18H), 7.61 (dd, J = 10.7, 4.5 Hz, 1.71H), 7.53–7.39 (m, 4.99H), 7.38–7.25 (m, 1.97H), 7.23–7.12 (m, 0.33H), 7.09–6.98 (m, 1.97H), 6.79 (td, J = 6.9, 3.2 Hz, 1.83H), 6.59 (dd, J = 7.9, 1.0 Hz, 0.82H). 13C NMR (CD3CN), δ, ppm: 163.3, 155.4, 151.9, 149.3, 135.1, 135.0, 134.9, 130.9, 129.6, 129.5, 129.4, 127.8, 127.6, 126.8, 126.2, 124.5, 123.7, 120.0, 116.8, 111.4, 110.9. C20H14N2O2, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.9809(7), b = 10.7407(4), c = 14.9870(10) Å, α = 73.451(5)°, β = 78.454(6)°, γ = 86.790(4)°, V = 1508.92 Å3, Z = 4, R1 = 0.044, CCDC 1538865.†
, a = 9.9809(7), b = 10.7407(4), c = 14.9870(10) Å, α = 73.451(5)°, β = 78.454(6)°, γ = 86.790(4)°, V = 1508.92 Å3, Z = 4, R1 = 0.044, CCDC 1538865.†
[Cu(L1)2](Cl)2 (C1). A methanol solution containing 500 mg (3 mmol) of CuCl2·2H2O was mixed with a methanol solution containing 1.38 mg (6 mmol) of L1. The mixture was mixed for 1 h and filtered. The formed dark brown complex solution was left for crystallization at r.t. for several days to form small prismatic crystals of [Cu(L1)2]Cl2. Yield: 90%. MW = 606.99. Calculated for C30H24Cl2CuN4O2– C (59,36), H (3,99), N (9,23); estimated – C (59,36), H (3,99), N (9,23). IR (KBr) ν, cm−1: 1728 (νC=O), 1659 (νC
N). ESI (MeOH), m/z: 608.06 [M]+, 570.088 [M − Cl]+, 535.11 [M − 2Cl]+. C30H24Cl2CuN4O2, P, a = 8.0870(8), b = 9.2417(10), c = 9.6103(10) Å, α = 75.944(9)°, β = 75.005(9)°, γ = 79.112(9)°, V = 666.903 Å3, Z = 1, R1 = 0.028, CCDC #1538868.†
[Cu2(μ-Cl)2(L7)2] (C2). A methanol solution containing 500 mg (3 mmol) of CuCl2·2H2O was mixed with a MeOH solution containing 820 mg (3 mmol) of L7. To the resulting brown colored solution, 1.55 mL of 10% iPrSH (3 mmol) methanol solution was gradually (to prevent copper(I) thiolate formation) added to the complex C1. The color changed from brown to dark green. The mixture was stirred for 1 h and filtered. The resulting solution was allowed to evaporate for 24 h, resulting in dark green crystalline product formation of binuclear complex [Cu2(μ-Cl)2(L7)2]. MW = 754.59. IR (KBr) ν, cm−1: 1718 (νC
O). C36H36Cl2Cu2N4O2, P21/c, a = 15.435(8), b = 8.1831(15) c = 14.316(6) Å, β = 114.64(5)°, V = 1643.55 Å3, Z = 2, R1 = 0.145, CCDC #1538877.†
[Cu2(μ-Br)2(L7)2] (C3). Th complex was prepared by two methods:
(A) A MeCN solution containing 500 mg of CuBr (3.5 mmol) was mixed with acetonitrile solution containing 1 g (3.5 mmol) of L7, and the resulting solution was stirred for 1 h, filtered and allowed to slowly evaporate. In 24 h, thin plate crystals of [Cu2(μ-Br)2(L7)2] were formed on the walls of the beaker. MW = 843.60. Calculated for C36H36Br2Cu2N4O2 – C (51,25), H (4,30), N (6,64); estimated – C (50,67), H (3,36), N (6,71). IR (KBr) ν, cm−1: 1717 (νCO). C36H36Br2Cu2N4O2, P21/c, a = 15.4205(6), b = 7.92427(19), c = 14.7380(5) Å, β = 112.819(4)°, V = 1659.98 Å3, Z = 2, R1 = 0.033, CCDC #1538873.†
(B) To a methanol solution containing 500 mg (2.2 mmol) of CuBr2, L7 dissolved in MeOH was gradually added under slight heating. Reaction resulted in the formation of dark-green crystals of the binuclear complex with the same crystal structure parameters.
[Cu2(μ-Br)2(L3)2] (C4). The complex was prepared in a similar way (both methods worked) to C3. MW = 815.54. Calculated for C34H32Br2Cu2N4O2 – C (50,07), H (3,95), N (6,87); estimated – C (50.12), H (3,94), N (7,36). IR (KBr) ν, cm−1: 1717 (νC
O). ESI (MeOH), m/z: 591.1803 [M − 2Br]+, 735.0265 [M − Br]+. C34H32Br2Cu2N4O2, P2/c, a = 26.9791(11), b = 8.2871(3), c = 14.1074(5) Å, β = 94.972(3)°, V = 3142.24 Å3, Z = 4, R1 = 0.039, CCDC #1538881.†
[Cu2(μ-Br)2(L4)2] (C5). The complex was prepared in a similar way as that described for C3. MW = 927.76. Calculated for C42H48Br2Cu2N4O2 – C (54,37), H (5,21), N (6,04); estimated – C (57.21), H (5,9), N (5,9). IR (KBr) ν, cm−1: 1722 (νC
O). ESI (MeOH), m/z: 703.3065 [M − 2Br]+, m/z 424.14 {Cu(L4)MeCN}+. C21H24Br0.3Cu0.3N2O, C21H24Br0.7Cu0.7N2O2, P, a = 11.0842(4), b = 11.7852(5), c = 17.1333(5) Å, α = 99.747(3), β = 108.723(3), γ = 102.672(3)°, V = 1997.24 Å3, Z = 2, R1 = 0.049, CCDC #1538878.†
[Cu2(μ-Br)2(L5)2] (C6). The complex was prepared similarly to C3 from CuBr and L5. MW = 917.23 IR (KBr) ν, cm−1
:1724 (νCO). ESI (MeOH), m/z: 690.9406 [2L5 + Cu]+. C30H22Br4Cu2N4O2, P, a = 8.5542(4), b = 9.5043(6), c = 10.1464(4) Å, α = 90.501(4), β = 96.474(4), γ = 115.484(6)°, V = 738.402 Å3, Z = 1, R1 = 0.034, CCDC #1538883.†
[Cu2(μ-Br)2(L6)2] (C7). The complex was prepared similarly to C3 from CuBr and L6. MW = 787.49 IR (KBr) ν, cm−1: 1717 (νC
O). ESI (MeOH), m/z: 706.9965 [M − Br]+, 563.1490 [M − 2Br]+. C32H28Br2Cu2N4O2, P, a = 8.6213(4), b = 9.3911(5), c = 10.1721(4) Å, α = 90.122(4), β = 97.663(3), γ = 115.574(5)°, V = 734.605 Å3, Z = 1, R1 = 0.035, CCDC #1538876.†
[Cu(L6)2]Cl2 (C8). The complex was prepared similarly to C1. MW = 635.04. IR (KBr) ν, cm−1: 1729 (νC
O), 1657 (νCN). C32H28Cl2CuN4O2, P, a = 8.9050(7), b = 9.2277(4), c = 9.8071(7) Å, α = 74.790(5), β = 65.400(7), γ = 76.151(5)°, V = 699.242 Å3, Z = 1, R1 = 0.039, CCDC #1538871.†
[Cu(L1)2]Br (C9). The complex was prepared similarly to C1. MW = 615.99. IR (KBr) ν, cm−1: 1718 (νC
O). ESI (MeOH), m/z: 299.083 [Cu(L1)]+, 535.118 [M − Br]+. XRD: C2/c, a = 17.9841(19), b = 6.9520(8), c = 21.036(2) Å, β = 91.011(2), V = 2629.63 Å3, Z = 4, R1 = 0.039, CCDC #1538874.†
[Cu(L1)2]I (C10). An acetonitrile solution containing 100 mg (4 mmol) of CuSO4 was mixed with 190 mg (8 mmol) of L1. To the obtained brown-colored complex solution, 70 mg (4 mmol) of KI in MeCN was added to reduce the Cu(II) ions. Upon the addition of I− ions, the reaction mixture turned a dark-magenta color; it was filtered and left for a week for product crystallization. Yield ∼ 80%. MW = 662.99. IR (KBr) ν, cm−1: 1717 (νC
O). ESI (MeOH), m/z: 535.121 [Cu(L1)2]+. XRD, C2/c, a = 17.9067(5), b = 6.9140(2), c = 21.1065(7) Å, β = 90.872(3)°, V = 2612.83 Å3, Z = 3, R1 = 0.031, CCDC #1538872.†
[Cu(L7)2(MeCN)](BF4) (C11) was prepared similarly to C1 from [Cu(MeCN)](BF4) and L7 in MeCN. MW = 748.10. IR (KBr) ν, cm−1: 1720 (νCO). C38H39BCuF4N5O2.3, P21/n, a = 12.8485(3), b = 14.2133(3), c = 21.2613(5) Å, β = 06.297(2)°, V = 3726.72 Å3, Z = 4, R1 = 0.049, CCDC #1538866.†
[Cu(L2)2]Br (C12) was prepared similarly to C1 from CuBr and L2 in MeOH. MW = 615.99. Calculated for C30H24BrCuN4O4 – C (58,49), H (3,93), N (9,10); estimated – C (59,36), H (3,99), N (9,23). IR (KBr) ν, cm−1: 1728 (νCO). ESI (MeOH), m/z: 669.01 [M − Br − Na]+. C30H24BrCuN4O4, C2/c, a = 17.9161(9), b = 6.9866(5), c = 21.0116(9) Å, β = 91.316(5)°, V = 2629.38 Å3, Z = 4, R1 = 0.076, CCDC #1538870.†
[Cu(μ-I)(L9)]n (C13). The complex was obtained by a gradual mixture of MeCN solutions containing equimolar amounts of CuI and L9. The resulting magenta colored solution was filtered and allowed to evaporate and form prismatic crystals of a binuclear complex. MW = 488.77. IR (KBr) ν (νC
O), cm−1: 1742, 1730, 1718. C20H14CuIN2O, P21/c, a = 12.7900(8), b = 17.0969(8), c = 7.9810(5) Å, β = 102.964(7)°, V = 1700.72(18) Å3, Z = 4, R1 = 0.051, CCDC #1538882.†
[Cu2(μ-I)2(L7)2] (C14). The complex was obtained similarly to C13. The resulting magenta colored solution was filtered and allowed to evaporate and form prismatic crystals of a binuclear complex. MW = 937.60. Calculated for C36H36Cu2I2N4O2 – C (46,11), H (3,87), N (5,98); estimated – C (45.54), H (3,86), N (6,11). IR (KBr) ν, cm−1: 1715 (νC
O). ESI (MeOH), m/z: 619.2110 [M − 2l]+. C36H36Cu2I2N4O2, P21/c, a = 15.5090(16), b = 7.8740(9), c = 15.023(2) Å, β = 111.451(14)°, V = 1707.5 Å3, Z = 2, R1 = 0.071, CCDC #1538875.†
[Cu2(μ-I)2(L1)2] (C15). The complex was prepared similarly to C14. MW = 853.45. IR (KBr) ν, cm−1: 1720 (νC
O). ESI (MeOH), m/z: 724.9643 [M − I]+, 535.1201 [M − 2I], 299.02 {[Cu(L1)]}+. XRD: P, a = 8.5695(9), b = 9.3493(7), c = 10.6986(11) Å, α = 74.782(9), β = 75.057(9), γ = 63.915(10)°, V = 732.754 Å3, Z = 2, R1 = 0.033, CCDC #1538879.† IR (KBr) ν, cm−1: 1717 (νCO). 1H NMR, δ, ppm: 7.64 (m, 0.23H), 7.53–7.27 (set of multiplets, 4.44H), 7.13 (m, 0.35H), 7.01–6.95 (m, 3.28), 6.75 (td, J = 7.7, 0.8 Hz, 9H). 13C NMR, δ, ppm: 163.9, 155.7, 151.5, 149.5, 135.0, 130.6, 129.5, 126.5, 126.3, 125.6, 123.9, 123.3, 119.9, 118.6, 116.7, 110.8, 110.1, 26.8, 26.3.
[Cu2(μ-I)2(L3)2] (C16). The complex was prepared similarly to C14. MW = 909.54. Calculated for C34H32Cu2I2N4O2 – C (44,90), H (3,55), N (6,16); estimated – C (45.02), H (3,12), N (6.47). IR (KBr) ν, cm−1: 1713 (νC
O). ESI (MeOH), m/z: 591.1807 [M − 2Cl]+. C34H32Cu2I2N4O2, C2/c, a = 27.2980(4), b = 8.22746(15), c = 14.3942(3) Å, β = 93.2951(13)°, V = 3227.49 Å3, Z = 4, R1 = 0.035, CCDC 1538880.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education and Science of the Russian Federation (14.B25.31013), by the Russian Foundation for Basic Research (16-34-50250) and by the Portugal Foundation for Science and Technology (UID/QUI/00100/2013 project). Physicochemical studies were performed at the Saint-Petersburg State University Centers for Magnetic Resonance, X-ray Diffraction Studies, and Chemical Analysis and Materials Research.Notes and references
- N. B. Gwilherm Evano, Copper-Mediated Cross-Coupling Reactions, John Wiley & Sons, Inc., 2013, pp. i–xxxv Search PubMed.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, in Advances in Organometallic Chemistry, ed. J. P. Pedro, Academic Press, 2015, vol. 63, pp. 91–174 Search PubMed.
- J. Kaur, S. S. Chimni, S. Mahajan and A. Kumar, RSC Adv., 2015, 5, 52481–52496 RSC.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- J. Jacquet, E. Salanouve, M. Orio, H. Vezin, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Chem. Commun., 2014, 50, 10394–10397 RSC.
- P. Davidovich, V. Aksenova, V. Petrova, D. Tentler, D. Orlova, S. Smirnov, V. Gurzhiy, A. L. Okorokov, A. Garabadzhiu, G. Melino, N. Barlev and V. Tribulovich, ACS Med. Chem. Lett., 2015, 856–860 CrossRef CAS PubMed.
- O. Fedorova, A. Daks, V. Petrova, A. Petukhov, L. Lezina, O. Shuvalov, P. Davidovich, D. Kriger, E. Lomert, D. Tentler, V. Kartsev, B. Uyanik, V. Tribulovich, O. Demidov, G. Melino and N. Barlev, Cell Cycle, 2018, 17, 1917–1930 CrossRef CAS PubMed.
- L. M. Kara, L. Vine, J. M. Locke and D. Skropeta, Adv. Anticancer Agents Med. Chem., 2013, 2, 254–312 Search PubMed.
- J. Jacquet, P. Chaumont, G. Gontard, M. Orio, H. Vezin, S. Blanchard, M. Desage-El Murr and L. Fensterbank, Angew. Chem., 2016, 128, 10870–10874 CrossRef.
- G. Cerchiaro, P. L. Saboya, A. M. da Costa Ferreira, D. M. Tomazela and M. N. Eberlin, Transition Met. Chem., 2004, 29, 495–504 CrossRef CAS.
- P. Davidovich, D. Novikova, V. Tribulovich, S. Smirnov, V. Gurzhiy, G. Melino and A. Garabadzhiu, J. Mol. Struct., 2014, 1075, 450–455 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- D. R. Brkić, A. R. Božić, A. D. Marinković, M. K. Milčić, N. Ž. Prlainović, F. H. Assaleh, I. N. Cvijetić, J. B. Nikolić and S. Ž. Drmanić, Spectrochim. Acta, Part A, 2018, 196, 16–30 CrossRef PubMed.
- A. S. Smirnov, D. N. Nikolaev, V. V. Gurzhiy, S. N. Smirnov, V. S. Suslonov, A. V. Garabadzhiu and P. B. Davidovich, RSC Adv., 2017, 7, 10070–10073 RSC.
- A. Ercag, S. O. Yildirim, M. Akkurt, M. U. Ozgur and W. F. Heinemann, Chin. Chem. Lett., 2006, 17, 243–246 CAS.
- M. F. N. N. Carvalho, M. T. Duarte, T. A. Fernandes, A. M. Galvão and A. M. Botelho do Rego, Inorg. Chem., 2010, 49, 10330–10337 CrossRef CAS PubMed.
- S. Anga, M. Paul, K. Naktode, R. K. Kottalanka and T. K. Panda, Z. Anorg. Allg. Chem., 2012, 638, 1311–1315 CrossRef CAS.
- G. Speier, J. Csihony, A. M. Whalen and C. G. Pierpont, Inorg. Chim. Acta, 1996, 245, 1–5 CrossRef CAS.
- S. Periyaraja, P. Shanmugam and A. B. Mandal, Eur. J. Org. Chem., 2014, 954–965 CrossRef CAS.
- T. G. Gray and J. P. Sadighi, in Molecular Metal–Metal Bonds, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 397–428 Search PubMed.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- C. Mukherjee, U. Pieper, E. Bothe, V. Bachler, E. Bill, T. Weyhermüller and P. Chaudhuri, Inorg. Chem., 2008, 47, 8943–8956 CrossRef CAS PubMed.
- C. Mukherjee, T. Weyhermüller, E. Bothe and P. Chaudhuri, Inorg. Chem., 2008, 47, 2740–2746 CrossRef CAS PubMed.
- P. Naumov and F. Anastasova, Spectrochim. Acta, Part A, 2001, 57, 469–481 CrossRef CAS.
- A. V. Belyakov, K. O. Nikolaenko, P. B. Davidovich, A. D. Ivanov, A. I. Ponyaev, A. N. Rykov and I. F. Shishkov, J. Mol. Struct., 2017, 1132, 44–49 CrossRef CAS.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–47 Search PubMed.
- N. M. R. Martins, S. Anbu, K. T. Mahmudov, R. Ravishankaran, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, A. A. Karande and A. J. L. Pombeiro, New J. Chem., 2017, 41, 4076–4086 RSC.
- L. M. Slaughter, J. P. Collman, T. A. Eberspacher and J. I. Brauman, Inorg. Chem., 2004, 43, 5198–5204 CrossRef CAS PubMed.
- A. Sabbatini, L. M. D. R. S. Martins, K. T. Mahmudov, M. N. Kopylovich, M. G. B. Drew, C. Pettinari and A. J. L. Pombeiro, Catal. Commun., 2014, 48, 69–72 CrossRef CAS.
- C. C. Lu, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, Inorg. Chem., 2007, 46, 7880–7889 CrossRef CAS PubMed.
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS.
- F. Marken, A. Neudeck and A. M. Bond, in Electroanalytical Methods: Guide to Experiments and Applications, eds. F. Scholz, A. M. Bond, R. G. Compton, D. A. Fiedler, G. Inzelt, H. Kahlert, Š. Komorsky-Lovrić, H. Lohse, M. Lovrić, F. Marken, A. Neudeck, U. Retter, F. Scholz and Z. Stojek, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 57–106 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1412864, 1538859, 1538860, 1538858, 1538861, 1538862, 1538863, 1538865, 1538868, 1538877, 1538873, 1538881, 1538878, 1538883, 1538876, 1538871, 1538874, 1538872, 1538866, 1538870, 1538882, 1538875, 1538879 and 1538880. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj02718h |
| ‡ Current address: Trinity College, Dublin 2, Ireland. |
| § Here and onwards, hydrogen atoms are omitted for clarity. |
| ¶ Potentially, these compounds could be used in the diastereoselective syntheses of spiroazetidinimine-2-oxindoles; the three component one-pot synthesis of these probes from an ISBD, p-toluenesulfonyl azide and phenyl acetylene in the presence of 10 mol% CuI in MeCN was reported earlier; where, most probably, CuI and the ISBD form a binuclear complex analogue to C14 that reacts with two other components forming spiroazetidinimine-2-oxindole.21 |
| || Reactions of copper(I) iodide in MeCN with L3 and L7 yielded analogues C14–C16, complexes of binuclear structure. |
| ** Peaks C′ probably correspond to the reduction of intermediates and will not be discussed. |
| †† The presence of the double reduced L42− species was confirmed by negative mode MS analysis (L42−·H3O+ MW = 339.21) of the reaction between L4 and metal sodium. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |