
Synthesis of a SN38 prodrug grafted to amphiphilic phosphorylcholine polymers and their prodrug miceller properties†
Fan
Chen
 a,
Yuanyuan
Cai
a,
Lei
Huang
a,
Yuanwei
Chen
*a and
Xianglin
Luo
*ab
a,
Yuanyuan
Cai
a,
Lei
Huang
a,
Yuanwei
Chen
*a and
Xianglin
Luo
*ab
aCollege of Polymer Science and Engineering, Sichuan University, Chengdu 610065, P. R. China
bState Key Lab of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: luoxl_scu@126.com
First published on 17th November 2018
Abstract
Polymer prodrug micelles, combining the advantages of prodrugs and polymer micelles, can greatly improve the solubility, permeability and stability of drugs. In this work, amphiphilic block copolymers poly(α-azide caprolactone-co-caprolactone)-b-poly(2-methacryloyloxyethyl phosphorylcholine), P(ACL-co-CL)-b-PMPC, were synthesized, and an antineoplastic drug 7-ethyl-10-hydroxy camptothecin (SN38) was grafted to the amphiphilic copolymers to obtain a prodrug and their micelles. The preparation of P(ACL-co-CL)-b-PMPC was proceeded by ring-opening polymerization of ε-caprolactone/α-bromide caprolactone, converting the bromide to an azide, and atom transfer radical polymerization (ATRP). The camptothecin derivative SN38-alkyne was bonded to the polymer to obtain P(CL/CL-g-SN38)-b-PMPC by a “click reaction”. The obtained polymer–drug complex can easily self-assemble to form prodrug micelles with the shell composed of PMPC and the core composed of PCL and SN38. The micelles presented characteristics including improved drug loading efficiency and stabilization of the drug. In vivo, the retention time of the prodrug micelles in blood could be tripled, and the clearance was delayed compared with the SN38 solution. The in vivo anti-tumor experiments demonstrated that the prodrug micelles had a better treatment effect and low toxicity to 4T1-bearing BALB/C mice.
1. Introduction
7-Ethyl-10-hydroxycamptothecin (SN38) is an anti-cancer drug against many malignancies including colorectal carcinoma, lung tumors, gastric, cervical and ovarian cancers, and malignant lymphoma. Acting as a potent topoisomerase I inhibitor,1–3 SN38 belongs to the class of 20(S)-camptothecin (CPT) compounds, and it is up to 200–2000-fold more potent than irinotecan (CPT-11) against several tumor cell lines.4 However, the water solubility of SN38 is about 11–38 μg mL−1, much worse than that of CPT-11. Thus, SN38 cannot be used directly for clinical applications.CPT-11 has been used in clinics and shown to exhibit anti-tumor activity clinically5 using an active metabolite, SN38, which possesses a broad-spectrum and high-potency therapeutic effect. The in vivo conversion of CPT-11 to SN-38 proceeds via carboxylesterase (CE)-mediated de-esterification in the human liver.6 However, the influence of genetic and environmental factors on CE activity was up to 10 folds.7 Owing to the great inter-individual differences of the CE activity and the complexity of CPT-11 metabolism, interpatient variability after administration of CPT-11 may be very high in both efficacy and toxicity. And the conversion of CPT-11 to SN38 in humans is very inefficient, only about 2–8% of the administered dose. Besides, in the presence of human plasma albumin, the lactone ring of CPT-11 possessing pharmacological importance is readily converted to the inactive carboxylate form.8 Furthermore, the clinical utility of CPT-11 results in severe gastrointestinal toxicity and myelosuppression. These have spawned considerable interest in the direct administration of SN38, which may have a significant clinical advantage and overcome the drawbacks of CPT-11 in drug activation, metabolism and elimination.
As we have described, SN38 cannot be directly administered because of its extremely low water solubility in any physiologically compatible and pharmaceutically acceptable solvents and oils, such as DMSO (0.5%, m/m), formic acid and so on. Therefore, in order to improve the water solubility and bioavailability of SN38, it has been extensively investigated to maintain the active form of SN38 through a drug delivery system.9–11 Several approaches using drug carriers have been proposed, such as incorporation into liposomes,12,13 nanoparticles,14–16 polymeric micelles,17–20 nanogels21 and conjugation to water-soluble polymers such as polyethylene glycol.22
Among drug carriers, polymeric micelles have been developed for anticancer drug delivery in recent years due to their tailorable molecule structures. The drug-loaded micelles show improved drug solubility, a prolonged circulation time by avoiding rapid clearance by the renal and reticuloendothelial systems (RES) and passive targeting to tumor tissues via the enhanced permeability effect (EPR).23 When hydrophilic or amphiphilic polymers are combined with SN38, polymer–SN38 conjugates, i.e. prodrugs, are obtained, and SN38 prodrug micelles can be formed. The micelles of polymer–SN38 conjugates have several advantages,22 including enhanced therapeutic efficacy, reduced side effects, decreased drug administration and improved patient compliance. SN38 conjugates are generally formed by the attachment of SN38 to amphiphilic or hydrophilic polymers such as methoxypoly(ethylene glycol)-b-poly(lactide) (mPEG-PLA),24 polyglutamic acid,10 poly HPMA,25 or polyethylene glycol polymeric micelles.13 Among hydrophilic polymers, PEG is widely recognized as a “stealth” material. But with more in vivo investigations, it has been found that PEG is not perfect. However, in the recent few decades, it has been found that micelles with phosphatidylcholine surfaces have good biocompatibility, long blood circulation times, and high enrichment of drugs in tumor tissue.26–28 Phosphorylcholine polymeric micelles are attracting more and more widespread attention.29–33 However, it can be seen that there are few reports on SN38 conjugates with phosphorylcholine polymers.
In this work, we propose the synthesis of amphiphilic block copolymers poly(α-azide caprolactone-co-caprolactone)-b-poly(2-methacryloyloxyethyl phosphorylcholine), P(ACL-co-CL)-b-PMPC, by enabling the bonding of SN38–alkyne by a click reaction to obtain SN38 conjugates, P(CL/CL-g-SN38)-b-PMPC. The successful synthesis of P(CL/CL-g-SN38)-b-PMPC was confirmed by 1H NMR and FT-IR. Meanwhile, their physiochemical properties were characterized. The conjugates can easily self-assemble to form prodrug micelles with the shell composed of PMPC and the core composed of PCL and SN38. The structure of the SN38 prodrug micelles was able to avoid the loss of pharmacological effects due to opening the loop of SN38 upon contact with water molecules. The micellar properties including drug release, cytotoxicity, pharmacokinetics and in vivo anti-tumor effect were studied. Our data demonstrated that the SN38 prodrug micelles presented characteristics including improved drug loading efficiency, prolonged in vivo retention time and clearance, enhanced therapeutic effect and reduced toxic and side effects.
2. Materials and experiments
2.1 Materials and reagents
ε-Caprolactone (ε-CL) from Aldrich (New Jersey, USA) was dried with calcium hydride (CaH2) and distilled under reduced pressure. 2-Methacryloyloxyethyl phosphoryl choline (MPC) was offered by Aladdin (USA) and used as received. 2,2′-Dithiodiethanol, stannous 2-ethylhexanoate [Sn(Oct)2, 95%], 2-bromoisobutyryl bromide (BIBB, 97%), 2,2′-bipyridyl (Bpy), sodium azide (NaN3), (DIPC) N,N′-diisopropylcarbodiimide and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (USA). Cuprous bromide (CuBr, 99%) was stirred in acetic acid overnight, filtered, washed five times with ethanol, and finally washed with acetone until the solvent turned colorless, and was then dried under vacuum. Toluene and tetrahydrofuran (THF) were purified by refluxing over sodium before use. Triethylamine (TEA), dichloromethane (CH2Cl2) and N,N-dimethylformamide (DMF) were purified by distillation under vacuum over CaH2 before utilization. Liquid bromine (Br2), CaH2 and all solvents were purchased from Chengdu KeLong Chemical Co. Ltd, China. Monomer α-BrCL (α-bromide caprolactone) was synthesized according to ref. 29.2.2 Synthesis of P(CL/CL-g-SN38)-b-PMPC copolymers
Poly(α-bromide caprolactone-co-caprolactone) (P(BCL-co-CL)) was synthesized through the ring-opening polymerization of ε-caprolactone and α-BrCL using dodecanol as the initiator and Sn(Oct)2 as the catalyst. The polymerization of P(BCL-co-CL) with degree 29 was as an example. Briefly, laurinol initiator (0.7426 g, 3.98 mmol), α-bromide caprolactone (2.3 g, 11.94 mmol) and ε-caprolactone (9.0855 g, 79.6 mmol) were quickly added into a round bottom flask (25 mL) and it was connected to a Schlenk line. After adding SnOct2 (1.8 mg, 4.44 mmol) in 100 μL dry toluene, an exhaustion–refilling process with nitrogen for the flask was repeated three times to remove residual water and toluene, and then the round bottom flask was put into an oil bath at 110 °C for 48 h. After cooling the reaction flask to room temperature, the crude product was diluted with 5 mL CH2Cl2, and poured dropwise into 100 mL cold methanol under vigorous stirring. A white precipitate was obtained and then dried under vacuum overnight to a constant weight. The white powder polymer P(BCL-co-CL)29 (the percentage of α-BrCL was 10%) was synthesized. Yield: 10.10 g (83.11%)Poly(α-azido caprolactone-co-caprolactone) (P(ACL-co-CL)) was obtained through the conversion of α-bromide in P(BCL-co-CL) by NaN3. A typical procedure is as follows. P(BCL-co-CL)29 (8.6973 g, 2.33 mmol) was added into a round bottom flask with 25 mL DMF and sodium azide (4.3979 g, 67.65 mmol). The reaction was carried out at room temperature for 8 hours. After then, 40 mL CH2Cl2 was added into the flask to dissolve the polymer and washed with 30 mL water twice. The organic layer was separated and the solvent was volatilized by rotary evaporation. The white powder polymer was dried under vacuum overnight to a constant weight. Yield: 5.30 g (63.34%).
The macroinitiator P(ACL-co-CL)-Br for the ATRP of MPC was prepared by the esterification of the end hydroxyl groups of P(ACL-co-CL) with 2-bromoisobutyryl bromide. P(ACL-co-CL)29 (5.2106 g, 1 mmol) and triethylamine (0.44 g, 0.010 mol) were dissolved in methylbenzene. 2-Bromoisobutyryl bromide (0.9935 g, 4 mmol) was added dropwise to the abovementioned solution under N2 at −20 °C. After that, the reaction was performed at room temperature for 48 h. The reaction mixture was filtered through a short layer of neutral alumina to remove the quaternary ammonium salts. After condensing the filtrate by rotary evaporation, the product was precipitated in cold methanol and then dried under vacuum to a constant weight. The white powder polymer P(ACL-co-CL)29-Br was obtained. Yield: 5.38 g, 91.86%.
2.3 Characterization of the polymers
1H NMR spectra were recorded on a spectrometer operating at 400 MHz (Unity Inova 400). CDCl3 and DMSO-d6 were used as the solvents. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet Magna 560 spectrometer using the KBr disk method. The UV-Vis absorption spectra were recorded on a UV-Vis spectrophotometer (SPECORD PLUS 200, Analytik Jena). The average molecular weights and molecular weight distributions (PDIs) of the polymers were analyzed using a gel permeation chromatography (GPC) instrument (Waters 1515). The measurements were performed using THF as the eluent at a flow rate of 0.6 mL min−1 at 40 °C and a series of narrow polystyrene standards for the calibration of the columns. Differential Scanning Calorimetry (DSC) was performed on a DSC-100 instrument (Japan) at a heating rate of 10 °C min−1 in the −20 °C to 100 °C range under a steady flow of nitrogen.2.4 Preparation of the micelles
The micelles of the precursor copolymers and the SN38 prodrugs were all prepared through a solvent evaporation method. 10 mg of P(ACL-co-CL)-b-PMPC or P(CL/CL-g-SN38)-b-PMPC were dissolved in 2 mL solvent mixture of THF and methanol (CH3OH) (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The stock copolymer solution was added to 6 mL of ultra-purified water at a speed of 100 mL min−1. The solvent was evaporated overnight at room temperature and the final volume was readjusted to 10 mL, and the polymeric micelles (1 mg mL−1) were obtained.
:1). The stock copolymer solution was added to 6 mL of ultra-purified water at a speed of 100 mL min−1. The solvent was evaporated overnight at room temperature and the final volume was readjusted to 10 mL, and the polymeric micelles (1 mg mL−1) were obtained.
The critical micelle concentration (CMC) of the copolymers in water was estimated by using pyrene as a hydrophobic fluorescent probe. A series of micellar solutions with different concentrations were prepared. Fluorescence spectra of pyrene were recorded using a fluorescence spectrophotometer (F-7000, Hitachi Co., Ltd) at room temperature. Emission was carried out at 395 nm, and excitation spectra were recorded in the range of 240 nm to 360 nm. The CMC was estimated as the crosspoint when extrapolating the intensity ratios I337/I334 in low and high concentration regions.
Dynamic light scattering (DLS) measurements were performed to determine the micellar size and distribution using a Malvern Zetasizer Nano Series equipped with DLS software and a 4 mW He–Ne laser at 633 nm. Analysis was performed at an angle of 173° and a constant temperature of 25 °C. The morphologies of the micelles were observed using a transmission electron microscope (TEM). Specimens for TEM observation were prepared by dropping 10 μL of 0.02 mg mL−1 micelle suspension on the copper grid, followed by staining with phosphotungstic acid (1 wt%). Then the copper grid was frozen using liquid nitrogen, and finally the samples were freeze dried. The TEM images of the micelles were obtained using a Tecnai G2 F20 S-TWIN field emission transmission electron microscope operated at 200 kV.
The drug conjugation capacity (DCC) and drug grafting efficiency (DGE) were determined by ultraviolet spectroscopy and calculated using the following equations:
| DCC = Mass of the drug grafted in the micelles/mass of the micelles × 100% | (1) |
| DGE = Mass of the drug grafted in the micelles/mass of the drug used for the micelles × 100% | (2) |
2.5 In vitro SN38 release from P(CL/CL-g-SN38)-b-PMPC
The release of SN38 from the P(CL/CL-g-SN38)-b-PMPC micelles with an equivalent SN38 concentration of 50 μg mL−1 was investigated at 37 °C in two kinds of buffer solutions: (a) phosphate buffer solution (pH 7.4), and (b) phosphate buffer solution (pH 7.4) with 2 mM PLE (pig liver esterase).In brief, a P(CL/CL-g-SN38)-b-PMPC micelle solution (3 mL) was added into a dialysis bag. The bag was sealed and placed into 100 mL PBS (pH = 7.4 or pH = 7.4 + PLE). The experiment was performed at 37 °C with gentle shaking. At a pre-determined time interval, 0.2 mL of the micelle solution was taken out and dissolved in 1.8 mL solvent. The concentration of SN38 was analyzed using a UV/visible spectrophotometer at an absorbance wavelength at 370 nm. The drug released was calculated by the following formula:
| SN38 release (%) = (1 − Wr/Wt) × 100% | (3) |
2.6 In vitro cell cytotoxicity
L929, MCF-7 and 4T1 cells were obtained from Huaxi Medical Research Center of Sichuan University. The cells were cultured in RPMI 1640 culture medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C under a humidified atmosphere containing 5% CO2, and used in their growth state. A standard methyl thiazolyl tetrazolium (MTT) assay was used to evaluate the cytotoxicity of P(ACL-co-CL)-PMPC and to measure the half maximal inhibitory concentration (IC50) of P(CL/CL-g-SN38)-b-PMPC and free SN38.The cells were seeded into 96-well plates using 200 μL of 1.5 × 104 cells per mL per well and were cultured for 24 h. Then the culture medium was replaced with fresh culture medium or with fresh culture medium containing the micelles or free SN38 at the same concentration. At 48 h, 20 μL of an MTT solution (5 mg mL−1) was added to each well. After 4 h incubation at 37 °C, the MTT solution was replaced with 150 μL dimethyl sulfoxide (DMSO) per well, followed by 10 min shaking. The optical density (OD) of each well was determined using a microplate reader at a wavelength of 370 nm. The cells cultured without micelles were set as the blank control. The cell viability was calculated according to the following formula:
| Cell viability (%) = ODt/ODb × 100% | (4) |
2.7 Evaluation of pharmacokinetics in vivo
Sprague Dawley rats weighing 200 to 220 g were purchased from Chengdu Dossy Experimental Animals Co. Ltd (Chengdu, China). All animal experiments comply with the requirements of the Institute's Animal Care and Use Committee. Prior to the experiment, the mice were fasted for 12 hours but were allowed free access to water. Free SN38 was dissolved in a mixture solution (L(+)-arginine and normal saline) to form a free SN38 solution for injection, and the P(CL/CL-g-SN38)-b-PMPC micelles were solubilized in normal saline. The free SN38 solution and the SN38-conjugated micelles were administered intravenously via the tail vein at a dosage of 5 mg SN38 per kg (n = 3 for each group). At different time points (0.5, 1, 2, 4, 8, 12 and 24 h post-injection), 250 μL blood was collected via the tail vein into heparinized tubes. Then the samples were centrifuged immediately at 4000 rpm for 5 min at 4 °C to obtain plasma. 100 μL of plasma was added into 0.5 mL of chloroform/isopropanol (4:1, v/v) and was mixed by vortex for 120 s, and then centrifuged at 4 °C at 10000 rpm for 5 min. The organic phase was separated and dried under a nitrogen atmosphere at room temperature overnight. The residue was dissolved in 1 mL of DMSO. After centrifugation at 10000 rpm for 3 min, the supernatant was tested by fluorescence spectroscopy at excitation/emission wavelengths of 370/540 nm to calculate SN38 concentration. Non-compartmental pharmacokinetic analysis was done using the Drug and Statistics (DAS) software (DAS 2.0, Mathematical Pharmacology Professional Committee, People's Republic of China) by fitting to a two compartment model.
2.8 In vivo anti-tumor efficiency and histological assessment
The animal models were established by injecting 4T1 cells (1.5 × 106 cells suspended in 200 μL cell culture medium) subcutaneously into the lower left flanks of the BALB/C mice. After the volume of the tumor reached about 50–100 mm3, the mice were randomly divided into two groups (n = 6) and given intravenous administration of PBS and the SN38-conjugated micelles (5 mg per kg SN38). Mice were dosed every three days for a total of four times. The tumor volumes and bodyweights were monitored and recorded. At the end of the experiment, the mice were euthanized.2.9 Statistical analysis
Statistical analysis was performed using SPSS statistics software. One way ANOVA with Bonferroni multiple comparison tests was used to assess the statistical significance. All data are presented as mean ± SD, with p values of <0.05 indicating the statistical significance.3. Results and discussion
3.1 Synthesis and characterization of the block polymers
Although phosphatidylcholine polymeric micelles have gained much attention because of their good biocompatibility, resistance to protein adsorption and high efficiency of cellular uptake,28,29,33 drug conjugates with amphiphilic phosphorylcholine polymers have been scarcely been reported. So, in this work, poly(caprolactone/caprolactone-grift-SN38)-b-poly(2-methacryloyloxyethyl phosphorylcholine), P(CL/CL-g-SN38)-b-PMPC, was designed and synthesized (Scheme 1). The drug SN38 through its alkyne derivative was conjugated to the biodegradable polycaprolactone–polyphosphorylcholine polymer with azide groups (P(ACL-co-CL)-b-PMPC) by a “click reaction”. | ||
| Scheme 1 Synthetic route to P(CL/CL-g-SN38)-PMPC. | ||
P(ACL-co-CL)-PMPC was synthesized by the ATRP of MPC using P(ACL-co-CL)-Br as an initiator, and the latter was prepared by the esterification of poly(α-azido-caprolactone-co-caprolactone) (P(ACL-co-CL)) with 2-bromoisobutyryl bromide. P(ACL-co-CL) was obtained through the conversion of the bromine groups in poly(α-bromine caprolactone-co-caprolactone) (P(BCL-co-CL)) by NaN3, which was synthesized through the ring-opening polymerization of 3-caprolactone and α-BrCL using dodecanol as the initiator and Sn(Oct)2 as the catalyst.
The 1H NMR and FT-IR spectra of P(ACL-co-CL)-b-PMPC, SN38–alkyne and the final product P(CL/CL-g-SN38)-b-PMPC are shown in Fig. 1 and 2. The 1H NMR spectra and the FT-IR spectra of the precursors of the target copolymers are presented in Fig. S1 and S2 (ESI†).
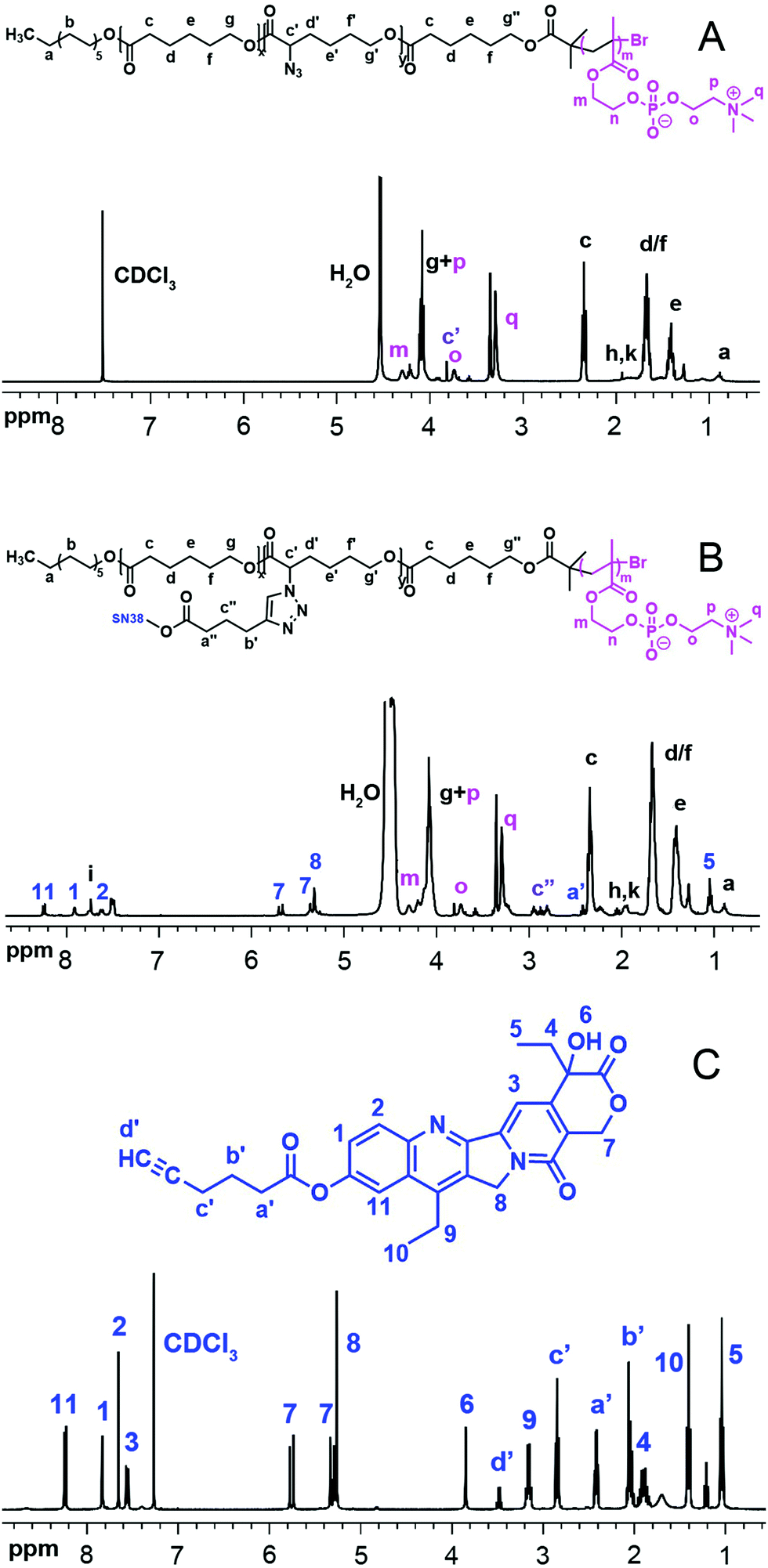 | ||
| Fig. 1
1H NMR of P(ACL-co-CL)-b-PMPC (A), P(CL-g-SN38)-PMPC (B) in CDCl3/CD3OD (V/V = 2:1) and SN38–alkyne (C). | ||
The total degree of polymerization (DP) for polycaprolactone in all the polymers was determined by 1H NMR, as shown in Fig. S1(A) (ESI†), from the integral ratio of peak g′′ at 3.65 ppm (methylene protons neighbouring OH in end CL) to peak c at 2.3 ppm (methylene protons neighbouring the carbonyl group in CL). The calculated DP of CL in the copolymers was 29 and 48, respectively. Peak g at 4.2 ppm was attributed to the methenyl proton of the α-BrCL moieties. Thus, the α-BrCL molar fraction was determined using the integral ratio of the peak at 4.2 ppm to peak c at 2.3 ppm to be around 10% of sum units. The molar fraction also would be the maximum molar fraction for conjugated SN38. As shown in Fig. S1(B) (ESI†), the movement of the peak of the methenyl proton of α-BrCL to 3.83 ppm from 4.2 ppm indicates the successful nucleophilic substitution reaction of P(BCL-co-CL)-OH with NaN3. The obtained macroinitiator P(ACL-co-CL)-Br used for the synthesis of P(ACL-co-CL)-b-PMPC was proved by complete disappearance of peak g′′ at 3.65 ppm and appearance of the m peak of the methyl protons at 1.94 ppm in Fig. S1(C) (ESI†). The FT-IR spectra in Fig. S2 (ESI†) were further used to determine the structures of these precursors.
After the ATRP of MPC using P(ACL-co-CL)-Br as an initiator, the 1H NMR spectrum of the final product was recorded and shown in Fig. 1. P(CL/CL-g-SN38)-b-PMPC (Fig. 1(B)), similar to its precursor P(CL-co-ACL)-b-PMPC (Fig. 1(A)), showed the characteristic peaks assigned to PMPC (δ 4.32, –COOC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2CH2O–; δ 4.21, –COOCH2C2O–; δ 3.74, –OC2CH2N+(CH3)3; δ 3.29, –OCH2CH2N+(C3)3). The unit number of MPC in both amphiphilic phosphorylcholine polymers was calculated using the integral ratio of peak o at 3.74 ppm to peak c at 2.3 ppm. The calculated DP of MPC in the copolymers was 10. Specifically, compared with the 1H NMR spectrum of P(CL-co-ACL)-b-PMPC, new peak i at δ 7.60 ppm in Fig. 1(B) appeared, attributed to the proton of the triazole ring, meanwhile, the characterization peaks of SN38 were clearly detected. The characterization peaks of SN38–alkyne are also shown in Fig. 1(C).
2CH2O–; δ 4.21, –COOCH2C2O–; δ 3.74, –OC2CH2N+(CH3)3; δ 3.29, –OCH2CH2N+(C3)3). The unit number of MPC in both amphiphilic phosphorylcholine polymers was calculated using the integral ratio of peak o at 3.74 ppm to peak c at 2.3 ppm. The calculated DP of MPC in the copolymers was 10. Specifically, compared with the 1H NMR spectrum of P(CL-co-ACL)-b-PMPC, new peak i at δ 7.60 ppm in Fig. 1(B) appeared, attributed to the proton of the triazole ring, meanwhile, the characterization peaks of SN38 were clearly detected. The characterization peaks of SN38–alkyne are also shown in Fig. 1(C).
The structure of the prodrug was further confirmed by FT-IR (Fig. 2). In the spectrum of P(CL/CL-g-SN38)-b-PMPC, there were strong peaks at ∼1728 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the ester group in the PCL block and absorptions at 1240, 1089 (–POCH2–) and 970 cm−1 (–N+(CH3)3) from PMPC, indicating the coexistence of the PCL and PMPC blocks, which was similar to that for P(ACL-co-CL)-b-PMPC. However, the νa peak at 2100 cm−1 of the azide in Fig. 2(B) disappeared in Fig. 2(A), hinting the conjugation of P(ACL-co-CL)-b-PMPC with clickable SN38–alkyne showing the absorption peak of the C
O of the ester group in the PCL block and absorptions at 1240, 1089 (–POCH2–) and 970 cm−1 (–N+(CH3)3) from PMPC, indicating the coexistence of the PCL and PMPC blocks, which was similar to that for P(ACL-co-CL)-b-PMPC. However, the νa peak at 2100 cm−1 of the azide in Fig. 2(B) disappeared in Fig. 2(A), hinting the conjugation of P(ACL-co-CL)-b-PMPC with clickable SN38–alkyne showing the absorption peak of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C group at 2122 cm−1 in Fig. 2(C).
C group at 2122 cm−1 in Fig. 2(C).
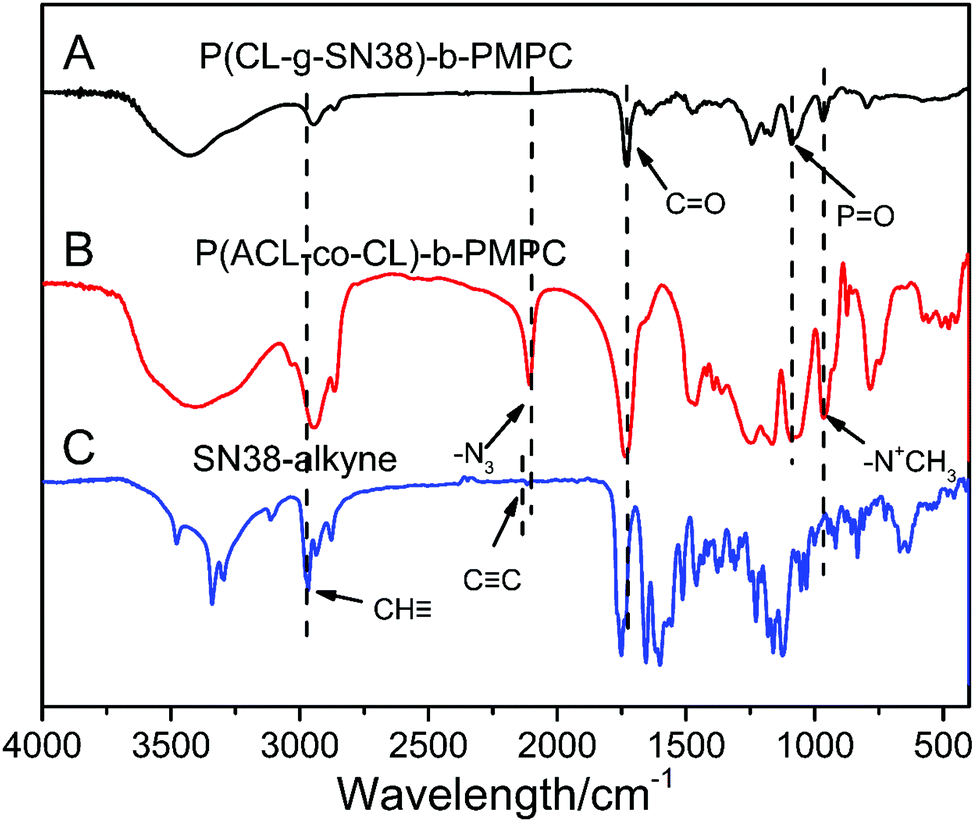 | ||
| Fig. 2 FT-IR of P(CL-g-SN38)-PMPC (A), P(ACL-co-CL)-b-PMPC (B) and SN38–alkyne (C). | ||
The GPC trace of P(CL-g-SN38)-PMPC was considered for measurement, however since the PCL block is hydrophobic and cannot dissolve in water, and the PMPC block is soluble only in water or alcohols and is not soluble in other organic solvents such as THF, DMF, CHCl3 and so on, GPC analysis did not proceed for P(ACL-co-CL)-b-PMPC or P(CL-g-SN38)-PMPC. Nonetheless, the macroinitiators P(CL-co-ACL)-Br were characterized by GPC (Fig. S3, ESI†). The results of GPC analysis revealed that P(CL-co-ACL)-Br presented a single peak and narrow distribution, in detail, Mn of P(CL-co-ACL)29-Br was 7609 and Mw/Mn was 1.29, meanwhile Mn of P(CL-co-ACL)48-Br was 11295 and Mw/Mn was 1.46.
In addition, the crystallization behaviors of these polymers were studied by DSC. The data are listed in Table 1 and DSC curves are shown in Fig. S4 (ESI†). The PCL block was semicrystalline, and its Tm (melting temperature) was around 60 °C. The P(BCL-co-CL)-OH copolymers showed two Tm values because of the different Tm values of α-BrCL and ε-CL. In addition, with a higher DP of the hydrophobic chain, the Tm and crystallinity were higher. The macroinitiators P(CL-co-ACL)-Br displayed lower Tm than precursor P(BCL-co-CL)-OH, but presented much higher crystallization degree than P(CL-co-ACL)-b-PMPC, since introducing a PMPC block into PCL limits the movement of the PCL block.34 When SN38 was grafted to P(CL-co-ACL)-b-PMPC, the Tm and crystallinity totally reduced since P(CL/CL-g-SN38) -b-PMC had large side SN38 groups.
| Structurea | T c (°C) | T m1 (°C) | T m2 (°C) | ΔHf (J g−1) | X c (%) |
|---|---|---|---|---|---|
| a Calculated from 1H NMR. b “—” means that the item does not exist or could not be measured for the sample. | |||||
| P(CL-co-BCL0.1)29-OH | 8.53 | 37.4 | 43.55 | 32.09 | 23.04 |
| P(CL-co-BCL0.1)48-OH | 10.06 | 39.11 | 45.51 | 55.97 | 40.18 |
| P(CL-co-ACL0.1)29-Br | 12.24 | 33.05 | 39.64 | 38.61 | 27.72 |
| P(CL-co-ACL0.1)48-Br | 7.37 | 35.36 | 44.52 | 49.3 | 35.39 |
| P(CL-co-ACL0.1)29-b-PMPC10 | 10.89 | 32.86 | 44.14 | 13.81 | 9.91 |
| P(CL-co-ACL0.1)48-b-PMPC10 | 6.71 | 35.7 | 44.82 | 20.11 | 14.44 |
| P(CL/CL-g-SN380.1)29-b-PMPC10 | —b | — | — | — | — |
| P(CL/CL-g-SN380.1)48-b-PMPC10 | — | — | — | — | — |
The above several characterization results can show the successful synthesis of the SN38 conjugates of amphiphilic phosphorylcholine polymers.
3.2 Characterization of the SN38 prodrug micelles
SN38 prodrug micelles were prepared by a solvent evaporation method. Briefly, 10 mg P(CL/CL-g-SN38)-b-PMPC or their precursors were dissolved in 2 mL solvent mixture (THF/CH3OH = 1/1) and the solution was dispersed in water by ultrasonication. The organic phase was evaporated in room temperature to obtain SN38 prodrug micelles.The critical micelle concentration (CMC) of P(CL-co-ACL)-b-PMPC and P(CL/CL-g-SN38)-b-PMPC was measured using their pyrene excitation spectra. As shown in Fig. 3 and in Table 2, the CMC values of all micelles were around 0.27–8 mg mL−1, indicating that the micelles could be easily formed. And the value of the P(CL-co-ACL)48-b-PMPC10 micelles was lower than that of the P(CL-co-ACL)29-b-PMPC10 micelles. This is due to the increase of the hydrophobicity/hydrophilicity ratio with more portions of PCL. The CMC values of both SN38 prodrug micelles were one-tenth lower than those of their precursor micelles. The reasonable explanation for this phenomenon was that the conjugation of hydrophobic SN38 has significantly increased the hydrophobicity of P(CL/CL-g-SN38)-b-PMPC.
 | ||
| Fig. 3 CMC curves for P(ACL-co-CL)29-b-PMPC10 (A), P(ACL-co-CL)48-b-PMPC10 (B), P(CL/CL-g-SN38)29-PMPC10 (C) and P(CL/CL-g-SN38)48-PMPC10 (D). | ||
| Sample | Zeta (mV) | Size (nm) | PDI | DCC/SN38a,d (%) | DGE/SN38b,d (%) | CMC (mg L−1) |
|---|---|---|---|---|---|---|
| a Drug conjugation capacity. b Drug grafting efficiencies. c “—” means that the item does not exist or could not be measured for the sample. d The theoretical values. | ||||||
| P(ACL-co-CL)48-PMPC10 | −15.5 ± 0.7 | 143.2 ± 3.1 | 0.10 ± 0.01 | —c | — | 5.37 |
| P(ACL-co-CL)29-PMPC10 | −10.4 ± 0.8 | 153.1 ± 2.9 | 0.19 ± 0.01 | — | — | 7.94 |
| P(CL/CL-g-SN38)48-PMPC10 | −13.9 ± 0.3 | 196.0 ± 7.8 | 0.20 ± 0.04 | 12.76/16.38 | 75.55/100 | 0.52 |
| P(CL/CL-g-SN38)29-PMPC10 | −8.4 ± 2.4 | 236.5 ± 6.1 | 0.16 ± 0.03 | 12.70/13.76 | 98.91/100 | 0.27 |
As shown in Table 2, both SN38 prodrug micelles showed more than 12% drug conjugation capacity (DCC) and higher than 75% drug grafting efficiencies (DGE). The DCC and DGE were almost the same or higher than the SN38 encapsulation efficiencies in PLGA nanoparticles.35 The high DCC and DGE may be because there are physically entrapped SN38 in the SN38 prodrug micelles and they are difficult to separate from the micelles. In any case, the method of conjugating drugs to amphiphilic polymers is suitable for SN38 with poor aqueous solubility and instability at physiological pH and low encapsulation in the amphiphilic polymers.
The size, size distribution and zeta-potentials were measured by DLS. The results showed that all the micelles were less than 240 nm, and the diameters of the precursor polymer micelles were lower than those of the drug-conjugate micelles, and their size distributions were narrow. These properties showed that the P(CL-co-ACL)-b-PMPC and P(CL/CL-g-SN38)-b-PMPC micelles were suitable to be used through intravenous injection.
Fig. 4 shows the typical size distribution peaks and the TEM images of the P(CL-co-ACL)-b-PMPC and P(CL/CL-g-SN38)-b-PMPC (0.2 mg mL−1 in water) micelles. Both micelles displayed single peak distribution, and the size of P(CL/CL-g-SN38)-b-PMPC was larger than that of P(CL-co-ACL)-b-PMPC. The morphologies of these micelles observed by TEM clearly exhibited spherical morphologies, and the micelles had almost uniform sizes, and the average size was in agreement with the results of the DLS experiment.
 | ||
| Fig. 4 The sizes and size distribution of the P(ACL-co-CL)-PMPC, and P(CL/CL-g-SN38)-PMPC micelles by DLS (up); the TEM images of the P(ACL-co-CL)-PMPC and P(CL/CL-g-SN38)-PMPC micelles (down). | ||
3.3 In vitro drug release
The drug release properties of the SN38 prodrug micelles at physiological pH (7.4) at 37 °C and at physiological pH (7.4) with PLE (pig liver esterase) were studied. As shown in Fig. 5, at pH 7.4,about 43% of SN38 was released from the P(CL/CL-g-SN38)29-b-PMPC10 and P(CL/CL-g-SN38)48-b-PMPC10 micelles within the first 12 h. After then, the accumulative drug release of SN38 from the two micelles was quite less, within 5% over 70 h. The fast release of SN38 may be due to the presence of the free drug embedded in the SN38 prodrug micelles as we mentioned before. The slight release after the initial burst was attributed to the conjugation of SN38 to the amphiphilic phosphorylcholine polymer.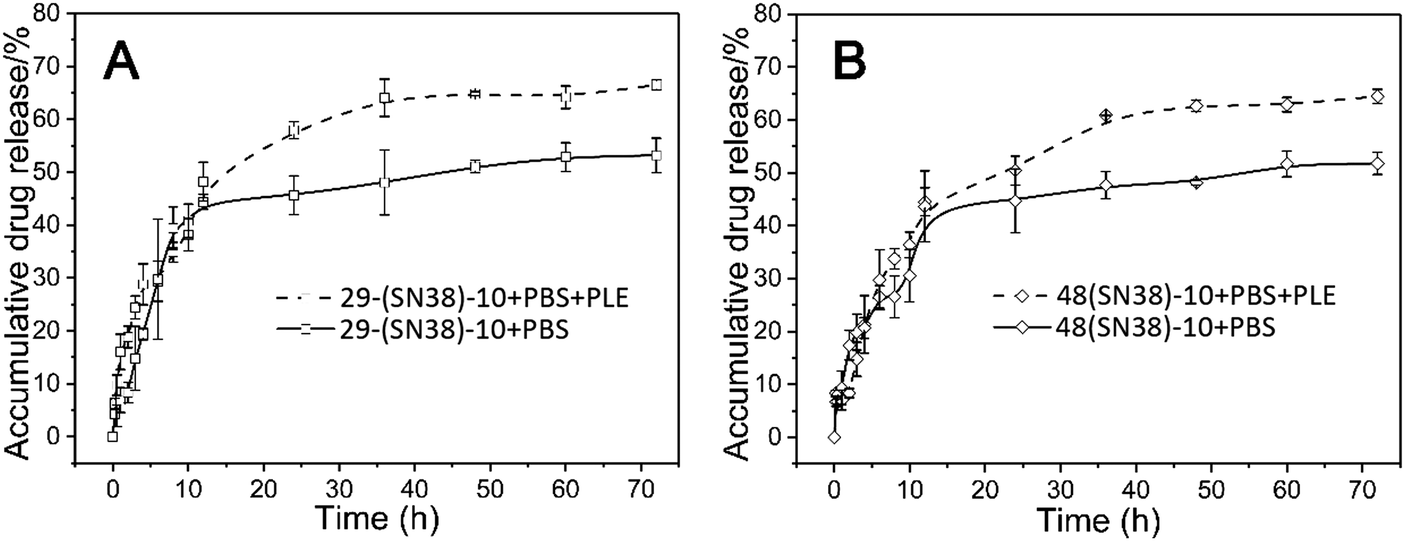 | ||
| Fig. 5 In vitro drug release of the P(CL/CL-g-SN38)29-b-PMPC10 (A) and P(CL/CL-g-SN38)48-b-PMPC10 (B) micelles in PBS and PBS + PLE (PLE: pig liver esterase). | ||
Esterases are known to be abundant in cytosol and lysosomes. Thus, although the ester linkage between SN38 and the amphiphilic phosphorylcholine polymer is stable during transport in blood circulation, once SN38 prodrug micelles enter into the cells, the ester linkage between SN38 and the polymer will be easily cleaved on account of exposure to intracellular esterase, resulting in the release of SN38. In order to provide a more realistic simulation of drug release, 0.2 mmol L−1 PLE (pig liver esterase) was added into PBS (pH 7.4), which led to the cleavage of the phenyl ester of SN38 in P(CL/CL-g-SN38)-b-PMPC and accelerated SN38 release from the amphiphilic phosphorylcholine polymers. As shown in Fig. 5, after addition of PLE, the accumulative drug release of SN38 from these two micelles reached about 70% in 70 h. Therefore, the conjugation of SN38 to the amphiphilic phosphorylcholine polymer was stable during transport in blood circulation, except the SN38 release of the free part embedded in the micelles was similar to that physically entrapped in traditional amphiphilic micelles.36,37
3.4 Cell cytotoxicity
The cytotoxicity of the SN38 prodrug micelles to mouse fibroblast cells (L929), human breast cancer cells (MCF-7) and mouse breast cancer cells (4T1) was evaluated by an MTT assay. Different from no cytotoxicity of their precursor micelles to normal cells (L929) or cancer cells (Fig. S5(A–C), ESI†), the SN38-conjugate (such as P(CL/CL-g-SN38)48-b-PMPC10) micelles displayed concentration dependent cytotoxicity, as shown in Fig. 6(A and B). With increasing concentration of SN38, the inhibition of the SN38 prodrug micelles towards cancer cells was stronger. The reason is that the main components of the micelles are PCL and PMPC, which have been confirmed to be highly biocompatible in previous studies,33,34 but SN38 demonstrates significant anti-tumor activity. The half-maximal inhibitory concentration (IC50) values are summarized in Table S1 (ESI†). The IC50 of the SN38 prodrug micelles for MCF-7 cells was low, around 0.27, while 24.06 for 4T1 cells. The reasonable interpretation for these values being higher than those of free SN38 is that SN38 conjugated to the polymer do not all cleave in 48 h cell incubation. Even so, the P(CL/CL-g-SN38)-b-PMPC micelles showed very strong lethality to the cancer cells. | ||
| Fig. 6 In vitro toxicity of the SN38 and SN38 prodrug micelles evaluated by MTT with MCF-7 cells (A) and 4T1 cells (B) under 48 h incubation. | ||
3.5 Evaluation of pharmacokinetics in vivo
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sichuan University and approved by the Animal Ethics Committee of West China Hospital, Sichuan University (Chengdu, China). To improve the systemic bioavailability of SN38 in vivo, SN38 prodrugs were designed, and the prodrug micelles were expected to be obviously superior to free SN38 solutions. SN38 has poor water solubility so that it cannot be directly used for the experiment. In order to conduct a pharmacokinetics study, arginine was used to solubilize SN38. A standard curve of SN38 in blood was obtained, and there was a good linear correlation (R2 = 0.9999) between the fluorescence intensity and the SN38 concentration in the range of 0–1 μg mL−1 (Fig. S6, ESI†).The pharmacokinetic study was performed by intravenous injection of 1 mg mL−1 free SN38 and the SN38 prodrug micelles at an equivalent SN38 dose of 5 mg kg−1. The profiles of the SN38 concentration in plasma of the Sprague Dawley rats versus time are shown in Fig. 7, and the pharmacokinetic parameters were calculated by DAS 2.0 software using a two-compartment model as listed in Table 3. According to the data, the SN38 prodrug micelles significantly prolonged the retention of SN38 in the circulation system in vivo.
 | ||
| Fig. 7 The pharmacokinetic profiles of the SN38 and SN38 prodrug micelles. | ||
| Parametere | Unit | SN38 | P(CL/CL-g-SN38)48-PMPC10 |
|---|---|---|---|
| a Elimination half-life. b Total volume of distribution. c Total body clearance. d Area under the plasma SN38 concentration–time curves. e These parameters were calculated using DAS 2.0 based on a two compartment model. | |||
| t 1/2α | h | 0.48 | 1.28 |
| t 1/2β | h | 2.29 | 6.64 |
| V1b | L kg−1 | 9.84 | 8.88 |
| CLc | L h−1 kg−1 | 1.64 | 0.96 |
| AUC (0–∞)d | mg L−1 h−1 | 3.06 | 5.23 |
| MRT (0–∞) | h | 19.45 | 27.84 |
| C max | mg L−1 | 0.35 | 0.44 |
After intravenous injection, the SN38 prodrug micelles provided a longer clearance half life (T1/2α) (2.64-fold) and distribution half life (T1/2β) (2.9-fold). The Cmax values of SN38 from the micelles and solution in plasma were 0.437 μg mL−1 and 0.347 μg mL−1, respectively. The area under the curve (AUC(0–∞)) value for the SN38 prodrug micelles was 5.23 mg L−1 h−1, and only 3.06 mg L−1 h−1 was detected in the free SN38 group. Similarly, the mean residence time (MRT(0–∞)) of the SN38 prodrug micelles was longer than that of free SN38, and they were 27.84 and 19.45 h, respectively. Meanwhile, the decreased clearance (CL) indicated that the SN38 prodrug micelles could prolong the acting time of SN38 in vivo. This result could be attributed to not only the conjugation of SN38 to the polymer but also to the protein-resistance properties of the PMPC corona of the micelles.28,38
3.6 Tumor inhibitory efficacy
The in vivo anti-tumor activity of the SN38 prodrug micelles was evaluated in 4T1 xenograft BALB/C mice. They were treated with P(CL/CL-g-SN38)48-PMPC10 with PBS as a control. The results obtained are summarized in Fig. 8. For the PBS group, the tumor volumes increased quite rapidly, while the tumors of the mice that received drug treatment grew constantly with different degrees of inhibition. At 21 days post treatment, the average tumor volumes of the mice treated with P(CL/CL-g-SN38)48-PMPC10 was 32% of those of the PBS control group (Fig. 8A). This result was almost consistent with the changing trend of the tumor weight stripped from the mice at the end of the experiment (Fig. 8B). In addition, the toxic and side effects of SN38 did not emerge for the SN38 prodrug micelle group, since the mice treated with the SN38 prodrug micelles had almost no significant weight loss compared to the PBS control group. In short, the SN38 prodrug micelles displayed good anti-tumor efficiency. | ||
| Fig. 8 The tumor volumes and body weight changes (A) of the mice treated with PBS and the SN38-conjugated micelles; tumor weights (B) of the excised tumors at the end of the experiment. (*p < 0.05, **p < 0.01). | ||
4. Conclusions
In summary, amphiphilic copolymers with capability of conjugating drugs, poly(α-azide caprolactone-co-caprolactone)-b-poly(2-methacryloyloxyethyl phosphorylcholine), P(ACL-co-CL)-b-PMPC were successfully synthesized by the combination of ROP, an azide reaction, end group conversion and ATRP. The camptothecin derivative SN38–alkyne was bonded to the polymer by a “click reaction” to obtain (PCL/CL-g-SN38)-b-PMPC with high drug conjugation capacity and drug grafting efficiency. The SN38-conjugated polymers could self-assemble into micelles of size around 190–240 nm. In vitro drug release showed that the conjugates of SN38 and the amphiphilic phosphorylcholine polymer were stable during transport in blood circulation. The MTT assay demonstrated that the P(ACL-co-CL)-b-PMPC micelles were biocompatible while the SN38 prodrug micelles inhibited MCF-7 and 4T1 cell proliferation effectively. Evaluation of pharmacokinetics in vivo showed that the SN38 prodrug micelles significantly prolonged the retention of SN38 in blood circulation. Finally, the SN38 prodrug micelles demonstrated an in vivo anti-tumor effect and low toxicity to 4T1-bearing BALB/C mice.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 51673129, 51473099). We would also like to thank our laboratory members for their generous help and gratefully acknowledge the Analytical and Testing Center at Sichuan University for testing.References
- Y.-H. Hsiang and L. F. Liu, Cancer Res., 1988, 48, 1722 CAS.
- Y.-H. Hsiang, M. G. Lihou and L. F. Liu, Cancer Res., 1989, 49, 5077 CAS.
- Y.-H. Hsiang, L. F. Liu, M. E. Wall, M. C. Wani, A. W. Nicholas, G. Manikumar, S. Kirschenbaum, R. Silber and M. Potmesil, Cancer Res., 1989, 49, 4385 CAS.
- Y. Kawato, M. Aonuma, Y. Hirota, H. Kuga and K. Sato, Cancer Res., 1991, 51, 4187 CAS.
- N. Saijo, Ann. N. Y. Acad. Sci., 2006, 922, 92–99 CrossRef.
- J. G. Slatter, P. Su, J. P. Sams, L. J. Schaaf and L. C. Wienkers, Drug Metab. Dispos., 1997, 25, 1157 CAS.
- V. Charasson, M.-C. Haaz and J. Robert, Drug Metab. Dispos., 2002, 30, 731 CrossRef CAS.
- N. F. Smith, W. D. Figg and A. Sparreboom, Toxicol. In Vitro, 2006, 20, 163–175 CrossRef CAS.
- A. Wang and S. Li, BMC Biotechnol., 2008, 8, 46 CrossRef.
- Y. Matsumura, Adv. Drug Delivery Rev., 2011, 63, 184–192 CrossRef CAS.
- N. Vijayalakshmi, A. Ray, A. Malugin and H. Ghandehari, Bioconjugate Chem., 2010, 21, 1804–1810 CrossRef CAS.
- J. A. Zhang, T. Xuan, M. Parmar, L. Ma, S. Ugwu, S. Ali and I. Ahmad, Int. J. Pharm., 2004, 270, 93–107 CrossRef CAS.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS.
- P. Ebrahimnejad, R. Dinarvand, A. Sajadi, M. R. Jaafari, A. R. Nomani, E. Azizi, M. Rad-Malekshahi and F. Atyabi, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 478–485 CrossRef CAS.
- K. K. Vangara, J. L. Liu and S. Palakurthi, Anticancer Res., 2013, 33, 2425–2434 CAS.
- N. Mosallaei, A. Mahmoudi, H. Ghandehari, V. K. Yellepeddi, M. R. Jaafari and B. Malaekeh-Nikouei, Eur. J. Pharm. Biopharm., 2016, 104, 42–50 CrossRef CAS.
- A. Takahashi, N. Ohkochi, M. Yasunaga, J. Kuroda, Y. Koga, H. Kenmotsu, T. Kinoshita and Y. Matsumura, Clin. Cancer Res., 2010, 16, 4822–4831 CrossRef CAS.
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202 CrossRef CAS.
- A. Carie, J. Rios-Doria, T. Costich, B. Burke, R. Slama, H. Skaff and K. Sill, J. Drug Delivery, 2011, 2011, 869027 Search PubMed.
- W. Ying, G. Miao, L. Yu, D. Li-Ying, R. Wen-Ting, L. Ya-Qing, S. Fei-Fei and Y. Shu-Qin, Nanotechnology, 2012, 23, 495103 CrossRef.
- X. Wang, C. Wang, X. Wang, Y. Wang, Q. Zhang and Y. Cheng, Chem. Mater., 2017, 29, 1370–1376 CrossRef CAS.
- A. Patnaik, K. P. Papadopoulos, A. W. Tolcher, M. Beeram, S. Urien, L. J. Schaaf, S. Tahiri, T. Bekaii-Saab, F. M. Lokiec, K. Rezaï and A. Buchbinder, Cancer Chemother. Pharmacol., 2013, 71, 1499–1506 CrossRef CAS.
- Y. Shi, T. Lammers, G. Storm and W. E. Hennink, Macromol. Biosci., 2016, 17, 1600160 CrossRef.
- L. Lu, Y. Zheng, S. Weng, W. Zhu, J. Chen, X. Zhang, R. J. Lee, B. Yu, H. Jia and L. Qin, Colloids Surf., B, 2016, 142, 417–423 CrossRef CAS.
- C. C. Williams, S. H. Thang, T. Hantke, U. Vogel, P. H. Seeberger, J. Tsanaktsidis and B. Lepenies, ChemMedChem, 2011, 7, 281–291 CrossRef.
- R. Matsuno and K. Ishihara, Nano Today, 2011, 6, 61–74 CrossRef CAS.
- J. P. Salvage, T. Smith, T. Lu, A. Sanghera, G. Standen, Y. Tang and A. L. Lewis, Appl. Nanosci., 2016, 6, 1073–1094 CrossRef CAS.
- Q. Jin, Y. Chen, Y. Wang and J. Ji, Colloids Surf., B, 2014, 124, 80–86 CrossRef CAS.
- J. Zhao, Y.-D. Chai, J. Zhang, P.-F. Huang, K. Nakashima and Y.-K. Gong, Acta Biomater., 2015, 16, 94–102 CrossRef CAS.
- J. P. Salvage, C. Thom, A. L. Lewis, G. J. Phillips and A. W. Lloyd, J. Mater. Sci.: Mater. Med., 2015, 26, 150 CrossRef.
- G. Liu, W. Zhuang, X. Chen, A. Yin, Y. Nie and Y. Wang, Polymer, 2016, 100, 45–55 CrossRef CAS.
- M. Cai, J. Cao, Z. Wu, F. Cheng, Y. Chen and X. Luo, Colloids Surf., B, 2017, 157, 268–279 CrossRef CAS.
- M. Cai, Z. Wu, Y. Li, J. Cao, Y. Chen and X. Luo, Mater. Sci. Eng. Carbon, 2018, 89, 401–412 CrossRef CAS.
- Z. Wu, M. Cai, J. Cao, J. Zhang and X. Luo, Int. J. Nanomed., 2017, 12, 487–500 CrossRef CAS.
- N. Sepehri, H. Rouhani, F. Tavassolian, H. Montazeri, M. R. Khoshayand, M. H. Ghahremani, S. N. Ostad, F. Atyabi and R. Dinarvand, Int. J. Pharm., 2014, 471, 485–497 CrossRef CAS.
- K. Duan, X. Zhang, X. Tang, J. Yu, S. Liu, D. Wang, Y. Li and J. Huang, Colloids Surf., B, 2010, 76, 475–482 CrossRef CAS.
- Q. Guo, P. Luo, Y. Luo, F. Du, W. Lu, S. Liu, J. Huang and J. Yu, Colloids Surf., B, 2012, 100, 138–145 CrossRef CAS PubMed.
- L. Liu, W. Li and Q. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 599–614 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj04908d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |