
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hot injection synthesis of CuInS2 nanocrystals using metal xanthates and their application in hybrid solar cells†
Verena
Perner
a,
Thomas
Rath
 *a,
Franz
Pirolt
a,
Otto
Glatter
a,
Karin
Wewerka
b,
Ilse
Letofsky-Papst
b,
Peter
Zach
c,
Mathias
Hobisch
d,
Birgit
Kunert
e and
Gregor
Trimmel
a
*a,
Franz
Pirolt
a,
Otto
Glatter
a,
Karin
Wewerka
b,
Ilse
Letofsky-Papst
b,
Peter
Zach
c,
Mathias
Hobisch
d,
Birgit
Kunert
e and
Gregor
Trimmel
a
aInstitute for Chemistry and Technology of Materials (ICTM), NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at
bInstitute for Electron Microscopy and Nanoanalysis and Center for Electron Microscopy, Graz University of Technology, NAWI Graz, Steyrergasse 17, 8010 Graz, Austria
cInstitute of Analytical Chemistry and Food Chemistry, NAWI Graz, Graz University of Technology, Graz, Austria, Stremayrgasse 9, 8010 Graz, Austria
dInstitute of Paper, Pulp and Fibre Technology, Graz University of Technology, Inffeldgasse 23, 8010 Graz, Austria
eInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
First published on 23rd November 2018
Abstract
Copper indium sulfide (CuInS2) nanocrystals with a size of 3–4 nm and a chalcopyrite crystal structure were synthesized from copper and indium xanthates as precursors in a hot injection synthesis performed at a temperature of 200 °C. Dioleamide molecules served as stabilizing ligands and were exchanged to 1-hexanethiol after the synthesis. TEM images reveal that after the ligand exchange process, the inter-particle distance is significantly reduced and the nanocrystals agglomerate slightly, which is also indicated by small angle X-ray scattering and AFM measurements. Information about charge transfer between the conjugated polymer PCDTBT and the nanocrystals was gained from photoluminescence quenching measurements. Furthermore, the prepared CuInS2 nanoparticles with an optical band gap of 1.57 eV were applied as acceptors in polymer/nanocrystal bulk heterojunction solar cells and their performance was evaluated. The obtained solar cells showed high open circuit voltages up to 730 mV and overall power conversion efficiencies of 0.23%.
1. Introduction
Copper containing chalcogenide nanocrystals became an exciting research topic because of manifold application possibilities based on their optical and electronic properties.1,2 In addition, their properties can be further tuned through quantum confinement effects based on variations in size and shape and as well through engineering of the chemical composition and the ligand environment.3–9 Copper indium sulfide (CuInS2) nanocrystals are particularly interesting, as they are a potential non-toxic alternative to cadmium or lead containing nanocrystals.1,2,10 Copper indium sulfide possesses a high absorption coefficient, a direct band gap of 1.5 eV11 and a large Stokes shift.12 Thus CuInS2 nanocrystals find applications in biolabelling,13,14 LEDs,15 photocatalysis16 and, in particular also, photovoltaics.17 Regarding solar cell applications, CuInS2 nanocrystals are used in mesoscopic semiconductor sensitized solar cells,8,18,19 nanocrystalline thin film solar cells,20,21 and bulk heterojunction polymer/nanoparticle solar cells.22–29However, compared to Cd- or Pb-containing sulfide or selenide nanocrystals, the application of CuInS2 nanocrystals in bulk heterojunction hybrid solar cells is less researched and the performance of CuInS2 nanocrystal based bulk heterojunction solar cells remained much lower. While power conversion efficiencies (PCEs) up to 5.5% or 6.3% are obtained with PbS,Se nanocrystal/polymer30 or CdTe/polymer31 bulk heterojunction absorber layers, respectively, PCEs lower than 0.1% are reached with ligand exchanged CuInS2 nanocrystals in combination with a conjugated polymer.22–26 Higher efficiencies up to 2.9% were only obtained with ligand-free CuInS2 nanocrystals synthesized directly in the conjugated polymer film.27,32,33 These results reveal that the non-toxic copper indium sulfide nanocrystals have a high potential in hybrid solar cells.
In recent years much research effort was devoted to exploring synthesis strategies and the investigation of the properties of CuInS2 nanocrystals. Synthetic approaches include colloidal synthesis routes (heat up and hot injection syntheses),34 hydrothermal and solvothermal methods35 including microwave assisted syntheses36 as well as in situ formation in polymeric matrices.32 In most cases molecules such as oleylamine,34 oleic acid, octadecene, trioctylphosphine or dodecanethiol are used as ligands and copper and indium salts (e.g. halides, acetates, acetylacetonates) in combination with a sulfur source (e.g. elemental sulfur, thiourea, dodecanethiol) as precursors.1 Furthermore, single source precursors37,38 and precursors which contain the metal and sulfur source in one compound (e.g. dithiocarbamates)39 are already investigated. In this regard, also metal xanthates have been proven to be very suitable.40–45 Examples for a hot injection synthesis of CuInS2 nanoparticles comprise the syntheses from the corresponding copper and indium ethyl xanthates using glycol38 or trioctylamine as solvent and oleylamine/trioctylphosphine as ligands.39 Alternatively, nanocrystals have been isolated via the decomposition of copper and indium xanthates in a mixture of o-dichlorobenzene as solvent and oleylamine/trioctylphosphine40 or poly(3-hexylthiophene) (P3HT) as polymeric ligand.41 Furthermore, a series of xanthates with different alkyl side chains was investigated for the synthesis of CuInS2 nanocrystals with hexagonal and cubic phase by Al-Shakban et al.37 Whereas the formation to the metal sulfide proceeds via the thermally activated Chugaev reaction, the reaction pathway might change in the presence of alkyl amines as we have recently shown for a room temperature synthesis of CuInS2 nanocrystals using oleylamine.42
In this study, we focus on the synthesis of CuInS2 nanocrystals from metal xanthates using dioleamide (oleyl oleamide, N-((Z)-octadec-9-en-1-yl)oleamide) as capping ligand and investigate the nanocrystals as well as the ligand exchange with hexanethiol via X-ray diffraction and scattering measurements, thermogravimetric analysis, IR spectroscopy, and transmission electron microscopy. Furthermore, we evaluate the performance of the ligand-exchanged nanocrystals in polymer/nanocrystal bulk heterojunction solar cells.
2. Experimental
2.1. Sample and solar cell preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone = 1:1 (volume ratio of 10:1 with regard to the ligand exchange solution). The nanoparticles were separated by centrifugation. The brownish supernatant was decanted and the remaining nanoparticles were dissolved in chloroform resulting in a black-brown solution.
:5, 1:9 and 1:15. The PCDTBT/CuInS2 absorber layers were spin coated at a speed of 1500 rpm. Finally, 80–90 nm thick silver cathodes were deposited in a vacuum chamber (8 × 10−6 mbar) via thermal evaporation through a shadow mask. The active area of the devices was 0.09 cm2.
:acetone = 1:1 (volume ratio of 10:1 with regard to the ligand exchange solution). The nanoparticles were separated by centrifugation. The brownish supernatant was decanted and the remaining nanoparticles were dissolved in chloroform resulting in a black-brown solution.
:5, 1:9 and 1:15. The PCDTBT/CuInS2 absorber layers were spin coated at a speed of 1500 rpm. Finally, 80–90 nm thick silver cathodes were deposited in a vacuum chamber (8 × 10−6 mbar) via thermal evaporation through a shadow mask. The active area of the devices was 0.09 cm2.
2.2. Characterisation techniques
X-ray diffraction measurements were conducted on a PANalytical Empyrean diffractometer in Bragg–Brentano configuration operated at 40 kV and 40 mA using Cu Kα radiation. UV-Vis absorption spectra were measured on a Shimadzu 1800 spectrophotometer. Thermogravimetric analyses (TGA) were performed on a Netzsch Jupiter STA 449C. All measurements were carried out in helium atmosphere and at a heating rate of 10 °C min−1 from room temperature to 550 °C. FT-IR spectra were acquired using a Bruker Alpha FT-IR spectrometer in attenuated total reflection (ATR) mode (films on glass substrates, spectral range between 4000 and 400 cm−1). 1H-NMR spectroscopy was carried out on a Bruker Avance 300 MHz spectrometer. CDCl3 was obtained from Cambridge Isotope Laboratories Inc.For transmission electron microscopy (TEM), samples were prepared by dropping a nanoparticle solution (solvent: chloroform) onto a Quantifoil TEM-grid with a carbon film and subsequent evaporation of the solvent at room temperature. TEM images were acquired on a Tecnai F20 microscope (FEI company) at 200 kV acceleration voltage, equipped with a Schottky emitter, an energy dispersive X-ray spectrometer, a monochromator and a Gatan Image Filter (GIF) with an UltraScanCCD camera.
The Small Angle X-Ray Scattering (SAXS) equipment consisted of a high-flux SAXSess camera (Anton Paar, Austria) connected to a Debyeflex 3003 X-ray generator (GE-Electric, Germany), operating at 40 kV and 50 mA with a sealed-tube Cu anode. The Goebel-mirror focused and Kratky-slit collimated X-ray beam was line shaped (17 mm horizontal dimension at the sample). The scattered radiation was measured in transmission mode and recorded by a one-dimensional MYTHEN-1k microstrip solid-state detector (Dectris, Switzerland) within a q-range (with q being the magnitude of the scattering vector) of 0.01 to 0.5 Å−1. Using Cu Kα radiation of a wavelength of 1.54 Å and a sample-to-detector distance of 307 mm, this corresponds to a total 2θ region of 0.14° to 7°, applying the conversion q = 4π(sinθ)/λ with 2θ being the scattering angle with respect to the incident beam and λ the wavelength of the X-rays. Capillaries of 1.0 mm diameter were used and the exposure was set to 60 s and repeated 5 times. The alignment of the apparatus allowed a resolution of 47.24 nm diameter (qmin = 0.0665 nm−1). 90% of the toluene curve was subtracted from the sample files and the resulting curves were binned to decrease the amount of data points. The treated data files were evaluated using the Indirect Fourier Transformation Method.46
Photoluminescence spectra were measured in ambient atmosphere on a FluoroLog 3 spectrofluorometer from Horiba Scientific equipped with a NIR-sensitive R2658 photomultiplier from Hamamatsu (300–1050 nm). The PL spectra were corrected for the spectral sensitivity of the detector.
JV curves of the solar cells were recorded in a glovebox using a Keithley 2400 source measure unit and a custom made LabView software. The samples were illuminated by a Dedolight lamp with a spectrum similar to the AM 1.5G spectrum at 100 mW cm−2. The intensity was calibrated with a Fraunhofer silicon reference solar cell. Layer thicknesses were measured using a Bruker Dektak XT surface profiler.
Atomic Force Microscopy (AFM) measurements were performed on a Veeco Multimode Quadrax MM atomic force microscope (Bruker) in tapping mode using non-coated Si-cantilevers (NCH-VS1-W, NanoWorld AG) with a resonance frequency of 291 kHz and a force constant of 42 N m−1. The measurements were acquired at room temperature under ambient conditions. All calculations and image processing was done with Nanoscope software (V7.30r1sr3, Veeco).
3. Results and discussion
The CuInS2 nanocrystals were prepared via the hot-injection method using copper and indium xanthates as precursors. These precursors are known to decompose rapidly at a temperature of approx. 130 °C.27 Therefore, the reactions were carried out in trichlorobenzene as solvent, which has a sufficiently high boiling point to conduct the synthesis at 200 °C. Regarding the choice of capping ligands, preliminary hot-injection experiments have shown that mixtures of oleic acid and oleylamine gave better results than experiments using solely oleylamine or oleic acid. In the latter case larger precipitates were observed, which were insoluble after the precipitation (primary crystallite size according to Scherrer equation: 16 nm; the X-ray diffraction pattern is shown in Fig. S1, ESI†). When only oleylamine was used as capping ligand, despite the good results we obtained in a room temperature synthesis,42 extremely small nanocrystals were synthesized, which could not be precipitated and separated from the reaction solution. However, by using a 1:1 mixture of oleylamine and oleic acid, dioleamide is formed directly in the reaction solution during the pre-heating phase, acting then as ligand during the nanoparticle synthesis. This dioleamide formation was already observed by D. G. Calatayud et al.,47 and was also confirmed by 1H-NMR spectroscopy (see ESI,† Fig. S2). After the preheating phase, the solution of copper and indium xanthates was swiftly injected into the solution of dioleamide in trichlorobenzene at 200 °C and the reaction solution was held at this temperature for 30 min before the nanoparticles were isolated by several precipitation steps (Fig. 1).
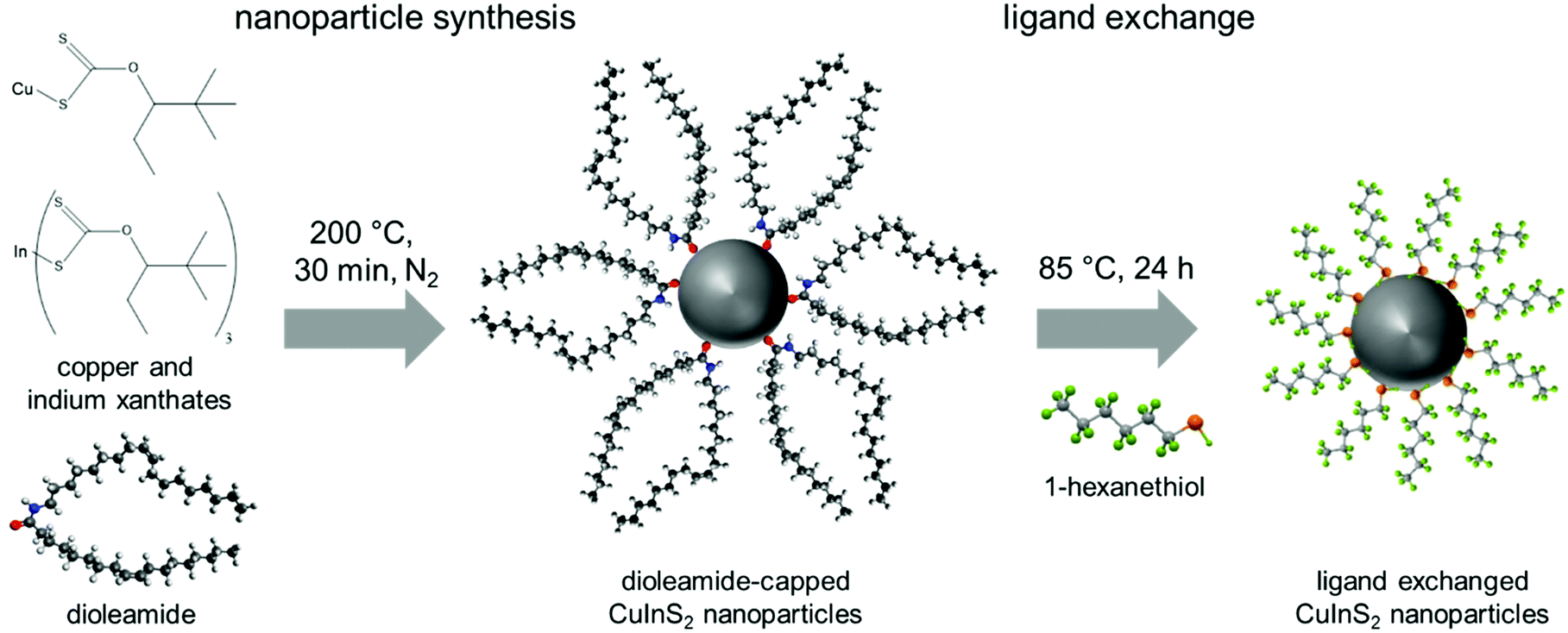 | ||
| Fig. 1 Scheme of the nanoparticle synthesis and ligand exchange with 1-hexanethiol. | ||
The X-ray diffraction pattern of the prepared CuInS2 nanocrystals is depicted in Fig. 2A and shows broad peaks at 28.0 (112), 46.6 (220), 55.0 (116/312) and 75° 2θ (316/322), which match well with the reference pattern of chalcopyrite CuInS2 (PDF 01-75-0208). From the broadening of the peaks and using Scherrer equation, an instrumental broadening of 0.12° 2θ and a Scherrer form factor of 0.9 (for spherical particles), an average primary crystallite size of 3.6 nm is estimated.
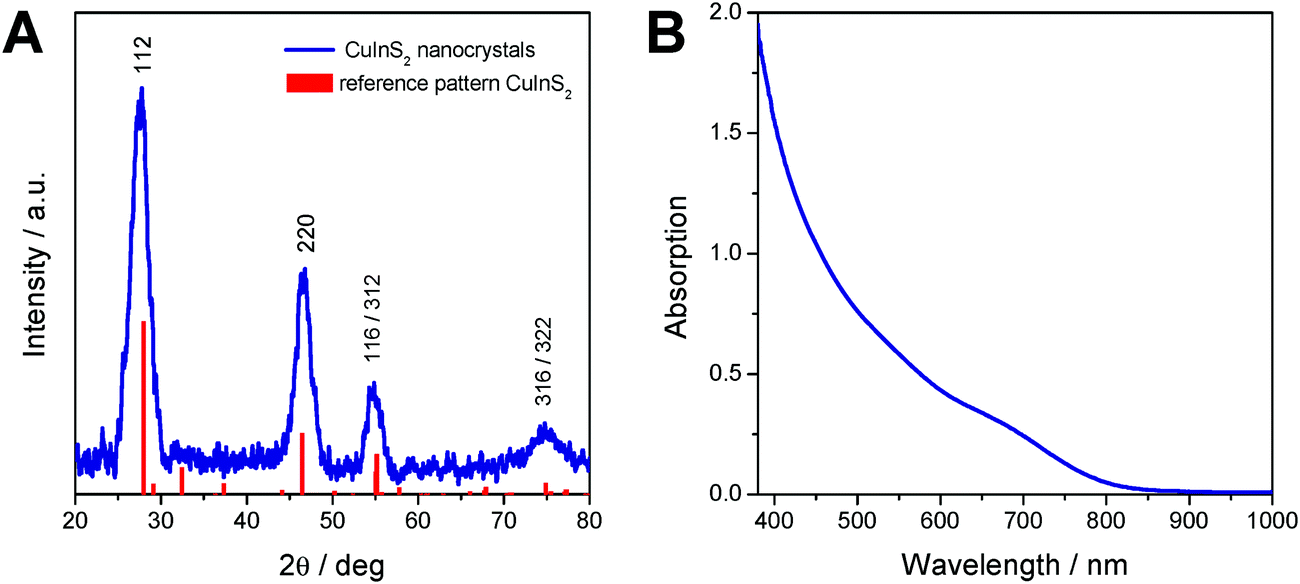 | ||
| Fig. 2 (A) X-ray diffraction pattern and (B) UV-Vis spectrum of the prepared CuInS2 nanocrystals. | ||
The UV-Vis absorption spectrum of the nanoparticles dissolved in chloroform shows the typical shape of CuInS2 nanocrystals10,12,48 and reveals an absorption onset at 830 nm, which corresponds to an optical band gap of 1.57 eV.
For the application of the CuInS2 nanocrystals in solar cells, the nature of the capping ligands is essential, as these critically influence properties such as solubility of the nanocrystals, wettability of the nanocrystal solutions as well as the interface between the nanocrystals in quantum dot sensitized solar cells or the interface of the nanocrystals and the conjugated polymer in polymer/nanocrystal hybrid solar cells. Thiols are known as good capping ligands for many metal sulfide nanocrystals. Thus, in this work, the bulky dioleamide ligands were exchanged with 1-hexanethiol by stirring the dioleamide stabilized nanocrystals in 1-hexanethiol at 80 °C for 24 h. After the ligand exchange, the solubility of the nanocrystals in toluene, chloroform and chlorobenzene is slightly reduced.
To evaluate the efficiency of the ligand exchange and to determine the amount of capping ligand present in the nanocrystal samples before and after ligand exchange, thermogravimetric analysis was performed. The TGA traces of a sample before and after ligand exchange with 1-hexanethiol are presented in Fig. 3A. The mass loss of the nanocrystal sample is reduced from 32.2% before ligand exchange to 17.1% after the ligand exchange process. Moreover, for the sample before ligand exchange, a one-step decomposition with an onset (5% mass loss) at 310 °C was observed. This mass loss stems most likely from the decomposition of the dioleamide. The mass loss of the ligand exchanged sample displays a 2-step mass loss with onsets at 200 °C and 310 °C. We assume that this first mass loss stems from the evaporation or decomposition of hexanethiol followed by the decomposition of remaining dioleamide in the second mass loss step. Based on these measurements, we can conclude that a major part of the dioleamide ligands has been removed or exchanged by 1-hexanethiol, however, a minor part is still present in the nanocrystal sample.
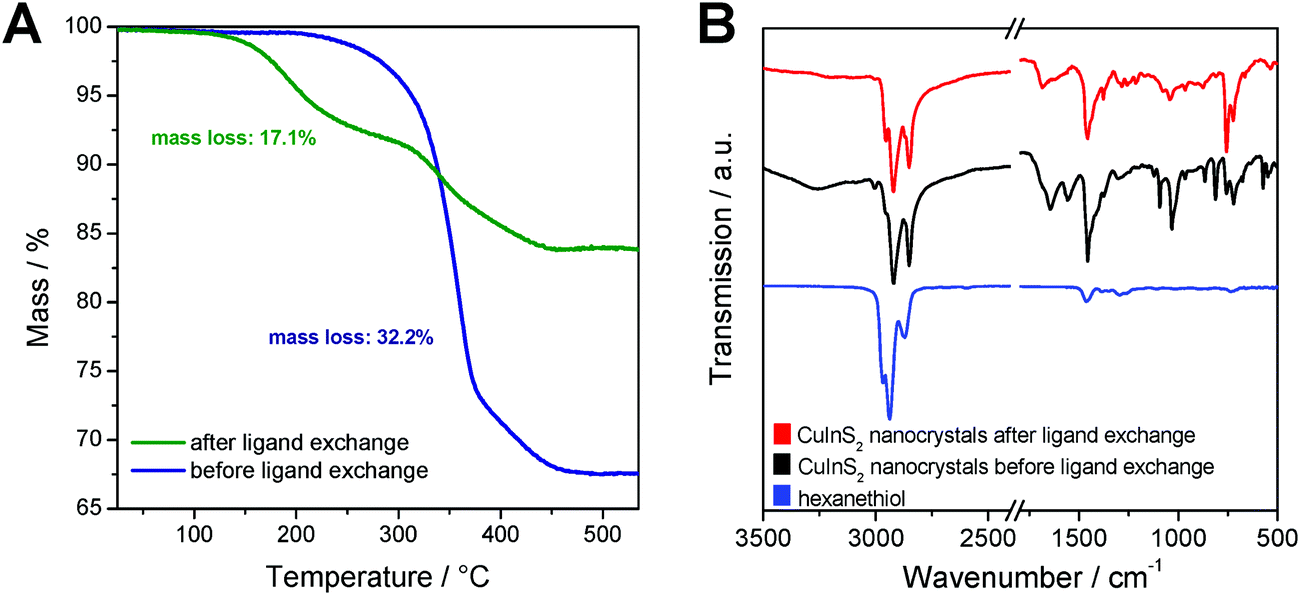 | ||
| Fig. 3 (A) Thermogravimetric analysis of non-ligand exchanged and ligand exchanged nanoparticle samples; (B) FT-IR spectra of 1-hexanethiol and the CuInS2 nanoparticle sample before and after ligand exchange. | ||
Moreover, the ligand exchange was characterized by FT-IR spectroscopy, which confirms the results from the TGA analysis. The IR spectra of the nanocrystals before and after ligand exchange and of pristine 1-hexanethiol are shown in Fig. 3B. In the spectrum of the non-ligand exchanged nanocrystals, distinct bands at 1558 and 1644 cm−1 are visible, which correspond to the bending vibration of the N–H bond and to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, respectively. The band at 1644 cm−1 can also be ascribed to the bending vibration of the CC bonds. Additionally, there are bands at 3005 cm−1 (overtone of amide II band) and at around 3260 cm−1 (stretching vibration of the N–H group) observable. All these bands, which can be attributed to dioleamide, are significantly reduced in the spectrum of the ligand-exchanged CuInS2 nanocrystals.
O bond, respectively. The band at 1644 cm−1 can also be ascribed to the bending vibration of the CC bonds. Additionally, there are bands at 3005 cm−1 (overtone of amide II band) and at around 3260 cm−1 (stretching vibration of the N–H group) observable. All these bands, which can be attributed to dioleamide, are significantly reduced in the spectrum of the ligand-exchanged CuInS2 nanocrystals.
The bands at 1458, 2852, 2921 and 2954 cm−1 are visible in both nanocrystal samples and correspond to the scissor vibration of the C–H-bond and the asymmetric and symmetric stretching vibration of the C–H bond. Due to the aliphatic structure of dioleamide and 1-hexanethiol, these two ligands cannot be unambiguously distinguished based on these bands.
The bands corresponding to the aliphatic bonds in the IR spectrum of pristine 1-hexanethiol are shifted to slightly lower wavenumbers when hexanethiol is coordinated to the CuInS2 nanocrystals, which can be clearly seen in Fig. 3B. The unique band for 1-hexanethiol at 2594 cm−1, which corresponds to the stretching vibration of the S–H bond is only of weak intensity and is not observable in the ligand-exchanged nanocrystal sample.49
Furthermore, the size, shape and agglomeration behavior of the CuInS2 nanocrystals before and after the ligand exchange process were examined by transmission electron microscopy (TEM). Fig. 4 shows TEM images of the nanocrystal samples in different magnifications. It can be seen in Fig. 4A and B that the non-ligand exchanged nanoparticles are well separated and homogeneously distributed over the TEM grid with a certain distance between the nanoparticles. After the ligand exchange with hexanethiol, the inter-particle distance decreases significantly and they tend to agglomerate (see Fig. 4C and D). The nanocrystals have an average diameter of approx. 3–4 nm with a comparably narrow particle size distribution and show a globular shape. In the high resolution images (insets in Fig. 4B and D) lattice fringes are visible and an interplanar distance of 0.32 nm is observed, which corresponds to the spacing distance of the (112) planes of the chalcopyrite crystal structure.
 | ||
| Fig. 4 TEM images of the nanoparticles in different magnifications before (A and B) and after ligand exchange (C and D). | ||
In Fig. 5A, the SAXS curves of the CuInS2 nanocrystals dissolved in toluene are depicted. The SAXS data exhibit a slight upturn at low q values (q < 0.5 nm−1), which indicates the presence of larger particles, such as aggregates or agglomerates in the sample. Therefore, a polydispersity analysis was performed resulting in size distributions weighted by volume (DV(r)), see Fig. 5B. The particle size distribution weighted by volume reveals that the majority of the samples consist of populations of primary particles of around 3 nm in diameter. In addition, the presence of small amounts of larger aggregates with a size of approx. 12 nm is visible in the particle size distribution. Moreover, as already seen in the TEM images, the size distribution of the primary nanocrystals stays constant during the ligand exchange process, however, the size distributions of the nanoparticle aggregates changes slightly.
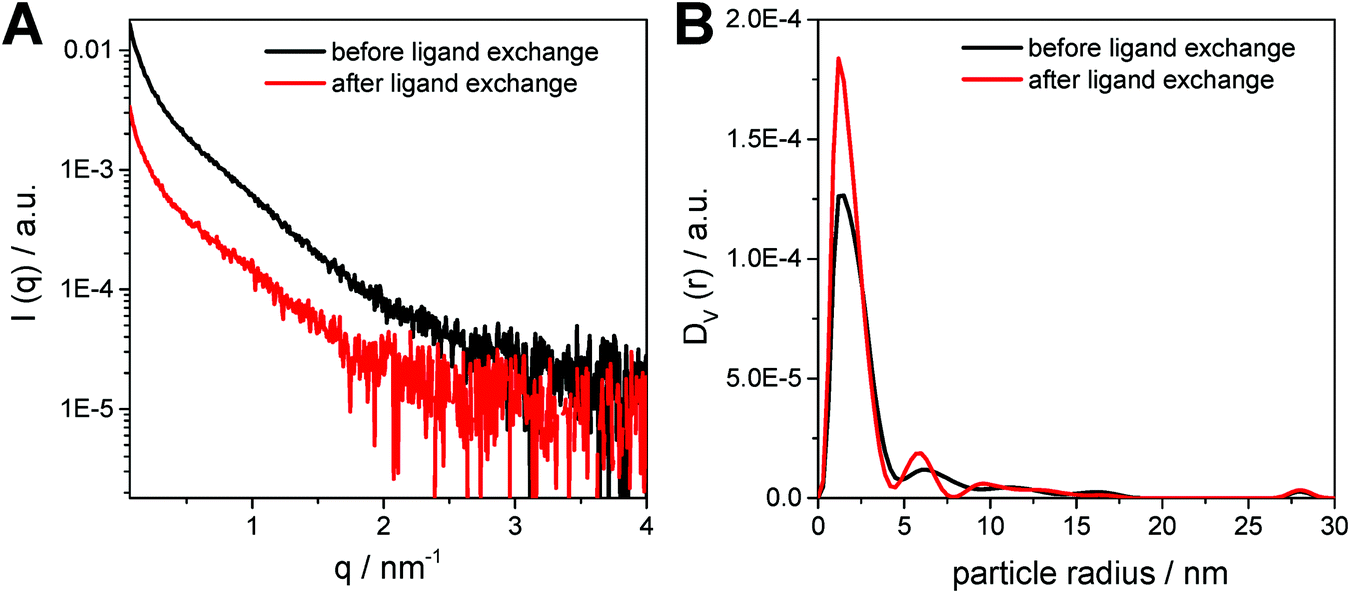 | ||
| Fig. 5 (A) SAXS curves and (B) the corresponding particle size distributions weighted by volume for the nanoparticle samples before and after ligand exchange. | ||
In a next step, the ligand exchanged CuInS2 nanocrystals were applied in polymer/nanocrystal hybrid solar cells using PCDTBT as polymer component. The choice of polymer is based on our previous studies with in situ prepared CuInS2 nanocrystals showing that PCDTBT or PSiF-DBT led to the best device performances.27,33 PCDTBT has a LUMO level of −3.6 eV50 and the conduction band of CuInS2 nanocrystals lies slightly deeper at approx. −3.9 to −4.0 eV (see also Fig. S3, ESI†).24,51 This makes the CuInS2 nanocrystals a well suited electron acceptor in this material combination. To obtain information about possible electron transfer from the conjugated polymer to the nanocrystals, photoluminescence (PL) quenching measurements were conducted. Fig. 6A shows the PL spectrum of a pristine PCDTBT film and PCDTBT nanocomposite films with polymer/CuInS2 weight ratios of 1:5, 1:9 and 1:15. The conjugated polymer shows a photoemission peak with a maximum at 725 nm and for the polymer/CuInS2 sample with a weight ratio of 1:5, this photoemission is already quenched to about 46% of its initial value. For the polymer/nanoparticle films with increased CuInS2 nanocrystal content (1:9, w:w) the photoluminescence is decreased to 22% where it remains almost unchanged when going to higher polymer/CuInS2 ratios (see Fig. 6B). From these data it is apparent that the PL quenching is not as efficient as for nanocomposite layers containing CuInS2 nanocrystals prepared in situ in the conjugated polymer matrix.27,52,53 A possible reason for the non-complete quenching of the photoluminescence of the polymer in the films might be polymer domains in the nanocomposite layer in which the polymer is not well blended with the CuInS2 nanocrystals.46 This assumption is corroborated by the AFM images (see Fig. 7), which reveal a significant increase of roughness in the PCDTBT/CuInS2 absorber layer compared to the pristine polymer film. The increased roughness stems most probably from a partial agglomeration of the ligand exchanged nanocrystals. This leads in turn also to polymer domains which are not well mixed with the CuInS2 nanocrystals. The phase image also indicates phase separation, however, at least at the surface of the film, the phase separation is much finer as would be expected based on the topographic AFM measurements.
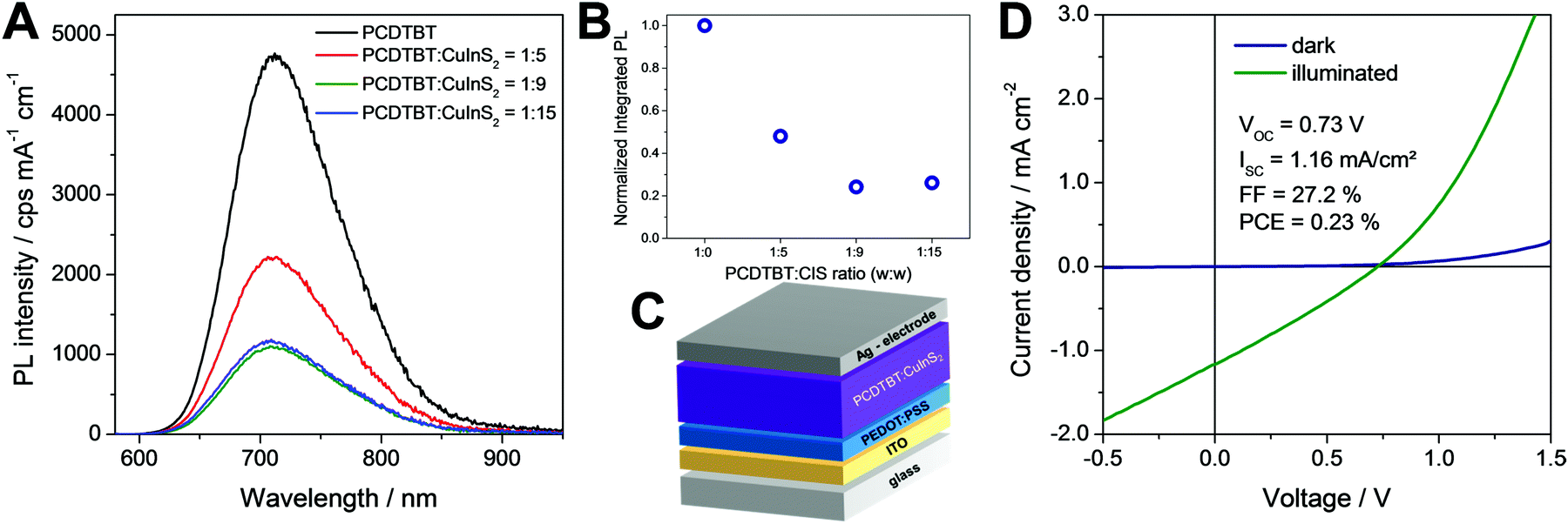 | ||
| Fig. 6 (A and B) Photoluminescence quenching data, (C) scheme of the device architecture and (D) JV curves of a typical PCDTBT/CuInS2 based solar cell with a polymer/nanoparticle weight ratio of 1:9 in the dark and under 100 mW cm−2 illumination. | ||
 | ||
| Fig. 7 AFM images of a pristine PCDTBT film and a PCDTBT/CuInS2 (1/9, w/w) nanocomposite film (A and C: topography images, B and D: phase images). | ||
Furthermore, to investigate the photovoltaic properties of the hybrid layers, we prepared solar cells in the architecture glass/ITO/PEDOT:PSS/PCDTBT-CuInS2/Ag (see Fig. 6C) with the ligand exchanged nanoparticles. The thickness of the hybrid absorber layer was 90–100 nm. In addition, we intended to prepare solar cells with non-ligand exchanged CuInS2 nanocrystals to gain additional information about the influence of the ligand exchange on the solar cell performance. However, the polymer/nanoparticle solution did not wet the PEDOT:PSS layer resulting in very poor film quality and only partial coverage of the PEDOT:PSS layer with the absorber layer, which impeded the fabrication of solar cells with the non-ligand exchanged nanocrystals.
The JV characteristics of typical solar cells prepared using the ligand exchanged nanoparticles with different polymer/nanoparticle weight ratios are presented in Fig. 6D and Fig. S4 (ESI†). For the solar cells with weight ratios of 1:5 and 1:15, the PCEs remained far below 0.1%. We assume that the low performance of the devices with a weight ratio of 1:5 is due to a too low amount of nanocrystals in the hybrid films leading to less charge generation, inefficient electron transport in the nanocrystal phase and in turn to a low JSC. The films with a weight ratio of 1:15 appeared very rough and inhomogeneous, leading most likely to a partial short circuiting of the device.
In contrast to that, the solar cells with a polymer/nanocrystal weight ratio of 1:9 exhibited PCEs of up to 0.23%. The open circuit voltage was at 730 mV, which is significantly higher than the VOC values, which can be reached with solar cells based on absorber layers with in situ prepared ligand-free CuInS2 nanocrystals in the conjugated polymer (typical values up to 540 mV).27,33 However, the JSC (1.16 mA cm−2) and the FF (27.2%) remained low. This is most likely due to the remaining ligands on the CuInS2 nanocrystals, the agglomeration of the nanocrystals or the rough interface between the absorber layer and the metal electrode. The average values and standard deviation of the five best solar cells prepared in this study are given in Table 1. Annealing of the absorber layer after spin coating did not positively affect the solar cell performance. Typically, the PCE values were slightly reduced when an annealing step was applied (see Fig. S5, ESI†).
:9. The average values and standard deviations were calculated from the five best devices
| V OC/mV | I SC/mA cm−2 | FF | PCE/% |
|---|---|---|---|
| 0.716 ± 0.035 | 1.09 ± 0.07 | 0.27 ± 0.01 | 0.21 ± 0.02 |
Even though these devices cannot compete with the PCEs of in situ prepared polymer/CuInS2 solar cells, they are more efficient than polymer/CuInS2 hybrid solar cells reported with separately synthesized CuInS2 nanoparticles so far.22,23,25,29 The highest PCE reported in previous studies is 0.08% and was obtained with P3HT/CuInS2 hybrid solar cells.29
4. Conclusions
In summary, the investigated hot injection synthesis route using copper and indium xanthates as precursors and dioleamide as capping ligands leads to comparably small CuInS2 nanocrystals with sizes of only 3–4 nm. The nanocrystals are well soluble in solvents such as chloroform, toluene and chlorobenzene. As expected, after ligand exchange with 1-hexanethiol, the solubility of the nanocrystals is slightly reduced. Thermogravimetric analyses revealed that after ligand exchange 17 wt% of the overall sample mass is remaining capping ligand and FT-IR measurements indicate that the majority of the dioleamide ligands are removed and exchanged to hexanethiol during the ligand exchange process. TEM images illustrate that the distance between the nanoparticles is significantly smaller after the ligand exchange and slight agglomeration of the nanocrystals is observed. In addition, SAXS measurements confirmed the size of the nanocrystals found in the TEM images. Moreover, the nanocrystals were implemented into the absorber layer of polymer/nanocrystal hybrid solar cells. It was found that these devices showed higher photovoltage compared to in situ (ligand-free) prepared polymer/CuInS2 hybrid solar cells, however, the photocurrent remained significantly lower. With the ligand exchanged nanocrystals, solar cells with PCEs of 0.23% were obtained. Even though the PCEs generally remained low, they are considerably higher compared to other polymer/CuInS2 hybrid solar cells with separately synthesized CuInS2 nanoparticles reported before.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Josefine Hobisch for performing the TGA analyses and Manuel Hollauf for assistance with the NMR measurements.References
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- W. van der Stam, A. C. Berends and C. de Mello Donega, ChemPhysChem, 2016, 17, 559–581 CrossRef CAS PubMed.
- Y. Min, G. D. Moon, C.-E. Kim, J.-H. Lee, H. Yang, A. Soon and U. Jeong, J. Mater. Chem. C, 2014, 2, 6222–6248 RSC.
- J. Jie, W. Zhang, I. Bello, C.-S. Lee and S.-T. Lee, Nano Today, 2010, 5, 313–336 CrossRef CAS.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- D. Aldakov, A. Lefrançois and P. Reiss, J. Mater. Chem. C, 2013, 1, 3756–3776 RSC.
- M. V. Kolvalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef PubMed.
- D. H. Jara, S. J. Yoon, K. G. Stamplecoskie and P. V. Kamat, Chem. Mater., 2014, 26, 7221–7228 CrossRef CAS.
- S. Shen and Q. Wang, Chem. Mater., 2013, 25, 1166–1178 CrossRef CAS.
- J. Kolny-Olesiak and H. Weller, ACS Appl. Mater. Interfaces, 2013, 5, 12221–12237 CrossRef CAS PubMed.
- B. Tell, J. L. Shay and H. M. Kasper, Phys. Rev. B: Solid State, 1971, 4, 2463 CrossRef.
- A. D. P. Leach and J. E. Macdonald, J. Phys. Chem. Lett., 2016, 7, 572–583 CrossRef CAS PubMed.
- C. Zhao, Z. Bai, X. Liu, Y. Zhang, B. Zou and H. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 17623–17629 CrossRef CAS PubMed.
- K. Yu, P. Ng, J. Ouyang, M. B. Zaman, A. Abulrob, T. N. Baral, D. Fatehi, Z. J. Jakubek, D. Kingston, X. Wu, X. Liu, C. Hebert, D. M. Leek and D. M. Whitfield, ACS Appl. Mater. Interfaces, 2013, 5, 2870–2880 CrossRef CAS PubMed.
- Z. Bai, W. Ji, D. Han, L. Chen, B. Chen, H. Shen, B. Zhou and H. Zhong, Chem. Mater., 2016, 28, 1085–1091 CrossRef CAS.
- S. Khanchandani, S. Kumar and A. K. Ganguli, ACS Sustainable Chem. Eng., 2016, 4, 1487–1499 CrossRef CAS.
- O. Stroyuk, A. Raevskaya and N. Gaponik, Chem. Soc. Rev., 2018, 47, 5354–5422 RSC.
- Z. Pan, I. Mora-Seró, Q. Shen, H. Zhang, Y. Li, K. Zhao, J. Wang, X. Zhong and J. Bisquert, J. Am. Chem. Soc., 2014, 136, 9203–9210 CrossRef CAS PubMed.
- N. Guijarro, E. Guillén, T. Lana-Villarreal and R. Gómez, Phys. Chem. Chem. Phys., 2014, 16, 9115–9122 RSC.
- J. E. Halpert, F. S. F. Morgenstern, B. Ehrler, Y. Vaynzof, D. Credgington and N. C. Greenham, ACS Nano, 2015, 9, 5857–5867 CrossRef CAS PubMed.
- L. Li, N. Coates and D. Moses, J. Am. Chem. Soc., 2010, 132, 22–23 CrossRef CAS PubMed.
- N. Radychev, D. Scheunemann, M. Kruszynska, K. Frevert, R. Miranti, J. Kolny-Olesiak, H. Borchert and J. Parisi, Org. Electron., 2012, 13, 3154–3164 CrossRef CAS.
- C. Krause, R. Miranti, F. Witt, J. Neumann, D. Fenske, J. Parisi and H. Borchert, Sol. Energy Mater. Sol. Cells, 2014, 124, 241–246 CrossRef CAS.
- E. Arici, N. Sariciftci and D. Meissner, Adv. Funct. Mater., 2003, 13, 165–171 CrossRef CAS.
- W. Yue, S. Han, R. Peng, W. Shen, H. Geng, F. Wu, S. Tao and M. Wang, J. Mater. Chem., 2010, 20, 7570–7578 RSC.
- S. Jindal and S. M. Giripunje, Mater. Res. Express, 2017, 4, 115506 CrossRef.
- T. Rath, M. Edler, W. Haas, A. Fischereder, S. Moscher, A. Schenk, R. Trattnig, M. Sezen, G. Mauthner, A. Pein, D. Meischler, K. Bartl, R. Saf, N. Bansal, S. A. Haque, F. Hofer, E. J. W. List and G. Trimmel, Adv. Energy Mater., 2011, 1, 1046–1050 CrossRef CAS.
- M. Arar, M. Gruber, M. Edler, W. Haas, F. Hofer, N. Bansal, L. X. Reynolds, S. A. Haque, K. Zojer, G. Trimmel and T. Rath, Nanotechnology, 2013, 24, 484005 CrossRef PubMed.
- C. Krause, D. Scheunemann, J. Parisi and H. Borchert, J. Appl. Phys., 2015, 118, 205501 CrossRef.
- Z. Liu, Y. Sun, J. Yuan, H. Wie, X. Huang, L. Han, W. Wang, H. Wang and W. Ma, Adv. Mater., 2013, 25, 5772–5778 CrossRef CAS PubMed.
- X. Du, Q. Zeng, G. Jin, F. Liu, T. Ji, Y. Yue, Y. Yang, H. Zhang and B. Yang, Small, 2017, 13, 1603771 CrossRef PubMed.
- T. Rath and G. Trimmel, Hybrid Mater., 2014, 1, 15–36 Search PubMed.
- C. Fradler, T. Rath, S. Dunst, I. Letofsky-Papst, R. Saf, B. Kunert, F. Hofer, R. Resel and G. Trimmel, Sol. Energy Mater. Sol. Cells, 2014, 124, 117–125 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- W.-C. Huang, C.-H. Tseng, S.-H. Chang, H.-Y. Tuan, C.-C. Chiang, L.-M. Lyu and M. H. Huang, Langmuir, 2012, 28, 8496–8501 CrossRef CAS PubMed.
- A. Pein, M. Baghbanzadeh, T. Rath, W. Haas, E. Maier, H. Amenitsch, F. Hofer, C. O. Kappe and G. Trimmel, Inorg. Chem., 2011, 50, 193–200 CrossRef CAS PubMed.
- S. L. Castro, S. G. Bailey, R. P. Raffaelle, K. K. Banger and A. F. Hepp, J. Phys. Chem. B, 2004, 108, 12429–12435 CrossRef CAS.
- J. J. Nairn, P. J. Shapiro, B. Twamley, T. Pounds, R. von Wandruszka, T. R. Fletcher, M. Williams, C. Wang and M. G. Norton, Nano Lett., 2006, 6, 1218–1223 CrossRef CAS PubMed.
- D. Pan, D. Weng, X. Wang, Q. Xiao, W. Chen, C. Xu, Z. Yang and Y. Lu, Chem. Commun., 2009, 4221–4223 RSC.
- M. Al-Shakban, P. D. Matthews, X. L. Zhong, I. Vitorica-Yrezabal, J. Raftery, D. J. Lewis and P. O'Brien, Dalton Trans., 2018, 47, 5304–5309 RSC.
- D. P. Dutta and G. Sharma, Mater. Lett., 2006, 60, 2395–2398 CrossRef CAS.
- S. H. Lu, T. F. Chen, A. J. Wang, D. Zheng, Y. L. Li and Y. S. Wang, Mater. Sci. Eng., B, 2016, 203, 19–26 CrossRef CAS.
- A. Kharkwal, K. Jain, S. B. Tyagi, A. K. Singh, S. N. Sharma and M. Kharkwal, Colloid Polym. Sci., 2014, 292, 2913–2926 CrossRef CAS.
- A. Kharkwal, S. N. Sharma, K. Jain, L. Arora, P. Chawla, A. K. Singh and S. Chand, Colloid Polym. Sci., 2013, 291, 2607–2617 CrossRef CAS.
- C. Buchmaier, T. Rath, F. Pirolt, A.-C. Knall, P. Kaschnitz, O. Glatter, K. Wewerka, F. Hofer, B. Kunert, K. Krenn and G. Trimmel, RSC Adv., 2016, 6, 106120–106129 RSC.
- O. Glatter, J. Appl. Crystallogr., 1980, 13, 7–11 CrossRef CAS.
- D. G. Calatayud, T. Jardiel, M. Rodríguez, M. Peiteado, D. Fernández-Hevia and A. C. Caballero, Ceram. Int., 2013, 39, 1195–1202 CrossRef CAS.
- D. So and G. Konstantatos, Chem. Mater., 2015, 27, 8424–8432 CrossRef CAS.
- G. Socrates, Infrared characteristic group frequencies: tables and charts, Wiley, 2nd edn, 1998 Search PubMed.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- H. Zhong, S. S. Lo, T. Mirkovic, Y. Li, Y. Ding, Y. Li and G. D. Scholes, ACS Nano, 2010, 4, 5253–5262 CrossRef CAS PubMed.
- A.-C. Knall, A. O. F. Jones, B. Kunert, R. Resel, D. Reishofer, P. W. Zach, M. Kirkus, I. McCulloch and T. Rath, Monatsh. Chem., 2017, 148, 855–862 CrossRef CAS PubMed.
- M. Jäger, R. Trattnig, M. Postl, W. Haas, B. Kunert, R. Resel, F. Hofer, A. Klug, G. Trimmel and E. J. W. List, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1400–1410 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra and additional XRD and solar cell data. See DOI: 10.1039/c8nj04823a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |