
Cascade recognition of Hg2+ and cysteine using a naphthalene based ESIPT sensor and its application in a set/reset memorized device†
Navneet
Kaur
 *a,
Gitanjali
Jindal
a,
Sukhvinder
b and
Subodh
Kumar
b
*a,
Gitanjali
Jindal
a,
Sukhvinder
b and
Subodh
Kumar
b
aDepartment of Chemistry, Panjab University, Chandigarh 160014, India. E-mail: neet_chem@yahoo.co.in; neet_chem@pu.ac.in
bDepartment of Chemistry, Guru Nanak Dev University, Amritsar 143005, India
First published on 19th November 2018
Abstract
An optical ESIPT sensor for Hg2+ and cysteine based on a naphthalene platform (1) was designed and synthesized by a one step reaction and characterized by using common spectroscopic techniques. Upon addition of Hg2+ to a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) solution of 1, the highly fluorescent probe becomes weakly fluorescent, showing a color change from colorless to dark yellow visible to the naked eye and from fluorescent blue to light yellow when irradiated with 365 nm light. The sensing mechanism has been supported by DFT and 1H NMR titration studies. The in situ generated 1–Hg2+ complex has been used for effectively sensing Cys. Owing to the stronger binding affinity of the sulfhydryl group to Hg2+, Cys can extract Hg2+ from the 1–Hg2+ complex, resulting in the release of 1 and revival of the emission intensity. Sensor 1 has also been successfully applied for the detection of Hg2+ in real water samples with good recovery. The alternate addition of Hg2+ and Cys ions generated “on–off–on” fluorescence cycles, which enabled 1 to be used as a reversible and reconfigurable set/reset memorized device at the molecular level.
:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) solution of 1, the highly fluorescent probe becomes weakly fluorescent, showing a color change from colorless to dark yellow visible to the naked eye and from fluorescent blue to light yellow when irradiated with 365 nm light. The sensing mechanism has been supported by DFT and 1H NMR titration studies. The in situ generated 1–Hg2+ complex has been used for effectively sensing Cys. Owing to the stronger binding affinity of the sulfhydryl group to Hg2+, Cys can extract Hg2+ from the 1–Hg2+ complex, resulting in the release of 1 and revival of the emission intensity. Sensor 1 has also been successfully applied for the detection of Hg2+ in real water samples with good recovery. The alternate addition of Hg2+ and Cys ions generated “on–off–on” fluorescence cycles, which enabled 1 to be used as a reversible and reconfigurable set/reset memorized device at the molecular level.
Introduction
Now-a-days, the quest to design sensitive and selective analytical methods for the sensing of environmentally and biologically associated heavy and transition metal ions is attracting a considerable amount of attention.1,2 In the field of chemo- and biosensors, the construction of selective chemosensors has emerged as an important research area.3,4 Metal ions play harmful and/or vital roles in both biological and environmental processes, the roles being strictly regulated by their respective concentration levels. Metal ions show extremes in either being beneficial or toxic if present in the requisite or an uncontrolled amount, respectively.5 For instance, the most deleterious heavy metal element, mercury, poses threats to the environment by accumulating due to its non-degradable nature and causes many health problems in the brain, the kidneys and the central nervous system.6,7 In addition, the existence of mercury in different forms like metallic, ionic and as a part of organic salts and complexes further enhances the problem. Due to the lipid solubility and easy absorptivity of methylmercury species, they are the most toxic species and can lead to prenatal brain damage, various cognitive and motion disorders, vision and hearing impairment and Minamata disease.8 On the other hand, cysteine (Cys), one of the thiol containing amino acids, plays a pivotal role in many biological processes such as redox reactions and cellular detoxification.9,10 The intracellular level of cysteine (Cys) has been linked to lots of diseases, such as Alzheimer's disease, AIDS and cancers.11–13 Thus, effective analytical approaches aimed at simultaneous monitoring of cellular free Hg2+ ion and cysteine (Cys) are desirable to elucidate their behavior in healthy and disease status.14Amongst various types of chemosensors, fluorescent sensors are more advantageous, as with the fluorescence technique, no reference is required and the analyte can be easily reused. Further, the instrumentation is very easy and it enables a faster response time.15,16 Recently, excited-state intramolecular proton transfer (ESIPT) has emerged as a renowned photophysical process responsible for unusually large Stokes shifts in benzazole derivatives. The main characteristic of an ESIPT-based chemosensor is the formation of an intramolecular hydrogen bond in the ground state with the adjacent hydrogen-bond acceptor17 and this phenomenon has been employed for the formation of new probes.18–21
Photonic driven systems and networks that function as molecular level logic gates are the successful outcomes of the development of supramolecular chemistry.22–24 With the development of molecular logic gates25,26 and their integration into working automation27 and arithmetic systems,28,29 chemists have come closer to the realization of a molecular scale calculator i.e. moleculator. These molecular logic gates operate in a wireless mode and thus they have the potential for computation on a nanometer scale that silicon-based devices cannot address.30
In the present work, as an outcome of excited state intramolecular proton transfer (ESIPT), cascade recognition of Hg2+ and Cys has been demonstrated in a 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) solution. Here, the secondary analyte (Cys) is sensed and detected by the cooperative interaction of sensor molecule 1 and the primary analyte (Hg2+). Also, alternate addition of Cu2+ and CN− ions resulted in reversible and reproducible “on–off–on” cycles and enabled 1 to exhibit ‘read–erase–write–read’ behavior at the molecular level. Earlier, we have shown that a similar imidazole possessing chemosensor devoid of the o-OH group responsible for ESIPT phenomenon showed optical changes with only Cu2+ ions.31
Results and discussion
Synthesis of chemosensor 1
The synthesis of chemosensor 1 is depicted in Scheme 1. Chemosensor 1 was synthesized by refluxing benzil, 2-hydroxy-1-naphthaldehyde and ammonium acetate in hot glacial acetic acid. | ||
| Scheme 1 Synthesis of chemosensor 1. | ||
Chemosensor 1 was obtained in quantitative yield and characterized by several techniques such as 1H NMR, 13C NMR and HRMS spectroscopy (Fig. S1–S3, ESI†). The spectral investigations gave consistent data for the structure of 1.
Colorimetric and UV-vis spectral response
In order to evaluate the sensing performance towards miscellaneous metal ions including Na+, K+, Mg2+, Al3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+ and Cd2+ ions, the naked-eye experiment (Fig. 1a) and UV-vis absorption spectroscopy analysis (Fig. 1b) of chemosensor 1 (5 μM) in a 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) were performed. A remarkable color change from colorless to dark yellow color was observed with addition of only Hg2+ ions. In addition, the absorption spectral response of 1 was also studied upon addition of 100 equiv. of the above mentioned metal ions in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer). The UV-vis spectrum of sensor 1 shows two peaks at 308 nm and 356 nm. With the addition of Hg2+ ions, a broad new band was observed at 430 nm pointing to the binding of Hg2+ with sensor 1. The addition of other metal ions except Hg2+ did not show any color or absorption changes (Fig. 1b). So sensor 1 was a selective and sensitive sensor probe for Hg2+ ions. However, the increased absorbance due to the Cu2+ ions below 300 nm was due to its own absorbance in this region and for the same reason, any actual changes observed with the Cu2+ ions cannot be identified and measured.
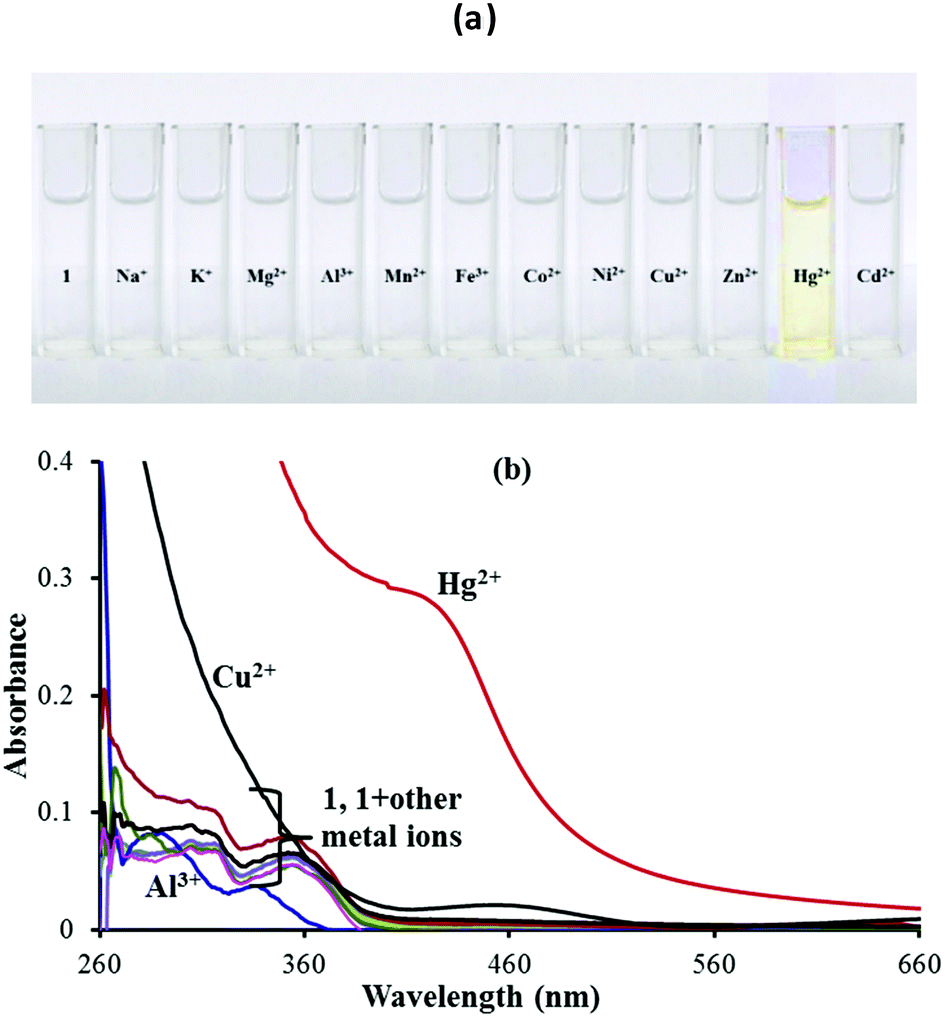 | ||
| Fig. 1 (a) Colorimetric analysis and (b) UV-vis spectral response of 1 in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) upon addition of 100 equiv. of different metal ions. | ||
Based on UV-vis spectrophotometric titrations, the sensing properties of chemosensor 1 (5 μM) were examined by adding Hg2+ to the 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) solution of 1. Upon incremental addition of Hg2+ ions, a new absorption band started forming at 430 nm, resulting in a color change to dark yellow (Fig. S4, ESI†). The spectral fitting of the absorption titration data using SPECFIT showed the formation of a 1:1 stoichiometry with logβ11 = 4.24 ± 0.0082. However, below 300 nm, the increased absorbance is due to the inherent absorbance of Hg2+ in this region and due to the same reason, any actual changes observed with Hg2+ ions were identified at 430 nm only.
Fluorescence emission spectral response
Sensor 1 exhibits a strong emission at 454 nm (λex: 350 nm) due to the presence of the intramolecular conjugated system and the presence of an excited state intramolecular proton transfer (ESIPT) induced by the “keto–enol tautomerism” of 1. The photonics of 1 was further explored with the addition of other metal ions viz. Na+, K+, Mg2+, Al3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+ and Cd2+ ions. Among the various anions and metal ions investigated, addition of Hg2+, Al3+ and Cu2+ ions to the solution of 1 (5 μM; 9:1 (v/v) aqueous CH3CN; pH 7.0 HEPES buffer) quenched its fluorescence intensity (Fig. 2a). This fluorescence quenching was accompanied by a color change from dark fluorescent blue to light yellow color, when irradiated with light of wavelength 365 nm (Fig. 2b). The other tested metal ions exhibited no fluorescence quenching response under the same spectroscopic conditions (Fig. 2a).
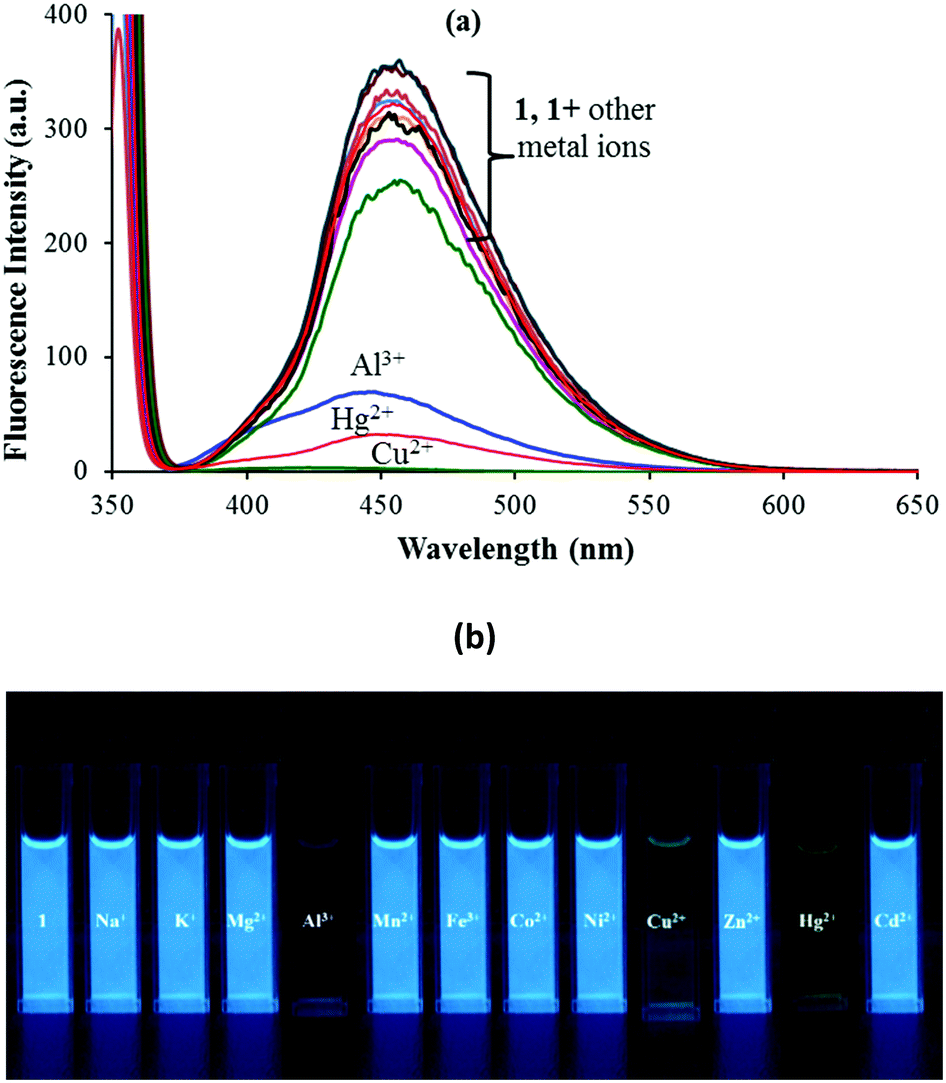 | ||
| Fig. 2 (a) Fluorescence spectral response and (b) fluorescence color change of 1 in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) upon addition of 100 equiv. of different metal ions. | ||
The interactions between 1 and Mn+ (Hg2+, Cu2+ and Al3+) were further investigated through fluorescence titration methods by adding standard solutions of the corresponding ions to a solution of sensor molecule 1 (5 μM). As depicted in Fig. 2, upon gradual addition of Hg2+, the fluorescence intensity started quenching steadily with a negligible spectral shift (Fig. 3). The fluorescence quenching of the probe upon complexation with Hg2+ may be attributed to the heavy atom effect followed by the electron transfer.
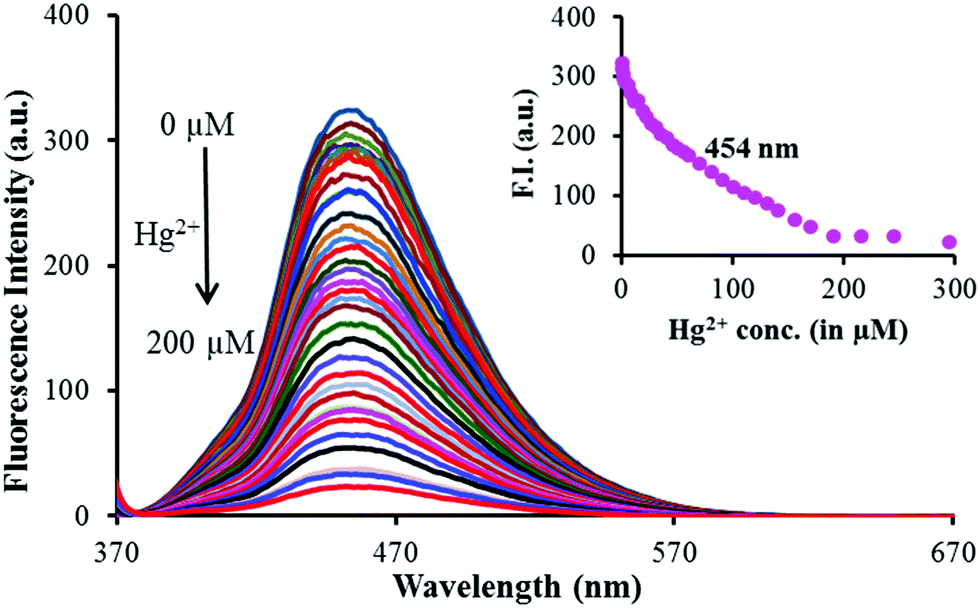 | ||
| Fig. 3 Fluorescence titration spectra of 1 (5 μM) in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer) in the presence of various concentrations of Hg2+ excited at 350 nm. Inset: Plot of the fluorescence intensity at 454 nm versus the concentration of the added Hg2+ ions. | ||
The detection limit was calculated using equation LOD = 3σ/ρ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 32 and was estimated to be 3.2 μM. The spectral fitting of the fluorescence titration data using SPECFIT showed the formation of 1:1 (1:Hg2+) stoichiometry with logβ11 = 4.14 ± 0.0009 (Fig. S5, ESI†). This binding constant is comparable to the one calculated via UV-vis titration.
32 and was estimated to be 3.2 μM. The spectral fitting of the fluorescence titration data using SPECFIT showed the formation of 1:1 (1:Hg2+) stoichiometry with logβ11 = 4.14 ± 0.0009 (Fig. S5, ESI†). This binding constant is comparable to the one calculated via UV-vis titration.
Similar fluorescence quenching was observed with addition of Cu2+ and Al3+ ions (Fig. S6, ESI†). The spectral fitting of the fluorescence titration data for the Cu2+ and Al3+ ions using SPECFIT showed the formation of a 1:1 stoichiometry in both the cases with logβ11 = 5.36 ± 0.06 and 5.41 ± 0.07, respectively (Fig. S7, ESI†).
From the changes in the Cu2+ or Al3+ dependent fluorescence intensity, the detection limit was estimated to be 2.9 μM and 4.3 μM, respectively.
Effect of pH
In order to test the application extent of 1 as a sensor, the influence of pH on 1 as well as 1–Hg2+ complex was evaluated in 9:1 (v/v) aqueous acetonitrile via both UV-vis and fluorescence titrations. The absorbance of 1 and its Hg2+ complex remained, by and large, unaffected between pH 5.0 and 8.5 (Fig. S8, ESI†). Significantly, upon increasing the pH from 7.0, the increase in the absorption intensity was observed, attributed to the formation of a phenoxide ion at basic pH. In the fluorescence spectra, both sensor 1 and the 1–Hg2+ complex themselves are quite stable in the pH range of 6.0–8.5. At lower pH values (2–5), the quenching of the fluorescence intensity was observed along with the addition of the acid. This quenching of fluorescence could be attributed to the protonation of the –NH and –OH functionalities of sensor 1. In the basic medium, the fluorescence intensity remains, by and large, the same, although it increased to a small extent beyond pH 10.0 (Fig. S9, ESI†).
1H NMR titration
To explore the nature of interactions between sensor 1 and Hg2+ ions, a 1H NMR titration experiment was performed (Fig. 4). With the addition of just 0.05 equiv. of Hg2+ ions, the signal of free sensor 1 at δ 12.02 ppm due to –OH firstly broadened and then vanished at 0.35 equiv. without displaying any downfield shift; while the naphthyl doublet at δ 8.21 ppm and the aryl triplet at δ 7.91 ppm (corresponding to Ha and Hb) being closer to more basic imidazolyl–N and OH broadened and finally merged into a single peak at δ 8.01 ppm. These changes indicated the formation of a supramolecular complex followed by deprotonation between receptor 1 and a Hg2+ ion. Before the addition of Hg2+ ions, the Ha and Hb protons of naphthyl and aryl, respectively, were in the same environment and hence exhibited two separate peaks, whereas upon adding Hg2+ ions, the protons closer to –N and OH got involved in the interaction process and could not be resolved separately. Thus, they displayed a broad signal with higher added concentration of Hg2+ ions (0.35 equiv.). | ||
| Fig. 4 1H NMR titration of 1 (3.94 × 10−2 M) with incremental addition of Hg2+ in DMSO-d6. | ||
DFT calculations
The SPECFIT calculations revealed the formation of a 1:1 stoichiometry between sensor 1 and the sensed cations (Hg2+, Cu2+ and Al3+ ions). The computational studies of sensor 1 were done with Hg2+ ions to figure out the structural features of the complex and to support the proposed mechanism and the experimental results. Chemosensor 1 and the 1–Hg2+ complex (1:1) were optimized by the DFT method and in order to further understand the behaviour of Hg2+ ions with chemosensor 1, DFT calculations were carried out. The structural optimization of the keto and enol forms of chemosensor 1 and its 1–Hg2+ complex was done using the Gaussian 03 package using the exchange correlation function B3LYP and the basis set 6-31G (d,p) for C, H, N and O atoms and LANL2DZ for Hg atoms.33 The optimized structures of 1 and its 1–Hg2+ complex are shown in Fig. S10 (ESI†). As can be seen in Fig. S10a (ESI†), the distance between the H of the hydroxyl and N in the enol form is 1.63 Å, satisfying the excited state intramolecular proton transfer (ESIPT) requirements.34 Moreover, the Hg–O and Hg–N distances in the 1–Hg2+ optimized structure (Fig. S10c, ESI†) are well within the sum of van der Waals radii of the corresponding atoms (Hg 1.55–2.23 Å; O 1.52 Å; N 1.54 Å).35,36
The energy comparison diagram (Fig. 5) shows that, upon complexation of 1 with Hg2+ ions, the calculated energy gap of the HOMO and the LUMO decreased with respect to the free chemosensor, indicating the formation of a stable complex. In 1, only one Hg2+ ion binds to imidazole–N and naphthyl–OH with simultaneous longer distance overlap between the orbitals of the metal ion and the orbitals of the π-system of the naphthyl fluorophore, justifying the experimental 1:1 stoichiometric ratio. The electron density of the HOMO of 1 lies on the electron withdrawing part of the respective ligands and for the LUMO, the electron density lies on the electron donating part. The contour plots of the HOMO after the addition of Hg2+ ions showed that the electron density is over the electron-withdrawing part of chemosensor 1, however the electron density of the LUMO was spread over both the electron donating and electron withdrawing parts (Fig. 5).
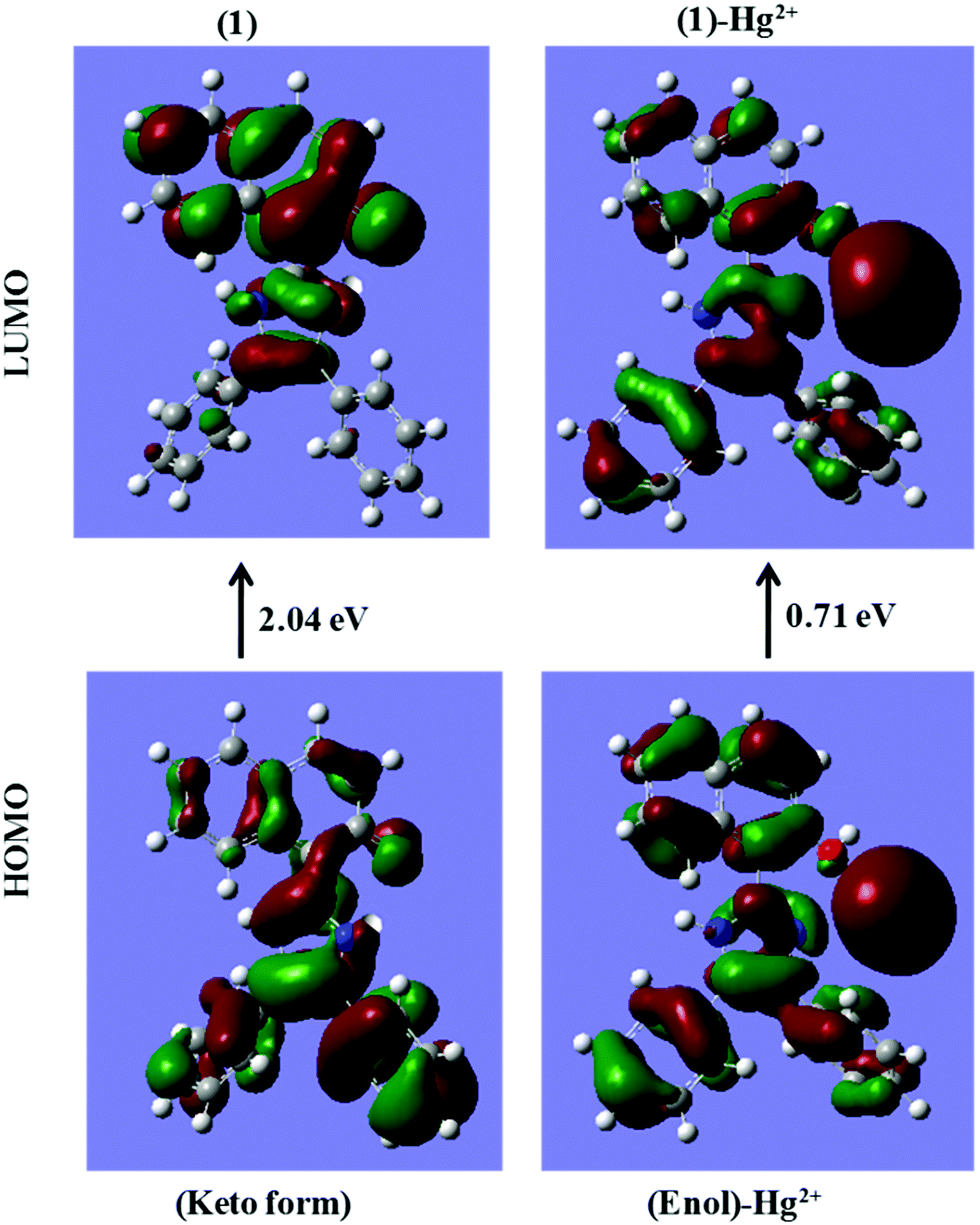 | ||
| Fig. 5 DFT computed LUMO and HOMO diagrams of sensor 1 and sensor 1 with Hg2+ [sensor 1 is in its keto form in sensor 1; while it is in the enol form in the 1 + Hg2+ complex as proposed in Scheme 2]. | ||
Selective response of sensor 1 to cysteine (Cys)
In recent research studies, Hg2+ based complexes have been exploited for the sensing of sulphur containing amino acids because of their high affinity.37,38 To test the feasibility of this strategy for the quantification of sulphur containing amino acids i.e. cysteine (Cys), methionine (Met) and cystine (Cyst), the in situ generated 1–Hg2+ complex was verified in the presence of the mentioned amino acids viz. Cys, Met and Cyst. As cystine (Cyst) becomes soluble in an acidic medium and both 1 and 1–Hg2+ complexes are pH dependent at lower pH values (below pH 5.0), actual fluorescence changes caused by cystine could not be identified and measured. Upon addition of cysteine to the solution of the in situ generated 1–Hg2+ complex, the intensity of the emission band at 454 nm greatly increased; while addition of methionine did not induce any obvious change (Fig. 6a). As shown in Fig. 6b, the emission intensity of 1–Hg2+ at 454 nm starts increasing steadily with the increase in the Cys concentration, thus restoring the emission profile of sensor 1 (Fig. 6b). The spectral fitting of the fluorescence titration data of the in situ generated 1–Hg2+ complex for Cys using SPECFIT showed the formation of a 1:1 stoichiometry with logβ11 = 5.11 ± 0.19 (Fig. S11, ESI†). The LOD of the 1–Hg2+ complex for Cys was measured to be 1.3 μM. The binding constant for the Hg2+–Cys complex is higher than that of the 1–Hg2+ complex, so, the dissociation of 1–Hg2+ leads to the enhancement of the fluorescence.39 Very recently, Zhang and co-workers exploited a Cu2+ based complex for the selective detection of Cys.40 In addition, as indicated in Fig. 2a, sensor 1 underwent fluorescence quenching upon addition of Cu2+. So, the fluorescence changes in the 1–Cu2+ complex were checked with gradual addition of Cys. But, only 20% recovery of Cu2+ was observed with addition of Cys to the 1–Cu2+ complex (Fig. S12, ESI†).
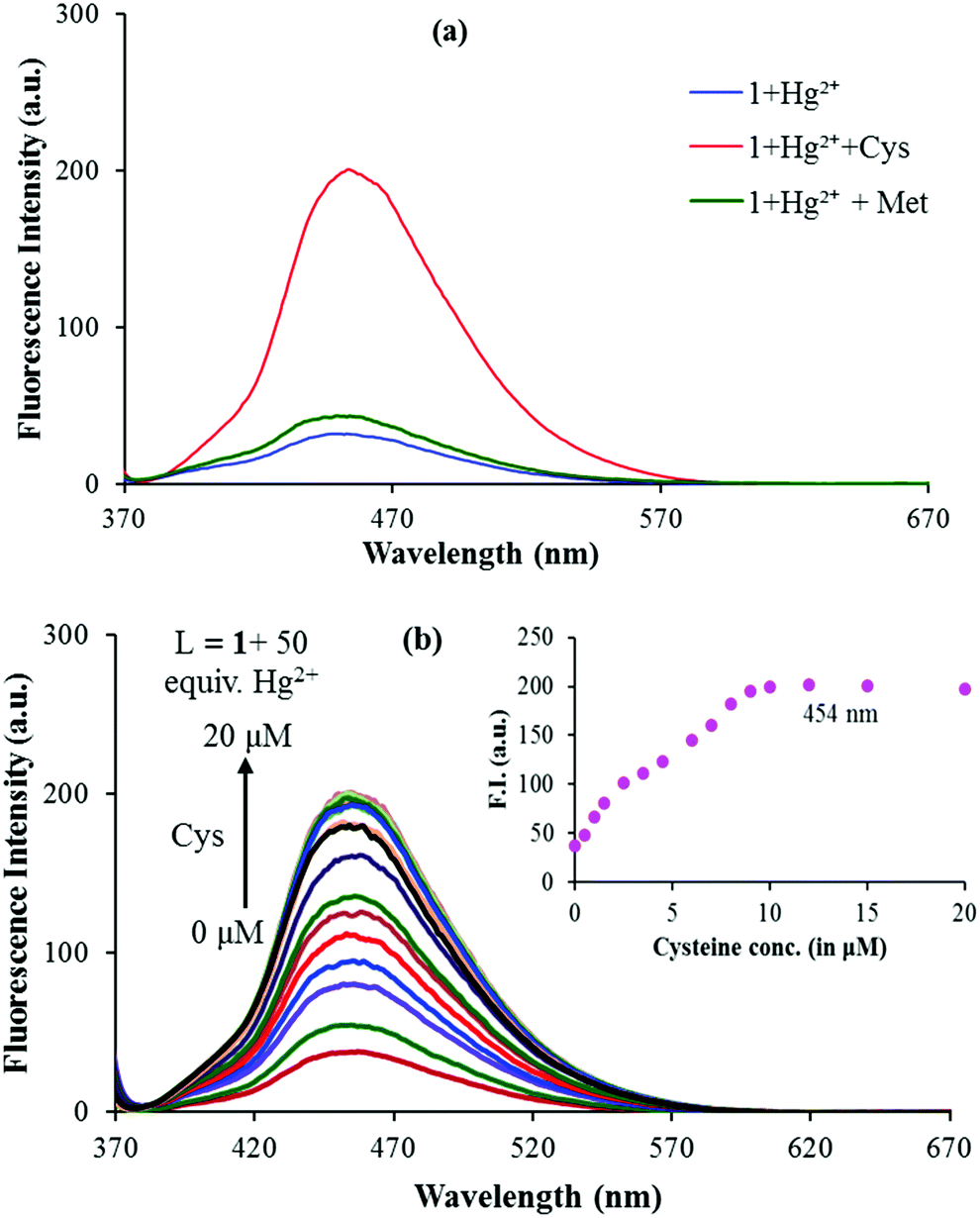 | ||
| Fig. 6 (a) Fluorescence spectral response of 1 (5 μM) + 50 equiv. of Hg2+ upon addition of 100 equiv. of Cys and Met and (b) fluorescence titration spectra of the 1–Hg2+ complex in the presence of various concentrations of Cys excited at 350 nm in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer). Inset: Plot of the fluorescence intensity at 454 nm versus the concentration of added Cys. | ||
As reversibility is an important aspect for a chemical sensor to be widely used for the detection of specific analytes, the reversibility of the fluorescence response of 1 was further verified during three cycles of alternate addition of Hg2+ and Cys to a solution of 1. The fluorescence emission of the tested solutions showed alternating enhancement and quenching emissions at 454 nm as shown in Fig. 7.
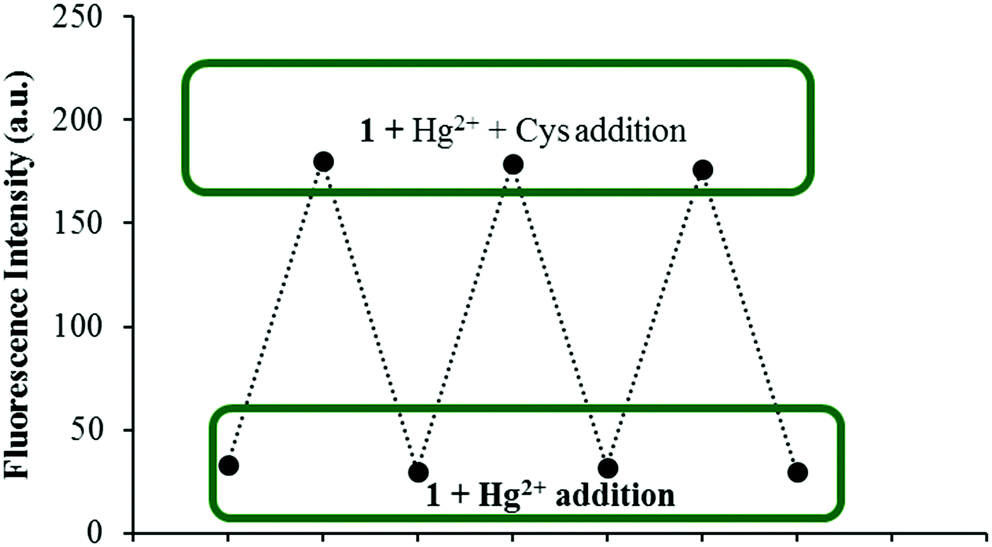 | ||
| Fig. 7 Fluorescence spectra showing the reversible behavior between 1 (5 μM) and Hg2+ (50 equiv.) upon addition of Cys ions (50 equiv.). | ||
A plausible sensing model for chemosensor 1
Due to the presence of the hydroxyl unit as a functional group in sensor molecule 1, the tautomerization process can be observed in it, which leads to the existence of the ESIPT (excited state intramolecular proton transfer) phenomenon within sensor 1. The equilibrium between the two tautomeric forms i.e. the “keto” and “enol” forms is hampered in the presence of Hg2+ ions, resulting in partial shifting of the equilibrium towards the ‘enol’ form and due to this fluorescence quenching was observed.20,21Scheme 2 demonstrates the plausible sensing mechanism on the basis of the SPECFIT calculations, DFT computational calculations and 1H NMR titration studies. Here, the active participation of the imidazole nitrogen and oxygen of the –OH group might be contributing to cation binding corresponding to a 1:1 stoichiometry (Scheme 2).
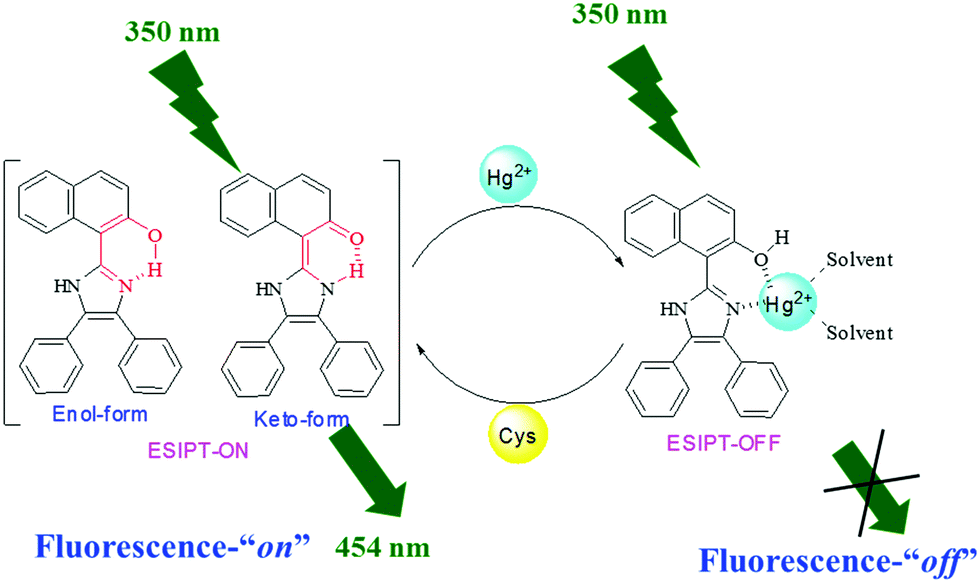 | ||
| Scheme 2 Proposed binding mechanism for chemosensor 1 with Hg2+ in solution. | ||
Application in real samples
To investigate the reliability of the proposed chemosensor, 1, various water samples like drinking water, tap water and river water samples were collected for the estimation of Hg2+ concentrations. All the suspended insoluble particles present in the collected samples were removed with the help of a syringe-filter. To keep a steady system at a given solvent ratio and pH, HEPES buffer was added to maintain the pH at 7.0. Increasing concentrations of Hg2+ were added to these samples to obtain the spiked samples and their absorption spectra were recorded. The concentration of Hg2+ was determined using the regression graphs of chemosensor 1 (5 μM) (Table 1). The obtained results are found to be in good accord with the concentration of the Hg2+ ions in the system, which displayed good recovery. Therefore, it reveals the effectiveness of chemosensor 1 in the detection of Hg2+ in actual samples.| Sample | Hg2+ added/mol L−1 | Hg2+ found/mol L−1 | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Drinking water | 0 | Not detected | — | — |
| 5.00 × 10−6 | 4.89 × 10−6 | 97.8 | 0.86 | |
| 10.00 × 10−6 | 9.78 × 10−6 | 97.8 | 0.45 | |
| 15.00 × 10−6 | 14.90 × 10−6 | 99.33 | 0.31 | |
| 40.00 × 10−6 | 39.70 × 10−6 | 99.25 | 0.42 | |
| 60.00 × 10−6 | 59.42 × 10−6 | 99.03 | 0.36 | |
| Tap water | 0 | Not detected | — | — |
| 5.00 × 10−6 | 4.45 × 10−6 | 89 | 1.7 | |
| 10.00 × 10−6 | 9.36 × 10−6 | 93.6 | 2.2 | |
| 15.00 × 10−6 | 15.20 × 10−6 | 101.3 | 0.4 | |
| 40.00 × 10−6 | 39.97 × 10−6 | 99.92 | 0.08 | |
| 60.00 × 10−6 | 58.99 × 10−6 | 98.31 | 0.63 | |
| River water | 0 | Not detected | — | — |
| 5.00 × 10−6 | 5.20 × 10−6 | 104 | 1.2 | |
| 10.00 × 10−6 | 9.94 × 10−6 | 99.4 | 1.1 | |
| 15.00 × 10−6 | 15.30 × 10−6 | 102 | 1.1 | |
| 40.00 × 10−6 | 40.20 × 10−6 | 100.5 | 0.3 | |
| 60.00 × 10−6 | 59.99 × 10−6 | 99.98 | 0.2 | |
“Set–reset” memorized device
Recently, the development of sequential logic devices based on the conversion of chemically encoded information into optical (fluorescence/absorbance) signals has attracted tremendous attention. Sequential circuits have the ability to store information and usually operate through feedback loops, where one of the outputs of the device functions as an input and is memorized as a “memory element”.41,42 We found that the “on–off–on” behaviour of sensor 1 observed here was reversible (Fig. 7). The fluorescence emission of sensor 1 was repeatedly quenched and regenerated by the sequential addition of Hg2+ and Cys. This “on–off–on” switching process could be repeated several times with little fluorescence efficiency loss. Currently, reversible switches are of immense significance due to their considerable impact on the realm of information technology. By the use of the current fluorescence “on–off–on” system, a sequential logic circuit displaying a “Write–Read–Erase–Read” sequence in line with binary logic functions was developed. In the present molecular memory device, the two chemical inputs are Hg2+ (In1) and Cys (In2). The truth table (Fig. 8C) represents “set/reset flip-flop” corresponding to the memory device, as shown in Fig. 8A, where different output values were observed at 454 nm upon sequential addition of Hg2+ and Cys. The system is in a ‘write’ (off) state in the presence of Hg2+ (In1) and memorized as a ‘1’ binary set. This “off” written information could be erased by the second Input (In2, Cys) (‘erase’ state) because of the regeneration of the fluorescence emission, which is memorized as a ‘0’ binary set. This reversible fluorimetric switching between the “off” and “on” emission states in the feedback loop enabled sensor 1 to display “read–erase–write–read” behavior through the output signal at 454 nm with the help of reversible logic operations (Fig. 8B).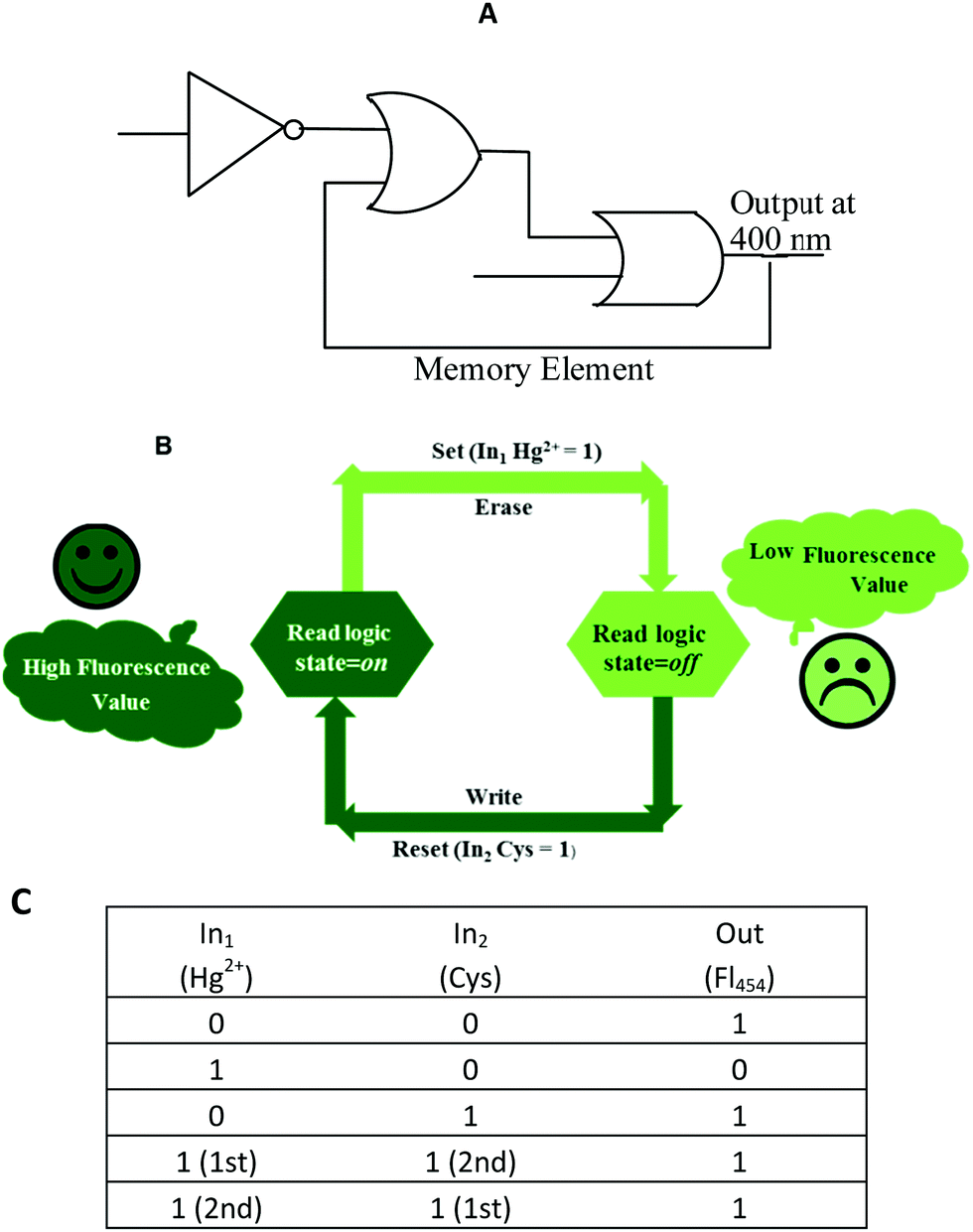 | ||
| Fig. 8 (A) Logic symbol; (B) schematic representation and (C) truth table of the reversible logic operations for the memory element possessing “write–read–erase–read” functions. | ||
Conclusions
In summary, we have successfully developed a naphthalene-based ESIPT fluorescent chemosensor (1) for the detection of Hg2+ in 9:1 (v/v) aqueous CH3CN (pH 7.0 HEPES buffer). Addition of Hg2+ to sensor 1 shows large fluorescence quenching, which was recovered by using cysteine. Job's plot, DFT calculations and 1H NMR studies confirmed the 1:1 stoichiometry as well as the sensing mechanism. This reversible detection monitoring process can be cycled multiple times. The sensor was successfully used to determine Hg2+ ions in real water samples. In addition, using Hg2+ and Cys as chemical inputs and observing fluorescence changes as the output signal at 454 nm, the reversible sequences of set/reset logic operations in a feedback loop demonstrated the “read–erase–write–read” memory functions in sensor 1 at the molecular level.
Experimental
Materials and methods
Benzil, 2-hydroxy-1-naphthaldehyde, cysteine and methionine amino acids and perchlorate salts of various metal ions were purchased from Aldrich. All other chemicals were used as received without further purification. Acetonitrile (CH3CN) was of HPLC grade. Melting points were determined in capillary tubes and are uncorrected. 1H and 13C NMR spectra were recorded on BRUKER AVANCE 400 and 100 MHz instruments using tetramethylsilane as an internal standard. Various metal ions such as Na+, K+, Mg2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+ and Cd2+ were added as their perchlorate salts for UV-vis and fluorescence experiments. Aliquots of the metal ions and amino acids under investigation were then injected into the sample solution through a rubber septum in the cap. The solutions were allowed to stabilize after each addition and were then scanned.General procedure for UV-vis and fluorescence experiments
UV-vis and fluorescence titrations were conducted on a 5 μM solution of 1 in a 9:1 (v/v) aqueous acetonitrile (pH 7.0 HEPES buffer) solution. All the UV-vis experiments were carried out on a Shimadzu UV-240 spectrophotometer; while fluorescence spectra were recorded using a HITACHI-7000 spectrophotometer equipped with a 220–240 V Xe lamp with a quartz cell of 1 cm width and 3.5 cm height. The excitation was carried out at 350 nm for sensor 1 with 5 nm excitation and emission slit widths in a fluorometer. The stock solution of sensor 1 (1 × 10−2 M) was prepared in DMSO and was diluted with an aqueous CH3CN (v/v 9:1; pH = 7.0) solution for further different spectroscopic experiments. All absorption scans were saved as ACS II files and further processed in Excel(tm) to produce all graphs shown.
General procedure for 1H NMR experiments
For 1H NMR titrations, two stock solutions were prepared in DMSO-d6, one of them containing the host (1 of 3.94 × 10−2 M conc.) only and the other containing an appropriate concentration of the guest (Hg2+). Aliquots of the two solutions were mixed directly in NMR tubes, which then were diluted to 0.5 mL with DMSO-d6 if required.General procedure for SPECFIT calculations
The spectral data obtained by UV-Vis or fluorescence titration of the molecular probes with the analytes (Hg2+, Cu2+, Al3+ and cysteine) were analysed for their stoichiometries and binding constant using SPECFIT programme version 3.0.36. The programme was written by Robert A. Binstead, Andreas D. Zuberbuhler and Bernhard Jung and was available commercially. The programme performs global analysis of equilibrium and kinetic systems with a singular value of decomposition and nonlinear-regression modelling by the Levenberg–Marquardt method. The programme simulates the absorption or fluorescence data obtained experimentally. The spectra obtained using the UV-Vis and fluorescence machines were converted to an ASCII file and were transferred to MS-Excel spread sheets. These spread sheets were converted to a text file. The file was imported into SPECFIT/32 software and the data were simulated using different stoichiometric models. The stoichiometry of the species formed, distribution of the species and their association constants were determined through a fit model.Synthesis of 1-(4,5-diphenyl-1H-imidazol-2-yl)-naphthalen-2-ol (1)
Benzil (1 g, 4.75 mmol) and ammonium acetate (10.56 g, 4.75 mmol) were dissolved in 10 mL of hot glacial acetic acid. To this mixture, a solution of 2-hydroxy-1-naphthaldehyde (0.5044 mL, 4.75 mmol) in 10 mL of glacial acetic acid was added dropwise. The mixture was heated at 90 °C for 4 h and the progress of the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was poured into 200 mL of water. The solution was neutralized with ammonium hydroxide to pH = 7 and was then cooled to room temperature. The contents were filtered to yield crude precipitates, which were then washed with large portions of water. Recrystallization of the crude product from a mixture of methanol and chloroform resulted in the formation of a pure product (1). Light yellow solid; 82% yield; m.p. (°C) 186; FT-IR (neat, ν cm−1): 3238 (O–Hstr.), 3019 (Ar-Hstr.), 1583 (Ar-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cstr.), 1617 (–CNstr.), 1234 (–C–Nstr.); 1H NMR (400 MHz, DMSO-d6, ppm): 12.02 (brs, 2H, –NH, –OH), 8.21 (d, 1H, Ar-H, J = 8 Hz), 7.88 (d, 1H, Ar-H, J = 8.2 Hz), 7.91 (d, 1H, Ar-H, J= 8.3 Hz), 7.59 (d, 4H, Ar-H), 7.50 (td, J = 8 Hz, 1H, Ar-H), 7.41–7.29 (m, 8H, Ar-H); 13C NMR (100 MHz, DMSO-d6, ppm): 154.5, 142.9, 132.2, 130.6, 128.4, 128.1, 127.9, 127.6, 127.1, 126.9, 124.4, 123.0, 118.3, 109.1; HRMS: m/z (relative abundance (%), assignment) = 363.15 [100, (M + 1)+].
Cstr.), 1617 (–CNstr.), 1234 (–C–Nstr.); 1H NMR (400 MHz, DMSO-d6, ppm): 12.02 (brs, 2H, –NH, –OH), 8.21 (d, 1H, Ar-H, J = 8 Hz), 7.88 (d, 1H, Ar-H, J = 8.2 Hz), 7.91 (d, 1H, Ar-H, J= 8.3 Hz), 7.59 (d, 4H, Ar-H), 7.50 (td, J = 8 Hz, 1H, Ar-H), 7.41–7.29 (m, 8H, Ar-H); 13C NMR (100 MHz, DMSO-d6, ppm): 154.5, 142.9, 132.2, 130.6, 128.4, 128.1, 127.9, 127.6, 127.1, 126.9, 124.4, 123.0, 118.3, 109.1; HRMS: m/z (relative abundance (%), assignment) = 363.15 [100, (M + 1)+].
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are greatly thankful to the SAIF, Panjab University, Chandigarh for recording the NMR spectra and are grateful to the DST PURSE-II (Grant no. 48/RPC) for the financial assistance.References
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS.
- B. Kaur, N. Kaur and S. Kumar, Coord. Chem. Rev., 2018, 358, 13–69 CrossRef CAS.
- K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh and S. Kumar, Coord. Chem. Rev., 2012, 256, 1992–2028 CrossRef CAS.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS.
- S. Devaraj, D. Saravanakumar and M. Kandaswamy, Sens. Actuators, B, 2009, 136, 13–19 CrossRef CAS.
- W. F. Fitzgerald, C. H. Lamgorg and C. R. Hammerschmidt, Chem. Rev., 2007, 107, 641–662 CrossRef CAS PubMed.
- B. K. Rani and S. A. John, J. Hazard. Mater., 2018, 343, 98–106 CrossRef CAS.
- E. Cernichiari, G. J. Myers, N. Ballatori, G. Zareba, J. Vyas and T. Clarkson, NeuroToxicology, 2007, 28, 1015–1022 CrossRef CAS PubMed.
- E. Weerepana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef PubMed.
- C. Yin, F. Huo, J. Zhang, R. M. Manez, Y. Yang, H. Lv and S. Li, Chem. Soc. Rev., 2013, 42, 6032–6059 RSC.
- J. Reiser, B. Adair and T. Reinheckel, J. Clin. Invest., 2010, 120, 3421–3431 CrossRef CAS.
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC.
- X. Zhou, X. Jin, G. Sun and X. Wu, Chem. – Eur. J., 2013, 19, 7817–7824 CrossRef CAS.
- B. Guo, X. Pan, Y. Liu, L. Nie, H. Zhao, Y. Liu, J. Jing and X. Zhang, Sens. Actuators, B, 2018, 256, 632–638 CrossRef CAS.
- A. T. Wright and E. V. Anslyn, Chem. Soc. Rev., 2006, 35, 14–28 RSC.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- A. Helal, N. T. T. Thao, S. Lee and H.-S. Kim, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 87–94 CrossRef CAS.
- G. Dhaka, N. Kaur and J. Singh, J. Photochem. Photobiol., A, 2017, 335, 174–181 CrossRef CAS.
- N. Kaur, G. Kaur and P. Alreja, J. Photochem. Photobiol., A, 2018, 353, 138–142 CrossRef CAS.
- R. B. Orfao Jr., F. D. Carvalho, P. H.-d.-M. Mello and F. H. Bartoloni, J. Braz. Chem. Soc., 2017, 28, 1896–1904 Search PubMed.
- R. B. Orfao Jr., J. Alves and F. H. Bartoloni, J. Fluoresc., 2016, 26, 1373–1380 CrossRef.
- J. Andreasson and U. Pischel, Chem. Soc. Rev., 2010, 39, 174–188 RSC.
- K. Szacilowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS.
- N. Kaur, Curr. Org. Chem., 2014, 18, 2892–2909 CrossRef CAS.
- J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS.
- V. Balzani, M. Venturi and A. Credi, Molecular devices and Machines: A Journey into the Nanoworld, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- M. N. Stojanovic and D. Stefanovic, Nat. Biotechnol., 2003, 21, 1069–1074 CrossRef CAS.
- F. Remacle, R. Weinkauf and R. D. Levine, J. Phys. Chem. A, 2006, 110, 177–184 CrossRef CAS.
- X. F. Guo, D. Q. Zhang, G. X. Zhang and D. B. Zhu, J. Phys. Chem. B, 2004, 108, 11942–11945 CrossRef CAS.
- D. Margulies, G. Melman and A. Shanzer, J. Am. Chem. Soc., 2006, 128, 4865–4871 CrossRef CAS.
- N. Kaur and P. Alreja, J. Chem. Sci., 2015, 127, 1253–1259 CrossRef CAS.
- J. D. Winefordner and G. L. Long, Anal. Chem., 1983, 55, 712A–724A Search PubMed.
- S. R. Patil, J. P. Nandre, D. Jadhav, S. Bothra, S. K. Sahoo, M. Devi, C. P. Pradeep, P. P. Mahulikar and U. D. Patil, Dalton Trans., 2014, 43, 13299–13306 RSC.
- S. Sahana, G. Mishra, S. Sivakumar and P. K. Bharadwaj, Dalton Trans., 2015, 44, 20139–20146 RSC.
- S.-Z. Hu, Z.-H. Zhou and B. E. Robertson, Z. Kristallogr., 2009, 224, 375–383 CAS.
- S. C. Nyburg and C. H. Faerman, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 274–279 CrossRef.
- T. Lou, Z. Chen, Y. Wang and L. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 1568–1573 CrossRef CAS.
- H. Xu and M. Hepel, Anal. Chem., 2011, 83, 813–819 CrossRef CAS.
- W. Huang, C. Song, C. He and C. Duan, Inorg. Chem., 2009, 48, 5061–5072 CrossRef CAS.
- B. Guo, X. Pan, Y. Liu, L. Nie, H. Zhao, Y. Liu, J. Jing and X. Zhang, Sens. Actuators, B, 2018, 242, 849–856 Search PubMed.
- N. Kaur, G. Dhaka and J. Singh, New J. Chem., 2015, 39, 6125–6129 RSC.
- S. Ozlem and E. U. Akkaya, J. Am. Chem. Soc., 2009, 131, 48–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj03909g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |