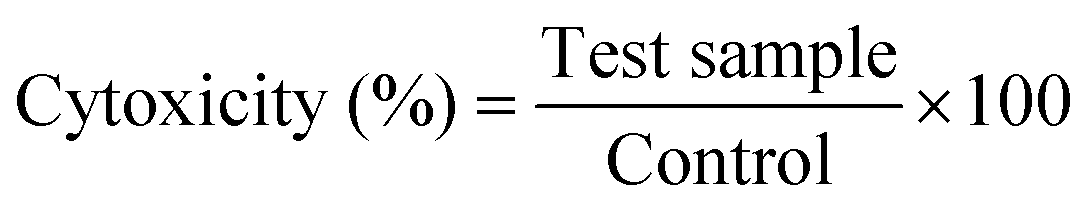DOI:
10.1039/C7NJ03143B
(Paper)
New J. Chem., 2018,
42, 725-734
Development of self-repair nano-rod scaffold materials for implantation of osteosarcoma affected bone tissue†
Received
22nd August 2017
, Accepted 14th November 2017
First published on 14th November 2017
Abstract
Osteosarcoma is the most widely recognized fatal bone disease in children and young adults. The osteosarcoma affected places of bone implant materials lose their activity after a period of time due to the possibility of regenerating sarcoma cells. Hence, the complete recovery of this disease is very challenging. Subsequently, new helpful methodologies, including natural antioxidant loaded bone implant materials, are effectively used to treat osteosarcoma cells. In this regard, nano-hydroxyapatite reinforced with a xylitol based poly(xylitol sebacate) PXS co-polymer together with a capsaicin loaded scaffold was investigated on osteosarcoma cells. The physicochemical properties of the scaffold were evaluated by FT-IR (Fourier transform infrared spectroscopy), SEM (scanning electron microscopy), TEM (transmission electron microscopy), and XRD (X-ray diffraction). The in vitro release and antioxidant activity of the capsaicin loaded nHAP/PXS/CAP scaffold were evaluated by UV-Visible spectroscopy. Cytotoxicity against the Saos-2 cancer line and cell viability in the osteoblast cell MG63 are reported. Eventually, the composite enlarges the creation of reactive oxygen species (ROS) in Saos-2 cells.
1. Introduction
Osteosarcoma (OS) is the most common and prevalent form of a malignant neoplasm of bone. The yearly prevalence of OS is 8–11 cases per million in the age group of 15–19 years. Even with its paucity, it has been shown to be the second most dominant cause of cancer-associated death in children and young adults.1 One of the considerable goals to exchange a bone construct is the development of bone graft scaffolds to restore/connect large bone defects caused by trauma, tumors, and infection.2 Scaffolds play a vital role in osseous tissue engineering, and their primary function is to act as an appropriate template to facilitate cell growth. They also provide differentiation between the bone defect as well as providing structural and functional support for newly formed bone.3 The design of materials with particular structural morphologies, sizes, and chemical combinations has been attracting great attention because their physicochemical and biotic properties fluctuate in potential applications such as dynamic segments.4–6 Consequently, advancement of new methodologies to control the morphological aspects, size of the particles, structure of the materials and organization of the scaffolds has turned into a current research area in dental and orthopedic applications.7
Nano morphology is shown when a variety of hydroxyapatite (HAP) ingredients are inserted into a collagen network to form essential building elements for all types of bone material such as the bio-mineralized collagen component.8–10 The important properties of the osteoconductive nature of HAP have prompted its use across the board as a coating or added substance in bone material frameworks in dental and orthopedic implants.11–13 However, the points of interest offered by HAP remain to be fully exploited because of issues in its applications.14–16 Furthermore, the multifunctionality and properties of HAP polymer composites are required for bone tissue engineering because they help the structural requirement of the bone tissues, permit molecular communication with cells that promote cell adhesion and proliferation, and influence cell differentiation.17–20
In this regard, extensive research in the dental and orthopedic fields has been focused on achieving increased rates of osseointegration.21 On the other hand, research efforts devoted to inhibiting cancer regeneration and bacterial colonization are focused on cytotoxic agents or treatments that do not positively affect host tissues.22 The majority of approaches described in the literature tend to only focus on either improving cell adhesion or preventing bacterial infection but rarely explore a combined effect.23–25 Over the past few years, a significant number of synthetic biodegradable polymers have been incorporated with HAP such as gelatin,26 alginate polycaprolactone polypeptide and polylactide, and copolymers,27 graphene and carbon nanotubes28–30 have been added to HAP to improve their efficacy.
Exceptionally, the miscellaneous pharmacological and physiological properties of the scaffold materials upon the addition of a bioactive molecule are studied as a new method to improve the materials’ assets. Capsaicin has attracted dental and orthopedic antioxidant activity towards the generation of a reactive oxygen species (ROS).31 Capsaicin (8-methyl-N-vaillyl-6-nonenamide) is an active component of Capsicum annuam (Red chilli) plants. Moreover, capsaicin is utilized topically to treat various diseases like diabetic neuropathy, osteoarthritis, rheumatoid joint inflammation, and post therapeutic neuralgia. The anticancer role of capsaicin has been known for a long time.32,33 In this work, we have demonstrated the synthesis of a well-defined morphology of HAP and reinforced it with poly(xylitol sebacate) (PXS) co-polymeric materials. The aptitudes of the new HAP–PXS materials were evaluated for the loading of CAP and exhibiting antioxidant properties on osteosarcoma cells.
2. Materials and methods
2.1 Materials
Ammonium hydroxide solution (NH4OH), calcium chloride dihydrate (CaCl2·2H2O), diammonium hydrogen phosphate (NH4)2HPO4, ethyl alcohol (C2H5OH), phosphate buffer saline (PBS), sodium hydroxide (NaOH), and capsaicin (C18H23NO3) were received from Sigma-Aldrich, Chennai, India. Analytical grade chemicals were used throughout experiments without any refining. Deionized water was utilized all through the analysis.
2.2 Synthesis of hydroxyapatite (HAP)
Preparation of HAP was carried out in 0.05 M of CaCl2·2H2O dissolved in distilled water with the addition of 0.03 M (NH4)2HPO4 to the CaCl2·2H2O solution under magnetic stirring. The pH of the reaction solution was kept at pH 10.0 using ammonium hydroxide solution. After 30 minutes of stirring, the suspension was transferred into a sealed autoclave and heated to 180 °C for 2 h in a muffle furnace and then cooled to room temperature (27 °C). The precipitate was collected and washed alternately with ethyl alcohol and deionized water by centrifugation (5000 rpm, 10 min). Finally, the purified product was dispersed in deionized water to form an aqueous dispersion and the solution was freeze-dried in an SSIPL-LYF freeze drier at −40 °C. The synthesized HAP was calcinated at 800 °C for 6 h in a muffle furnace to obtain pure HAP nanorods.
2.3 Synthesis of the HAP/PXS nanocomposite
The PXS materials were synthesized by a polycondensation reaction.37 Xylitol and sebacic acid were mixed in a round bottom flask at 140 °C and stirred for 1 h under an atmosphere of nitrogen gas to form a prepolymer. The mixture was further heated up to 150 °C and condensation was extended for an additional 5 h to prepare the PXS polymers. The viscosity of the products was raised slowly with the increasing reaction time. The cross-linked elastomer was used for further characterization. The formation of an ester bond between the monomers of xylitol and sebacic acid was confirmed by FTIR. After confirmation, 5 wt% of polymer was added to synthesized pure HAP nanorods, followed by heating in a micro oven (2.5 GHz, LG, India) at 720 W for about 10 min to form the HAP/PXS composite.
2.4 Loading of capsaicin on the HAP/PXS composite
In order to load capsaicin (CAP) onto the HAP/PXS composite, CAP was dissolved in ethyl alcohol solution at 5 mg mL−1. CAP was added to the HAP/PXS composite and stirred using a magnetic stirrer for 30 min. Then, the mixture was sonicated for 30 min. Finally, the solution was freeze-dried to obtain the drug loaded HAP/PXS/CAP composite.
2.5 Physicochemical characterizations
2.5.1 Fourier transform infrared spectroscopy (FTIR).
The HAP, HAP/PXS, and HAP/PXS/CAP composites were characterized by a Bruker Tensor 27 Series FTIR spectrometer in the region of 400–4000 cm−1 with 2 cm−1 resolution and 16 scans. The test samples were prepared by grinding 0.2 g sample powder together with 1 g KBr and pressing into a transparent disc.
2.5.2 X-ray diffraction.
X-ray diffraction (XRD) analysis was carried out to investigate the phase composition and crystallinity of synthesized HAP, HAP/PXS, and HAP/PXS/CAP composites. This analysis was performed using a Bruker D8 Advance diffractometer with a monochromatic Cu Kα source operated at 40 kV and 30 mA. An acceleration voltage of 30 kV and a current of 15 mA were used. The analysis was performed over the 2θ range of 10 to 60° in step scan mode with a step size of 0.02° and at a scan rate of 0.02° min−1.
2.5.3 Scanning electron microscopy (SEM).
The morphology and microstructure of the HAP, HAP/PXS and HAP/PXS/CAP composites were examined using SEM (VEGA3 TESCAN) operated at an accelerating voltage of 10 kV equipped with energy dispersive X-ray analysis (EDAX).
2.5.4 Transmission electron microscopy.
The morphology and SAD images of synthesized HAP and HAP/PXS/CAP were investigated by transmission electron microscopy using high-resolution transmission electron microscopy (HR-TEM, TECNAI F30). For HR-TEM analysis, the synthesized nanoparticles and their composites were dispersed in ethanol by ultrasonication for 15 min and afterwards were loaded onto a carbon coated copper mesh.
2.5.5
In vitro release studies.
The in vitro release behavior of CAP from the HAP/PXS/CAP composite was investigated by a dialysis method in a phosphate buffer solution at pH 7.4. In brief, 50 mg of the HAP/PXS/CAP composite was sealed in dialysis bags (MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) and incubated in 100 mL of the PBS at pH 7.4 at 37 °C under 100 rpm stirring for different intervals of days. At discrete time intervals the supernatant solution of the reaction medium was taken and checked for the concentration of CAP in the solution at the λmax value of 280 nm using UV-Spectroscopy,33 and the solution was replenished with an equal volume of fresh medium. The percentage of drug release was calculated using the following formula
000 Da) and incubated in 100 mL of the PBS at pH 7.4 at 37 °C under 100 rpm stirring for different intervals of days. At discrete time intervals the supernatant solution of the reaction medium was taken and checked for the concentration of CAP in the solution at the λmax value of 280 nm using UV-Spectroscopy,33 and the solution was replenished with an equal volume of fresh medium. The percentage of drug release was calculated using the following formula
| Drug release (%) = AR/AC × 100 |
where AR is the absorbance of capsaicin released from the composite and AC is the total amount of capsaicin loaded onto the composite.
2.5.6 Antioxidant activity of CAP.
The ability of CAP to scavenge H2O2 was determined by UV-Spectrophotometry. A 40 mM solution of H2O2 was prepared in PBS (pH 7.4) at 25 °C. The H2O2 concentration was determined using a UV-Spectrophotometer at the λmax value of 230 nm. The molar extinction coefficient for H2O2 was checked with the addition of CAP in ethanol in a H2O2 solution at various concentrations such as 0.0, 20.0, 40.0, 60.0, 80.0, 100.0, 120.0 and 150.0 μg mL−1 at 25 °C. After 10 min the H2O2 concentration was determined by a UV-Spectrometer. The blank solution of H2O2 without CAP was also analyzed. The percentage of hydrogen peroxide is calculated by the following equation
| H2O2 scavenging activity (%) = [AC − AS]/AC × 100 |
where AC is the absorbance of the control and AS is the absorbance of the test sample of CAP.
2.5.7 Cell viability.
The cell viability of the HAP/PXS/CAS combined composites was surveyed utilizing human osteoblast-like cells (MG63) bought from the National Center for Cell Science (NCCS), Pune, India, cultured in Dulbecco's modified Eagle medium (Hi Media Laboratories) supplemented with 10% fetal ox-like serum, streptomycin (100 U mL−1) and penicillin (100 U mL−1). The medium was renewed every 2 days. Cells were preserved in a humidified environment of CO2 at 37 °C. The samples were disinfected in an autoclave at 120 °C for 120 min and after that were aliquoted into 96-well cell development plates. The cell development medium was expelled from all culture wells following 1, 3, and 7 days of brooding, and the cells were then transferred to new 96-well development plates at a thickness of 5 × 104 cells for all wells. Then, a tetrazolium salt 3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) suspension at 10 mg mL−1 was included. The cells were left to grow for 4 h and at that point the MTT suspension was expelled. 10% (200 μL) of dimethyl sulfoxide (DMSO) was added to each well. The cell suitability was evaluated at 570 nm on a spectrophotometric microplate reader. The engendering velocity of cells was assessed by measuring the optical thickness (OD) at 570 nm. Control cells contained the development medium and the cell viability (%) was assessed in view of the average of 5 reproductions, utilizing the following formula.
2.5.8 Cytotoxicity effect on bone tumor Saos-2 cells.
Human osteosarcoma (Saos-2) cells were acquired from the National Center for Cell Sciences (Pune, India). Cells were developed in Dulbecco's modified Eagle medium with 20% fetal bovine serum and upgraded with 2% penicillin and streptomycin. The Saos-2 cells were incubated at 27 °C (RT) in a 10% CO2 climate in a humidified incubator. The cell practicality was controlled by MTT examination. Quickly, 5 × 104 cells from every well in the medium were exchanged to new 96-well development plates and allowed to grow overnight. Cells were treated with nanoparticles for 1, 3, and 7 days at 27 °C and the cell cytotoxicity was resolved using the following.
2.5.9 Capsaicin on inducing cell apoptosis.
Saos-2 cells were cultured in DMEM and treated with 10 μg mL−1 of HAP/PXS/CAP for 1, 3, and 7 days, and washed with cold 1× PBS. Later the cells were stained with a mixture of AO and EtBr (100 μg mL−1) at room temperature for 10 min. The stained cells were washed twice with 1× PBS and observed under a fluorescence microscope using a blue filter.
2.5.10 Determination of reactive oxygen species.
Saos-2 cells were investigated with different concentrations of a stock solution of 10 μg mL−1 of HAP/PXS/CAP for 1, 3, and 7 days and were incubated in 5% CO2 at 37 °C for 24 h. The treated cells were loaded with 100 μM DCFH–DA (diluted in DMEM + 1% FBS) for 1 h (at 37 °C, in 5% CO2). Cells were then washed with 1× PBS, and untreated cells were considered as controls. DCF fluorescence images were recorded under a fluorescent microscope.
2.5.11 Hoechst staining.
Saos-2 cells were investigated with 10 μg mL−1 of HAP/PXS/CAP at 1, 3, and 7 days after 24 hours. The cells were washed with PBS and fixed in 70% ethyl alcohol for 30 minutes and then incubated with 10 mg mL−1 bisbenzimide trihydrochloride (Hoechst 33258) staining solution for 30 minutes. After treatment, cells were washed with PBS and afterwards were observed under a fluorescence microscope.
2.5.12 Statistical analysis.
All of the experiments were carried out individually and replicated three times. The one way analysis of variance (ANOVA) was used as the statistical analysis tool. At the point when the general ANOVA F-test broke down, Tukey's test was utilized as a post hoc examination. Data are accounted for as mean ± standard commitments (SD) with a centrality level of P < 0.005.
2.5.13 Measurement of mitochondrial membrane potential (MMP).
The mitochondrial membrane potential was determined based on the uptake of the cationic fluorescent dye rhodamine 123 (Rh123). The cells treated with 10 μg mL−1 of HAP/PXS/CAP for 1, 3, and 7 days were harvested and washed twice with PBS. The cells were then resuspended in 2 mL fresh medium containing rhodamine 123 (2.0 μM) and were incubated at 37 °C for 10 min with gentle shaking. The cells were collected by centrifugation and washed twice with PBS. Finally, the obtained cells were analyzed by fluorescence microscopy.
3. Results and discussion
3.1 FTIR characterizations
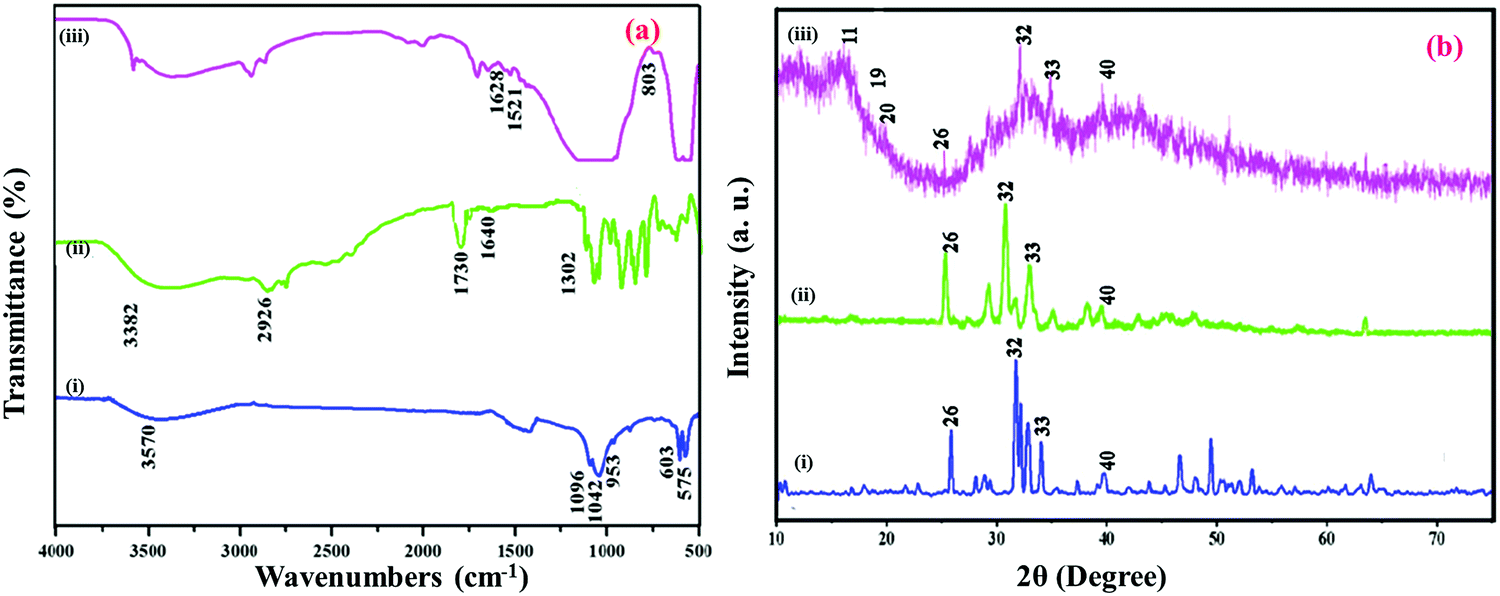
The development of self-repair bone implant materials HAP/PXS and HAP/PXS/CAP nanocomposites for cancer affected bone was investigated. Primary structural analysis of the nanocomposites was determined by FTIR spectroscopy. Fig. 1(a) shows the FTIR spectrum of the formation of HAP, the HAP conjugated PXS polymer and CAP loaded HAP/PXS composites. Fig. 1a(i) shows the HAP spectrum and the peaks detected at 3570 cm−1 and 634 cm−1 corresponded to stretching and bending of the hydroxyl group of HAP. The spectral peaks found at 1096 and 1042 cm−1 are attributed to the triply degenerate asymmetric stretching vibration of the hydroxyl (γ3) mode of phosphate groups. The peak at 953 cm−1 corresponds to a nondegenerate symmetric stretching vibration (γ1) of the phosphate groups. The peaks found at 603 cm−1 and 575 cm−1 represent the symmetric P–O stretching vibration (γ4). Consequently, all the peaks revealed in this spectrum strongly confirm the formation of HAP. The presence of TCP is also revealed by the stretching frequency of a phosphate group at 870 cm−1. Fig. 1a(ii) depicts the confirmation of the synthesized polymer PXS by the formation of an ester bond between xylitol and sebacic acid at 1730 cm−1. The stretching frequency of a free acid carbonyl group (C–O) was observed at 1302 cm−1. A broad spectrum observed at 3382 cm−1 recognized the hydroxyl groups of the PXS polymer, and the peak found at 1640 cm−1 (Ca+–COO−) indicates the formation of a bond to PXS.35 The existence of this calcium PXS complex confirms the formation of the HAP/PXS composite. The peak found at 2926 cm−1 indicates the presence of the C–H stretching vibration of PXS and is illustrated in the spectrum of the HAP/PXS composite in Fig. 1a(ii). Furthermore, in Fig. 1a(iii) characteristic peaks show that the two bands of CAP appearing at 1628 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 803 cm−1 occur for NH plane bending and the peak at 1521 cm−1 corresponds to an aromatic (CC) bond. The peak appearing at 3400 cm−1 indicates the intramolecular interaction of a hydroxyl group between CAP and the HAP/PXS composite.
O) and 803 cm−1 occur for NH plane bending and the peak at 1521 cm−1 corresponds to an aromatic (CC) bond. The peak appearing at 3400 cm−1 indicates the intramolecular interaction of a hydroxyl group between CAP and the HAP/PXS composite.
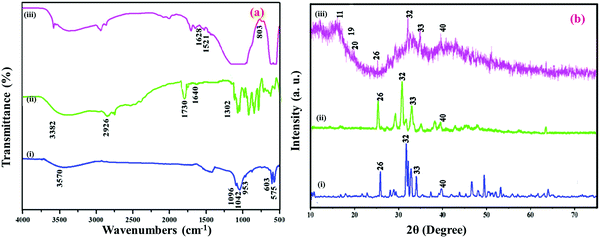 |
| | Fig. 1 FTIR (a) spectrum of HAP (i); HAP/PXS (ii); HAP/PXS/CAP (iii) and XRD (b) spectrum of HAP (i); HAP/PXS (ii); HAP/PXS/CAP (iii). | |
3.2 XRD analysis
XRD patterns of the HAP, HAP/PXS, and HAP/PXS/CAP composites and the nanoparticles synthesized are shown in Fig. 1b. For HAP, shown in Fig. 1b(i), the nanocrystals identified at 2θ = 26, 32, 33, and 40 are attributed to the (002), (211), (300), and (310) planes, respectively, and the results are well correlated with standard data (JCPDS No. 09-0432).34,35Fig. 1b(ii) illustrates the prepared PXS polymer with the HAP composite (HAP/PXS). Fig. 1b(iii) shows that following the addition of CAP onto the HAP/PXS composite, crystalline peaks at values of 11, 19, and 20 were recorded.33,35 The corresponding peaks indicate that the composite exists as a crystalline material. Usually, HAP with little crystalline character is essential for biomedical use owing to its high in vivo restorable property. Hence, the addition of 10% (w/v) polymer significantly reduces the crystalline character of the synthesized HAP nanorods.36
3.3 SEM analysis
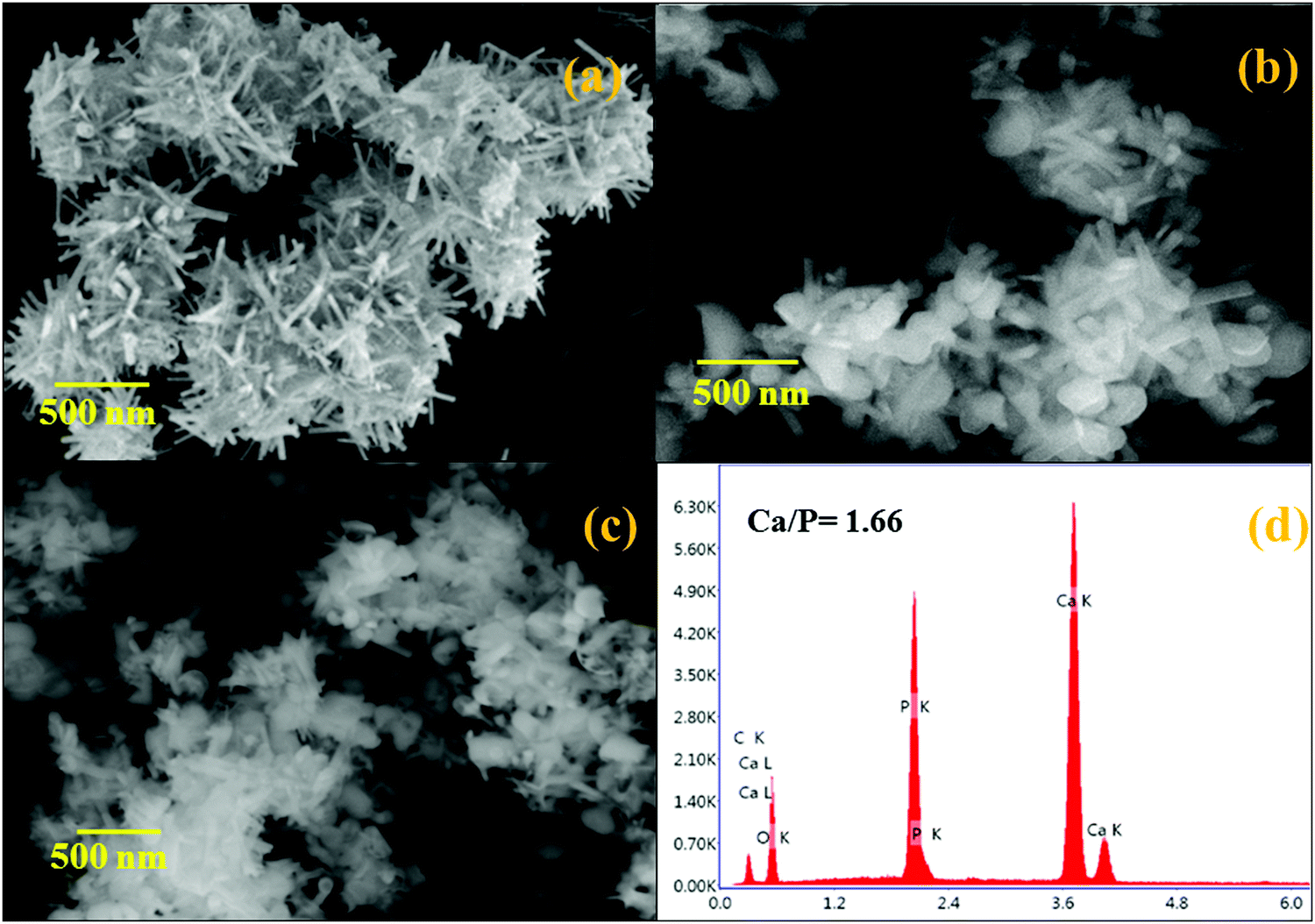
The influence of the morphology of hydrothermally prepared HAP, HAP/PXS, and HAP/PXS/CAP composites was investigated by SEM. The results are displayed in Fig. 2. Fig. 2(a) clearly indicates the formation of HAP rods and the bundles of HAP rods could be prepared by a hydrothermal method.30Fig. 2(d) corresponds to the EDAX spectrum of HAP. The EDAX spectrum specifies that the Ca/P molar ratio is 1.66, which is slightly less than a stoichiometric composition of 1.67. Fig. 2(b) indicates the HAP/PXS (5%) composition and we confirm that in the presence of the polymer, it wraps around HAP for its ideal shape. Fig. 2(c) corresponds to HAP/PXS/CAP and it exhibits a similar structure to HAP/PXS (5%).
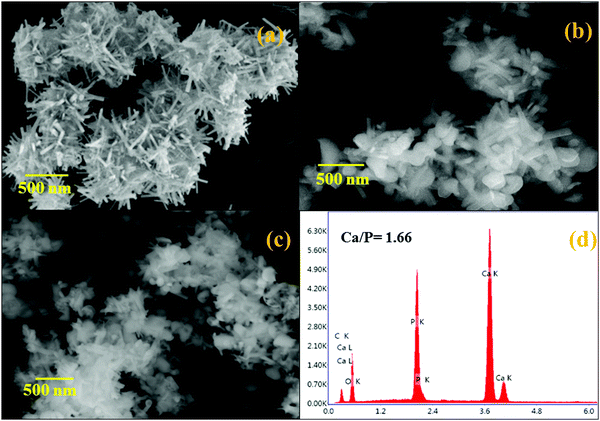 |
| | Fig. 2 SEM images of HAP (a); HAP/PXS (b); HAP/PXS/CAP (c); EDAX of pattern of HAP/PXS/CAP (d). | |
3.4 TEM analysis
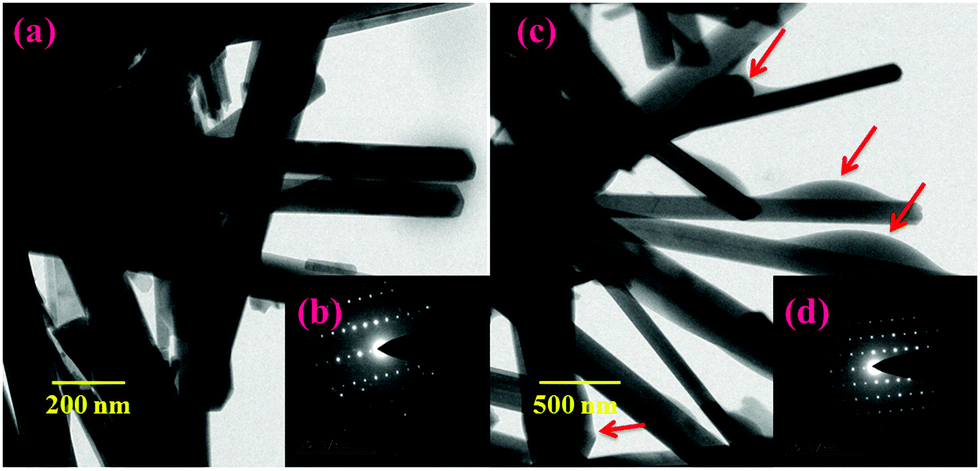
Fig. 3(a and b) shows the morphologies of the prepared HAP and HAP/PXS/CAP composites. The samples were composed of agglomerated clusters of rod-shaped hydroxyapatite nanocrystals. After hydrothermal treatment, longer rod-shaped hydroxyapatite nanocrystals were obtained as shown in Fig. 3(a and c). These results suggest that the prolonged hydrothermal treatment could promote the preferential growth of hydroxyapatite along the c-axis. Fig. 3(b and d) correspond to SADE images of HAP and HAP/PXS/CAP, respectively, and represent the (002), (211), (300), and (310) planes of HAP, which are well correlated with the XRD patterns.
 |
| | Fig. 3 TEM images of HAP/PXS (a) and HAP/PXS/CAP (c); SADE images of HAP/PXS (inset) (b) and HAP/PXS/CAP (inset) (d); the red colored arrows indicate the entrapment of the drug on the HAP nanorods. | |
3.5
In vitro release studies
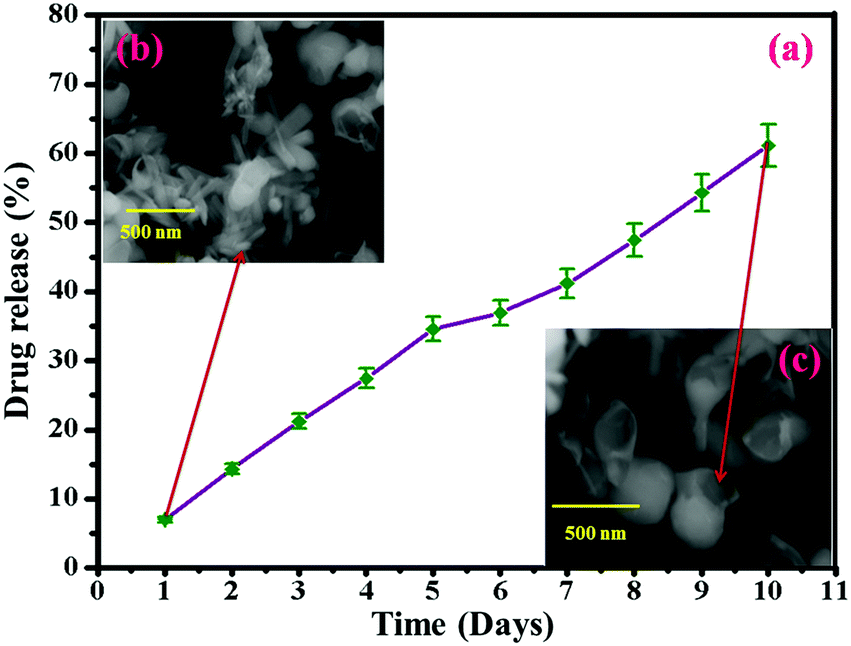
The in vitro release behavior of HAP/PXS/CAP in a release medium was investigated by a dialysis method over 24 hours. In brief, 50 mg of the HAP/PXS/CAP composite, placed and sealed in dialysis bags was incubated in 100 mL of the release medium at 37 °C under 100 rpm stirring for 24 h, with PBS (pH 7.4) as the release medium. At discrete time intervals (1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 days), 3 mL of the release medium was extracted and replenished with an equal volume of fresh PBS solution. The drug content was determined by comparing with the standard value of capsaicin (Fig. S1, ESI†). The in vitro release behaviors of HAP/PXS/CAP in PBS (pH 7.4) are shown in Fig. 4(a). It was observed that the release profiles of CAP and HAP/PXS/CAP showed the sustained release of CAP. Approximately 61% of CAP was released from the composite after 10 days. Fig. 4(a) shows the in vitro drug release and Fig. 4(b and c) represent the SEM images after the drug release, which confirm that the drug was released. The capsaicin concentration in the collected samples was measured using UV/Visible spectroscopy (Shimadzu 1600, Japan), which was used for CAP quantification at a fixed wavelength of 280 nm. The reference cuvette contained 5 mg of capsaicin dissolved in 3 mL of ethyl alcohol.
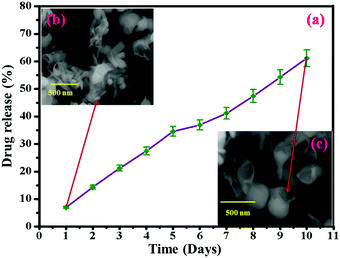 |
| | Fig. 4
In vitro drug release behavior of HAP/PXS/CAP (a); SEM image of drug released HAP/PXS/CAP (b) and SEM image of the degraded polymer (c). | |
3.6 Cell viability
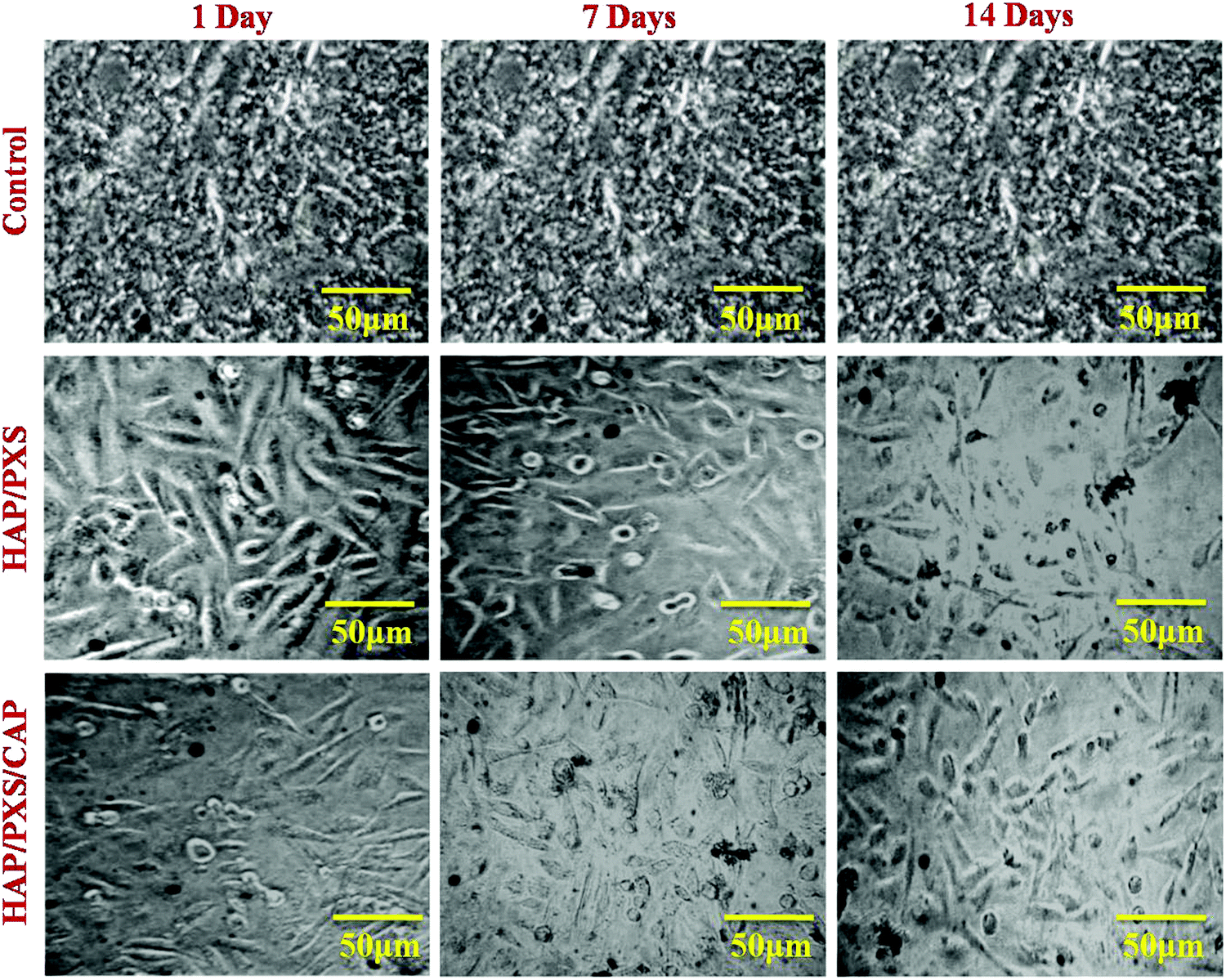
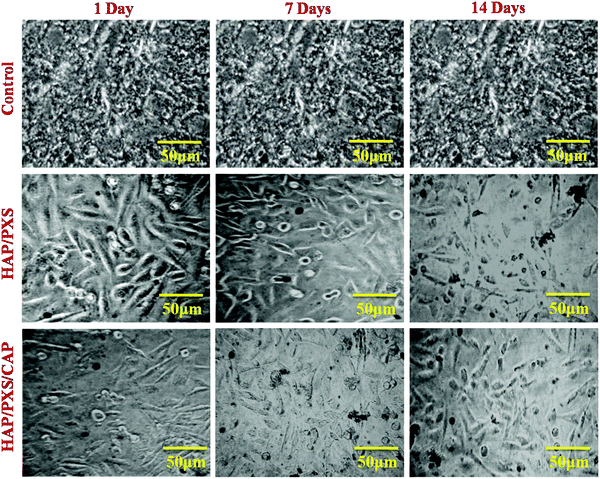
In this present study, cell viability of the synthesized HAP/PXS/CAP nanocomposite was monitored on osteoblast cell-like MG63 cells received from the National Center for Cell Science (NCCS), Pune, India along with the complementary assay of MTT (3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyltetrazolium bromide). Osteoblast cells were seeded in a 24-well plate at a density of 4 × 104 cells per well and were cocultured with the prepared three samples of the HAP, HAP/PXS, and HAP/PXS/CAP composites. After incubating for 1, 3, and 7 days, the sample solutions were separated, and 100 μL MTT solution (5 mg mL−1) was added in 1 mL culture medium to every well dish and it was then incubated at 37 °C for 4 h. After that, 1 mL dimethyl sulfoxide (DMSO) was added to the solution (after the supernatant medium had been removed). Finally, the solution was centrifuged to remove the HAP/PXS and HAP/PXS/CAP composites before being transferred into a 96-well plate and the absorbency OD values were recorded at a wavelength of 490 nm. The viability of the MG63 cells was investigated with the as-synthesized samples of HAP/PXS and CAP loaded HAP/PXS as shown in Fig. 5 and Fig. S2 (ESI†), which represent the cell viability, that is the proliferation of the cells on the materials. The concentration and the time (days) increased when the cell proliferation significantly increased. The results show that MG63 cells can proliferate in the presence of the HAP/PXS composite, which indicates that the composite is non-toxic in nature. Moreover, the extension of high biocompatibility of the HAP/PXS composite may be explained based on the properties of the as-synthesized HAP/PXS nanorods. Also, CAP loaded on HAP/PXS exhibits significantly slightly more cell viability behavior. It can be explained by the fact that the bone implant material becomes immune compromised and vulnerable to attack by any facultative opportunistic microbes to invade the implanted area during the course of therapy. Thus, the addition of an antioxidant on HAP/PXS is explored for additional cell proliferation activity in the growth of the osteoblast cells.38
 |
| | Fig. 5 Morphology of MG63 cells cultured on the as-synthesized HAP and HAP/pectin composites as investigated by optical microscopy on the 1st, 3rd, and 7th days. | |
3.7 Cytotoxicity effect against sarcoma cells (Saos-2)
The cytocompatibility of HAP/PXS, HAP/PXS/CAP, and CAP on human osteosarcoma cells (Saos-2) was tested using an MTT assay method. In this analysis, the cytotoxicity of the CAP, HAP/PXS, and CAP loaded HAP/PXS composites has different profiles. Free CAP and CAP loaded HAP/PXS have more of a cytotoxicity effect against human osteosarcoma cells compared to pure HAP/PXS nanocomposites. It was shown that osteosarcoma human bone cells died and their survival on the corroding surface of the CAP loaded HAP/PXS composite was difficult. Capsaicin is an efficient drug in killing numerous cancer cells as the mechanism includes an increase in cell-cycle arrest and apoptosis, but the exact cellular mechanism is still not completely understood.40 Apoptosis is an essential barrier against cancer development and progression and loss of apoptotic signaling is highly associated with malignancy. Capsaicin exhibits antigrowth activity against the Saos-2 cancer cell line, because CAP exhibits diverse inhibitory effects against cancer initiation, promotion, progression and metastasis.41 Cytotoxicity results suggest that capsaicin is able to kill the cancer cells. MTT assay results are shown in Fig. 6 and Fig. S3 (ESI†). Capsaicin was shown to induce apoptosis in the Saos-2 cancer cell line, so we can confirm that CAP can be a significant anticancer agent and we demonstrated that capsaicin dose-dependently inhibited the viability of osteosarcoma cell lines (Saos-2). Meanwhile, the consequence of the presence of capsaicin in osteosarcoma cells was duration dependent, and the effect of time (days) from 1 to 7 days provided a significant variation in cell death observed on CAP and the CAP/HAP/PXS composites. However, there is no effect on pure HAP/PXS, and this is due to there being no cytotoxicity effect of the composite against sarcoma cells.
 |
| | Fig. 6 Morphology of the cytotoxicity effect against sarcoma cells (Saos-2) cultured on the as-synthesized HAP and HAP/pectin composite as investigated by optical microscopy on the 1st, 3rd, and 7th days. | |
3.8 Antioxidant activity
Capsaicin demonstrated scavenging action against hydrogen peroxide where the activity increased by increasing the concentration of capsaicin, and it also demonstrated antioxidant activity where 0.25 mg/180 μL of the capsaicin indicated 75.5% scavenging activity (Fig. S4, ESI†). The cancer preventing action of the capsaicin indicated small movements in the expansion of the centralization of the specimens. Capsaicin demonstrated an unrivaled H2O2 scavenging capacity. This offers the likelihood that hydrogen peroxide is the fundamental ROS in bone tumor improvement. Tumors emerging in bone and delicate tissues share regular trademark highlights due to normal mesenchymal cell formation as well as anatomical encompassing. The greater part of bone sarcomas is bicompartmental in their introduction, obliterating the cortex and spreading specifically into the delicate tissue partners. In any case, aside from the known hazards, the capacity of oxidative worry in essential bone and delicate tissue sarcomas remains to be investigated further.
3.9 Cell apoptosis analysis
There are significant differences among the cell numbers and morphology of Saos-2 on each substrate of pure HAP/PXS, CAP/HAP/PXS and the control after 1, 3 and 7 days of seeding (Fig. 7). The morphology of the Saos-2 cells was observed to be substantially dissimilar to the increasing number of days. Fig. 7(b–d) show the AO/EtBr stained images of HAP/PXS sample seed sarcoma cells and that when the time (days) increases from 1 to 7 days the process of apoptosis is increased. It is clearly shown that the color changes from green to red after 7 days. At 7 days, the maximum cell death occurred, due to the time contingent on the activity of the CAP loaded nanocomposites. CAP shows and employs dual effects as an anti-tumor agent and a normal cell promoting agent, which are dependent on the (in the way of molecular targeting) cellular targets of CAP and the initiation of its molecular mechanisms. In this investigation, the CAP loaded nanocomposites on osteoblast cells promote cell growth due to the presence of CAP, and in a sarcoma cell line it inhibits cell development. The CAP/HAP/PXS nanocomposite could be a potential scaffold as a sarcoma cellular target and for anti-tumor effects in bone tissue engineering. As shown in Fig. 7(d–f) the treatment of HAP/PXS/CAP on Saos-2 cells was observed using blue fluorescent Hoechst 33342. The blue dye is recognized by chromatin condensation and fragmentation of the sarcoma nuclei from the apoptotic cells. The fluorescence intensity of the cells treated with the CAP loaded nanocomposite is increased with increasing time (days) because of the synergistic impact and utilization of low amounts of CAP in the implant material. This combination improves the viability and lessens the dangerous effects and resistance caused by utilizing higher dosages of CAP compounds. Various investigations highlighted that the novel combination of the treatment of various diseases with different phytochemicals and other chemopreventive operators might be applied to antitumor action through an added substance or synergistic activity.39,40
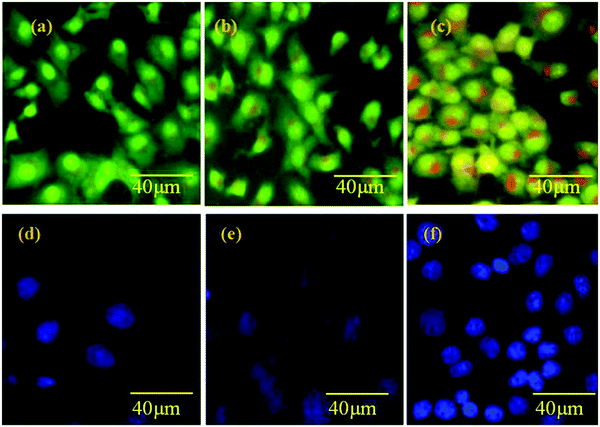 |
| | Fig. 7 Apoptosis of the Saos-2 cell line control (a); with HAP/PXS (b); with HAP/PXS/CAP (c) and apoptosis in a normal cell line control (d); with HAP/PXS (e); with HAP/PXS/CAP (f). | |
3.10 Detection of intracellular ROS generation and mitochondrial membrane potential analysis
ROS is the unmistakable agent in controlling different biochemical capacities in our bodies, where any deviation in their movement prompts the advancement of neurotic conditions including a tumor. The part of the nanoparticles which affect cell ROS creation for anti-cancer potential is well reported. Capsaicin considerably induces reactive oxygen species production concurrently with mitochondria-dependent apoptosis, resulting in decreased growth of osteosarcoma cells (Saos-2) Fig. 8(d–f). In the current study, the ROS production of CAP loaded HAP/PXS composites was observed using a DCFH-DA assay based on the intracellular peroxide-dependent oxidation of 2′7′-dichlorofluorescein (DCF). It is a profoundly sensitive, quantifiable, and ongoing assessment of ROS creation. ROS production was increased with an increasing time duration at a constant concentration (Fig. 8). Upon comparison with untreated control cells, the treated CAP/HAP/PXS cells displayed a higher generation of intracellular ROS, which means more significant harm to the cell, particularly to DNA, lipids, mitochondria, and cell proteins. The mitochondrion is the essential origin of ROS generation, and exorbitant deposition of ROS in internal tissue has a major harmful impact to its mtDNA and external layer potential. This is well correlated with the apoptosis of CAP/HAP/PXS seeded Saos-2 cells (Fig. 8). These divergent actions of CAP/HAP/PXS on the death of a cell in Saos-2 cells are reflected in the effect on ROS generation. There are significant differences among the cell numbers and morphology of Saos-2 on each day of seeding.
 |
| | Fig. 8 ROS of the Saos-2 cell line control (a); with HAP/PXS (b); with HAP/PXS/CAP (c) and a mitochondrial study in a normal cell line control (d); with HAP/PXS (e); with HAP/PXS/CAP (f). | |
Since we affirm the impacts of ROS generation on mitochondrial layer possibilities, the Rh 123 color was utilized to evaluate the charge depolarization in the external layer of mitochondria. From Fig. 8(d–f) it is obviously observed that upon seeding with the CAP loaded HAP/PXS composites, the intensity of the fluorescence decreased, as the Rh123 color is known for its capacity to be effectively taken up by and react with mitochondria. The untreated control cells demonstrated higher fluorescence intensity in fluorescence microscopy compared to the CAP/HAP/PXS treated cells. Hence, the CAP/HAP/PXS treated cells showed a lower rate of fluorescence because of the depolarization of the permeability transition pore complex (PTPC), which controls the penetrability of the mitochondrial surface. The depolarized PTPC encourages the inundation of solutes upon substantial ROS generation and the ROS particles harm the mtDNA. In this way, we prompt different critical changes in the transcriptional level and physiology of the cell.42
4. Conclusion
2D models are a promising approach for researching the mechanism and interactions of cancer within the bone material microenvironment. PXS is one of the polymers for controlled drug delivery because of its biocompatibility and biodegradable nature. From the results it was concluded that the dose of the capsaicin loaded in the HAP/PXS/CAP nanocomposite is a better candidate for applications in tissue engineering. Also, the analysis of cellular uptake, apoptosis, and ROS revealed dependent toxicity of CAP towards cancer cells. Synthesized nanorods of HAP were found to possess similar chemical composition and crystallinity to mineral bones. The biocompatibility test indicated that the MG63 osteoblast cell line had better adhesion, spreading and multiplication on HAP/PXS/CAP through the nanorods. The excellent in vitro bioactivity and biocompatibility of HAP/PXS/CAP proves that it has great prospects in tissue engineering.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
MR acknowledges major financial support from the Department of Science and Technology, Science and Engineering Research Board (Ref. YSS/2015/001532; New Delhi, India) and also acknowledges the PURSE program for the purchase of SEM and FT-IR, and UPE programs for the purchase of TEM. The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No. RG-1438-074.
References
- Z. Ma, E. Troussard and J. A. Bokhoven, Appl. Catal., A, 2012, 423–424, 130–136 CrossRef CAS.
- K. Ronan and M. Bobby, ACS Sustainable Chem. Eng., 2017, 5, 2237–2245 CrossRef CAS.
- V. Anna Li, S. Cinzia, C. Valeria Vetri and L. Gennara Mariano, ACS Appl. Mater. Interfaces, 2017, 9, 14453–14469 Search PubMed.
- G. Nowsheen, K. Behnam, S. Marc, B. L. Archana, J. Ulrich, P. Ullrich, J. Dhanjay and S. Holger, Biomacromolecules, 2017, 18, 1563–1573 CrossRef PubMed.
- C. M. Zhang, C. X. Li, S. H. Huang, Z. Y. Hou, Z. Y. Cheng, P. P. Yang, C. Peng and J. Lin, Biomaterials, 2010, 31, 3374–3383 CrossRef CAS PubMed.
- M. Selvakumar, H. Singh Pawar, N. K. Francis, B. Das, S. Dhara and S. Chattopadhyay., ACS Appl. Mater. Interfaces, 2016, 8(9), 5941–5960 CAS.
- X. Guo, W. Wang, G. Wu, J. Zhang, C. Mao, Y. Deng and H. Xia, New J. Chem., 2011, 35, 663–671 RSC.
- Z. Ding, H. Han, Z. Fan, H. Lu, Y. Sang, Y. Yao, Q. Chemg, Q. Lu and D. L. Kaplan, ACS Appl. Mater. Interfaces, 2017, 9(20), 16913–16921 CAS.
- J. C. Newcomb, R. K. Sturat, D. E. Spoerke and I. S. Samuel, Chem. Rev., 2008, 108(11), 4754–4783 CrossRef PubMed.
- S. V. Dorozhkin, Calcium orthophosphates, J. Mater. Sci., 2007, 42, 1061–1095 CrossRef CAS.
- D. Govindaraj, M. Rajan, A. Munusamy, A. Alarfaj and S. Suresh Kumar, New J. Chem., 2017, 1–33 Search PubMed.
- C. Du, F. Z. Cui, W. Zhang, Q. L. Feng, X. D. Zhu and K. D. Groot, J. Biomed. Mater. Res., 2000, 15:50(4), 518–527 CrossRef.
- N. Razavi and B. Akhlaghinia, New J. Chem., 2016, 40, 447–457 RSC.
- B. S. Goodman, Z. Yao, K. Michael and Y. Fan, Biomaterials, 2013, 34, 3174–3183 CrossRef PubMed.
- L. Kaili, Z. Yanling, Z. Yue, Q. Haiyun, C. Feng, Z. Yingjie and C. Jiang, J. Mater. Chem., 2011, 21, 16558–16565 RSC.
- J. Raphel, M. Holodniy, S. B. Goodman and S. C. Heilshorn, Biomaterials, 2016, 84, 301–314 CrossRef CAS PubMed.
- K. Zawisza, A. Strzep and J. Wiglusz, New J. Chem., 2017, 1–23 Search PubMed.
- W. Ke-Wei, Z. Ying-jie, C. Xiao-Yan, Z. Wan-Yin, W. Qi, C. Feng, C. Jiang and D. You-Rong, Chem. – Asian J., 2010, 5(12), 2477–2482 CrossRef PubMed.
- X. E. Renlong and Y. Kaifeng, J. Mater. Sci., 2009, 44, 4205–4209 CrossRef.
- L. Kaili, C. Jiang, Z. Yingjie, W. Wei, C. Guofeng, Z. Yi and R. Meiling, Cryst. Growth Des., 2009, 1, 177–181 Search PubMed.
- M. Guo Ma, Int. J. Nanomed., 2012, 7, 1781–1791 Search PubMed.
- X. Cui, Y. Koujima, H. Seto, T. Murakami, Y. Hoshino and Y. Miura, ACS Biomater. Sci. Eng., 2016, 2, 205–212 CrossRef CAS.
- M. Jevtić, M. Mitric, S. Skapin, B. Jancar, N. Ignjatovic and D. Uskokovic, Cryst. Growth Des., 2008, 8(7), 2217–2222 Search PubMed.
- K. Lin, L. Xia, J. Gan, Z. Zhang, H. Chen, X. Jiang and J. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 8008–8017 CAS.
- M. Jevtic, M. Mitric, S. Skapin, B. Jancar, N. Ignjatovic and D. Uskokovic, Cryst. Growth Des., 2008, 8(7), 2217–2222 CAS.
- H. N. Mireia, V. Ferran, G. Maria-Pau, M. José María, F. Javier Gil, R. J. Carlos Mas-Moruno, M. Holodniy, S. B. Goodman and S. C. Heilshorn, Biomaterials, 2016, 84, 301–314 CrossRef PubMed.
- M. A. Fernandez-Yague, S. A. Abbah, L. Mcnamara, D. I. Zeugolis, A. Pandit and M. J. Biggs, Adv. Drug Delivery, 2015, 84, 1–29 CrossRef CAS PubMed.
- S. J. Florczyk, G. Liu, F. M. Kievit, A. M. Lewis, J. D. Wu and M. Zhang, Adv. Healthcare Mater., 2012, 1(5), 590–599 CrossRef CAS PubMed.
- C. Sharma, A. K. Dinda, P. D. Potdar, C. F. Chou and N. C. Mishra, Mater. Sci. Eng., C, 2016, 64, 416–427 CrossRef CAS PubMed.
- G. Ciardelli, V. Chiono, G. Vozzi, M. Pracella, A. Ahluwalia, N. Barbani, C. Cristallini and P. Giusti, Biomacromolecules, 2005, 6(4), 1961–1976 CrossRef CAS PubMed.
- P. Zi, X. Yang, H. Kuang and L. Yu, Int. J. Pharm., 2008, 358, 151–158 CrossRef CAS PubMed.
- P. Maija, V. Kirish, H. Mia, V. Paula, S. Haikkai, H. Matti, P. Markku and T. Kuitunen, Front. Pharmacol., 2010, 1(140), 1–12 Search PubMed.
- M. Piming, L. Ting, W. Wei, S. Dongjian, D. Fang, B. Huiyu, D. Weifu and C. Mingqing, Front. Pharmacol., 2014, 110, 50–55 Search PubMed.
- M. Sumathra, D. Govindraj, M. Jeyaraj, A. Abdulla, M. A. Munusamy, S. Suresh Kumar and M. Rajan, Sustainable Chem. Pharm., 2017, 5, 46–53 CrossRef CAS.
- K. Anselme, Biomaterials, 2000, 21(7), 667–681 CrossRef CAS PubMed.
- H. Tanaka, M. Futaoka, R. Hino, K. Kandori and T. Ishikawa, J. Colloid Interface Sci., 2005, 283(2), 609–612 CrossRef CAS PubMed.
- E. L. S. Fong, S.-E. Lamhamedi-Cherradi, E. Burdett, V. Ramamoorthy, A. J. Lazar, F. K. Kasper, M. C. Farach-Carson, D. Vishwamitra, E. G. Demicco and B. A. Menegaz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(16), 6500–6505 CrossRef CAS PubMed.
- G. Benítez, M. Gonzalez-Tejero and J. Molero-Mesa, J. Ethnopharmacol., 2010, 129(1), 87–105 CrossRef PubMed.
- A. Lewinska, P. Jarosz, J. Czech, I. Rzeszutek, A. Bielak-Zmijewska, W. Grabowska and M. Wnuk, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2015, 779, 23–34 CrossRef CAS PubMed.
- D. Weief, L. Ting, X. Shunanfei, M. Piming and Q. Ming, Bioelastomer, Chem. Polym., 2013, 5, 1339–1351 Search PubMed.
- R. Clark and S.-H. Lee, Anticancer Res., 2016, 36, 834–837 Search PubMed.
- S. Changyun, Y. Xinghao, W. Yan, Z. Jinwen and C. Chen, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 263–274 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: In vitro drug release, viability, cytotoxicity and scavenging activity. See DOI: 10.1039/c7nj03143b |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Sami A.
Alyahya
b,
Naiyf S.
Alharbi
c,
Shine
Kadaikunnan
c and
S. Suresh
Kumar
d
*a,
Sami A.
Alyahya
b,
Naiyf S.
Alharbi
c,
Shine
Kadaikunnan
c and
S. Suresh
Kumar
d