
A computational analysis of pro-angiogenic therapies for peripheral artery disease†
Lindsay E.
Clegg‡
 a and
Feilim
Mac Gabhann
*ab
a and
Feilim
Mac Gabhann
*ab
aInstitute for Computational Medicine, Institute for NanoBioTechnology, and Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA. E-mail: feilim@jhu.edu
bDepartment of Materials Science and Engineering, Johns Hopkins University, Baltimore, MD, USA
First published on 3rd January 2018
Abstract
Inducing therapeutic angiogenesis to effectively form hierarchical, non-leaky networks of perfused vessels in tissue engineering applications and ischemic disease remains an unmet challenge, despite extensive research and multiple clinical trials. Here, we use a previously-developed, multi-scale, computational systems pharmacology model of human peripheral artery disease to screen a diverse array of promising pro-angiogenic strategies, including gene therapy, biomaterials, and antibodies. Our previously-validated model explicitly accounts for VEGF immobilization, Neuropilin-1 binding, and weak activation of VEGF receptor 2 (VEGFR2) by the “VEGFxxxb” isoforms. First, we examine biomaterial-based delivery of VEGF engineered for increased affinity to the extracellular matrix. We show that these constructs maintain VEGF close to physiological levels and extend the duration of VEGFR2 activation. We demonstrate the importance of sub-saturating VEGF dosing to prevent angioma formation. Second, we examine the potential of ligand- or receptor-based gene therapy to normalize VEGF receptor signaling. Third, we explore the potential for antibody-based pro-angiogenic therapy. Our model supports recent observations that improvement in perfusion following treatment with anti-VEGF165b in mice is mediated by VEGF-receptor 1, not VEGFR2. Surprisingly, the model predicts that the approved anti-VEGF cancer drug, bevacizumab, may actually improve signaling of both VEGFR1 and VEGFR2 via a novel ‘antibody swapping’ effect that we demonstrate here. Altogether, this model provides insight into the mechanisms of action of several classes of pro-angiogenic strategies within the context of the complex molecular and physiological processes occurring in vivo. We identify molecular signaling similarities between promising approaches and key differences between promising and ineffective strategies.
Insight, innovation, integrationInducing angiogenesis to form hierarchical networks of perfused vessels remains an unmet challenge. We use a computational systems pharmacology model to screen multiple promising pro-angiogenic strategies, highlighting the dependence of therapy action on the complex underlying molecular and physiological processes occurring in vivo. We show that biomaterial-based delivery of VEGF engineered for increased affinity to the extracellular matrix maintains VEGF close to physiological levels and extends the duration of VEGFR2 activation without saturating VEGFR2. We also explore antibody-based pro-angiogenic therapy, demonstrating accurate model prediction of signaling in mice following treatment with anti-VEGF165b. Intriguingly, the model predicts that an approved anti-VEGF drug, bevacizumab, may improve signaling of both VEGFR1 and VEGFR2 via a novel ‘antibody swapping’ effect that we demonstrate here. |
Introduction
Inducing angiogenesis, the growth of new vessels from the existing vasculature, in order to establish collateral blood flow, has long been a therapeutic goal in ischemic disease.1,2 Peripheral artery disease (PAD), the manifestation of systemic atherosclerosis in the legs, leads to pain, limited mobility, and elevated risk of amputation. PAD is characterized by skeletal muscle ischemia without induction of sufficient angiogenesis to restore normal perfusion.3,4 The molecular mechanisms underlying this insufficient vascular remodeling have not been fully elucidated.5 Delivery of vascular endothelial growth factor (VEGF), a key angiogenic factor, via gene- or cell-based therapy has been tested in multiple clinical trials for PAD, but no constructs have proceeded to Phase III trials or regulatory approval, due to lack of efficacy at improving patient outcomes as well as occurrence of edema in some patients.5,6 This failure, which could be attributed in part to inefficient, spatially heterogeneous, short duration gene delivery,7–10 has motivated development of newer therapeutic strategies to better induce and regulate angiogenesis in ischemia. Strategies to form functional vessel networks in thick engineered tissues are also of high interest.11 Key to success of these strategies is a more in-depth understanding of both the underlying cause(s) of impaired angiogenic signaling in PAD, and the effect of these molecular mechanisms on therapy effectiveness.The VEGF family consists of five ligands, with VEGFA (hereafter VEGF) considered the primary pro-angiogenic isoform, three receptors (VEGFR1–3), and the Neuropilins as co-receptors.12 Both ligands and receptors can be alternatively spliced, the latter resulting in production of soluble receptors, most notably soluble VEGFR1 (sR1),13 and the former resulting in VEGF isoforms with different binding affinities for HSPGs in the extracellular matrix (ECM) and for Neuropilin-1 (NRP1).14,15 VEGF splicing varies by tissue,16,17 with the most prominent isoforms in humans being VEGF121, VEGF165, and VEGF189.12 Expression of single VEGF isoforms in mice or tumors leads to different vascular phenotypes; non-ECM-binding VEGF121 promotes production of wide diameter vessels with few branch points, while expression of ECM-binding VEGF165 alone leads to phenotypically normal vessels, and strong ECM-binding VEGF189 induces networks of thin, highly branched vessels.18–26 Our recent computational work quantified the contributions to VEGF receptor family signaling in vivo of both VEGF sequestration in the ECM and binding of ECM-bound VEGF to VEGF receptors.26
An alternate set of VEGF isoforms – the “b” isoforms – with the same numbers of amino acids, but a switch in the last six amino acids (from exon 8a to exon 8b) have recently been discovered and characterized.27,28 Despite very similar sequences, these isoforms, the most-studied being VEGF165b (as opposed to VEGF165a), do not bind to NRP1 or to HSPGs, and induce only weak phosphorylation of VEGFR2,28–30 despite binding to the receptor with the same affinity as other VEGF isoforms. While VEGF is found at normal levels in resting PAD-afflicted skeletal muscle,31,32 and is elevated in the plasma of patients with PAD,33–36 splicing of VEGF is altered in PAD37,38 (and other diseases). Specifically, expression of VEGF165b increases in tissue, likely accompanied by reduced expression of VEGF165a (to maintain unchanged total VEGF levels); secretion of VEGF165b in the bloodstream by peripheral blood mononuclear cells (e.g. monocytes) also increases substantially.38 Despite these observations, quantitative measurements of absolute VEGF165a and VEGF165b levels in healthy and ischemic tissue remain very limited.39
Several different approaches to pro-angiogenic therapy for PAD, tissue engineering, and wound healing are currently under development, which leverage different aspects of the VEGF system. The first uses biomaterials for tunable, extended release of VEGF protein and other growth factors in vivo.11 The groups of Jeff Hubbell and Andrea Banfi engineered VEGF forms for increased affinity to the ECM or covalent binding in fibrin gels with tunable proteolytic release to deliver low VEGF and/or PDGF doses over the course of weeks.40,41 These constructs, which can be delivered in injectable fibrin gels, improved wound healing, reduced permeability, enhanced angiogenesis without subsequent vessel regression, and/or improved perfusion recovery in rodent models, compared to delivery of wild-type VEGF165a protein. These constructs were designed to control VEGF release over time; the impact of engineering VEGF immobilization on dynamic VEGF receptor activation in vivo remains poorly understood, as it is difficult to quantify without computational tools.11,25 Scientists are also improving gene delivery tools to induce highly efficient transfection, with spatially homogeneous and finely controlled gene expression of tunable duration, targeted to specific cell types.42,43 Such optimized control of gene delivery, tuned to match the underlying physiological system, would likely address many of the failures observed in past clinical trials designed to induce angiogenesis via VEGF overexpression.7,8 Finally, Kikuchi et al. recently showed that treatment with an antibody that binds specifically to the VEGF165b isoform improves perfusion recovery in murine hindlimb ischemia models,38 an observation confirmed by Ganta et al.31 The mechanism of action of this therapeutic was predicted by Kikuchi et al. to involve an increase in VEGFR2 phosphorylation, consistent with the prevailing hypothesis in the field that ‘weak-activating’ VEGF165b competes with ‘strong-activating’ VEGF165a for binding to VEGFR2, reducing VEGFR2 phosphorylation. However, Ganta et al. showed that VEGFR2 Y1175 phosphorylation was unchanged following antibody treatment, while VEGFR1 Y1333 phosphorylation increased. Our previous model of VEGF165b in PAD confirmed that, while VEGF165a and VEGF165b both bind to VEGFR2, they do not compete for VEGFR2 in physiological conditions. Interestingly, the model predicted that VEGF165b does compete for binding to VEGFR1, consistent with the Ganta et al. study.31
Objective
Our objective was to leverage a previously-developed systems pharmacology model of human peripheral artery disease to screen promising protein-, gene-, and antibody-based pro-angiogenic therapeutics. Our systems pharmacology model, which was built using, and validated against, in vitro, ex vivo, in vivo, and clinical data, bridges from detailed molecular biology measured in vitro to in vivo human physiology (Fig. 1A).26,44 This integrated multiscale framework allows us to: (1) ask questions that are difficult or impossible to answer experimentally, (2) provide insight into the mechanisms of action of therapies, and (3) identify potentially non-intuitive side effects, toxicity, or challenges to efficacy that merit further study. Specifically, we can predict free VEGF concentrations and VEGFR2 phosphorylation in diseased tissue following therapy administration, accounting for differences in growth factor distribution and VEGFR2 signaling as a function of VEGF isoform ECM- and NRP1-binding properties. By simulating failed therapies and those that show promise in mice within a single quantitative framework, we identify potential molecular drivers of therapy success or failure,45 explicitly accounting for the critical yet oft unconsidered effects of molecular and physiological processes on therapy action. In elucidating these key rules, we hope to identify strategies most likely to be effective in the context of human disease, working within, and potentially leveraging, the underlying biology. | ||
| Fig. 1 Overview of model structure and therapy implementation. (A) Structure of multi-scale whole body compartment model of human PAD, including three compartments: the bulk of tissue (Main Body Mass), a calf muscle with PAD (PAD Calf Muscle), and the blood.44 Antibody delivery is simulated as a 90 minute intravenous infusion,46 while gene therapy is simulated to target either parenchymal cells (targeting ligands) or endothelial cells (receptors) specifically in the PAD Calf Muscle. Biomaterial-based VEGF delivery assumes delivery into the extracellular space (ECM) of the PAD Calf Muscle. (B) Overview of key PAD-specific features in model: VEGF165b properties (left),28–30 and changes in VEGF splicing and secretion occurring in PAD (right).31,32,38 The binding and VEGFR2 phosphorylation properties of VEGF165b are explicitly included in the model equations, and VEGF secretion is altered from the healthy baseline model to reflect changes in VEGF distribution observed in PAD.44 | ||
Results
Implementing diverse therapeutic strategies in a whole body compartment model of human PAD
The systems pharmacology model used in this study accurately captures VEGF family signaling in human PAD and murine hindlimb ischemia.44 Therapies are directly incorporated into this multiscale quantitative framework in as realistic a manner as possible. Briefly, biomaterial-based VEGF delivery is simulated by adding a bolus of ECM-bound VEGF protein to the extracellular space in the PAD Calf Muscle at a specified time, with no change to the underlying model equations. VEGF gene therapy is implemented by altering the secretion rate for VEGF by parenchymal cells within the model equations, and receptor-based gene therapy is mimicked by changing the endothelial receptor production rates in the PAD Calf Muscle. Antibody-based therapy is simulated as a bolus injection into the bloodstream at a specified time, as done previously.46 Details on model structure and parameters for therapy administration can be found in the Methods.Quantitative insight into design and dosing for biomaterial-based VEGF delivery in PAD
We used our computational systems pharmacology model (Fig. 1A) to perform in silico screening of VEGF protein delivery strategies, including native VEGF165a (which has failed to produce benefit in clinical trials), and two engineered constructs that have shown promise in mice: VEGF engineered for “super affinity” to the ECM,40 and VEGF covalently bound to fibrin with tunable proteolytic release41 (Fig. 2A and B). Calibrating against experimental release data in mice (see Methods), our model gives as output detailed pharmacodynamics: the predicted local VEGF concentration and VEGFR2 phosphorylation following therapy (Fig. 2B) in the context of human PAD. We used allometric scaling to adjust the dose for our human model. We found that, unlike native VEGF165a (in red), the VEGF constructs engineered for increased ECM affinity (purple & green) maintain free VEGF levels within a physiological range of no more than 3–5-fold baseline levels (based on changes from baseline in wounds,47 exercise,48,49 and disease32,38,47) (Fig. 2B, center), likely reducing induction of permeability following treatment. The engineered constructs also elevate VEGFR2 phosphorylation for at least 2 weeks, a duration close to the range shown experimentally to prevent vessel regression.7,8 Simulations also predict increased ligation of endothelial VEGFR1 by all three constructs, but to a smaller extent than the increase on VEGFR2 (Fig. S1G and H, ESI†). For this analysis, we assumed high local secretion of the ‘weak’ VEGFR2-activating VEGF165b as a worst-case scenario; the action of these therapies was similar regardless of endogenous VEGF165b secretion (Fig. S2, ESI†). | ||
| Fig. 2 ECM-binding affinity and dosing are key design considerations for effective VEGFR activation following biomaterial-based VEGF delivery. (A) Summary of experimentally-tested VEGF constructs delivered in biomaterials and observed results in mice.40,41 (B) Calibrating against experimental data (left), we simulated the predicted magnitude and duration of increased local VEGF concentration (middle) and VEGFR2 phosphorylation (right) following delivery of 354 μg of each VEGF construct (based on allometric scaling from mouse experiments) delivered in a fibrin gel at the same dose, and in the same system. The teal box indicates the physiological range of VEGF expression, which typically varies no more than 3–5× in physiological and pathological conditions.47 (C) Simulation of delivery of the “Covalent VEGF with Proteolysis” construct at different doses. | ||
We next examined the important and challenging question of appropriate VEGF dosing. In ref. 41, low doses of the “Covalent VEGF with Proteolysis” construct stimulated stable angiogenesis, while high doses promoted formation of angiomas. We simulated these experiments to probe the molecular mechanisms underlying this switch from constructive angiogenesis to angioma formation. Our simulations predict that VEGF doses leading to effective angiogenesis without angioma formation in mice correspond to sub-saturating VEGF receptor activation (Fig. 2C). Indeed, the lowest dose tested in ref. 41, 0.01 μg mL−1, which still induced stable angiogenesis by 3 months after gel implantation, is predicted to elevate VEGFR2 phosphorylation by only about 30%. This result suggests that small increases in VEGFR2 signaling, if sustained for weeks, are sufficient to induce and sustain therapeutic angiogenesis. Interestingly, only the highest dose (100 μg mL−1) was predicted to saturate endothelial surface VEGFR1 (Fig. S3G, ESI†). These predictions suggest that receptor saturation and VEGF-ECM binding affinity are both important considerations in dosing and translation of pro-angiogenic biomaterials.
Gene therapy effectiveness depends on target and magnitude, as well as optimized delivery
We next examined delivery of VEGF family-related genes to the PAD Calf Muscle. We examined whether, with sufficient improvements in gene delivery, VEGF family gene therapy would hold similar potential to biomaterial-based VEGF delivery to improve angiogenic signaling in PAD. Thus, we assumed ideal delivery and expression: instant, 100% efficient, spatially homogeneous transfection of myocytes at a constant expression rate for the duration of the experiment. While several trials in humans have delivered VEGF at levels sufficient to increase plasma or serum VEGF,50–52 based on our above results for VEGF protein delivery, we chose to examine smaller increases in VEGF expression, which do not induce any detectable systemic effects (Fig. 3, top row). We found that expression of VEGF165a at a level roughly 5-fold higher than endogenous VEGF165 secretion resulted in changes to Free VEGF and VEGFR2 phosphorylation in the PAD Calf Muscle (Fig. 3) in the same range as that induced by the engineered VEGF constructs in Fig. 2, while 20-fold increased expression was sufficient to saturate VEGFR2 (Fig. S5G and H, ESI†). Interestingly, expression of the engineered super affinity VEGF construct increased predicted VEGFR2 phosphorylation more than VEGF165a expression (Fig. 3, bottom row), with a smaller increase in tissue free VEGF (Fig. 3, top row), suggesting that this construct would improve efficacy delivered as either a protein or a gene construct. This difference in signaling following identical delivery and expression of VEGF165a and super affinity VEGF highlights the role of ECM-binding not only in regulating VEGF pharmacokinetics, but also in directly impacting receptor-level activation (pharmacodynamics), a mechanism of action predicted by our models but not commonly considered in therapy design. | ||
| Fig. 3 Distinct patterns of VEGF distribution and receptor activation predicted following different gene therapy approaches. Panels show fold change from baseline six days after treatment with one of six different forms of gene therapy (across top). Gray dashed lines are provided for reference to more easily compare changes between the different approaches. Top row: Changes in free VEGF (not bound to ECM, BM, or sR1) in the Blood, Main Body Mass, and PAD Calf Muscle. Middle row: Predicted occupancy of endothelial VEGF receptors in the PAD Calf Muscle. Bottom row: VEGFR2 phosphorylation in the PAD Calf Muscle. | ||
We also tested several other approaches, to compare the effect of targeting VEGF family ligands vs. receptors. First, we simulated induction of a splicing switch for VEGF165, from all VEGF165b to all VEGF165a in the PAD Calf Muscle, an approach of current interest in the field.38 This strategy increased pR2 slightly (∼80%, Fig. 3, bottom row), but decreased predicted free VEGF and VEGFR1 occupancy, unlike direct delivery of VEGF165a gene (Fig. 3). Finally, we tested receptor-based therapy, over-expressing VEGFR2 or knocking down expression of sR1, a soluble receptor that modulates VEGFR ligation. Increased VEGFR2 expression increased predicted total pR2, but decreased the fraction of VEGFR2 phosphorylated (pR2/R2, Fig. 3, bottom row), as well as decreasing VEGFR1 ligation and free VEGF in the PAD Calf Muscle (Fig. 3) to a larger extent than the splicing switch. The magnitude of this effect is limited by available VEGF, and effectiveness depends on: (1) specific targeting of gene therapy to endothelial cells, and (2) the assumption that total pR2, as opposed to relative pR2/R2, is a key driver of signaling. Conversely, while blocking sR1 in mice has shown promise, in human PAD minimal effect is predicted, suggesting that this is not an effective strategy. These simulation results highlight the potential of several approaches to gene therapy for PAD, as our clinical and experimental gene delivery toolkit begins to allow for the consistent and controllable expression levels (in specific in vivo cell populations) necessary to achieve the physiologically-reasonable signaling profiles described here.42,43
Non-intuitive, systemic effects with anti-VEGF treatment for peripheral artery disease
First, we compared model predictions following anti-VEGF165b treatment to experimental measurements in murine hindlimb ischemia, in order to confirm that our model is predictive of therapeutic response. We found that the model is consistent with a lack of change in endothelial VEGFR2 phosphorylation following antibody treatment (Fig. 4A). Owing to a lack of mechanistic understanding in the field, our model does not directly predict VEGFR1 phosphorylation. However, consistent with previous work,26 the model predicts increases in PlGF and non-VEGF165b VEGF isoforms binding to VEGFR1 (Fig. 4A and B), while VEGF165b–VEGFR1 decreases. This aligns with the results of Ganta et al.,31 showing that VEGF165b appears not to induce phosphorylation of VEGFR1 on Y1333, and that VEGFR1 phosphorylation, presumably by PlGF and/or other VEGF isoforms, increases following antibody treatment (Fig. 4B). These results provide confidence in our model predictions of signaling in response to anti-VEGF165b treatment, and support the conclusion that improved perfusion following anti-VEGF165b is likely mediated by VEGFR1, not VEGFR2. Interestingly, the model predicts that VEGF165b secretion into the bloodstream by monocytes – included in our model though it is not yet clear whether serum VEGF165b is free in plasma or confined to formed elements – would have minimal impact on response to anti-VEGF165b treatment in the PAD Calf Muscle (Fig. 4C). This result suggests that the question of blood VEGF165b source is not critical to understand patient response to therapy.
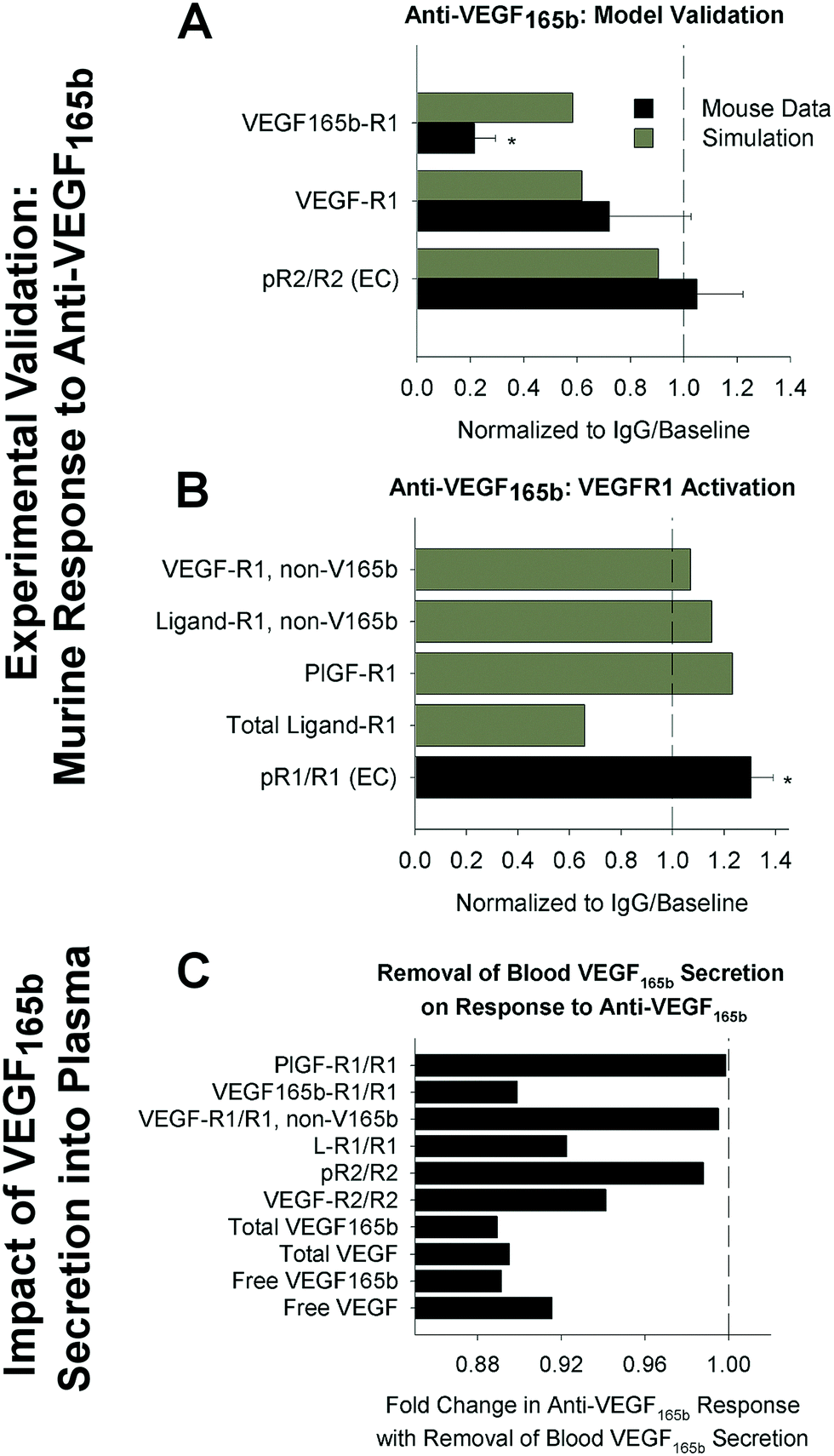 | ||
| Fig. 4 Model accurately predicts experimental response to anti-VEGF165b in mice. (A and B) Model-predicted response to anti-VEGF165b treatment, compared to measurements in mouse hind-limb ischemia, from an analysis of the data set presented in Ganta et al.,31 using the same simulation protocol used in ref. 44. Simulation outputs (black) shown for the PAD Calf Muscle. Mouse measurements (gray) are taken from gastrocnemius muscle after femoral artery ligation and antibody treatment, and represent total tissue measurements (receptor-bound ligand and VEGF protein) or CD31+ cells (pR2/R2, pR1/R1), normalized by equivalent quantities in IgG-treated controls. Asterisks denote significance using an unpaired, two-tailed t-test with p < 0.05. n = 3 for pR2/R2, n = 5 for IgG group and n = 7 for treatment group for VEGF binding to VEGFR1, and n = 5 for pR1/R1. (A) Model validation. (B) Comparison of model-predicated ligand-VEGFR1 binding to experimental VEGFR1 phosphorylation on Y1333. (C) Model predictions of changes in PAD Calf Muscle in response to anti-VEGF165b treatment in the absence of VEGF165b secretion into the bloodstream by monocytes. | ||
Pre-treatment, our model predicts that tissue VEGF receptor signaling is driven by locally-secreted VEGF.44 In contrast, the model predicts important systemic effects following antibody administration. This occurs as a result of two antibody properties: (1) VEGF-antibody binding is reversible, and (2) the in vivo half-life of bevacizumab is 21 days,46 similar to that of other monoclonal antibodies. As such, the antibody does not simply bind to VEGF and remove it from the system, but rather continues to circulate. In tissue compartments with high target (VEGF165b) concentration, the antibody will tend to bind to target. However, upon transport to a tissue with lower target concentration, the antibody-target complex will tend to dissociate, facilitating ‘shuttling’ of target (VEGF165b) between compartments, and reducing the concentration difference between tissues (Fig. 5A).
 | ||
| Fig. 5 Mechanism of action of VEGF-targeting antibodies in PAD. (A) Schematic of antibody “shuttling” effect. When the antibody is in a compartment with high target concentration, it tends to bind to target. Upon moving (via vascular permeability or lymphatic drainage) to a compartment with lower target concentration, mass action kinetics dictate that the target-antibody complexes will tend to dissociate. Thus, the antibody acts to reduce the concentration difference between two compartments. (B and C) Predicted fold change in free VEGF in the PAD Calf Muscle on Day 6 following treatment with anti-VEGF165b (B) or a non-isoform-specific anti-VEGF (C), as a function of the local fractional secretion of VEGF165b in the PAD Calf Muscle (x-axis) and the Main Body Mass (y-axis). Black indicates an increase in Free VEGF (desired for treating PAD), while red indicates a decrease. | ||
PAD patients are expected to have high VEGF165b in the PAD Calf Muscle and low VEGF165b in the Main Body Mass. In simulating anti-VEGF165b treatment in this quadrant, local free VEGF in the PAD Calf Muscle was predicted to decrease (to as low as 14% of baseline), as the antibody binds to VEGF165b (Fig. 5B, lower right corner). Levels of free VEGF165a in the PAD Calf Muscle were essentially unchanged (Fig. S6, ESI†). Conversely, free VEGF was predicted to increase in the PAD Calf Muscle if the conditions were reversed: low VEGF165b secretion in PAD Calf Muscle and high VEGF165b in Main Body Mass (Fig. 5B, upper left corner). This is a result of movement (shuttling) of VEGF165b by the antibody from the high concentration environment of the Main Body Mass into the low VEGF165b environment in the PAD Calf Muscle.
We also simulated treatment with a non-isoform-specific antibody (anti-VEGF), similar to bevacizumab. Because the antibody dose of 10 mg kg−1 is in excess of available VEGF, the ‘antibody shuttling’ effect occurs independently for each VEGF isoform. As total free VEGF levels are similar in both tissue compartments, treatment with anti-VEGF is predicted to have a much smaller effect on total free VEGF levels in the PAD Calf Muscle; the antibody ‘swaps’ VEGF165a and VEGF165b between tissues (range for total free VEGF: 65–103% of baseline) (Fig. 5C). VEGF165b transport is predicted to be essentially identical for anti-VEGF165b and anti-VEGF at all relative VEGF165b levels; the observed difference in free VEGF is due to shuttling of VEGF165a by anti-VEGF, but not by anti-VEGF165b (Fig. S6, ESI†). As such, anti-VEGF, a traditionally anti-angiogenic therapy, is predicted to bring additional, endogenous VEGF165a into the PAD Calf Muscle when VEGF165b is higher in the PAD Calf Muscle than the Main Body Mass, the conditions we expect in PAD patients. Due to the large size of the Main Body Mass, little effect on free VEGF in this compartment is predicted following treatment with either antibody (Fig. S7, ESI†). Predicted changes in plasma VEGF following anti-VEGF165b treatment track with fractional VEGF165b secretion in the large Main Body Mass, but are not predictive of VEGF165b levels in the smaller PAD Calf Muscle (Fig. S7, ESI†).
Next, we examined the effect of anti-VEGF165b or anti-VEGF treatment on endothelial VEGFR1 and VEGFR2 ligation and activation. The model predicts that anti-VEGF165b treatment would not increase VEGFR2 phosphorylation under any conditions, and may even decrease pR2 when local VEGF165b secretion is high, as expected in PAD-afflicted muscle (Fig. 6D). This is because removal of VEGF165b, a weak activator of VEGFR2, does not lead to increased VEGFR2 ligation by other VEGF isoforms (Fig. S9 and S10, ESI†). The model predicts much larger changes in endothelial VEGFR1 ligation (Fig. 6I); when VEGF165b is high in the PAD Calf Muscle and low in the Main Body Mass, cell surface VEGFR1 ligation is predicted to decrease to as low as 24% of baseline, with increased ligation of VEGFR1 by other VEGF isoforms (to 118% of baseline) and especially by PlGF (160% of baseline) (Fig. S11, middle column, ESI†).
 | ||
| Fig. 6 Effect of VEGF-targeting antibodies on endothelial VEGFR signaling in vivo. (Top) Predicted values of pR2/R2 (A–C) and fold change in pR2 (D and E) in the PAD Calf Muscle on Day 6 following treatment with control (left), anti-VEGF165b (middle) or a non-isoform-specific anti-VEGF (right), as a function of the local fractional secretion of VEGF165b in the PAD Calf Muscle (x-axis) and the Main Body Mass (y-axis). (Bottom) Predicted changes in total endothelial surface VEGFR1 ligation (F–H) and fold change in surface VEGFR1 ligation (I and J) in the PAD Calf Muscle on Day 6 following systemic antibody treatment. Black indicates an increase in receptor activation, while red indicates a decrease. | ||
Interestingly, treatment with the non-isoform-specific anti-VEGF is predicted to increase VEGFR2 phosphorylation in the PAD Calf Muscle (up to 366% of baseline) when VEGF165b secretion is higher in the PAD Calf Muscle than in the Main Body Mass (Fig. 6E, lower right corner). This occurs due to the increased VEGF165a (which is a strong activator of VEGFR2) brought into the calf by anti-VEGF as VEGF165b, the weak VEGFR2-activator, is shuttled out of the calf muscle. Additionally, in these conditions expected in PAD patients, anti-VEGF is predicted to reduce VEGFR1 ligation (to as low as 44% of baseline), with increased binding by PlGF (up to 150% of baseline) and other VEGF isoforms (up to 534%), similar to anti-VEGF165b. This occurs because VEGF165a binds preferentially to VEGFR2 over VEGFR1, due to its NRP1-binding properties.26 Thus, while anti-VEGF165b is predicted to act via VEGFR1, anti-VEGF treatment is predicted to improve signaling via both VEGFR2 (increased phosphorylation) and VEGFR1 (decreased ligation) under conditions of higher VEGF165b in the PAD Calf Muscle than in other tissues.
Antibody treatment induces qualitatively different effects than VEGF gene or protein delivery
Finally, we leveraged our ability to simulate disparate therapeutic strategies in a single framework to compare the dynamics of therapy response following biomaterial-based protein therapy, gene therapy, or antibody therapy. We chose a promising representative therapy from each category: the ‘Covalent VEGF with Proteolysis’ biomaterial construct; delivery of ‘Super Affinity’ VEGF gene at 5-fold normal VEGF expression levels; and the non-isoform-specific anti-VEGF. As the magnitude of change in signaling depends on the dose of gene or protein delivered, we were interested in the relative trends and time-courses of VEGF redistribution and endothelial signaling, more so than the relative magnitudes of change following therapy. It is worth noting, though, that the antibody treatment simulations represent saturating levels of antibody, and thus the maximum potential response for this strategy.While protein and gene therapy are predicted to increase free VEGF levels in the PAD Calf Muscle but have little-to-no systemic effects at low, physiologically reasonable doses, anti-VEGF is predicted to reduce free VEGF levels in the PAD Calf Muscle and slightly in the Main Body Mass, but increase free VEGF in the blood (Fig. 7A–C). These results are very similar to the changes predicted following anti-VEGF treatment in cancer.46 For all three treatments, phosphorylation of VEGFR2 is predicted to increase and remain elevated for more than two weeks (Fig. 7D), though the magnitude of increase is lower with anti-VEGF than could be achieved via VEGF delivery. However, while delivery of matrix-binding VEGF gene or protein is predicted to increase VEGFR1 ligation, anti-VEGF is predicted to reduce VEGFR1 ligation (Fig. 7F). Our currently limited understanding of VEGFR1 signaling make it difficult to predict which of these signaling profiles would more effectively stimulate angiogenesis in PAD.
 | ||
| Fig. 7 Anti-VEGF induces different VEGF distribution and endothelial VEGFR activation than biomaterial-based protein delivery or VEGF gene therapy. Therapies compared are biomaterial-based delivery of the ‘Covalent VEGF with Proteolysis’ construct to the PAD Calf Muscle at the dose used in Fig. 2B (green); therapeutic over-expression of the ‘Super Affinity VEGF’ construct by 5-fold (blue); and intravenous injection of anti-VEGF (red). (A–C) Changes in free VEGF (not bound to ECM, BM, sR1, or antibody) in the PAD Calf Muscle (A), blood (B), and Main Body Mass (C) over time following therapy administration. (D–F) VEGFR2 phosphorylation (D), endothelial cell surface VEGFR2 occupancy (E), and endothelial cell surface VEGFR1 occupancy (F) following treatment. | ||
Discussion
In silico screening of therapeutics in a physiological context
In this paper, we used a systems pharmacology model of VEGF isoforms and VEGFR signaling in human peripheral artery disease to screen three types of promising therapies in silico: biomaterial-based VEGF delivery; gene therapy; and pro-angiogenic antibodies. The simulations provide insight into the mechanisms of action of these therapies, identifying molecular signaling differences between promising and ineffective approaches to predict drivers of therapy success or failure. The model also highlights key questions and considerations for design, optimization, and translation of these therapeutics into humans (Table 1), which may also be relevant to other pro-angiogenic applications, including wound healing and tissue engineering. Along the way, we unearthed common trends and addressed open questions about the underlying biology that drives response to therapy. For example, a key unanswered question in dosing studies is: how much increase in pR2 is optimal? We showed that, while many past study designs have resulted in fully ligated VEGFR2, either throughout a tissue or in local areas, this saturation appears to be unnecessary, and likely detrimental for formation of stable collateral vessels. Rather, the model supports the hypothesis of the Banfi group that sufficient duration of VEGFR2 activation is a critical factor,7 and one which can potentially be achieved using biomaterial-based protein delivery, gene therapy, or antibody treatment with optimized therapy design and delivery.| Biomaterial-based VEGF delivery | Gene therapy | Pro-angiogenic antibodies | |
|---|---|---|---|
| Note: insights in bold are derived from this computational model, and insights in bold italics are confirmed by the model. This highlights the contributions of this study to therapy design & translation, within the context of other knowledge in the field (shown in italics). | |||
| Pros | • No systemic effects | • With proper controlled dosing, no systemic effects predicted | • Leverage underlying biology |
| • No dependence on underlying VEGF expression or splicing | • Multiple potential targets: VEGF, VEGFR2 | • Single factor, with easy (IV) delivery | |
| • Tunable, controlled duration and spatiotemporal patterning | • Therapeutically inducing a VEGF splicing switch may be a relative safe, effective option without the magnitude control concerns of other approaches. | • Long half-life | |
| • Lower dose = lower cost | • Precedent for approved anti-VEGF therapy | ||
| • Likely applicable to multiple diseases (PAD, wound healing, etc.) | • Lower local VEGF levels than VEGF protein or gene delivery, so no concern about inducing VEGFR2 saturation | ||
| • Challenging to deliver to large tissues with chronic, diffuse disease | • Precise control of magnitude & duration critical for success | • Potential for systemic effects | |
| Cons | • Cost of growth factor is high, especially for repeated delivery to large tissues. | • Challenging to deliver to large tissues with chronic, diffuse disease | • Efficacy depends on underlying VEGF splicing in healthy & diseased tissue, and relative size of tissues with high vs. low VEGF165b |
| • Growth factor + gel both require regulatory approval | • Concerns over safety of long-term or permanent gene therapy | ||
| Mechanism of action | • Low initial free VEGF spike (more physiological → less permeability, minimize angioma formation) | • VEGF: elevate local VEGF protein | • Antibody ‘shuttling’ between tissues with different VEGF isoform expression |
| • Extended pR2 elevation vs. native protein delivery (may prevent vessel regression) | • VEGF splicing: elevate local “strong” VEGFR2-activating VEGF165a | • Improves VEGFR1 signaling and could increase pR2 (anti-VEGF only) | |
| • VEGFR2: increase number of VEGFR2 to be activated per endothelial cell | |||
| Key open questions | • Need to understand in vivo receptor levels in each therapeutic case or experimental system of interest to select doses that avoid receptor saturation. | • Can induction of stable, high efficiency (but not permanent), spatially homogenous expression, targeting specific cell types be achieved? | • What are the absolute levels of VEGF165aand VEGF165bin healthy and ischemic tissue? In obese subjects and those with extensive CV disease, what fraction of tissue expresses high VEGF165blevels? |
| • How can VEGF splicing be tuned therapeutically? | • Need to better understand VEGFR1 signaling. | ||
Dosing and delivery leading to extended, sub-saturating VEGFR2 activation is key to successful biomaterial-based VEGF delivery
In simulating biomaterial-based VEGF delivery approaches leveraging engineered VEGF-ECM interactions, our model showed that the engineered constructs maintain more physiological free VEGF levels, likely accounting for reduced induction of permeability, and sustain VEGFR2 phosphorylation for weeks, promoting vessel stabilization following angiogenesis. We also simulated different doses of VEGF, finding that receptor saturation may be causative for angioma formation at high VEGF doses. This result aligns with the pathogenic C482R VEGFR2 mutant, which is constitutively active in the absence of ligand, and induces hemangioma formation in infants,54 similar to the phenotype observed with delivery of high VEGF doses. It also provides insight into the need for tight natural regulation of endogenous VEGF secretion; VEGF haploinsufficiency is lethal,23,55 as is even 2–3× global overexpression during development.56 Saturation could induce angioma formation via several different mechanisms: (1) saturation prevents cells from sensing VEGF gradients, allowing for cell proliferation but not directed migration, (2) VEGFR2 signaling is different at saturation than in sub-saturating regimes, or (3) high VEGF levels lead to depletion of available endothelial receptors, leading to changes in signaling. Interestingly, the first hypothesis is supported by recent predictions from an agent-based model of angiogenesis, showing formation of blob-like structures with high proliferation but low migration of endothelial cells.57The appropriate VEGF dose to stimulate angiogenesis without saturating VEGFR2 will likely vary depending on the species and tissue, as local capillary density (number of endothelial cells), interstitial VEGF concentration, and expression of receptors and proteases vary. Nonetheless, quantification of these variables is feasible, and can be used with computational analyses such as this to inform appropriate doses for experiments in mice and initial trials in humans with varying disease states. It should be noted that the threshold between therapeutic angiogenesis and angioma formation in ref. 41 varied by model system (mouse vs. rat, no injury vs. hindlimb ischemia vs. wound healing), from >0.5 μg mL−1 to <25 μg mL−1. The threshold appears to occur at the lowest end of this range in our model (∼0.5 μg mL−1), perhaps owing to molecular, physiological, and size differences,58 and the approximate nature of allometric dose scaling between mouse and human. VEGFR1, which is predicted to saturate at higher doses, could also play a role. Additionally, we set up our simulations to predict signaling within and in close proximity to the gel, the area imaged in the experimental study. To study gradients of VEGF concentration father from the gel, a spatial model including diffusion would be required. While diffusion would alter the shape of the release curves, the general match of simulations to the experimental release rate makes trends in the magnitude and duration of signaling, as well as the mechanisms of action highlighted by this modeling work, valuable and instructive. In the future we can build models with variable capillary density and receptor expression to further explore this issue. We can also build parallel mouse and human models to more effectively identify doses that achieve similar signaling between species, and potentially predict divergent effects in mice and humans.
Effective gene therapy depends on tight control of delivery and chosen target gene
It is believed that past VEGF gene therapy trials in PAD and CAD have failed largely due to insufficient and/or poorly controlled VEGF expression. Here, we asked: (1) what fold increase in expression would be effective at inducing pR2 without saturating VEGFR2, (2) what would the resulting changes in free VEGF and endothelial VEGFR signaling be, and (3) would the results be different if other VEGF-pathway targets were selected? We showed that approximately 5-fold over-expression of VEGF165 or equivalent expression of a super affinity VEGF construct produced similar signaling to the promising engineered VEGF protein constructs simulated above. It is of note that von Degenfeld et al.8 achieved effective angiogenesis following delivery of a myoblast clone that uniformly expresses VEGF at approximately 5-fold higher levels than control myocytes. Interestingly, inducing a splicing switch for VEGF165, from all VEGF165b to all VEGF165a in the PAD Calf Muscle shows promise. While the induction of pR2 was modest (180% of baseline), the success of even low doses of biomaterial-based VEGF delivery in inducing angiogenesis suggests that this small increase in pR2, coupled with a predicted reduction in VEGF165b–VEGFR1 and increased VEGFR1 binding by PlGF and other VEGF isoforms (Fig S3A, ESI†) may be promising. Alternative splicing of VEGFxxxa and VEGFxxxb is known to be regulated by IGF1, TNFα, TGFβ,59 as well as non-canonical Wnt5a signaling38 and ECM stiffness,60 though the mechanisms behind splicing changes in disease remain largely unknown. Here we assumed a splicing switch only in the PAD Calf Muscle; our previous work predicts that blood-produced VEGF has minimal effect on signaling in tissue, suggesting that targeting to a specific tissue may not be critical. However, VEGF165b levels decrease in several types of cancer (renal,27 colon,61 metastatic melanoma62) and in diabetic retinopathy,63 so detailed measurements of the ratio of “a” to “b” isoforms in healthy tissue will be necessary to determine the appropriate amount to shift splicing for optimal safety and efficacy.‘Anti-angiogenic’ anti-VEGF treatment predicted to improve endothelial VEGFR signaling in PAD via a novel ‘Antibody Shuttling’ effect
Finally, we simulated treatment of PAD with VEGF-binding antibodies. Our model supports the results from an extended analysis of the data in Ganta et al. that perfusion recovery in murine hindlimb ischemia following anti-VEGF165b treatment is mediated by VEGFR1, not VEGFR2.31 This result is consistent with model predictions that VEGF isoforms compete for binding to VEGFR1, but not VEGFR2.44 This appears to be the case in both murine hindlimb ischemia, where VEGF is elevated, and human PAD, where VEGF levels are unchanged, suggesting key similarities in response to anti-VEGF165b across species. This result motivates further study of VEGFR1 signaling in PAD, in order to better understand the most effective way to therapeutically alter VEGFR1 activation. While this model considers only endothelial VEGFR1, VEGFR1 expressed on monocytes and macrophages may also be relevant to VEGFR1 signaling in PAD.The model predicts that, due to reversible VEGF-antibody binding and the long half-life of monoclonal antibodies, a non-isoform-specific VEGF antibody would ‘shuttle’ VEGF isoforms between tissues with varying levels of VEGF isoform expression. This surprising mechanism of action has been computationally predicted and clinically observed in cancer following bevacizumab treatment, where a seemingly paradoxical increase in plasma VEGF can occur following treatment.46 This, combined with accurate model prediction of response to anti-VEGF165b treatment in mice, gives us confidence that this mechanism, while non-intuitive, is feasible. Due to this ‘antibody shuttling’ effect, and the relatively high VEGF165b levels in ischemic PAD tissue, the model predicts that treatment with ‘anti-angiogenic’ anti-VEGF would improve signaling via both VEGFR1 and VEGFR2, potentially providing more therapeutic benefit than anti-VEGF165b. This prediction can be tested in mice in the future. Note that, while the largest predicted change in pR2 occurs with highest local VEGF165b secretion, the absolute quantity of pR2 is still highest with low local VEGF165b (and high local VEGF165a), reinforcing the potential appeal of a therapeutic agent that can ‘fix’ impaired signaling in PAD by reverting splicing towards VEGF165a.
Our counter-intuitive prediction of signaling changes following anti-VEGF treatment suggests that differences in the underlying biology in cancer and ischemic disease may lead to opposite responses to the same therapy; VEGF levels are substantially elevated in cancer, while in PAD a splicing switch occurs, leading to relative differences in VEGF165a and VEGF165b between healthy and ischemic tissue. Interestingly, even in cancer, anti-VEGF is predicted to alter VEGFR1 ligation more than VEGFR2.46 Additionally, VEGF splicing alters prognosis and response to anti-VEGF treatment,64 though anti-VEGF-mediated redistribution of VEGF splice isoforms has not been investigated in this context. Note that the efficacy and safety of anti-VEGF treatment in PAD depends on a relatively small quantify of diseased tissue expressing VEGF165b at higher levels than healthy tissue. This motivates further quantitative measurement of absolute VEGF isoform levels in healthy and ischemic tissue, and suggests that this approach may not be effective in patients with extensive ischemic disease and/or large quantities of white adipose tissue, where VEGF165b expression is also elevated.17 Further work to establish the effect of antibody treatment on exercise-mediated increases in VEGF expression and signaling is also merited. More broadly, this prediction shows that consideration of systemic antibody-mediated growth factor redistribution may be a key, yet understudied, consideration for design of an effective antibody-based therapeutics.
Key similarities across promising therapeutic strategies
Comparing the therapeutic strategies simulated here, we saw two distinct profiles of signaling (Fig. 8) following treatment with promising therapeutics: (1) elevated local free VEGF, pR2, and VEGFR1 ligation produced by delivery of matrix-binding VEGF protein or gene, and (2) reduced local free VEGF, moderate increases in pR2, and reduced endothelial VEGFR1 ligation produced by anti-VEGF, inducing a splicing switch from VEGF165b to VEGF165a, or VEGFR2 gene delivery. Further experiments are necessary to fully determine whether one or both of these profiles results in sustained improvements in perfusion in human PAD. The common theme across both of these profiles is small, non-saturating improvements in VEGFR signaling that are sustained for weeks. We propose that this is a key criteria to drive design and dosing of these therapeutics in experimental systems and human subjects, taking into account the underlying physiology, molecular biology, and ligand and receptor expression levels in each system during dose selection.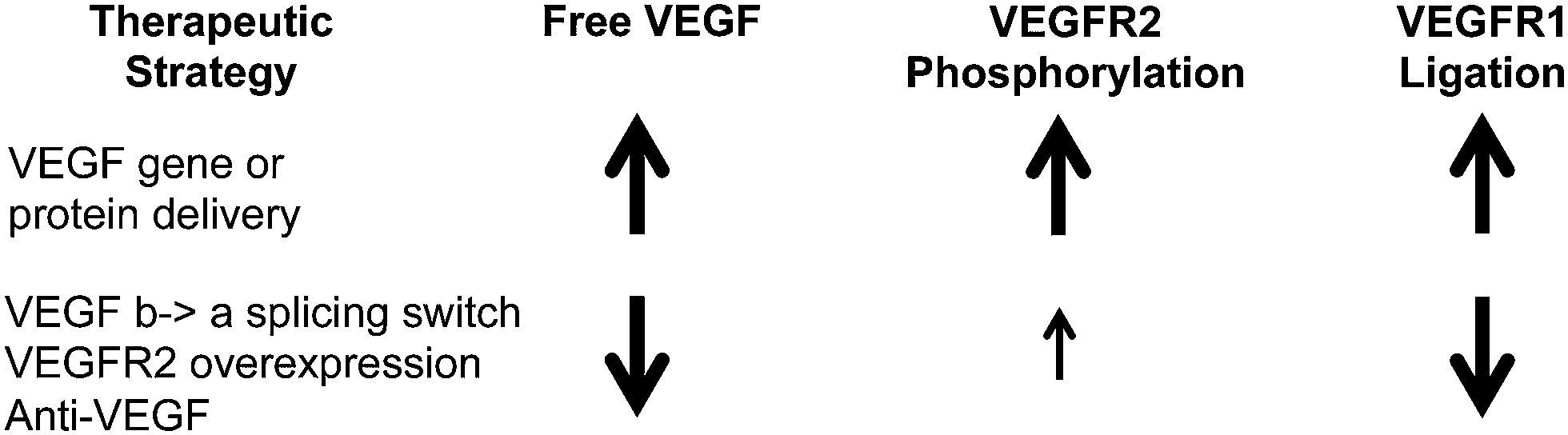 | ||
| Fig. 8 Summary of two promising therapeutic profiles. These molecular signatures were found in this analysis to be common across promising therapeutic strategies. | ||
Interpretation of model conclusions for therapy design
This model captures important trends in observed data in vitro, in vivo, and in the clinic, and provides valuable and informative mechanistic insight into the effects of underlying biology on therapy action, which could not have been obtained with experiments alone. It is important to remember that, due to differences between our model system, the experimental systems, and real diverse patient populations, along with assumptions used to simplify the model or account for lack of complete biological understanding, precise numeric match between simulations and observed experimental and clinical outcomes is challenging and should not be expected. Computational models have the advantage of easily implementing theoretical treatments that can be very challenging to effectively design. While this analysis does not address the practical or logistical issues associated with therapy production or delivery, the insight provided does facilitate decision-making about the feasibility of promising strategies, and highlight key questions that must be addressed for effective therapy translation (Table 1).Critical role for computational models in understanding effects of complex, multiscale biology on therapy action
The predictions presented here could not have been made based on cell culture experiments or measurements in mice alone, but are nonetheless consistent with observed signaling data and understanding of antibody properties, and are therapeutically relevant and actionable. This highlights the need for complex pharmacological models such as this to integrate detailed molecular interactions and signaling properties measured in vitro, observations in animal models, and knowledge of therapy pharmacokinetics into a single consistent framework, in order to predict the complex and sometimes surprising mechanisms of action and off-target effects of therapeutics in human subjects. Additionally, computational models allow comparison of diverse therapeutic approaches, leading to identification of key rules that translate between distinct approaches (Table 1), and moving towards more effective, generalizable therapy design criteria (Fig. 8).Specifically, the key rules identified here can inform design of novel therapeutic strategies not explicitly examined here, as well as predict responses to combinations of the three approaches simulated here. The modular model structure allows for incorporation of any therapeutic strategy or combination for which we have available data to model the drug pharmacokinetics and mechanism of action. As such, this modeling framework can be further validated and updated iteratively with future experimental developments to further evaluate and optimize translation of a diverse range of VEGF system-targeted therapeutics.
Methods
The whole body compartment model leveraged for this analysis was developed and validated previously.26,44 It accounts for molecular and physiological processes, including growth factor secretion in tissues and blood, growth factor and soluble receptor binding to endothelial receptors and HSPGs in the ECM and basement membranes, trafficking and tyrosine site-specific phosphorylation of endothelial VEGFR2, transport between compartments via vascular permeability and lymphatic drainage, and clearance of growth factor and sR1 in the blood (via the liver and kidneys). Each tissue compartment is approximated using the properties of skeletal muscle, and contains physiological proportions of endothelial cells, ECM, interstitial space, and basement membrane, as well as molecular expression levels adjusted to account for measured changes in PAD, compared to healthy skeletal muscle (Fig. 1). By incorporating VEGF and PlGF isoform-specific ECM- and NRP1-binding properties, the model can predict the effect of splicing changes, such as those occurring in PAD, on signaling. Additionally, the ability to predict the effect of VEGF-ECM binding on VEGFR2 signaling makes our model an excellent platform to study biomaterial-based VEGF delivery. These model capabilities have been validated against in vitro, ex vivo, murine, and human data from multiple groups in several previous studies. For detailed model formulation, please see ref. 44.Brief overview of model validation
The model used in this analysis is the result of iterative addition of biological and physiological detail, with each component validated upon addition, and the overall model checked for consistency with previous predictions and known biology at each step, over the course of over 10 years.25,26,44,65–68 While the overall model structure contains hundreds of equations, many fewer unique parameters exist (e.g. VEGFR2 trafficking and dephosphorylation are assumed to not depend explicitly on the VEGF isoform bound to VEGFR2, but equations for each isoform must be included in the model). Additionally, only a small number of parameters were added in any step before additional validation of the model.25,44 For example, adding detailed VEGFR2 trafficking to the in vitro version of the model required six new parameters,25 which we fit to one experimental study69 and validated against two additional, independent studies70,71 to ensure that the “trafficking module” was well-constrained. Similarly, after validation against in vitro data, the trafficking and phosphorylation detail was then incorporated into a previously-validated whole-body model of VEGF pharmacokinetics and pharmacodynamics, and again validated to ensure physiologically-reasonable behavior.26 Finally, in ref. 44, we modified this existing structure to specifically incorporate the VEGF165b isoform, and compared its distribution and signaling properties against all available data on VEGF165b in peripheral artery disease. This iterative process is necessary to produce a complex, multi-scale computational model that accurately captures the required VEGF system biology and physiology to study the mechanisms of actions of diverse therapeutic strategies, as presented here.Model implementation
The model is simulated in Fortran using the Livermore Solver for Ordinary Differential Equations with Automatic method switching for stiff and nonstiff problems (LSODA) on a laptop PC, using a relative error tolerance of 10−6. Simulation run time is less than 10 minutes for all scenarios examined.Therapy implementation in computational model
To simulate biomaterial-based protein therapy, gene therapy, or antibody therapy, we mimicked therapy delivery in experimental and clinical studies closely, making model predictions as realistic and meaningful as possible.Implementation of biomaterial-based VEGF delivery
The first therapeutic strategy we simulated was use of biomaterials to control and extend VEGF delivery. For these simulations, we modeled experimental constructs engineered by the groups of Jeff Hubbell and Andrea Banfi (Fig. 2A). In the Martino 2014 study, a dose of 200 ng was delivered to a 5–6 mm diameter punch biopsy wound.40 To deliver a roughly equivalent dose to the PAD Calf Muscle of our simulated 70 kg human, we used allometric scaling, with an exponent of 0.75 to account for differences in metabolic rate between mice and humans, then adjusted this value to account for differences in the size of the wound vs. PAD Calf Muscle relative to total body size. This resulted in a human dose of 354 μg VEGF, which was used for all three constructs in Fig. 2B, to provide an equivalent comparison. We used VEGF-HSPG affinity measurements from ref. 40 for VEGF165 and the engineered “Super Affinity” VEGF (VEGFSA) binding to ECM proteins (Table 2). Then, we tuned the model by adjusting the number of ECM binding sites added along with the VEGF (i.e. in the fibrin gel), in order to match experimentally measured kinetics for VEGF release from the implanted gels in vivo (Table 2). We matched simulation outputs to the fraction of VEGF retained in or within 2 mm of the gel when VEGF was delivered with fibrin. This adjustment helped to account for diffusion effects, which are not explicitly included in this model. As proteases are also not explicitly included in this model, we modeled release of the “Covalent VEGF with Proteolysis” construct as a reversible VEGF-ECM bond, reducing the koff rate constant from Super Affinity VEGF until the release kinetics matched experimental data from ref. 41. For these simulations, we assumed 100% local fractional VEGF165b secretion, a ‘worst-case’ scenario.| Protein construct | KD(V·M) in nM | k on(V·M) in M−1 s−1 | k off(V·M) in s−1 | [ECM] added (×VEGF dose) | Ref. |
|---|---|---|---|---|---|
| Note: all constructs assumed to bind NRP1, VEGFR1, VEGFR2, & sR1 with the same affinities as VEGF165. | |||||
| VEGF165 | 60.9 | 1.64 × 105 | 1.0 × 10−2 | 425 | 40 |
| Super affinity VEGF | 5.9 | 1.69 × 105 | 1.0 × 10−3 | 150 | 40 |
| Covalent VEGF with proteolysis | 0.002 | 1.69 × 105 | 3.3 × 10−7 | 150 | Tuned to match ref. 41 |
For the dosing study presented in Fig. 2C, we assumed uniform delivery of the VEGF-containing fibrin gel to the entire PAD Calf Muscle, at the same concentrations (in total tissue volume) used in the experimental study, ranging from 0.01 μg mL−1 to 100 μg mL−1,41 using the same added number of ECM sites as for Fig. 2B for all doses. To demonstrate similar therapy action regardless of underlying VEGF165b secretion, these simulations were run with no local VEGF165b secretion (0%).
Implementation of gene therapy
Next, we simulated gene-based pro-angiogenic therapies, including delivery of VEGF165 gene at moderate levels, VEGF engineered for increased affinity to the ECM (VEGFSA), increased VEGFR2 expression on endothelial cells, and decreased expression of sR1 (summarized in Table 3). We also examined the impact of switching VEGF165 splicing from all VEGF165b to all VEGF165a, another therapeutic strategy of interest. For the splicing switch, we assumed no change to VEGF165b secretion by monocytes into the blood. This has minimal effect on tissue signaling (Fig. 4C). We assume ideal delivery: 100% transfection efficiency, spatially homogeneous delivery, with expression occurring instantly, at a constant level for an infinite duration, and only in the target tissue (PAD Calf Muscle). We tested these therapies assuming high (100%) fractional secretion of VEGF165b.| Targeted gene | Targeted cells in PAD Calf Muscle | Fold change |
|---|---|---|
| VEGF165 | Parenchymal (e.g. skeletal muscle) | 5× basal VEGF165 secretion |
| VEGF165 | Parenchymal (e.g. skeletal muscle) | 20× basal VEGF165 secretion |
| Super affinity VEGF | Parenchymal (e.g. skeletal muscle) | 5× basal VEGF165 secretion |
| VEGF165 b → a switch | Parenchymal (e.g. skeletal muscle) | From 100% VEGF165b to 0% |
| VEGFR2 | Endothelial cells | 5× basal VEGFR2 production |
| sR1 | Endothelial cells | 0.1× basal sR1 secretion |
Implementation of antibody therapy
We modeled intravenous infusion of anti-VEGF165b in a single bolus over 90 minutes, at a dose of 10 mg kg−1 (Table 4), using the same binding and pharmacokinetic properties as previously used for bevacizumab.46 To provide a frame of reference against a similar approved antibody, we simulated a non-isoform-specific antibody (anti-VEGF) using the same protocol and binding properties, assuming an equivalent binding affinity for all VEGF isoforms. Additionally, as done before, we assumed the antibodies bind only to free VEGF, not VEGF bound to HSPGs, sR1, or endothelial receptors. We also neglected potential multimeric binding of VEGF by antibodies and therapy-induced tissue remodeling.| Parameter | Description | Value | Units | Ref. |
|---|---|---|---|---|
| Notes: (1) all parameters assumed same for anti-VEGF and anti-VEGF165b, aside from set of VEGF isoforms bound by ligand. (2) MBM = main body mass, PCM = PAD calf muscle. | ||||
| K D(V·AB) | Binding affinity for VEGF to AB | 2.2 | nM | 46 and 72 |
| k on(V·AB) | On-rate constant for V·AB | 9.2 × 104 | M−1 s−1 | 46 |
| k off(V·AB) | Off-rate constant for V·AB | 2.0 × 10−4 | s−1 | 46 |
| k perm(AB) | Vascular permeability of AB | 3 × 10−8 | cm s−1 | 46 |
| k perm(V·AB) | Vascular permeability of V·AB | 3 × 10−8 | cm s−1 | 46 |
| k lymph(AB), MBM | Lymphatic drainage of AB from MBM | 0.1418 | cm2 s−1 | 68 |
| k lymph(V·AB), MBM | Lymphatic drainage of V·AB from MBM | 0.1418 | cm2 s−1 | 68 |
| k lymph(AB), PCM | Lymphatic drainage of AB from PCM | 0.0022 | cm2 s−1 | 68 |
| k lymph(V·AB), PCM | Lymphatic drainage of V·AB from PCM | 0.0022 | cm2 s−1 | 68 |
| k CL(AB) | Clearance of AB from blood | 3.2 × 10−7 | s−1 | 46 |
| k CL(V·AB) | Clearance of V·AB from blood | 3.2 × 10−7 | s−1 | 46 |
| Bolus size | Dose (70 kg human subject) | 10 | mg kg−1 | 46 |
| Infusion duration | 90 | min | 46 | |
Simulations of human PAD and murine hindlimb ischemia
Simulations use 75% secretion of VEGF165b in the PAD Calf Muscle and 25% VEGF165b secretion in the Main Body Mass, to roughly mimic the 3-fold increase in ischemic tissue VEGFxxxb measured experimentally, as done in our previous analysis.44 Outputs shown are quantities measured in the PAD Calf Muscle 6 days after antibody administration.Simulations comparing different classes of therapies
We compared biomaterial-based delivery of the ‘Covalent VEGF with Proteolysis’ construct to the PAD Calf Muscle at the dose used in Fig. 2B, therapeutic expression of the ‘Super Affinity VEGF’ construct at 5-fold the normal expression level for VEGF165 in the PAD Calf Muscle, and intravenous injection of a non-isoform specific antibody to VEGF at a dose of 10 mg kg−1. All therapies are delivered with fractional VEGF165b secretion of 100% in the PAD Calf Muscle and 0% in the Main Body Mass for consistency.Funding
This work was funded by: NDSEG Fellowship (LEC), R01HL101200 (FMG), R00HL093219 (FMG), and a Sloan Research Fellowship (FMG). No funders had a role in study design.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Dr Brian Annex and Dr Vijay Ganta of the University of Virginia for helpful discussions and access to original data sets related to their recent publication.References
- L. E. Clegg and F. Mac Gabhann, Integr. Biol., 2015, 7, 498–512 RSC.
- D. J. Collinson and R. Donnelly, Eur. J. Vasc. Endovasc. Surg., 2004, 28, 9–23 CrossRef CAS PubMed.
- L. Norgren, W. R. Hiatt, J. A. Dormandy, M. R. Nehler, K. A. Harris, F. G. R. Fowkes, K. Bell, J. Caporusso, I. Durand-Zaleski, K. Komori, J. Lammer, C. Liapis, S. Novo, M. Razavi, J. Robbs, N. Schaper, H. Shigematsu, M. Sapoval, C. White, J. White, D. Clement, M. Creager, M. Jaff, E. Mohler, 3rd, R. B. Rutherford, P. Sheehan, H. Sillesen, K. Rosenfield and T. I. W. Group, European journal of vascular and endovascular surgery: the official journal of the European Society for Vascular Surgery, 2007, 33(Suppl 1), S1–75 CrossRef PubMed.
- J. A. Dormandy and R. B. Rutherford, J. Vasc. Surg., 2000, 31, S1–S296 CrossRef CAS PubMed.
- B. H. Annex, Nat. Rev. Cardiol., 2013, 10, 387–396 CrossRef CAS PubMed.
- A. Grochot-Przeczek, J. Dulak and A. Jozkowicz, Gene, 2013, 525, 220–228 CrossRef CAS PubMed.
- C. R. Ozawa, A. Banfi, N. L. Glazer, G. Thurston, M. L. Springer, P. E. Kraft, D. M. McDonald and H. M. Blau, J. Clin. Invest., 2004, 113, 516–527 CrossRef CAS PubMed.
- G. von Degenfeld, A. Banfi, M. L. Springer, R. A. Wagner, J. Jacobi, C. R. Ozawa, M. J. Merchant, J. P. Cooke and H. M. Blau, FASEB J., 2006, 20, 2657–2659 CrossRef CAS PubMed.
- S. Yla-Herttuala, J. E. Markkanen and T. T. Rissanen, Trends Cardiovasc. Med., 2004, 14, 295–300 CrossRef PubMed.
- S. Yla-Herttuala, T. T. Rissanen, I. Vajanto and J. Hartikainen, J. Am. Coll. Cardiol., 2007, 49, 1015–1026 CrossRef PubMed.
- P. S. Briquez, L. E. Clegg, M. M. Martino, F. M. Gabhann and J. A. Hubbell, Nat. Rev. Mater., 2016, 1, 15006 CrossRef CAS.
- F. Mac Gabhann and A. S. Popel, Microcirculation, 2008, 15, 715–738 CrossRef CAS PubMed.
- F. T. H. Wu, M. O. Stefanini, F. Mac Gabhann, C. D. Kontos, B. H. Annex and A. S. Popel, J. Cell. Mol. Med., 2010, 14, 528–552 CAS.
- S. Koch, S. Tugues, X. Li, L. Gualandi and L. Claesson-Welsh, Biochem. J., 2011, 437, 169–183 CrossRef CAS PubMed.
- S. Koch and L. Claesson-Welsh, Cold Spring Harbor Perspect. Med., 2012, 2, a006502 Search PubMed.
- Y. S. Ng, R. Rohan, M. E. Sunday, D. E. Demello and P. A. D'Amore, Dev. Dyn., 2001, 220, 112–121 CrossRef CAS PubMed.
- D. T. M. Ngo, M. G. Farb, R. Kikuchi, S. Karki, S. Tiwari, S. J. Bigornia, D. O. Bates, M. P. LaValley, N. M. Hamburg, J. A. Vita, D. T. Hess, K. Walsh and N. Gokce, Circulation, 2014, 130, 1072–1080 CrossRef CAS PubMed.
- P. Vempati, A. S. Popel and F. Mac Gabhann, Cytokine Growth Factor Rev., 2014, 25, 1–19 CrossRef CAS PubMed.
- J. E. Park, G. A. Keller and N. Ferrara, Mol. Biol. Cell, 1993, 4, 1317–1326 CrossRef CAS PubMed.
- C. Ruhrberg, H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz and D. T. Shima, Genes Dev., 2002, 16, 2684–2698 CrossRef CAS PubMed.
- S. Lee, S. M. Jilani, G. V. Nikolova, D. Carpizo and M. L. Iruela-Arispe, J. Cell Biol., 2005, 169, 681–691 CrossRef CAS PubMed.
- J. Grunstein, J. J. Masbad, R. Hickey, F. Giordano and R. S. Johnson, Mol. Cell. Biol., 2000, 20, 7282–7291 CrossRef CAS PubMed.
- P. Carmeliet, V. Ferreira, G. Breier, S. Pollefeyt, L. Kieckens, M. Gertsenstein, M. Fahrig, A. Vandenhoeck, K. Harpal, C. Eberhardt, C. Declercq, J. Pawling, L. Moons, D. Collen, W. Risau and A. Nagy, Nature, 1996, 380, 435–439 CrossRef CAS PubMed.
- P. Carmeliet, Y. S. Ng, D. Nuyens, G. Theilmeier, K. Brusselmans, I. Cornelissen, E. Ehler, V. V. Kakkar, I. Stalmans, V. Mattot, J. C. Perriard, M. Dewerchin, W. Flameng, A. Nagy, F. Lupu, L. Moons, D. Collen, P. A. D'Amore and D. T. Shima, Nat. Med., 1999, 5, 495–502 CrossRef CAS PubMed.
- L. W. Clegg and F. Mac Gabhann, PLoS Comput. Biol., 2015, 11, e1004158 Search PubMed.
- L. E. Clegg and F. Mac Gabhann, PLoS Comput. Biol., 2017, 13, e1005445 Search PubMed.
- D. O. Bates, T. G. Cui, J. M. Doughty, M. Winkler, M. Sugiono, J. D. Shields, D. Peat, D. Gillatt and S. J. Harper, Cancer Res., 2002, 62, 4123–4131 CAS.
- R. Delcombel, L. Janssen, R. Vassy, M. Gammons, O. Haddad, B. Richard, D. Letourneur, D. Bates, C. Hendricks, J. Waltenberger, A. Starzec, N. E. Sounni, A. Noel, C. Deroanne, C. Lambert and A. Colige, Angiogenesis, 2013, 16, 353–371 CrossRef CAS PubMed.
- H. Kawamura, X. Li, S. J. Harper, D. O. Bates and L. Claesson-Welsh, Cancer Res., 2008, 68, 4683–4692 CrossRef CAS PubMed.
- S. C. Suarez, M. Pieren, L. Cariolato, S. Arn, U. Hoffmann, A. Bogucki, C. Manlius, J. Wood and K. Ballmer-Hofer, Cell. Mol. Life Sci., 2006, 63, 2067–2077 CrossRef CAS PubMed.
- V. C. Ganta, M. Choi, A. Kutateladze and B. H. Annex, Circ. Res., 2017, 120, 282–295 CrossRef CAS PubMed.
- B. Hoier, M. Walker, M. Passos, P. J. Walker, A. Green, J. Bangsbo, C. D. Askew and Y. Hellsten, J. Appl. Physiol., 2013, 115, 1777–1787 CrossRef CAS PubMed.
- F. M. Belgore, A. D. Blann and G. Y. Lip, Clin. Sci., 2001, 100, 567–575 CrossRef CAS PubMed.
- A. D. Blann, F. M. Belgore, C. N. McCollum, S. Silverman, P. L. Lip and G. Y. H. Lip, Clin. Sci., 2002, 102, 187–194 CrossRef CAS PubMed.
- A. J. Makin, N. A. Y. Chung, S. H. Silverman and G. Y. H. Lip, Clin. Sci., 2003, 104, 397–404 CrossRef CAS PubMed.
- T. M. Lee, S. F. Su, C. H. Tsai, Y. T. Lee and S. S. Wang, Clin. Sci., 2001, 101, 305–311 CrossRef CAS PubMed.
- W. S. Jones, B. D. Duscha, J. L. Robbins, N. N. Duggan, J. G. Regensteiner, W. E. Kraus, W. R. Hiatt, A. O. Dokun and B. H. Annex, Vasc. Med., 2012, 17, 94–100 CrossRef PubMed.
- R. Kikuchi, K. Nakamura, S. MacLauchlan, N. Doan Thi-Minh, I. Shimizu, J. J. Fuster, Y. Katanasaka, S. Yoshida, Y. Qiu, T. P. Yamaguchi, T. Matsushita, T. Murohara, N. Gokce, D. O. Bates, N. M. Hamburg and K. Walsh, Nat. Med., 2014, 20, 1464–1471 CrossRef CAS PubMed.
- D. O. Bates, A. Mavrou, Y. Qiu, J. G. Carter, M. Hamdollah-Zadeh, S. Barratt, M. V. Gammons, A. B. Millar, A. H. J. Salmon, S. Oltean and S. J. Harper, PLoS One, 2013, 8, e68399 CAS.
- M. M. Martino, P. S. Briquez, E. Güç, F. Tortelli, W. W. Kilarski, S. Metzger, J. J. Rice, G. A. Kuhn, R. Müller, M. A. Swartz and J. A. Hubbell, Science, 2014, 343, 885–888 CrossRef CAS PubMed.
- V. Sacchi, R. Mittermayr, J. Hartinger, M. M. Martino, K. M. Lorentz, S. Wolbank, A. Hofmann, R. A. Largo, J. S. Marschall, E. Groppa, R. Gianni-Barrera, M. Ehrbar, J. A. Hubbell, H. Redl and A. Banfi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6952–6957 CrossRef CAS PubMed.
- J. F. Curtin, M. Candolfi, W. Xiong, P. R. Lowenstein and M. G. Castro, Mol. Cancer Ther., 2008, 7, 439–448 CrossRef CAS PubMed.
- C. J. Buchholz, T. Friedel and H. Buening, Trends Biotechnol., 2015, 33, 777–790 CrossRef CAS PubMed.
- L. Clegg, V. Ganta, B. Annex and F. Mac Gabhann, CPT: Pharmacometrics Syst. Pharmacol., 2017, 6, 833–844 CrossRef CAS PubMed.
- L. E. Clegg and F. Mac Gabhann, Pharmacol. Res., 2015, 99, 149–154 CrossRef CAS PubMed.
- M. O. Stefanini, F. T. H. Wu, F. Mac Gabhann and A. S. Popel, Cancer Res., 2010, 70, 9886–9894 CrossRef CAS PubMed.
- C. Kut, F. Mac Gabhann and A. S. Popel, Br. J. Cancer, 2007, 97, 978–985 CrossRef CAS PubMed.
- T. Gustafsson, A. Knutsson, A. Puntschart, L. Kaijser, A. C. S. Nordqvist, C. J. Sundberg and E. Jansson, Pfluegers Arch., 2002, 444, 752–759 CrossRef CAS PubMed.
- T. Gustafsson, H. Ameln, H. Fischer, C. J. Sundberg, J. A. Timmons and E. Jansson, J. Appl. Physiol., 2005, 98, 2137–2146 CrossRef CAS PubMed.
- K. G. Shyu, H. Chang, B. W. Wang and P. L. Kuan, Am. J. Med., 2003, 114, 85–92 CrossRef CAS PubMed.
- I. Baumgartner, A. Pieczek, O. Manor, R. Blair, M. Kearney, K. Walsh and J. M. Isner, Circulation, 1998, 97, 1114–1123 CrossRef CAS PubMed.
- J. M. Isner, I. Baumgartner, G. Rauh, R. Schainfeld, R. Blair, O. Manor, S. Razvi and J. F. Symes, J. Vasc. Surg., 1998, 28, 964–973 CrossRef CAS PubMed.
- J. C. Yang, L. Haworth, R. M. Sherry, P. Hwu, D. J. Schwartzentruber, S. L. Topalian, S. M. Steinberg, H. X. Chen and S. A. Rosenberg, N. Engl. J. Med., 2003, 349, 427–434 CrossRef CAS PubMed.
- S. Sarabipour, K. Ballmer-Hofer and K. Hristova, eLife, 2016, 5, e13876 Search PubMed.
- N. Ferrara, K. CarverMoore, H. Chen, M. Dowd, L. Lu, K. S. Oshea, L. PowellBraxton, K. J. Hillan and M. W. Moore, Nature, 1996, 380, 439–442 CrossRef CAS PubMed.
- L. Miquerol, B. L. Langille and A. Nagy, Development, 2000, 127, 3941–3946 CAS.
- K.-A. Norton and A. S. Popel, Sci. Rep., 2016, 6, 36992 CrossRef CAS PubMed.
- S. Yla-Herttuala and K. Alitalo, Nat. Med., 2003, 9, 694–701 CrossRef PubMed.
- D. G. Nowak, J. Woolard, E. M. Amin, O. Konopatskaya, M. A. Saleem, A. J. Churchill, M. R. Ladomery, S. J. Harper and D. O. Bates, J. Cell Sci., 2008, 121, 3487–3495 CrossRef CAS PubMed.
- F. Bordeleau, J. P. Califano, Y. L. Negrón Abril, B. N. Mason, D. J. LaValley, S. J. Shin, R. S. Weiss and C. A. Reinhart-King, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8314–8319 CrossRef CAS PubMed.
- A. H. R. Varey, E. S. Rennel, Y. Qiu, H. S. Bevan, R. M. Perrin, S. Raffy, A. R. Dixon, C. Paraskeva, O. Zaccheo, A. B. Hassan, S. J. Harper and D. O. Bates, Br. J. Cancer, 2008, 98, 1366–1379 CrossRef CAS PubMed.
- R. O. Pritchard-Jones, D. B. A. Dunn, Y. Qiu, A. H. R. Varey, A. Orlando, H. Rigby, S. J. Harper and D. O. Bates, Br. J. Cancer, 2007, 97, 223–230 CrossRef CAS PubMed.
- R. M. Perrin, O. Konopatskaya, Y. Qiu, S. Harper, D. O. Bates and A. J. Churchill, Diabetologia, 2005, 48, 2422–2427 CrossRef CAS PubMed.
- W. R. English, S. J. Lunt, M. Fisher, D. V. Lefley, M. Dhingra, Y.-C. Lee, K. Bingham, J. E. Hurrell, S. K. Lyons, C. Kanthou and G. M. Tozer, Cancer Res., 2017, 77, 2633–2646 CrossRef CAS PubMed.
- F. Mac Gabhann and A. S. Popel, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H153–H164 CrossRef CAS PubMed.
- F. Mac Gabhann and A. S. Popel, Am. J. Physiol.: Heart Circ. Physiol., 2007, 292, H459–H474 CrossRef CAS PubMed.
- M. O. Stefanini, F. T. Wu, F. Mac Gabhann and A. S. Popel, BMC Syst. Biol., 2008, 2 DOI:10.1186/1752-0509-2-77.
- F. T. Wu, M. O. Stefanini, F. Mac Gabhann, C. D. Kontos, B. H. Annex and A. S. Popel, Am. J. Physiol. Heart Circ. Physiol., 2010, 298, H2174–H2191 CrossRef CAS PubMed.
- K. Ballmer-Hofer, A. E. Andersson, L. E. Ratcliffe and P. Berger, Blood, 2011, 118, 816–826 CrossRef CAS PubMed.
- M. M. Martino, F. Tortelli, M. Mochizuki, S. Traub, D. Ben-David, G. A. Kuhn, R. Mueller, E. Livne, S. A. Eming and J. A. Hubbell, Sci. Transl. Med., 2011, 3, 100ra89 Search PubMed.
- S. M. Anderson, S. N. Siegman and T. Segura, Biomaterials, 2011, 32, 7432–7443 CrossRef CAS PubMed.
- W. C. Liang, X. M. Wu, F. V. Peale, C. V. Lee, G. Meng, J. Gutierrez, L. Fu, A. K. Malik, H. P. Gerber, N. Ferrara and G. Fuh, J. Biol. Chem., 2006, 281, 951–961 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ib00218a |
| ‡ Current address: Quantitative Clinical Pharmacology, Early Clinical Development, IMED Biotech Unit, AstraZeneca, Gaithersburg, MD. |
| This journal is © The Royal Society of Chemistry 2018 |