
Radical polymerization of biobased monomers in aqueous dispersed media
Samantha
Molina-Gutiérrez
a,
Vincent
Ladmiral
 a,
Roberta
Bongiovanni
b,
Sylvain
Caillol
a and
Patrick
Lacroix-Desmazes
*a
a,
Roberta
Bongiovanni
b,
Sylvain
Caillol
a and
Patrick
Lacroix-Desmazes
*a
aICGM, CNRS, ENSCM, Université de Montpellier, Montpellier, France. E-mail: patrick.lacroix-desmazes@enscm.fr
bDISAT, Politecnico di Torino, Torino, Italy
First published on 7th November 2018
Abstract
This review highlights the design and synthesis of biobased monomers from renewable resources for the preparation of latexes in aqueous dispersed media. Biobased polymers have been widely studied and are still a hot topic as environmental concerns and regulations require the use of green chemistry principles as guidelines for the synthesis of new polymer materials. Consideration should equally be given to green polymerization processes such as industrially relevant aqueous emulsion and suspension polymerizations. However, the synthesis and polymerization of biobased monomers through polymerization in aqueous dispersed media have not been sufficiently explored. Hence, constraints and opportunities arising from previous research work in this area will be presented focusing on aqueous (mini)emulsion and suspension polymerizations.
 Samantha Molina Gutiérrez | Samantha Molina Gutiérrez obtained her BSc in Chemistry in 2012 from the National Autonomous University of Mexico. She obtained her MSc in Chemistry in 2015 from the University of Lille 1 (France) and the University of Helsinki (Finland) where she worked on the synthesis of supramolecular structures. She is currently a PhD student at the National School of Chemistry in Montpellier under the direction of Dr Patrick Lacroix-Desmazes, where she is working on the synthesis of biobased monomers and their polymerization in aqueous dispersed media. |
 Vincent Ladmiral | Vincent Ladmiral graduated from Montpellier Chemistry School (France) in 1998 and received his PhD from the University of Warwick (UK) in 2006. He then worked as a postdoctoral associate at Kyoto University (Japan), at the University of Sydney (Australia), at the University of New South Wales (Australia) and at the University of Sheffield (UK). In 2012, he was recruited by CNRS as a research scientist at the Institute for Molecular Chemistry and Material Sciences in Montpellier (France). His research interests pertain to the study of polymer synthesis, self-assembly and colloidal structures, and to the synthesis and properties of fluorinated polymers. |
 Roberta Bongiovanni | Roberta Bongiovanni, after a degree in Chemistry at the University of Turin (Italy), received a M.Sc. in Colloid and Interface Science at the University of Bristol (UK) where later she obtained a Ph.D. in Physical Chemistry. After working for several years at ENI Central Laboratories in Milan (Italy), she joined the Politecnico di Torino where she is now a Full Professor of Chemistry. Her work focuses on photopolymerisation with a special interest for fluorinated photopolymers and photoinduced modification of polymers. |
 Sylvain Caillol | Sylvain Caillol was born in 1974 in Sète, France. He received his M. Sc. Degree in Chemistry from the National School of Chemistry in Montpellier. Then he received his PhD degree in 2001 from the University of Bordeaux. Subsequently he joined Rhodia Company, and then headed the Polymer Research Department at Aubervilliers. In 2007, he joined the CNRS at the University of Montpellier where he started a new research topic dedicated to Green Chemistry and Specialty Polymers. He won in 2010 the Innovative Techniques for Environment award. He is a co-author of several articles, patents and book chapters. |
 Patrick Lacroix-Desmazes | Patrick Lacroix-Desmazes graduated in 1992 from the National School of Chemistry in Montpellier (France) and received his MSc in Polymer Science from the University of Montpellier. He obtained his PhD in 1996 from the University of Lyon. After working at BP Chemicals (1997), he joined CNRS as a research scientist. His research interests cover reversible-deactivation radical polymerizations, including those in dispersed media, as well as the synthesis and use of polymers in supercritical carbon dioxide. He has been the deputy president of the French Polymer Group (2013–2016). Since 2015, he is the head of the team Engineering Macromolecular Architectures (IAM) in Montpellier. |
Introduction
Current depletion of conventional fossil fuels, instability of petroleum prices, environmental concerns and more stringent environmental regulations are encouraging the design and synthesis of biobased monomers and polymers from renewable stocks.1,2 Several goals are pursued: the increase of biogenic carbon in polymeric materials, the replacement of toxic and hazardous monomers and the production of materials with suitable chemical and mechanical properties which could mimic or even surpass the properties exhibited by their petrochemical counterparts. Molecules for this purpose can be selected from a vast biomass feedstock that includes terpenes, carbohydrates, lignin derivatives, proteins, vegetable oils and lipids (Fig. 1).3–5 Biobased materials are those composed of, or derived in whole or in part from, biological products obtained from biomass (including plant, animal, and marine or forestry materials).6 They are also defined by the American Standard for Testing and Materials (ASTM) as “an organic material in which carbon is derived from a renewable resource via biological processes. Bio-based materials include all plant and animal mass derived from CO2 recently fixed via photosynthesis, per definition of a renewable resource”.7 Acrylic acid, ethylene and styrene can be synthesized from renewable sources.8–13 Nevertheless, the syntheses of monomers usually derived from fossil fuels but which can now be prepared from renewable sources are beyond the scope of this review. The focus of this review is novel biobased monomers designed for free radical polymerization which could increase the biomass carbon content pursuing to equal or improve the performance of existing polymers from non-renewable sources. Certainly, effort should be made to replace toxic petroleum-based monomers or building blocks, such as styrene, bisphenol A, acrylonitrile and butadiene,14–17 in polymer formulations. | ||
| Fig. 1 Biomass classification. | ||
In the last few decades, most of the synthesized biobased monomers have been copolymerized by step growth polymerization in bulk or solution to yield thermosetting polymers such as phenolic compounds, polyepoxides, polyurethanes and a few thermoplastic polymers such as polyesters or polyamides.7,18–20 Indeed, the starting molecules obtained from biomass generally contain various functions (alcohols, phenols, acids, amines, etc.) allowing direct polycondensation, which combined with a high functionality leads easily to thermosets. In contrast, chain growth polymerization has been much less investigated. This is probably because biomass molecules rarely possess suitable reactive functions for radical polymerization. Indeed, the double bonds of vegetable oils are poorly reactive in radical polymerization, while biophenols behave as radical inhibitors.21,22 Therefore, synthesizing radically polymerizable biobased monomers is a real challenge.
In the pursuit of sustainable biobased polymers, green chemistry principles should be observed. Not only the design of safer monomers and the use of renewable feedstock must be achieved but also less hazardous chemical synthesis involving the use of safer solvents and reactants must be implemented.23,24 Reduction of volatile organic compounds (VOCs) can be attained through environmentally friendly polymerization methods such as aqueous emulsion and suspension polymerizations. The use of water as a continuous phase has several advantages, as it is an innocuous and non-flammable solvent. Moreover, it reduces the reaction medium viscosity and improves heat transfer enabling easier reaction temperature control. Aqueous polymerization in dispersed media includes several related processes such as emulsion polymerization, miniemulsion polymerization, microemulsion polymerization, dispersion polymerization and suspension polymerization.22,25–37 Emulsion and suspension polymerization processes are used at an industrial scale. Miniemulsion polymerization offers an alternative for very hydrophobic monomers; however, this technique has several constraints which hinder its use for industrial applications.28,38 Indeed, due to the variable solubilities of biomass in water, from non-polar oils to polar saccharides, using these molecules as building blocks for monomers to be used in radical polymerizations in aqueous dispersed media is also challenging.
Several recent reviews have presented the state of the art developments in the synthesis and polymerization of biobased monomers.7,18,19,39,40 Nevertheless, to the best of our knowledge, emulsion and suspension polymerizations of biobased monomers have not been reviewed to date. This article will thus present an overview of the different biobased building blocks that have been used in emulsion32,38 and suspension radical polymerizations.33 The synthesis of biobased surfactants and the emulsification of already formed biobased polymers are beyond the scope of the present review.
Vegetable oil- and lipid-based polymers
Vegetable oils and lipids are abundant, biodegradable and low toxic renewable feedstock for polymeric materials.41–44 The annual production of major vegetable oils was reported to be 177 million metric tons for 2015–2016 and increased to 189 million metric tons in 2016–2017.45 The most common vegetable oils are olive oil, soybean oil, linseed oil and sunflower oil.46Vegetable oils are composed of triglycerides (esters of glycerol and fatty acids). Fatty acids are carboxylic acids with long aliphatic chains which may be saturated, monounsaturated or polyunsaturated. To increase their reactivity towards radical polymerization, these molecules can be modified through their carboxylic acid function, alkene groups or allylic protons.47 However, most of the chemical modifications are performed on the carboxylic group.48 Fatty acids are indeed one of the main components of alkyd resins as binders.44 By virtue of their aliphatic chain, fatty acid monomers can be excellent plasticizers and facilitate coalescence during film formation. They are thus key biomass feedstocks to generate monomers for coatings and adhesive applications.49 Polymerization has been achieved via different functionalization reactions as exemplified in several reviews.7,39,48,50,51 Initially, they were incorporated into emulsion polymerization processes to produce alkyd–acrylic hybrid systems where the faster physical drying of acrylics could be combined with the oxidative curing of the alkyd leading to better chemical and water resistance properties.52–54 A decrease in the polymerization rate was observed, although the minimum film-forming temperature (MFFT) improved in comparison with cosolvent-free acrylic formulations. Long aliphatic chains from fatty acids are likely to have difficulties in diffusing through the aqueous continuous phase, from the monomer droplets to the nucleated polymer particles, in the emulsion polymerization process. This is due to their low water solubility, which can lead to long induction periods,55 even though surfactants may play an important role in solving this issue.56 Thus, miniemulsion polymerization may be a more suitable option for the polymerization of such highly hydrophobic monomers in dispersed aqueous media.57,58 Similar studies on miniemulsion polymerization showed that 20–30% of the double bonds in the aliphatic chain reacted.59,60 In addition, vegetable oils could also be used as co-stabilizers in miniemulsion polymerization for the preparation of nanocapsules for medical applications.61 Several examples of biobased latexes synthesized with solids contents typically ranging from 20 to 40% are presented in the next section.
Fatty acid emulsion polymerization
The unsaturation in fatty acids has traditionally been used for oxidative coupling reactions in alkyd resins for paint and varnish binders. However, Bunker and Wool62–64 functionalized methyl oleate through its chain double bonds by means of an epoxidation to obtain acrylated methyl oleate (AMO) (Fig. 2) and performed emulsion homopolymerization to prepare biobased pressure-sensitive adhesives (PSA). Very low conversion and low molar mass polymers were obtained due to the low AMO solubility in water (estimated to be 10−7 M). Therefore, AMO was copolymerized with hydrophilic acrylic acid to enhance nucleation. Likewise, Jensen et al.65 reported the emulsion copolymerization of AMO and styrene. They showed that the conversion and the average molar mass decreased as the concentration of AMO increased. Polymerizations conducted with more than 15 wt% of AMO resulted in an increase in the induction time and the decrease of particle nucleation and conversion. At 30 wt% of AMO, very low polymer concentrations were obtained, and large quantities of the initiator were necessary to increase the monomer conversion. Similar results were obtained by the same group in the emulsion copolymerization of acrylated soybean oil and methyl methacrylate (MMA).66 These results illustrate the difficulties encountered in the nucleation phase of emulsion polymerization of such hydrophobic monomers.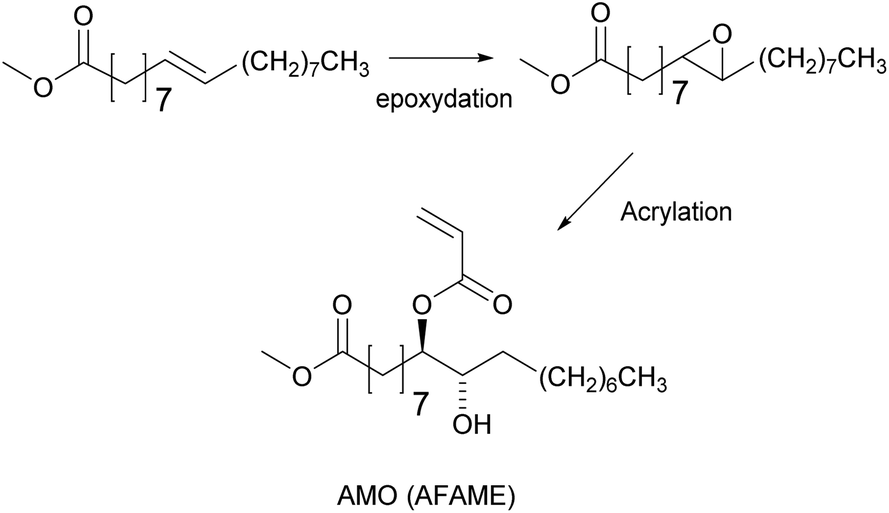 | ||
| Fig. 2 Synthesis of acrylated fatty acid methyl ester (AFAME) and acrylated methyl oleate (AMO). | ||
Vegetable oil derivatives with pendant acrylate functions easily undergo degradative chain transfer with the remaining allylic double bonds of the fatty acid, resulting in low conversion, high gel contents and branching.67 Indeed, Thames et al.68 reported the emulsion copolymerization of functionalized vegetable oil macromonomers (VOMMs) featuring at least one crosslinkable double bond in the aliphatic chain. The seeded emulsion copolymerization of MMA with soyamide monomers acrylated at their chain-end (after amidation of soybean oil with N-methyl ethanolamine)69,70 has also been reported (Fig. 3). In this case, the allylic functionalities were also preserved to further undergo auto-oxidation during the drying of the latex for ambient self-crosslinking. Emulsion copolymerizations of acrylamides (Fig. 3)71,72 derived from olive, soybean, sunflower and linseed oils with styrene73 and vinyl acetate46 were also studied. The copolymerization with styrene (up to 20 wt% of oil-based monomer) followed the Smith–Ewart theory, predicting the formation of latex particles via micellar nucleation and proportionally to the surfactant and initiator concentrations. Moreover, the reaction order of these copolymerizations was not affected by the degree of unsaturation of the monomers. However, an increase in the degree of unsaturation and in the proportion of biobased monomers in the reaction mixture led to copolymers of lower molar masses due to degradative chain transfer and allylic termination.
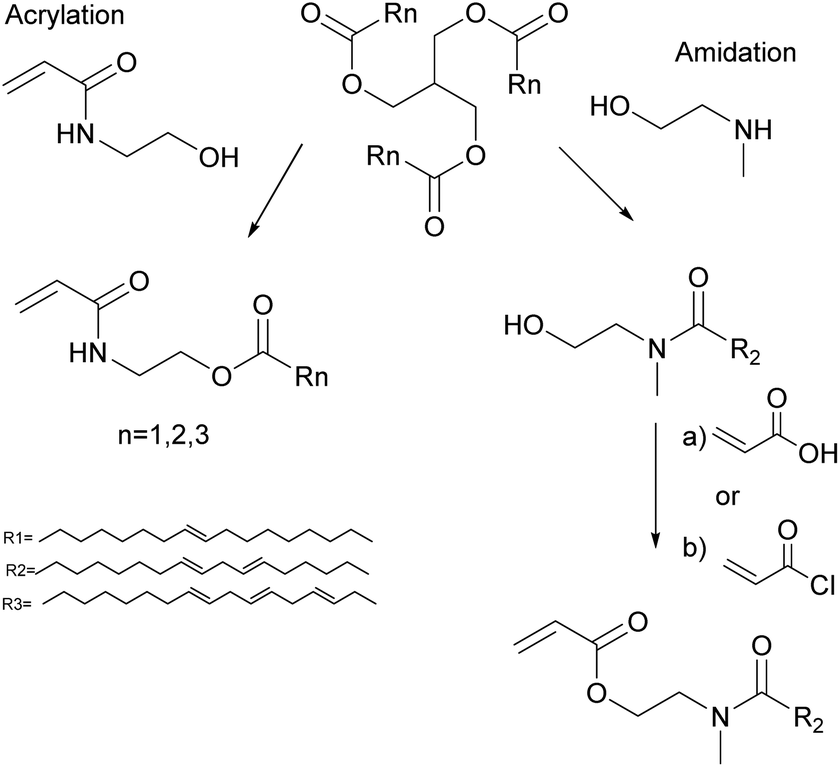 | ||
| Fig. 3 Synthesis of soyamide monomers and vegetable oil acrylamide monomers. | ||
Roberge and Dubé74 used cost-effective conjugated linoleic acid (CLA) with impurities (74% CLA, 13% oleic acid and 13% saturated fatty acid) to partially replace butyl acrylate (BA) in PSA formulations with styrene and BA, acrylic acid, n-dodecyl mercaptan as chain transfer agent (CTA) and divinyl benzene as a crosslinker. CLA was integrated into the polymer formulation at 16, 23 and 30 wt% relative to monomers. Two different factorial designs were used to study the performance of the obtained PSA. This work revealed that oleic acid, present as an impurity, helped in the introduction of CLA into the terpolymer. Moreover, as mentioned before, the increase in CLA (possessing allylic double bonds) reduced the polymerization rate and decreased the polymer molar mass.
Recently, in an attempt to synthesize a PSA with a higher biobased content, Badia et al.75 prepared waterborne pressure adhesives with biobased contents up to 72% using 2-octyl acrylate (OA, bio-content of 73%, Tg of −44 °C, derived from castor oil) and isobornyl methacrylate (IBOMA, bio-content of 71%, Tg of 150 °C, derived from pine resin) (Fig. 4). PSA formulations are composed mainly of a low Tg monomer, which provides tackiness, and a small quantity of a hard monomer, which provides cohesion to the system, combined with an unsaturated carboxylic acid for wettability and good peel and shear strength properties. The copolymerization (IBOMA, OA and methacrylic acid (MAA)) was performed through a seeded process under a starved monomer feed. The seed was composed of isobornyl acrylate (IBOA), 2-octyl acrylate and acrylic acid (AA). The peel, loop tack, work of adhesion, gel content, molar mass, shear and shear adhesion failure temperature, storage modulus and tan delta were assessed. The formulation of the PSA had to be adjusted and a chain transfer agent was added to achieve a microstructure that yielded adhesive performance as good as the petrol-based formulation.
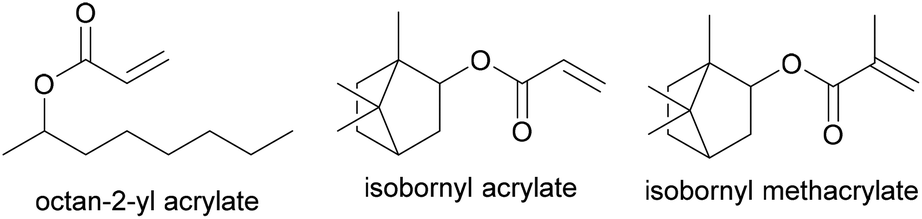 | ||
| Fig. 4 Biobased monomers OA, IBOA and IBOMA. | ||
Fatty acid miniemulsion polymerization
To improve the results obtained by the emulsion polymerization of AMO, Bunker et al.76 conducted a comparative study of the copolymerization of AMO (Fig. 2) with MMA, 1,4-butanediol diacrylate (BDDA) and 2-ethylhexyl acrylate (2-EHA) under emulsion and miniemulsion polymerization conditions. It was observed that the reaction time decreased from 18 h to 1 h and that the surfactant concentration could be reduced from 15 wt% to 2 wt% when using miniemulsion polymerization. Furthermore, copolymers with suitable properties for application as pressure sensitive adhesives (PSA) were obtained.Similarly, Quintero et al.58 used a soybean acrylate macromonomer (SAM)77 (Fig. 5) as a copolymerizable hydrophobe in miniemulsion polymerization. The stability of the miniemulsion droplets was confirmed with dynamic light scattering measurements. The SAM was copolymerized with MMA and BA. Gel content measurements showed that the unsaturation was preserved and could undergo oxidative curing during the drying process.
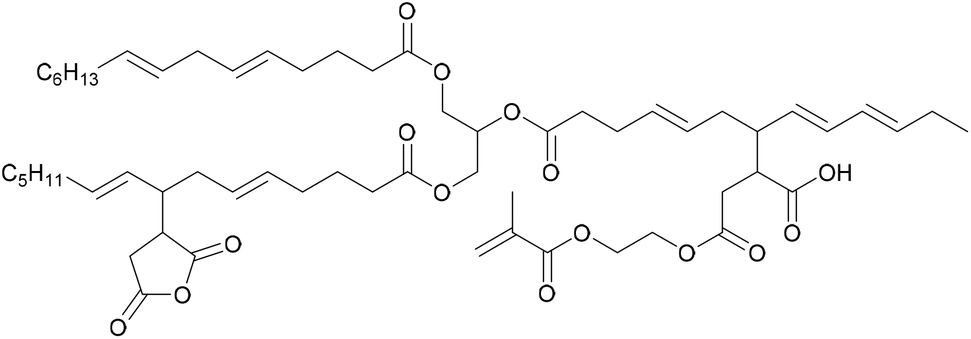 | ||
| Fig. 5 Soybean acrylated macromonomer (SAM) structure. | ||
Guo and Schork57 studied the miniemulsion copolymerization of MMA and BA with sunflower seed oil or high purity linoleic acid (97%) (Table 1) without any further functionalization. As the content of linoleic acid (two unsaturations in the aliphatic chain) increased the polymerization rate and the monomer conversion decreased. Moreover, molar mass distributions were broad due to chain transfer reactions leading to radical species with different reactivities in propagation and termination.
| Oil type | Fatty acids | ||||
|---|---|---|---|---|---|
| Saturated (wt%) | Unsaturated (wt%) | ||||
| Stearic C18H36O2 | Palmitic C16H32O2 | Oleic C18H34O2 | Linoleic C18H32O2 | Linolenic C18H30O2 | |
| Olive | 7.5–20 | 0.5–5 | 65–85 | 3.5–20 | 0–1.5 |
| Sunflower | 3–6 | 1–3 | 14–35 | 44–75 | 1–2 |
| Soybean | 7–11 | 2–6 | 22–34 | 43–56 | 7–10 |
| Linseed | 4–7 | 2.5 | 12–34 | 17–24 | 35–60 |
Copolymerization of styrene with transesterified soybean oil, acrylated through the double bond of the aliphatic chains, was also performed via miniemulsion polymerization.78 In this system, increasing the concentration of acrylated fatty acid methyl ester (AFAME) from soybean oil (Fig. 2) resulted in an increase of the latex particle size and a decrease of the total monomer conversion. This was explained by an increase of viscosity at an AFAME/styrene molar ratio higher than 5/95. Moreover, both the glass transition temperature and the thermal stability decreased whereas the gel fraction increased when the proportion of the AFAME was increased. Likewise, ricinoleic acid (castor oil) was also acrylated through the internal double bonds (Fig. 6), then copolymerized with MMA by miniemulsion polymerization to prepare a variety of polymers with Tg ranging from 50 to 124 °C.79 Similar drawbacks (bigger particle size, lower polymerization rates and lower molar masses) were encountered as the acrylated ricinoleic acid concentration increased.
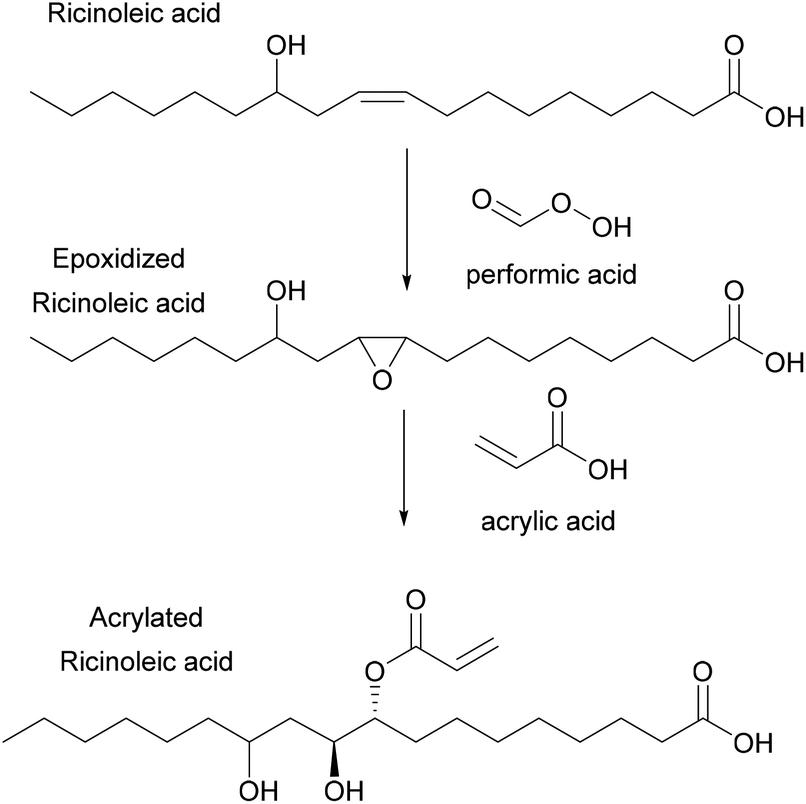 | ||
| Fig. 6 Synthesis of acrylated ricinoleic acid. | ||
To avoid the use of a chromium catalyst to synthesize acrylate derivatives from epoxidized vegetable oils,62 Maassen et al.80,81 described different synthesis pathways to suitable monomers for pressure-sensitive adhesives based on acrylated methyl oleate (AMO) and acrylated methyl erucate (Fig. 7). These monomers were copolymerized using solution and miniemulsion polymerizations. Shorter reactions times were possible using miniemulsion polymerization whereas the resulting modulus, tack, and peel values were better for the solution polymerization materials. The authors proposed that appropriate comonomers should be included to produce shorter segments between entanglements and thus improve the cohesive properties at high temperatures.
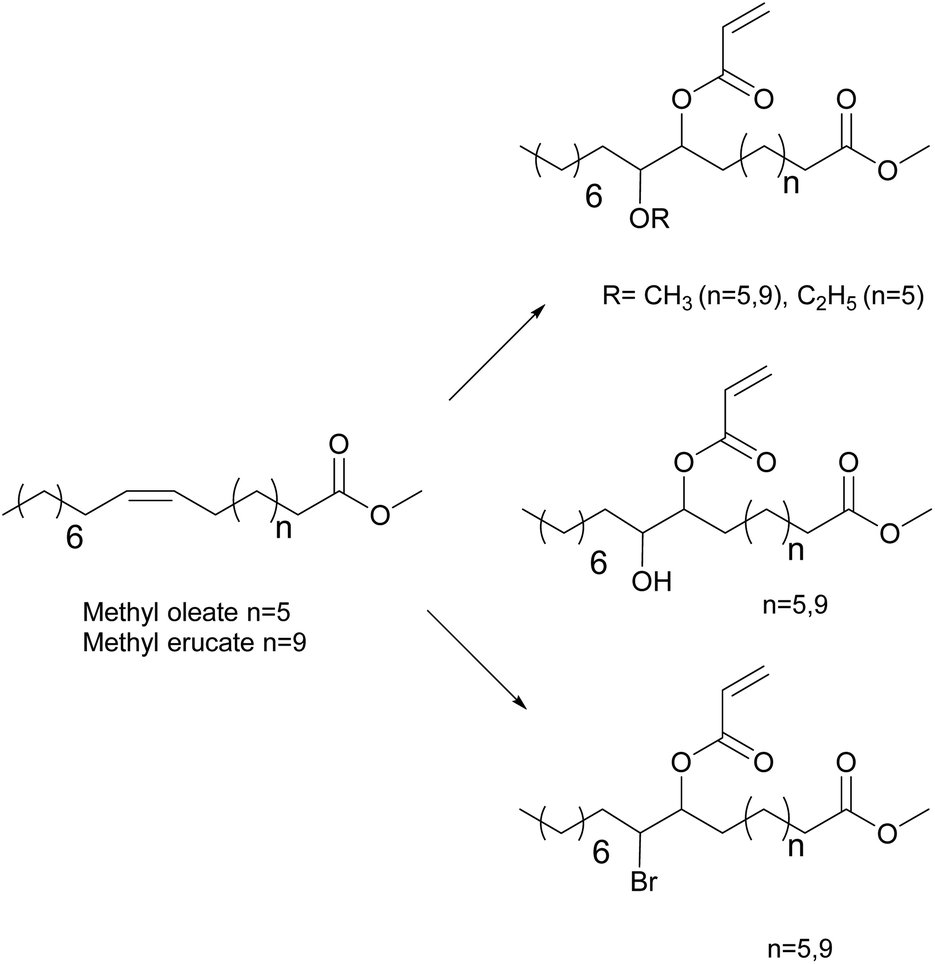 | ||
| Fig. 7 Synthesis of oleate and erucate derivatives. | ||
Biobased acrylamido monomers, obtained by condensation of N-(hydroxylethyl)acrylamide (Fig. 3) with fatty acids (soybean, sunflower, linseed and olive oils), were copolymerized with styrene in solution.46 The Q–e parameters of the biobased monomers were determined using the Alfrey–Price equations. These Q–e values were then applied to determine the reactivity ratios with MMA and vinyl acetate. Subsequently, copolymers of olive, soybean and linseed oils with MMA, and vinyl acetate were obtained by miniemulsion polymerization.46 The molar mass decreased with the increasing degree of unsaturation of the vegetable oil monomers due to degradative chain transfer. A type of seeded miniemulsion polymerization (a miniemulsion of a biobased monomer fed into another miniemulsion of MMA containing the initiator) was used to limit the extent of degradative chain transfer. In addition, copolymers of olive, soybean, linseed and hydrogenated soybean oils and styrene were prepared by miniemulsion polymerization.82 The crosslinking density increased proportionally with the unsaturation amount of the monomer feed and the latex properties could be adjusted by combining different oil-based monomers in the formulation.
Moreno et al.83,84 synthesized methacrylate monomers from oleic and linoleic acid by methacrylation of the carboxylic acid with glycidyl methacrylate thus preserving the double bonds on the long alkyl chain for further curing (Fig. 8).85 After methacrylation of the fatty acids, approximately 12–14 wt% of oleic acid and 13 wt% of linoleic acid remained unreacted. This unreacted carboxylic acid functional group was later reacted with a potassium hydroxide solution to produce surfactants in situ, able to form stable miniemulsions.86 Miniemulsion polymerizations were performed using thermal (potassium persulfate, KPS) and redox initiators (tert-butyl hydroperoxide and ascorbic acid, TBHP/AsA). The polymerization kinetics were slower for KPS as termination in the aqueous phase overtook propagation. Moreover, it was found that 30% of the double bonds on the aliphatic chain of the methacrylated oleic acid reacted during the miniemulsion polymerization.83 Furthermore, the TBHP/AsA initiation system promoted gel formation.84 This was ascribed to the lability of the two hydrogen atoms of the methylene group next to the double bonds (allylic protons). Indeed, the radical thus generated is stabilized by conjugation, less prone to propagation but still able to undergo termination. This effect was enhanced because under redox initiation, hydrophobic tert-butoxy radicals can improve droplet nucleation compared to the water soluble SO4−˙ radicals produced by KPS, leading to an increase of the radical availability, increasing both droplet nucleation and the rate of polymerization.87
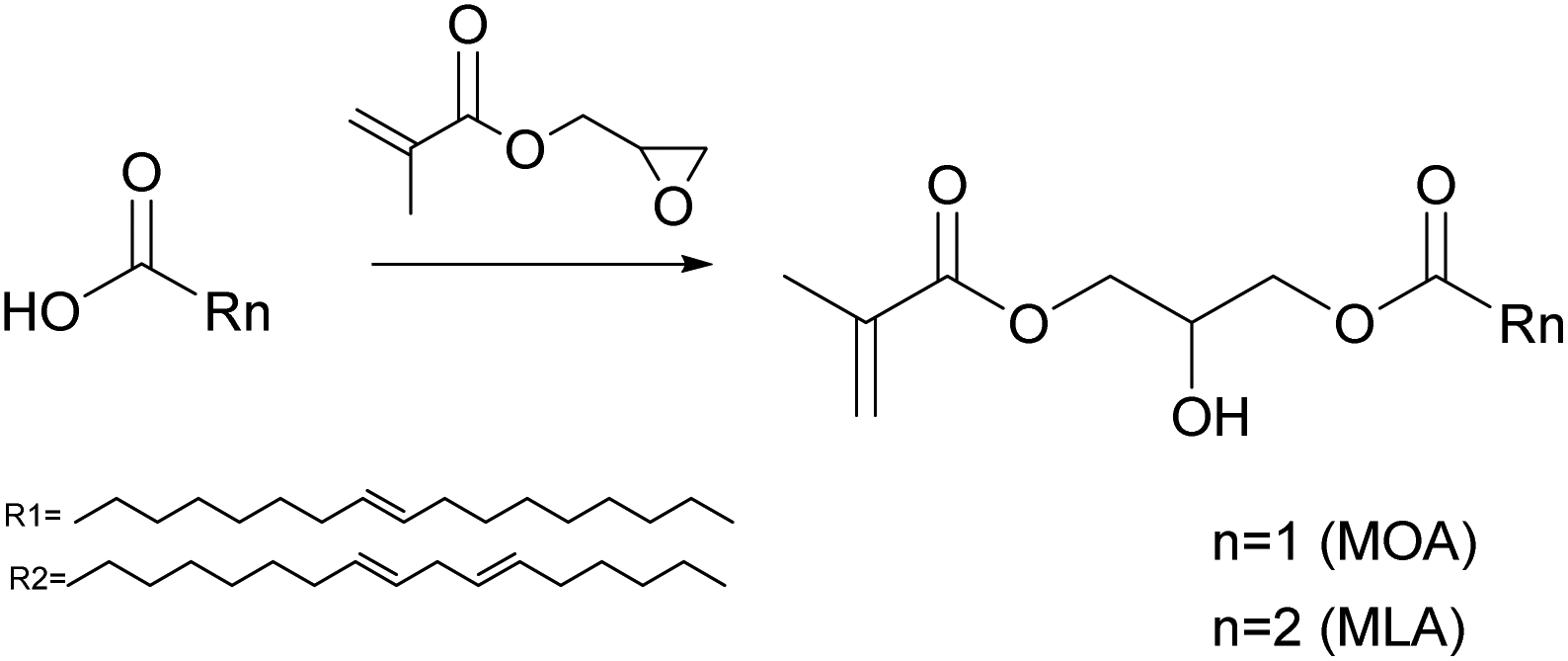 | ||
| Fig. 8 Synthesis of oleic and linoleic methacrylates. | ||
To develop polymers for coating applications, Moreno et al.88 synthesized latexes by miniemulsion polymerization from methacrylated oleic acid and α-methylene-γ-butyrolactone (Tulipalin-A). The homopolymer of α-methylene-γ-butyrolactone, a cyclic analogue of methyl methacrylate, has a Tg of 195 °C.89,90 Several concentrations of α-methylene-γ-butyrolactone (MBL) were copolymerized with methacrylated oleic acid (MOA) via miniemulsion copolymerization. MBL reacted slower than the methacrylated oleic acid leading to lower yields as its concentration in the feed increased. Moreover, higher gel contents were obtained as the amount of MOA increased due to the free reactive double bonds of this monomer. Finally, copolymers of MOA, methacrylated linoleic acid (MLA), MBL and MMA were reported and used in paint formulations.91 Properties such as hardness, gloss, rheological behaviour and open time were measured and compared with those of commercial samples.
Machado et al.92 reported the thiol–ene miniemulsion polymerization of dianhydro-D-glucityl diundec-10-enoate (DGU), synthesized from 10-undecenoic acid (derived from castor oil) and isosorbide, with 1,4-butanedithiol to produce a polymer with encapsulation properties for potential use in uterine and colon cancer treatments (Fig. 9). Different conditions were tested by varying the reaction time, temperature, emulsifier and amount of the initiator. Miniemulsion polymerization resulted in higher molar masses than bulk polymerization, due to radical compartmentalization where bimolecular termination was reduced (i.e. radicals remained active for a longer time). High viscosity at low reaction temperatures (60 °C, i.e. at a temperature lower than the melting temperature of the polymer, which is about 68 °C) produced low molar masses due to restricted molecular mobility. A reaction temperature of 80 °C provided the highest molar masses. Finally, organo-soluble azobisisobutyronitrile (AIBN) was preferred as the initiator over water-soluble KPS which may favour thiyl–thiyl termination in the water phase before thiyl radicals could enter the monomer droplets.
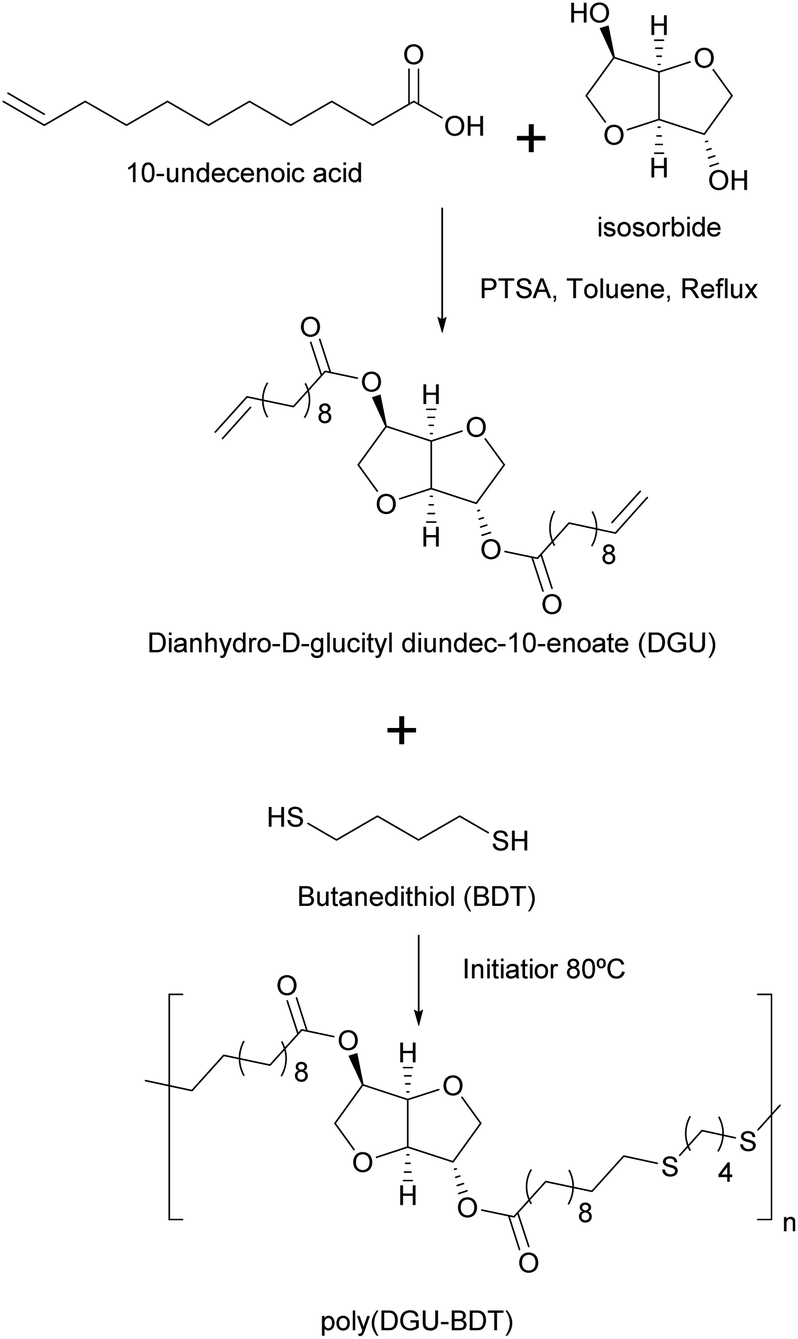 | ||
| Fig. 9 Synthesis of poly(dianhydro-D-glucityl diundec-10-enoate-co-butanedithiol). | ||
Likewise, 10-undecanoic acid was reacted with 1,3 propanediol to prepare a difunctionalized monomer which was later reacted with a dithiol such as mercaptoethyl ether (MEE) or 1,4-butanedithiol (BDT), via a thiol–ene miniemulsion polymerization (Fig. 10).93 Increasing the quantity of the initiator led to an increase of the molar mass due to the step-growth nature of the system. Lower molar masses were obtained when 2-mercaptoethyl was used. This could be due to the fact that MEE is more hydrophilic which induces a stoichiometric misbalance at the polymerization locus. Cytotoxic studies were also performed as the target applications for these polymers were temporary implants, tissue engineering or drug delivery systems.
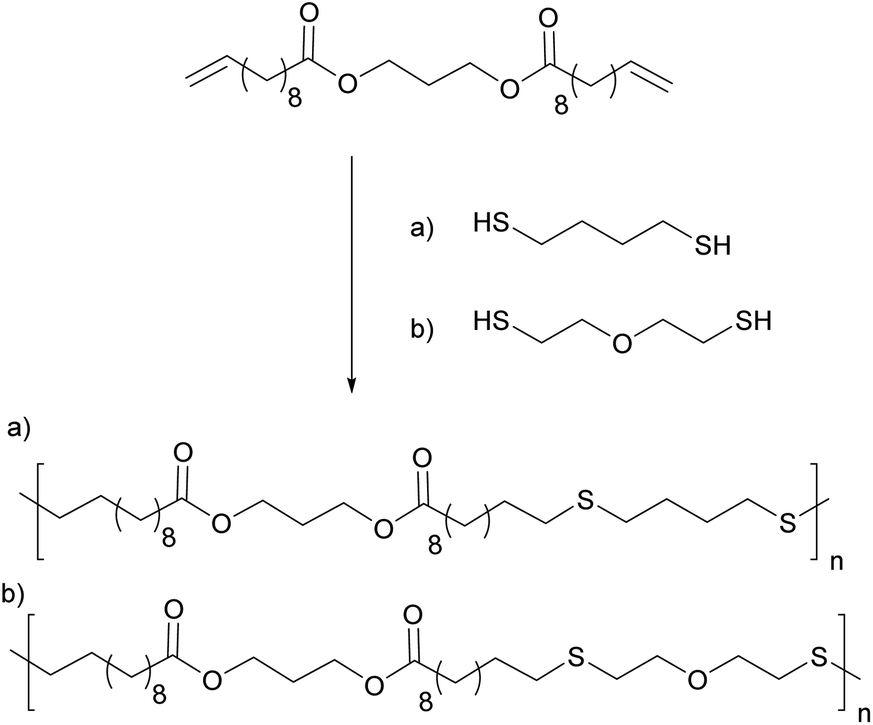 | ||
| Fig. 10 Thiol–ene miniemulsion polymerization from 10-undecanoic acid derivatives and dithiols. | ||
As mentioned before, due to their long aliphatic chains, vegetables oils do not diffuse easily through the aqueous phase. Indeed, only monomers possessing a water solubility greater than 10−7 wt% are capable of diffusing through the aqueous phase on a reasonable timescale.77 Thus, for vegetable oil derivatives, miniemulsion polymerization is preferred to emulsion polymerization. However, miniemulsion polymerization is not currently well suited to industrial processes; hence, the use of vegetable oils in polymerization in a large scale remains a challenge. Moreover, the double bonds within the aliphatic chains easily undergo degradative chain transfer due to the easy abstraction of allylic hydrogen atoms.94 These allylic transfer reactions are detrimental to the polymerization, and lead to low monomer conversion and high gel contents. Efforts have been made to understand these reactions and develop kinetic models for the preservation and exploitation of allylic double bonds.95–97 The modification of biobased precursors to introduce functional groups suitable for radical polymerization is often conducted using harmful chemicals and produces a considerable amount of waste (reducing the atom economy of the process). It is important to mention that although a percentage of vegetable oils is dedicated to industrial applications instead of food supply, preference should be given to non-edible vegetable oils for the production of biobased materials.98,99 In addition, the chemical and mechanical properties of polymers produced from vegetable oil monomers often do not match the performance of their petroleum-based counterparts. As a consequence, the use of monomer mixtures (petroleum-based and biobased monomers) is still necessary nowadays in new formulations to meet the requirements of commercial products.
Lipid emulsion polymerization
Recently, Ladmiral et al.100 reported for the first time the synthesis of a biobased latex from a cardanol derivative. Cardanol is a naturally occurring phenol extracted from Cashew Nut Shell Liquid (CNSL),101 which can be regarded as a valuable renewable material for the substitution of aromatic monomers such as styrene. Cardanol methacrylate (Fig. 11) was copolymerized with MMA by emulsion copolymerization and the resulting latex was cast into films which were photocrosslinked via thiol–ene chemistry. Gel formation during the copolymerization of cardanol methacrylate and MMA was ascribed to the hydrogen abstraction of the isopropyl alcohol proton group in the monomer or polymer (transfer reaction) by the highly electrophilic oxygen-centered sulfate radicals used as the initiator. Therefore, another synthetic approach was proposed to obtain a new cardanol-based methacrylate: hydroxyethylation of cardanol and further methacrylation (Fig. 12).102 In this synthesis, the use of epichlorohydrin was avoided. Due to the high hydrophobicity of the monomer, miniemulsion homo- and copolymerization were performed. The miniemulsion polymerization proceeded faster than the solution polymerization. Likewise, the miniemulsion copolymerization of cardanol methacrylate with MMA proceeded faster than homopolymerization, although higher dispersity in particle sizes was attained on increasing the MMA content, due to possible secondary nucleation. Furthermore, the gel content increased with the decreasing content of MMA and reached 83 wt% for the biobased homopolymer.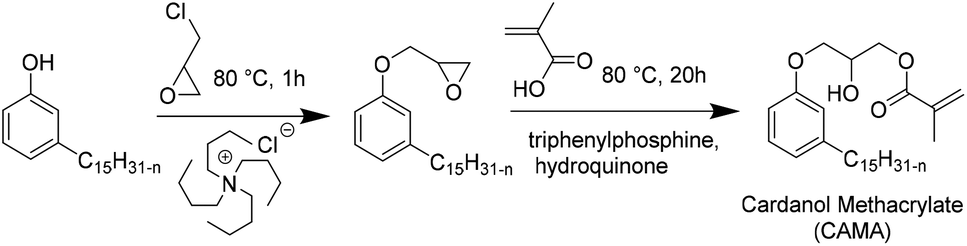 | ||
| Fig. 11 Synthesis of cardanol methacrylate. | ||
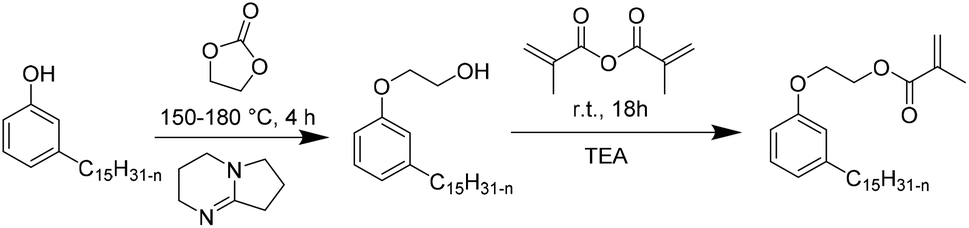 | ||
| Fig. 12 Alternative synthesis of cardanol methacrylate. | ||
Similar to vegetable oil derived monomers, lipid derived monomers show diffusion limitations in the aqueous phase. The miniemulsion polymerization technique is thus more suitable than emulsion polymerization. It is important to mention that cardanol is an abundant non-edible by-product of the cashew nut industry.103 The use of nonedible raw materials is preferred in the synthesis of new biobased monomers. Equally important is the new synthetic way (Fig. 12) which allowed avoiding the use of toxic reactants such as epichlorohydrin, thus making the biobased monomer synthesis more environmentally friendly. Still, efforts towards solvent-free and low energy consuming reactions (which can be performed at ambient temperature for example) should be made for the synthesis of novel biobased monomers.
Terpene-based polymers
Terpenes, terpenoids and rosin are hydrocarbon-based molecules with one or more isoprene (2-methyl-1,3-butadiene) units that comprise the largest single group of natural products.104–106 Terpenes have been particularly important for fine chemistry and the fragrance industry. Due to their low cost and ease of separation, α-pinene, β-pinene, limonene and myrcene have been studied relatively extensively as starting materials for the synthesis of polymers.107,108 Pinene has been used in cationic polymerization for adhesives, coatings and inks.109,110 Limonene has been used to prepare polyesters, polyamides and polyurethanes through thiol–ene functionalization.111 Several other examples of solution and condensation polymerizations with terpene derivatives have been discussed in recent articles and reviews.107,112,113 However, only a few examples of emulsion polymerization to produce latexes with 30% solids content exist, and they mostly concern acyclic terpenes such as myrcene and alloocimene. Myrcene is an acyclic monoterpene similar to some petrol-based unsaturated hydrocarbons such as 1,3-butadiene and isoprene. Myrcene can be obtained in small percentages from hop, celery, ginger root, rosemary, nutmeg and sage. However, it can be produced on a large scale by pyrolysis of β-pinene. Ocimene and alloocimene are isomers of myrcene and very sensitive to oxidation. Ocimene is prepared by thermal cracking of α-pinene, but it isomerizes to alloocimene at high temperatures.114Terpene emulsion polymerization
Sarkar and Bhowmick115 reported the synthesis of a β-myrcene homopolymer via free radical emulsion polymerization using either thermal or redox initiation. According to 1H NMR measurements, the thermal polymerization promoted the formation of 1,4-cis and 1,4-trans mixtures and 1,2 vinyl and 3,4 structures through side reactions (Fig. 13a), whereas in the redox initiated polymyrcene neither the 3,4 structure nor the 1,2 vinyl structure was formed and the microstructure predominantly contained 1,4 addition products.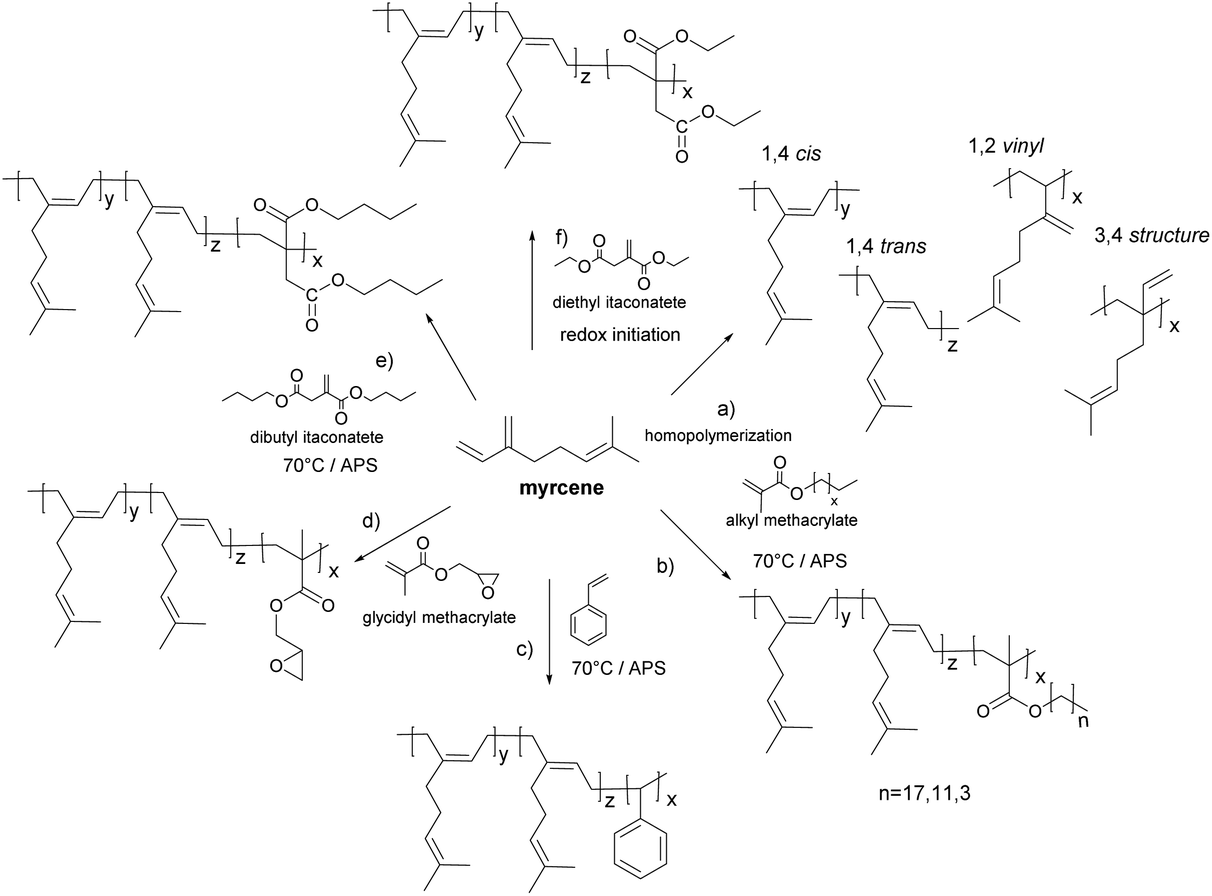 | ||
| Fig. 13 Polymerization of myrcene. | ||
At room temperature, side reactions and chain transfer effects were suppressed. The Tg value was found to be −73 °C for the thermal polymerization and −60 °C for the redox process. Similarly, free radical emulsion copolymerization of methacrylate molecules (stearyl, lauryl and butyl molecules) and β-myrcene has been reported (Fig. 13b).116 The rate of copolymerization and the molar mass decreased with the increase of the alkyl chain length of the methacrylic pendant group due to diffusion constraints. Moreover, only the myrcene-butyl methacrylate copolymer was devoid of 1,2 vinyl and 3,4 addition myrcene microstructures. This was related to the higher rate of copolymerization of butyl methacrylate which prevented side reactions. Furthermore, for the emulsion copolymerization of β-myrcene and styrene (Fig. 13c),117 a higher gel content and a lower rate of copolymerization were observed as the mass fraction of styrene decreased. Only 1,4-cis and 1,4-trans microstructures were found for those copolymers prepared with a 40 wt% or higher styrene content.
The synthesis of poly(myrcene-co-dibutyl itaconate) by emulsion copolymerization (Fig. 13e)118 showed a delayed nucleation process due to the polar character of dibutyl itaconate (DBI). As the DBI feed content increased, oligomers formed in the aqueous phase did not diffuse promptly into the micelles. In addition, DBI radicals are more stable than myrcene radicals, and thus less prone to chain propagation. Consequently, as DBI is introduced (10 wt% of the total monomer), the copolymer yield and molar mass decreased compared to the homopolymer of myrcene. However, upon increasing the content of DBI in the monomer feed, the yield and molar mass increased until 50 wt% of myrcene and 50 wt% of DBI in the copolymer mixture was reached, and then reductions in the yield and molar mass were observed. Moreover, the gel content increased with the content of myrcene. In addition, elastomers from polymyrcene and its copolymers with styrene, dibutyl itaconate and butyl methacrylate at a 30 wt% of the comonomer were compounded and vulcanized. Carbon black was used as a filler to improve the mechanical properties. Myrcene constituted 70 wt% of the total monomer feed mass in the formulation to maintain low Tgs. Properties such as tensile strength, elongation at break, crosslinking density, density, hardness and thickness were measured. The presence of aromatic rings in the copolymer of styrene and myrcene increased the stiffness of the polymer resulting in a higher Tg in spite of having a lower cross-link density than the polymyrcene homopolymer.119 However, no comparison with commercial products was given. Furthermore, the copolymerization of glycidyl methacrylate (GMA) (Fig. 13d) and myrcene was carried out to prepare a functional polymer capable of covalent interactions with silica for tyre reinforcement.120 The reaction temperature was maintained at 20 °C to avoid crosslinking reactions that occurred as the mass fraction of GMA increased in the comonomer mixture. Moreover, the copolymerization between myrcene and GMA corresponded to an azeotropic copolymerization.
Lei et al.121 performed the synthesis of a nanocomposite of myrcene and diethyl itaconate via redox emulsion polymerization employing sodium hydroxymethane sulfonate/Fe-EDTA/tert-butyl hydroperoxide as an initiating system at room temperature (Fig. 13f). Diethyl itaconate was used to improve the polarity of the resulting macromolecules. Only 1,4-cis and 1,4-trans microstructures of myrcene were found in the copolymers according to the 1H NMR spectra. Nano-silica was used as a filler instead of carbon black. The physical and mechanical properties of vulcanized silica/poly(myrcene-co-diethyl itaconate) elastomers were studied, achieving a tensile strength of 9.2 MPa and 443% of elongation at break for the 60 wt% myrcene formulation. Similar tendencies in the polymerization yield as the ones observed for dibutyl-itaconate118 were found. The highest yield was reached at a 70/30 weight ratio of a myrcene/diethyl itaconate comonomer mixture.
Sahu et al.122 reported the emulsion homopolymerization of alloocimene (Fig. 14) using two redox emulsion polymerization reaction systems: FeSO4·7H2O/TBHP and FeSO4·7H2O/Na2S2O5 at different temperatures (15, 25 and 35 °C). A rubber-type polymer was obtained in 20–25% yield. The authors claimed that, at 35 °C, the thermal decomposition of the polymer chains increased, leading to the reduction of the molar masses. The polymerization was thus preferably performed at room temperature to control side reactions and prevent cross-linking reactions.
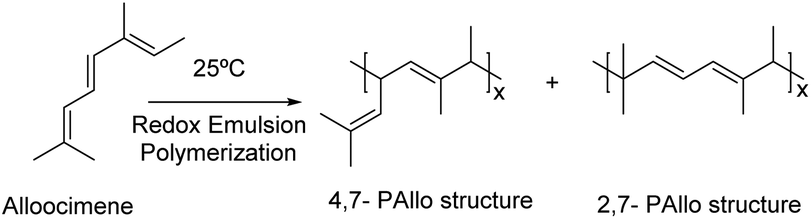 | ||
| Fig. 14 Alloocimene polymerization. | ||
Linear terpenes remain highly valuable monomers because of their resemblance to isoprene. The physicochemical properties of the resulting polymers are greatly influenced by the reaction conditions; thus, side reactions and chain transfer should be avoided to control the microstructure of the polymers. Stereoselective solution polymerization of myrcene at 70 °C has been performed succesfully.123 Likewise, this has been achieved in emulsion polymerization using low temperature redox initiation. Reactivity ratios of the biobased monomers should be studied and adequate copolymer mixtures should be formulated to reach high polymerization yields and suitable molar mass for the targeted applications. Miniemulsion polymerization remains a suitable option for monomers suffering from diffusion constraints although it might restrict their use in industrial processes. Seeded emulsion (co)polymerization with more hydrophilic monomers also offers a solution to overcome difficulties encountered in the nucleation step in ab initio emulsion polymerization.
Lignin derivative-based polymers
Lignin is the main renewable source of phenolic compounds, which provides a platform of aromatic chemicals that can be used for the production of polymers with different thermomechanical properties.124,125 Lignocellulosic biomass is mostly the cell wall material and is composed of three main kinds of molecules: cellulose (40–50 wt%), hemicellulose (20–30 wt%), and lignin (15–35 wt%). Hemicellulose and cellulose are polymers and copolymers of C5 and C6 sugars.126 Lignin constitutes 15–35% of dry lignocellulose and is the largest renewable source of aromatics on Earth. Lignin is a cross-linked amorphous copolymer produced from the radical polymerization of substituted phenyl propylene units: coumaryl, coniferyl, and sinapyl alcohols containing zero, one, and two methoxy groups, respectively.127,128 The depolymerization process of lignin has been widely studied.105,124,126 Interestingly, lignocellulose is the non-edible portion of the plant; therefore its use in producing polymer materials does not compete with food supply. Different small molecules such as vanillin, ferulic acid, eugenol, creosol and sinapyl alcohol derivatives can be isolated from lignin and can be used for the synthesis of biobased polymers. Several polymers have been prepared from these building blocks.18,129–131Lignin derivative suspension polymerization
Although the reactivity and functionalization of lignin derivatives have already been widely studied,127,129,131 emulsion polymerization reactions of lignin derivatives have not been reported so far. Exploring this kind of polymerization process taking advantage of the functionalization pathways already developed and building on the existing solution polymerization studies of molecules such as eugenol or vanillin is a valuable opportunity.132–134 The aqueous suspension polymerization of methacrylated eugenol (Fig. 15) in the presence of poly(vinyl alcohol) (PVA) as a steric stabilizer produced cross-linked microspheres with diameters ranging from 500 to 800 μm.135 These particles exhibited large oil absorbency and were targeted for applications in wastewater treatment. Concentrations of AIBN (initiator) and PVA (stabilizer) were found to be key factors of the reaction. The allylic double bond was also involved in the radical polymerization, thus cancelling the need for a cross-linking agent.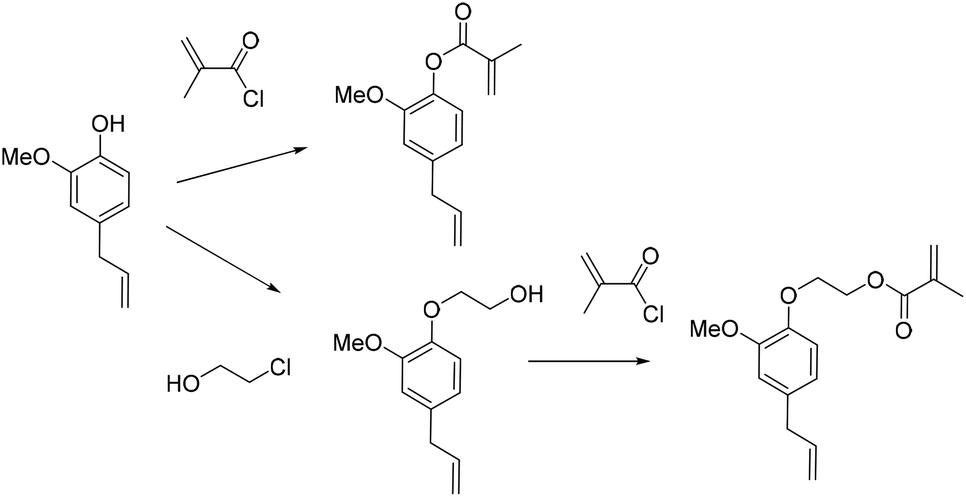 | ||
| Fig. 15 Eugenol methacrylation. | ||
Similarly, Zhang et al.136 reported the suspension polymerization of vanillin methacrylate to obtain porous microspheres also for wastewater treatment applications (Fig. 16). In this case, chloroform and toluene were used as cosolvents since vanillin methacrylate was in its solid state at the reaction temperature. The vanillin aldehyde group present on the resulting microspheres was reacted with glycine to form Schiff-bases with remarkable Cu2+ complexation capability. On the other hand, magnetic microspheres were prepared via suspension polymerization using vanillin methacrylate as the monomer and methacrylated-Fe3O4 magnetic nanoparticles as magnetic substrates.137 The aldehyde-containing magnetic microspheres were reacted with para-anisidine (used as a model amine) to determine their absorption capacity. The absorption of amines, by the formation of a Schiff base, was studied and a facile recovery of the microspheres was achieved thanks to their magnetic properties.
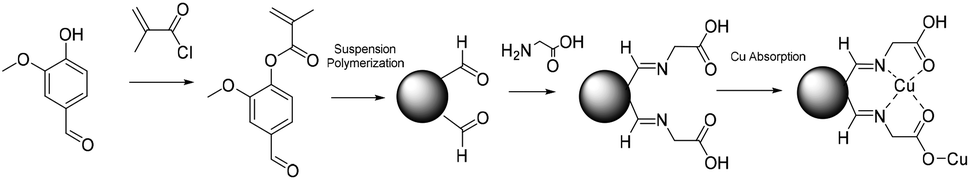 | ||
| Fig. 16 Vanillin methacrylate suspension polymerization. | ||
Currently, degradation of lignin into smaller molecules of interest has to be optimized.138 Molecules such as eugenol and vanillin are still prepared from few natural sources. Nevertheless, continuous research on the exploitation of natural feedstocks such as lignin could provide efficient and cost-effective synthetic routes to produce biobased building blocks in large quantities. The development of lignin-derived monomers and their polymerization processes is thus very timely.
Carbohydrate-based polymers
Carbohydrates represent roughly 75% of the annually renewable biomass (200 billion tons);139 thus they represent an important feedstock for the production of polymeric materials as presented in recent reviews.18,19,140,141 The synthesis of carbohydrate macromers for emulsion polymerization can be performed via hydroxyl, carboxylic acid and amine groups. Vinyl groups have been introduced by esterification of carbohydrates using (meth)acryloyl chlorides or (meth)acrylic anhydrides (Fig. 17a).142 Brune et al.143 prepared a starch based macromer by a condensation reaction between the hydroxyl groups from starch and bifunctional monomers such as N-methylolmethacrylamide, N-methylolacrylamide, hydroxyethyl methacrylate, or hydroxypropylmethacrylate in the presence of a catalyst. The resulting macromonomers were subsequently copolymerized with vinyl monomers such as styrene in emulsion polymerization to form starch-grafted latex particles. Another approach has been the free-radical polymerization through the “grafting from” technique using persulfate initiators (hydrogen abstraction from a C–H bond in the carbohydrate backbone) (Fig. 17b).144 This technique is employed to minimize the quantity of the carbohydrate in the continuous phase of the latex as it can induce flocculation or coagulation.145,146 Biopolymers containing amine functions, such as chitosan, have been copolymerized with acrylates using surfactant-free emulsion polymerization.147,148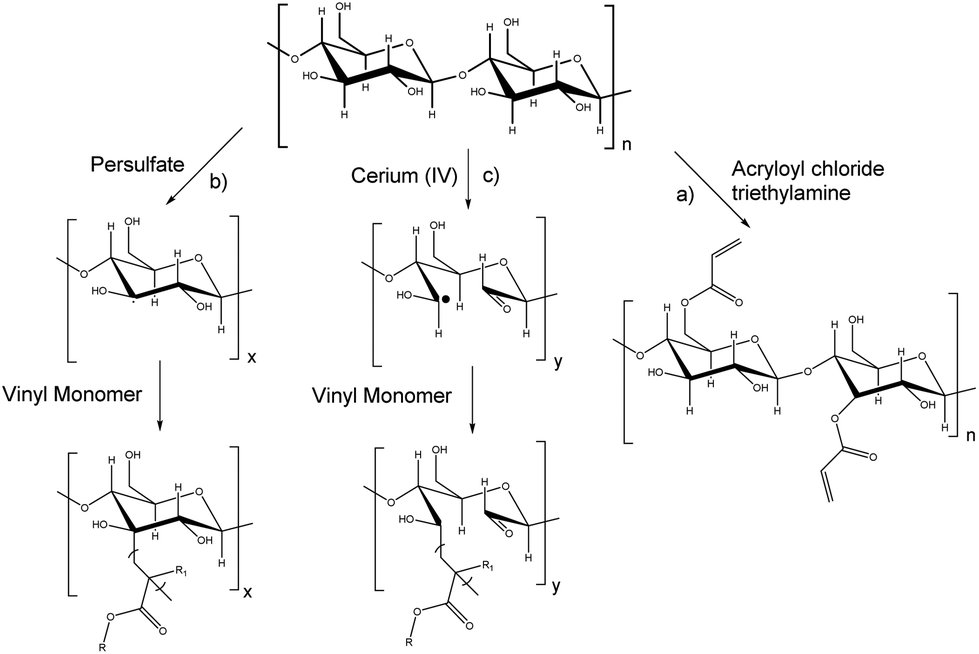 | ||
| Fig. 17 Grafting techniques on carbohydrates. | ||
Although more selective grafting techniques which involve the use of oxidant cations such as cerium, iron, manganese or copper ions able to react with the hydroxyl groups of carbohydrates (Fig. 17c)149 can be used (redox chemistry), they induce colouring in the final latexes. Consequently, the use of a persulfate is often preferred.
Möller and Glittenberg reported the emulsion copolymerization of styrene–butadiene in the presence of corn starches. The final hybrid latexes contained 50 pphm (parts per hundred monomer) of starch exhibiting improved performances in paper coating applications.150 A similar example was presented by Wang et al.151 where potato starch was degraded in situ using potassium persulfate to prepare latexes containing butyl acrylate and methyl methacrylate. Then, diacetone acrylamide was copolymerized as a functional monomer to produce a latex that could be further crosslinked by a reaction with adipic acid dihydrazide (ADH). These latexes exhibited humidity control properties, and could be used as indoor coatings. Similarly, Gaborieau et al.149 used a cerium(IV) redox initiator to produce core–shell particles from hydrophobic core-forming vinyl monomers and cationic carbohydrate shells. However, as Ce4+ ions can lead to the coloration of the latexes, hydrogen abstraction by alkyl hydroperoxides on amino groups of certain biopolymers has been reported by Li et al.147 As low molar mass polyamines can form redox pairs with alkyl hydroperoxide, a radical is generated on the nitrogen atoms in the amino group which subsequently initiated graft polymerization.152 Graft polymerization resulted in amphiphilic polymers that could self-assemble into micelle-like domains, leading to core–shell particle structures. Following a different approach, Smeets et al.37 reported the sonochemical homolytic chain scission of hydroxyethyl cellulose in the presence of hydrophobic monomers (such as butyl acrylate) to form functional hairy latex particles. Cummings et al.153 used dent corn sourced regenerated starch nanoparticles (RNSPs) modified with vinyl groups in a semi-batch, monomer-starved seeded emulsion copolymerization of butyl acrylate, methyl methacrylate and acrylic acid. Stable latexes were produced at 40 wt% solids and 25 wt% of RNSP loading (40 wt% incorporation into the latex). A comprehensive review of the use of carbohydrates as surfactants/stabilizers, macromonomers and transfer agents to prepare hybrid latex particles was recently published by Smeets et al.154
Abeylath et al.155 examined the emulsion copolymerization of ethyl acrylate and several monosaccharide acrylates and acrylamides. These sugar acrylates were derived from glucose, ribose, mannose, galactose and glucosamine and their hydroxyl groups were protected and unprotected to study the effects on the particle formation efficiency and size (Fig. 18). Hydroxyl groups were protected by acetonides in the case of glucofuranose, mannofuranose, galactofuranose and ribofuranose, and acetylated in the case of glucosamine. Moreover, ribofuranose hydroxyl groups were also protected with benzyl groups and glucosamine hydroxyl groups were protected with benzoyl groups. The copolymerizations were successful when the acetonide-protected derivatives were used and resulted in particles with an average diameter of 42 nm. However, the O-benzyl- and O-benzoyl-protected monosaccharide acrylates polymerized only with low conversions. This was explained by the effect of the bulky groups that may affect their reactivity. Emulsion polymerization of protected carbohydrate monomers resulted in a latex characterized by particles with an average diameter of 40 nm; however, unprotected monomers produce particles of approximately 80 nm diameter. The particle size increased with the proportion of the unprotected carbohydrate monomer in the copolymer (ethyl acrylate as a comonomer).
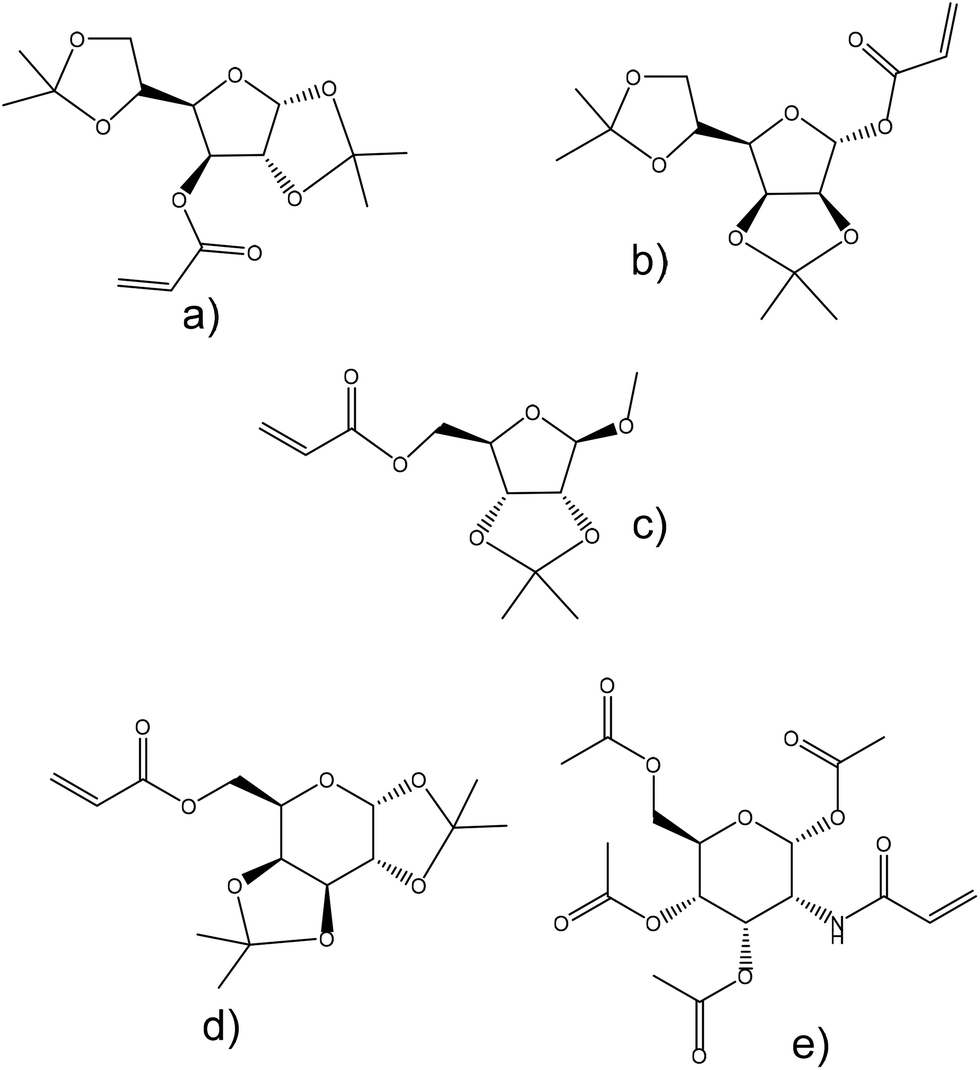 | ||
| Fig. 18 Carbohydrate monomers: (a) 3-O-acryloyl-1,2:5,6-di-O-isopropylidene-α-D-glucofuranose, (b) 1-O-acryloyl-2,3:5,6-di-O-isopropylidene-α-D-mannofuranose, (c) 5-O-acryloyl-1-methoxy-2,3-isopropylidene-β-D-ribofuranose, (d) 6-O-acryloyl-1,2:3,4-di-O-isopropylidene-α-D-galactopyranose, and (e) N-acryloyl-1,3,4,6-tetra-O-acetyl-β-D-glucosamine. | ||
Cellulose and hemicellulose can be degraded to obtain C5 and C6 sugar derivatives which can be used to synthesise several biobased building blocks.2 For example, levoglucosenone (LGO) can be produced by the pyrolysis of cellulose.156 Dihydro-5-hydroxyl furan-2-one (2H-HBO) is a LGO-derived compound produced by Baeyer–Villiger oxidation. It was methacrylated and used in emulsion homo- and copolymerization and reversible addition–fragmentation chain transfer (RAFT) emulsion polymerization to prepare latexes with 30% solids content.157 The yield of emulsion homopolymerization was 82% and the Tg of the resulting polymer was approximately 108 °C. Copolymerizations with commercially available monomers in water were also attempted. These experiments showed that the methacrylated 2H-HBO copolymerized well with polar monomers such as MMA and HEMA. However, low incorporation of the biobased monomer was achieved in copolymerization with styrene, producing copolymers with a low molar mass and rich in styrene.
Villanova et al.158 functionalized pectin with GMA (Fig. 19) to introduce methacrylic moieties and copolymerized the resulting monomer with MMA, BMA and ethyl acrylate (EA) via redox (FeSO4/APS/sodium dithionite) emulsion polymerization. The desired latexes were thought to have good mucoadhesive properties for use as pharmaceutical excipients, coating agents or matrix agents. The functionalized pectin monomer was introduced at 2, 3.5, 5 and 6 wt% in the total monomer feed. The increase in viscosity after the addition of mucin proved the formation of intermolecular interactions, which confirmed the potential use of these materials as bioadhesives.
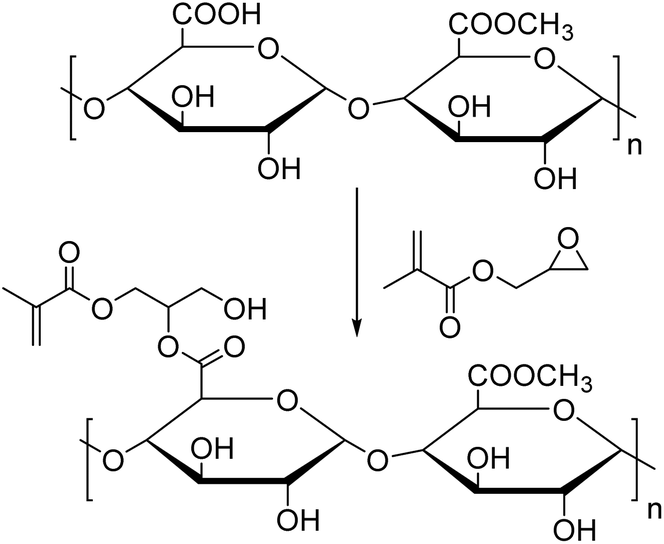 | ||
| Fig. 19 Functionalization of pectin. | ||
Another approach is the synthesis of methacrylated monomers using galactose and fructose-derived molecules (Fig. 20).159–161 A commercially available fructose-based monosaccharide bearing acetonide protecting groups was used in the synthesis of a methacrylated monomer, taking advantage of the peculiar reactivity of the primary alcohol in the anomeric position. The resulting methacrylate was used in emulsion homopolymerization and copolymerization with butyl acrylate, yielding up to 45% solids. The homopolymer glass transition temperature was reported to be approximately 115 °C. Under copolymerization conditions, polymerization rates increased with the increasing amount of fructose-methacrylate. Moreover, the gel content increased with the increase in the butyl acrylate content. Surprisingly, the gel content decreased with the decrease in the amount of potassium persulfate as the initiator. However, as gel content measurements were not performed systematically, this difference could be caused by the evolution of the latexes during storage.
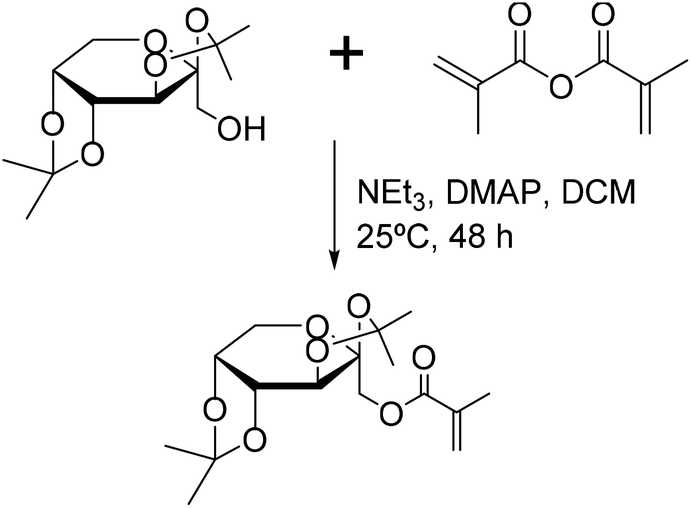 | ||
| Fig. 20 Methacrylation of protected fructose. | ||
Neqal et al.162 prepared acrylates and methacrylates from protected galactopyranose (1,2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3,4-di-O-isopropylidene-D-galactopyranose) (Fig. 21) and polymerized these monomers under emulsion polymerization conditions. The melting point of these glycomonomers was approximately 60 °C. Coagulation was observed when emulsion polymerization was carried out without any cosolvent. For the emulsion polymerization to proceed properly, the monomers had to be dissolved in toluene prior to the emulsification. However, coagulation was still not efficiently prevented; thus ethyl acetate was used instead of toluene. In addition, the initiator amount was increased and the solids content was decreased from 10 to 5 wt%. Moreover, due to the high tendency of acrylate glycomonomers to autopolymerize, the study was carried out only on the methacrylate glycomonomer as it could be safely stored. The polymerizations proceeded to full conversions. The polymers were deprotected using trifluoroacetic acid (TFA) and finally an N-alkylation reaction using dodecylamine was carried out. N-Dodecylgalactosylamine has been proven to possess antifungal properties.163 Nevertheless, in the case of the functionalized polymer particles, the antifungal activity was not as pronounced as in the case of N-dodecylgalactosylamine.
:3,4-di-O-isopropylidene-D-galactopyranose) (Fig. 21) and polymerized these monomers under emulsion polymerization conditions. The melting point of these glycomonomers was approximately 60 °C. Coagulation was observed when emulsion polymerization was carried out without any cosolvent. For the emulsion polymerization to proceed properly, the monomers had to be dissolved in toluene prior to the emulsification. However, coagulation was still not efficiently prevented; thus ethyl acetate was used instead of toluene. In addition, the initiator amount was increased and the solids content was decreased from 10 to 5 wt%. Moreover, due to the high tendency of acrylate glycomonomers to autopolymerize, the study was carried out only on the methacrylate glycomonomer as it could be safely stored. The polymerizations proceeded to full conversions. The polymers were deprotected using trifluoroacetic acid (TFA) and finally an N-alkylation reaction using dodecylamine was carried out. N-Dodecylgalactosylamine has been proven to possess antifungal properties.163 Nevertheless, in the case of the functionalized polymer particles, the antifungal activity was not as pronounced as in the case of N-dodecylgalactosylamine.
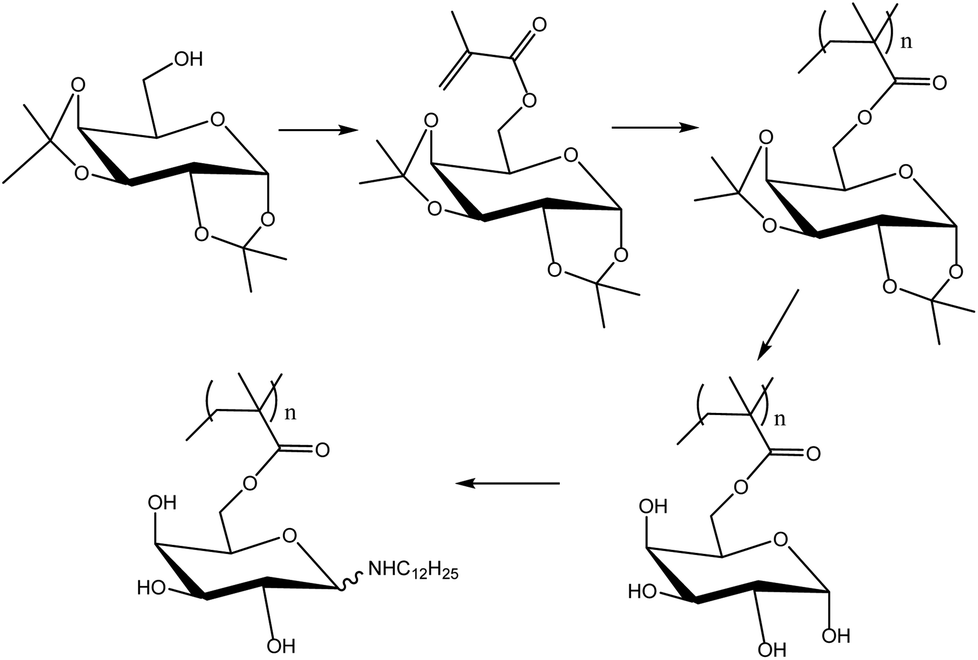 | ||
| Fig. 21 Synthesis of poly(6-O-methacryloyl-N-alkyl-β-D-galactosylamine). | ||
Due to the water solubility of saccharides and their lack of polymerizable functional groups able to undergo direct radical polymerization, several structural modifications are often required to obtain suitable monomers. These modifications sometimes involve the use of protecting groups which are undesirable according to green chemistry principles.23 In consequence, alternatives have been examined. For example, the use of grafting techniques is an option, although the control of these reactions is not straightforward. However, the water-solubility which is a source of problems when saccharides are used as monomers can be turned into an advantage if these saccharides are derivatized into surfactants or surfmers.164 Biobased surfactants and surfmers will not be presented here since they are beyond the scope of the present review.
Protein-based polymers
Milk proteins, casein and whey proteins are important for human nutrition. Moreover, they have found application in wood and paper adhesives.165 Casein, a protein derived from bovine milk, has good biocompatibility and biodegradability, high purity and low cost and exhibits a high Tg (∼180 °C).166 Therefore, it can form a hard phase in biphasic acrylic copolymers. By the addition of casein, binders employed in waterborne coatings can show a reasonable minimum film formation temperature (MFFT) and have an acceptable blocking resistance. Thus, casein in combination with soft acrylic monomers was employed. Emulsion polymerization of butyl acrylate via grafting to caprolactam-casein particles using persulfate initiators and in the absence of an emulsifier has been reported.167 Improvements in grafting efficiency resulted in higher elongation at break and tensile strength. To improve the mechanical properties, nano-silica was introduced into the copolymer,168 and core–shell particles were obtained. However, casein is easily oxidized and the resulting polymer acquired a yellow colour when persulfate initiators were used. As in the case of carbohydrates, the grafting of polymers on casein could be achieved using redox initiation based on amino functional groups (Fig. 22a). This approach was applied to casein, bovine serum albumin and gelatin.145 Thus, in pursuit of a hybrid acrylic–casein latex with potential applications in coatings, Picchio et al.166,169 investigated the emulsion polymerization of butyl acrylate and methyl methacrylate in the presence of casein at different concentrations, using a redox initiation system based on TBHP as the oxidant and the amine of casein as the reductant. As previously mentioned, TBHP radicals react with the amino groups of the protein leading to a grafting reaction with the methacrylates.145 The polymerization rate of the reaction increased as the content of casein increased due to the enhanced generation of amino radicals.169 However, the acrylic grafting efficiency decreased with the increasing casein concentration. Due to the presence of the ungrafted protein, particle size distributions of the resulting polymers showed two populations of particles (bimodal distribution) and the resulting films showed weak water resistance.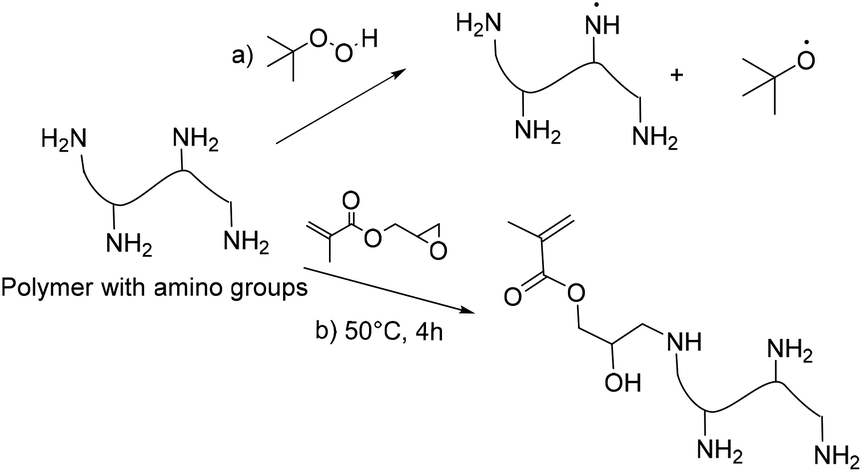 | ||
| Fig. 22 Casein functionalization. | ||
A different approach was presented for the copolymerization of casein and methacrylic monomers.170 Casein was functionalized with glycidyl methacrylate (Fig. 22b) to prepare methacrylated casein which was later copolymerized with methyl methacrylate and butyl acrylate in a surfactant-free emulsion copolymerization using TBHP as the initiator. The degree of grafting was determined using o-phthalaldehyde as a colouring agent in combination with UV spectroscopy. The maximum functionality reached was thirty-two vinyl double bonds per molecule of casein. In this case, the increase in methacrylic groups decreased the polymerization rate and the polymerization yield, because the initiation reaction is triggered by the reaction between the TBHP (oxidant) and the remaining amino groups in the casein.171
However, an increase in the degree of methacrylation allowed the adequate control of casein grafting efficiency,169 which varied from 20% to 76%. The transmission electron microscopy (TEM) micrographs showed dark regions corresponding to ungrafted casein in the polymers prepared with native casein, whereas polymers prepared with highly methacrylated casein exhibited a reduction in the free casein content, as a larger fraction of casein was grafted to the (meth)acrylic polymer. Moreover, copolymers prepared from methacrylated casein had better water and solvent (methyl ethyl ketone, MEK) resistance compared to copolymers prepared from native casein. Finally, a statistical study to optimize the formulation of casein-based clear coatings (i.e. without pigments) was carried out. The study revealed that the casein latexes prepared at 35% solids content exhibited good minimum film forming temperature (MFFT), blocking resistance, opacity and mechanical properties. However, low hardness and poor chemical resistance were observed in comparison with those of commercial binders.
Similar to carbohydrates, the use of proteins as building blocks for monomers in radical polymerization may require significant modification of their chemical structures. Nevertheless, they might also be thought of as potential surfmers due to their hydrophilic character. At present, it is still necessary to use them in a mixture with a petroleum-based monomer, but the biogenic carbon content could be increased in the polymeric materials, which is the starting point for the replacement of fossil-based polymers.
Conclusions
The vast range of available biomass provides a wide choice of molecules for the synthesis of novel biobased monomers. The ultimate goal of research in this field is to produce biobased monomers of lower toxicity which can be polymerized into polymer materials possessing mechanical and chemical properties at least equal to those of their petroleum-based counterparts. Nevertheless, to obtain a true green solution, environmentally friendly polymerization processes should also be employed. Heterogeneous polymerizations in aqueous dispersed media such as emulsion and suspension polymerizations are suitable to produce green latexes for coating applications and polymer beads for further polymer processing. However, several constraints remain to be overcome such as the design and synthesis of monomers with sufficient solubility in water to allow nucleation in ab initio emulsion polymerization. The control of the chain transfer reaction, gel formation (cross-linking) and grafting efficiency should also be carefully examined. Although it has been less explored so far, suspension polymerization of biobased monomers represents a valuable alternative to produce polymers in aqueous dispersed media. In addition, polymers produced via suspension polymerization are easily recovered by filtration. Besides obtaining stable latexes possessing the desired physicochemical characteristics for the targeted applications, monomers and polymers should fulfil specific requirements such as being non-edible, non-toxic, and produced via a cost- and atom-efficient facile synthesis with a straightforward purification pathway to comply with green chemistry principles.172 Most of the current synthetic pathways of biobased monomers still include the use of other chemicals and petroleum based reactants. For example, acrylate and methacrylate functionalization (which is the most common functionalization for emulsion polymerization) often comes from non-biobased sources. Nevertheless, acrylic acid173–176 and more recently methacrylic acid177,178 can be obtained from renewable resources. Additionally, many of the biobased monomers remain prohibitively expensive compared to their petroleum counterparts. Therefore, synthesis optimization will be a crucial step for these biobased monomers to have any significant industrial use and impact (once they are shown to give access to materials with matching or even better properties than non-biobased materials).Although the main objective is to replace petroleum-based monomers with biobased ones able to lead to materials with similar or better performance for the desired application, addressing the synthesis of novel biobased materials should be encouraged: stimuli responsive,179 nanostructured and/or self-healing materials180 could be engineered taking advantage of the functional groups present in the biomass. Other components in formulations such as initiators,181 surfactants,182,183 costabilizers, pigments, and so on, should not be neglected in the quest for greener latexes. In addition, the new latexes which will arise from the growing field of research on biobased latexes will possibly bring unexpected new properties (thanks to the combination of many new functional groups and structures, e.g. aromatics from lignocellulosic derivatives), which can in turn open the road to new applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Molina-Gutiérrez thanks SINCHEM Joint Doctorate Programme-Erasmus Mundus Action (framework agreement No. 2013-0037) for her PhD grant. The authors also thank Synthomer (UK) Ltd for financial support of their research on biobased latexes and for fruitful discussions (Dr Peter Shaw and Dr Renaud Perrin).Notes and references
- C. Williams and M. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CAS.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- J. Spekreijse, J. Le Nôtre, J. van Haveren, E. L. Scott and J. P. M. Sanders, Green Chem., 2012, 14, 2747 RSC.
- M. Iwamoto, Molecules, 2011, 16, 7844–7863 CrossRef CAS PubMed.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef CAS.
- R. S. Costa and M. A. P. da Silva, Braz. J. Chem. Eng., 2016, 33, 503–513 CrossRef CAS.
- R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 CrossRef CAS.
- Chemicals and Fuels from Bio-Based Building Blocks, ed. F. Cavani, S. Albonetti, F. Basile and A. Gandini, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016 Search PubMed.
- L. Birnbaum, Environ. Health Perspect., 1993, 101, 161–167 CrossRef CAS PubMed.
- K. C. Leibman, Environ. Health Perspect., 1975, 11, 115–119 CrossRef CAS PubMed.
- B. H. Neal, J. J. Collins, D. E. Strother and J. C. Lamb, Crit. Rev. Toxicol., 2009, 39, 589–612 CrossRef CAS PubMed.
- J. Łukaszczyk, B. Janicki and M. Kaczmarek, Eur. Polym. J., 2011, 47, 1601–1606 CrossRef.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2008, 33, 1199–1215 CrossRef CAS.
- S. Fujisawa and Y. Kadoma, Biomaterials, 1997, 18, 701–703 CrossRef CAS PubMed.
- A. Rigoussen, P. Verge, J.-M. Raquez and P. Dubois, Chemistry and Technology of Emulsion Polymerisation, John Wiley & Sons Ltd, Oxford, UK, 2013, vol. 6 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef PubMed.
- J. Ugelstad, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Polym. Lett. Ed., 1973, 11, 503–513 CrossRef CAS.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- J. M. Asua, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1025–1041 CrossRef CAS.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS.
- J. M. Sáenz and J. M. Asua, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1511–1521 CrossRef.
- C. S. Chern, Prog. Polym. Sci., 2006, 31, 443–486 CrossRef CAS.
- M. Save, Y. Guillaneuf and R. G. Gilbert, Aust. J. Chem., 2006, 59, 693 CrossRef CAS.
- S. C. Thickett and R. G. Gilbert, Polymer, 2007, 48, 6965–6991 CrossRef CAS.
- M. C. C. Pinto, J. G. F. Santos, F. Machado and J. C. Pinto, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013 Search PubMed.
- M. S. El-Aasser and P. A. Lovell, Emulsion polymerization and emulsion polymers, 1997 Search PubMed.
- B. Brooks, Chem. Eng. Technol., 2010, 33, 1737–1744 CrossRef CAS.
- J.-C. Daniel and C. Pichot, Les latex synthétiques: élaboration et applications, Tec&Doc-Lavoisier, 2006 Search PubMed.
- N. M. B. Smeets, M. E-Rramdani, R. C. F. van Hal, S. G. Santana, K. Quéléver, J. Meuldijk, A. M. Van Herk and J. P. A. Heuts, Soft Matter, 2010, 6, 2392 RSC.
- J. M. Asua, Prog. Polym. Sci., 2014, 39, 1797–1826 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Mater. Today, 2013, 16, 337–343 CrossRef CAS.
- P. F. H. Harmsen, M. M. Hackmann and H. L. Bos, Biofuels, Bioprod. Biorefin., 2014, 8, 306–324 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893 RSC.
- C. Zhang, T. F. Garrison, S. A. Madbouly and M. R. Kessler, Prog. Polym. Sci., 2017, 71, 91–143 CrossRef CAS.
- Qurat-ul-Ain, K. M. Zia, F. Zia, M. Ali, S. Rehman and M. Zuber, Int. J. Biol. Macromol., 2016, 93, 1057–1068 CrossRef CAS PubMed.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788 RSC.
- U. S. D. of A. O. World and M. and Trade, United States Department of Agriculture: Oilseeds: World Markets and Trade, https://apps.fas.usda.gov/psdonline/app/index.html#/app/downloads.
- Z. Demchuk, O. Shevchuk, I. Tarnavchyk, V. Kirianchuk, M. Lorenson, A. Kohut, S. Voronov and A. Voronov, ACS Omega, 2016, 1, 1374–1382 CrossRef CAS.
- S. F. Thames, O. W. Smith, J. M. Evans, S. Dutta and L. Chen, US Pat., 2005/0203246A1, 2005 Search PubMed.
- E. Sharmin, F. Zafar, D. Akram, M. Alam and S. Ahmad, Ind. Crops Prod., 2015, 76, 215–229 CrossRef CAS.
- R. Vendamme, N. Schüwer and W. Eevers, J. Appl. Polym. Sci., 2014, 131, 40669 CrossRef.
- M. A. Mosiewicki and M. I. Aranguren, Polym. Int., 2016, 65, 28–38 CrossRef CAS.
- F. Seniha Güner, Y. Yaǧci and A. Tuncer Erciyes, Prog. Polym. Sci., 2006, 31, 633–670 CrossRef.
- T. Nabuurs, R. A. Baijards and A. L. German, Prog. Org. Coat., 1996, 27, 163–172 CrossRef CAS.
- G. Booth, D. E. Delatte and S. F. Thames, Ind. Crops Prod., 2007, 25, 257–265 CrossRef CAS.
- P. Uschanov, N. Heiskanen, P. Mononen, S. L. Maunu and S. Koskimies, Prog. Org. Coat., 2008, 63, 92–99 CrossRef CAS.
- A. Matsumoto, N. Murakami, H. Aota, J. Ikeda and I. Capek, Polymer, 1999, 40, 5687–5690 CrossRef CAS.
- F. Boscán, M. Paulis and M. J. Barandiaran, Eur. Polym. J., 2017, 93, 44–52 CrossRef.
- J. Guo and F. J. Schork, Macromol. React. Eng., 2008, 2, 265–276 CrossRef CAS.
- C. Quintero, S. K. Mendon, O. W. Smith and S. F. Thames, Prog. Org. Coat., 2006, 57, 195–201 CrossRef CAS.
- S. T. Wang, F. J. Schork, G. W. Poehlein and J. W. Gooch, J. Appl. Polym. Sci., 1996, 60, 2069–2076 CrossRef CAS.
- X. Q. Wu, F. J. Schork and J. W. Gooch, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4159–4168 CrossRef CAS.
- J. P. Bigon, F. E. Montoro and L. M. F. Lona, Eur. J. Lipid Sci. Technol., 2018, 120, 1700130 CrossRef.
- S. P. Bunker and R. P. Wool, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 451–458 CrossRef CAS.
- J. La Scala and R. P. Wool, J. Am. Oil Chem. Soc., 2002, 79, 59–63 CrossRef CAS.
- R. P. Wool and S. P. Bunker, US Pat., 6646033B2, 2003 Search PubMed.
- A. T. Jensen, C. Sayer, P. H. H. Araujo and F. Machado, Eur. J. Lipid Sci. Technol., 2014, 116, 37–43 CrossRef CAS.
- G. R. Ferreira, J. R. Braquehais, W. N. da Silva and F. Machado, Ind. Crops Prod., 2015, 65, 14–20 CrossRef CAS.
- K. L. Diamond, R. B. Pandey and S. F. Thames, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 1164–1172 CrossRef CAS.
- S. F. Thames, K. G. Panjnani and O. S. Fruchey, US Pat., 6001913, 1999 Search PubMed.
- D. Delatte, E. Kaya, L. G. Kolibal, S. K. Mendon, J. W. Rawlins and S. F. Thames, J. Appl. Polym. Sci., 2014, 131, 40249 CrossRef.
- S. F. Thames, J. W. Rawlins, S. K. Mendon and D. Delatte, US Pat., 2009/0143527A1, 2009 Search PubMed.
- I. Tarnavchyk, A. Popadyuk, N. Popadyuk and A. Voronov, ACS Sustainable Chem. Eng., 2015, 3, 1618–1622 CrossRef CAS.
- Z. Demchuk, O. Shevchuk, I. Tarnavchyk, V. Kirianchuk, A. Kohut, S. Voronov and A. Voronov, ACS Sustainable Chem. Eng., 2016, 4, 6974–6980 CrossRef CAS.
- K. Kingsley, O. Shevchuk, Z. Demchuk, S. Voronov and A. Voronov, Ind. Crops Prod., 2017, 109, 274–280 CrossRef CAS.
- S. Roberge and M. A. Dubé, Int. J. Adhes. Adhes., 2016, 70, 17–25 CrossRef CAS.
- A. Badía, J. Movellan, M. J. Barandiaran and J. R. Leiza, Ind. Eng. Chem. Res., 2018, 57, 14509–14516 CrossRef.
- S. Bunker, C. Staller, N. Willenbacher and R. Wool, Int. J. Adhes. Adhes., 2003, 23, 29–38 CrossRef CAS.
- S. F. Thames, J. W. Rawlins and S. K. Mendon, in Miniemulsion Polymerization Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, vol. 57, pp. 139–171 Search PubMed.
- A. M. M. S. Medeiros, F. Machado, J. C. Rubim and T. F. L. McKenna, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1422–1432 CrossRef CAS.
- L. S. Laurentino, A. M. M. S. Medeiros, F. Machado, C. Costa, P. H. H. Araújo and C. Sayer, Chem. Eng. Res. Des., 2018, 137, 213–220 CrossRef CAS.
- W. Maassen, S. Oelmann, D. Peter, W. Oswald, N. Willenbacher and M. A. R. Meier, Macromol. Chem. Phys., 2015, 216, 1609–1618 CrossRef CAS.
- W. Massen, M. A. R. Meier and N. Willenbacher, Int. J. Adhes. Adhes., 2016, 64, 65–71 CrossRef.
- Z. Demchuk, A. Kohut, S. Voronov and A. Voronov, ACS Sustainable Chem. Eng., 2018, 6, 2780–2786 CrossRef CAS.
- M. Moreno, M. Goikoetxea and M. J. Barandiaran, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4628–4637 CrossRef CAS.
- M. Moreno, J. I. Miranda, M. Goikoetxea and M. J. Barandiaran, Prog. Org. Coat., 2014, 77, 1709–1714 CrossRef CAS.
- J. J. La Scala, J. M. Sands, J. A. Orlicki, E. J. Robinette and G. R. Palmese, Polymer, 2004, 45, 7729–7737 CrossRef CAS.
- M. Moreno, M. Goikoetxea and M. J. Barandiaran, Macromol. React. Eng., 2014, 8, 434–441 CrossRef CAS.
- L. I. Ronco, R. J. Minari and L. M. Gugliotta, Braz. J. Chem. Eng., 2015, 32, 191–200 CrossRef CAS.
- M. Moreno, M. Goikoetxea, J. C. de la Cal and M. J. Barandiaran, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3543–3549 CAS.
- J. T. Trotta, M. Jin, K. J. Stawiasz, Q. Michaudel, W.-L. Chen and B. P. Fors, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2730–2737 CrossRef CAS.
- M. K. Akkapeddi, Macromolecules, 1979, 12, 546–551 CrossRef CAS.
- M. Moreno, C. Lampard, N. Williams, E. Lago, S. Emmett, M. Goikoetxea and M. J. Barandiaran, Prog. Org. Coat., 2015, 81, 101–106 CrossRef CAS.
- T. O. Machado, P. B. Cardoso, P. E. Feuser, C. Sayer and P. H. H. Araújo, Colloids Surf., B, 2017, 159, 509–517 CrossRef CAS PubMed.
- P. B. Cardoso, T. O. Machado, P. E. Feuser, C. Sayer, M. A. R. Meier and P. H. H. Araújo, Eur. J. Lipid Sci. Technol., 2018, 120, 1700212 CrossRef.
- M. Black, J. Messman and J. Rawlins, J. Appl. Polym. Sci., 2011, 120, 1390–1396 CrossRef CAS.
- J. G. Tsavalas, Y. Luo and F. J. Schork, J. Appl. Polym. Sci., 2003, 87, 1825–1836 CrossRef CAS.
- L. Hudda, J. G. Tsavalas and F. J. Schork, Polymer, 2005, 46, 993–1001 CrossRef CAS.
- C. Costa, M. Wagner, A. Musyanovych, K. Landfester, C. Sayer and P. H. H. Araújo, Eur. J. Lipid Sci. Technol., 2016, 118, 93–103 CrossRef CAS.
- F. D. Gunstone, Eur. J. Lipid Sci. Technol., 2011, 113, 3–7 CrossRef CAS.
- A. E. Atabani, A. S. Silitonga, H. C. Ong, T. M. I. Mahlia, H. H. Masjuki, I. A. Badruddin and H. Fayaz, Renewable Sustainable Energy Rev., 2013, 18, 211–245 CrossRef CAS.
- V. Ladmiral, R. Jeannin, K. Fernandes Lizarazu, J. Lai-Kee-Him, P. Bron, P. Lacroix-Desmazes and S. Caillol, Eur. Polym. J., 2017, 93, 785–794 CrossRef CAS.
- C. Voirin, S. Caillol, N. V. Sadavarte, B. V. Tawade, B. Boutevin and P. P. Wadgaonkar, Polym. Chem., 2014, 5, 3142–3162 RSC.
- W. S. J. Li, C. Negrell, V. Ladmiral, J. Lai-Kee-Him, P. Bron, P. Lacroix-Desmazes, C. Joly-Duhamel and S. Caillol, Polym. Chem., 2018, 9, 2468–2477 RSC.
- S. Dworakowska, A. Cornille, D. Bogdał, B. Boutevin and S. Caillol, Eur. J. Lipid Sci. Technol., 2015, 117, 1893–1902 CrossRef CAS.
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408–414 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- N. I. Tracy, D. Chen, D. W. Crunkleton and G. L. Price, Fuel, 2009, 88, 2238–2240 CrossRef CAS.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed.
- S. Liu, L. Zhou, S. Yu, C. Xie, F. Liu and Z. Song, Biomass Bioenergy, 2013, 57, 238–242 CrossRef CAS.
- K. Satoh, A. Nakahara, K. Mukunoki, H. Sugiyama, H. Saito and M. Kamigaito, Polym. Chem., 2014, 5, 3222–3230 RSC.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- S.-S. Baek and S.-H. Hwang, Polym. Bull., 2016, 73, 687–701 CrossRef CAS.
- S.-S. Baek and S.-H. Hwang, Eur. Polym. J., 2017, 92, 97–104 CrossRef CAS.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072–1095 CrossRef CAS PubMed.
- P. Sarkar and A. K. Bhowmick, RSC Adv., 2014, 4, 61343–61354 RSC.
- P. Sarkar and A. K. Bhowmick, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2639–2649 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2016, 4, 5462–5474 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2016, 4, 2129–2141 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, Ind. Eng. Chem. Res., 2018, 57, 5197–5206 CrossRef CAS.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2018, 6, 6599–6611 CrossRef CAS.
- W. Lei, X. Yang, H. Qiao, D. Shi, R. Wang and L. Zhang, Eur. Polym. J., 2018, 106, 1–8 CrossRef CAS.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2017, 5, 7659–7669 CrossRef CAS.
- S. Loughmari, A. Hafid, A. Bouazza, A. El Bouadili, P. Zinck and M. Visseaux, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2898–2905 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- O. Y. Abdelaziz and C. P. Hulteberg, Waste Biomass Valorization, 2017, 8, 859–869 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843–1246843 CrossRef PubMed.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- E. Ten and W. Vermerris, J. Appl. Polym. Sci., 2015, 132, 42069 CrossRef.
- A. L. Holmberg, J. F. Stanzione, R. P. Wool and T. H. Epps, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- L. Rojo, B. Vazquez, J. Parra, A. López Bravo, S. Deb and J. San Roman, Biomacromolecules, 2006, 7, 2751–2761 CrossRef CAS PubMed.
- J. F. Stanzione, J. M. Sadler, J. J. La Scala and R. P. Wool, ChemSusChem, 2012, 5, 1291–1297 CrossRef CAS PubMed.
- J. Deng, B. Yang, C. Chen and J. Liang, ACS Sustainable Chem. Eng., 2015, 3, 599–605 CrossRef CAS.
- H. Zhang, X. Yong, J. Zhou, J. Deng and Y. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 2753–2763 CrossRef CAS PubMed.
- H. Zhang, J. Deng and Y. Wu, ACS Sustainable Chem. Eng., 2017, 5, 658–666 CrossRef CAS.
- P. Varanasi, P. Singh, M. Auer, P. D. Adams, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 14 CrossRef CAS PubMed.
- F. W. Lichtenthaler and S. Peters, C. R. Chim., 2004, 7, 65–90 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- H. Thomas, L. Tim and K. Andreas, Esterification of Polysaccharides, Springer-Verlag, Berlin/Heidelberg, 2006 Search PubMed.
- D. Brune and R. Von Eben-Worlée, US Pat., 6423775B1, 2002 Search PubMed.
- C. George, H. El Rassy and J. M. Chovelon, Int. J. Chem. Kinet., 2001, 33, 539–547 CrossRef CAS.
- P. Li, J. Zhu, P. Sunintaboon and F. W. Harris, Langmuir, 2002, 18, 8641–8646 CrossRef.
- P. Li, J. M. Zhun and F. W. Harris, US Pat., 2002/0143081A1, 2002 Search PubMed.
- W. Ye, M. F. Leung, J. Xin, T. L. Kwong, D. K. L. Lee and P. Li, Polymer, 2005, 46, 10538–10543 CrossRef CAS.
- J. H. Xin, P. Li and W. Ye, US Pat., 8349343B2, 2013 Search PubMed.
- M. Gaborieau, H. De bruyn, S. Mange, P. Castignolles, A. Brockmeyer and R. G. Gilbert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1836–1852 CrossRef CAS.
- D. Möller and K. Glittenberg, in TAPPI PaperCon symposium, 1990, pp. 85–91 Search PubMed.
- R.-M. Wang, X.-W. Wang, J.-F. Guo, Y.-F. He and M.-L. Jiang, Mater. Res., 2013, 16, 1246–1253 CrossRef CAS.
- K. M. Ho, W. Y. Li, C. H. Lee, C. H. Yam, R. G. Gilbert and P. Li, Polymer, 2010, 51, 3512–3519 CrossRef CAS.
- S. Cummings, M. Cunningham and M. A. Dubé, J. Appl. Polym. Sci., 2018, 135, 46485 CrossRef.
- N. M. B. Smeets, S. Imbrogno and S. Bloembergen, Carbohydr. Polym., 2017, 173, 233–252 CrossRef CAS PubMed.
- S. C. Abeylath and E. Turos, Carbohydr. Polym., 2007, 70, 32–37 CrossRef CAS PubMed.
- X. Sui, Z. Wang, B. Liao, Y. Zhang and Q. Guo, Bioresour. Technol., 2012, 103, 466–469 CrossRef CAS PubMed.
- P. Ray, T. Hughes, C. Smith, G. P. Simon and K. Saito, ACS Omega, 2018, 3, 2040–2048 CrossRef CAS.
- J. C. O. Villanova, E. Ayres and R. L. Oréfice, Carbohydr. Polym., 2015, 121, 372–381 CrossRef CAS PubMed.
- J. S. Desport, PhD thesis, University of the Basque Country, 2017.
- J. S. Desport, D. Mantione, M. Moreno, H. Sardón, M. J. Barandiaran and D. Mecerreyes, Carbohydr. Res., 2016, 432, 50–54 CrossRef CAS PubMed.
- J. Desport, M. Moreno and M. Barandiaran, Polymers, 2018, 10, 488 CrossRef.
- M. Neqal, A. Voisin, V. Neto, V. Coma and V. Héroguez, Eur. Polym. J., 2018, 105, 304–312 CrossRef CAS.
- V. Neto, A. Voisin, V. Héroguez, S. Grelier and V. Coma, J. Agric. Food Chem., 2012, 60, 10516–10522 CrossRef CAS PubMed.
- N. Pokeržnik and M. Krajnc, Eur. Polym. J., 2015, 68, 558–572 CrossRef.
- M. Guo and G. Wang, Polymers, 2016, 8, 324 CrossRef.
- M. L. Picchio, M. C. G. Passeggi, M. J. Barandiaran, L. M. Gugliotta and R. J. Minari, Prog. Org. Coat., 2015, 88, 8–16 CrossRef CAS.
- J. Ma, Q. Xu, J. Zhou, D. Gao, J. Zhang and L. Chen, Prog. Org. Coat., 2013, 76, 1346–1355 CrossRef CAS.
- Q. Xu, J. Ma, J. Zhou, Y. Wang and J. Zhang, Chem. Eng. J., 2013, 228, 281–289 CrossRef CAS.
- M. L. Picchio, R. J. Minari, V. D. G. Gonzalez, M. C. G. Passeggi, J. R. Vega, M. J. Barandiaran and L. M. Gugliotta, Macromol. Symp., 2014, 344, 76–85 CrossRef CAS.
- M. L. Picchio, S. J. Bohórquez, P. G. C. A. van den Berg, M. J. Barandiaran, L. M. Gugliotta and R. J. Minari, Ind. Eng. Chem. Res., 2016, 55, 10271–10277 CrossRef CAS.
- M. L. Picchio, M. C. G. Passeggi, M. J. Barandiaran, L. M. Gugliotta and R. J. Minari, Prog. Org. Coat., 2016, 101, 587–596 CrossRef CAS.
- M. A. Dubé and S. Salehpour, Macromol. React. Eng., 2014, 8, 7–28 CrossRef.
- C. V. Pramod, R. Fauziah, K. Seshan and J.-P. Lange, Catal. Sci. Technol., 2018, 8, 289–296 RSC.
- P. Venkitasubramanian, US Pat., 9234064B2, 2016 Search PubMed.
- J.-L. Dubois, C. Duquenne and W. Hölderich, WO, 2006/114506A1, 2006 Search PubMed.
- M. Shima and T. Takahashi, EP, 1710227A1, 2006 Search PubMed.
- J. C. Lansing, R. E. Murray and B. R. Moser, ACS Sustainable Chem. Eng., 2017, 5, 3132–3140 CrossRef CAS.
- J. Le Nôtre, S. C. M. Witte-van Dijk, J. van Haveren, E. L. Scott and J. P. M. Sanders, ChemSusChem, 2014, 7, 2712–2720 CrossRef PubMed.
- A. Kawamura, Y. Hata, T. Miyata and T. Uragami, Colloids Surf., B, 2012, 99, 74–81 CrossRef CAS PubMed.
- D. H. Turkenburg, Y. Durant and H. R. Fischer, Prog. Org. Coat., 2017, 111, 38–46 CrossRef CAS.
- E. M. S. van Hamersveld, J. J. G. S. van Es and F. P. Cuperus, Colloids Surf., A, 1999, 153, 285–296 CrossRef CAS.
- J. Hu, Z. Jin, T.-Y. Chen, J. D. Polley, M. F. Cunningham and R. A. Gross, Macromolecules, 2014, 47, 113–120 CrossRef CAS.
- C. Hazra, D. Arunbabu, D. Kundu, A. Chaudhari and T. Jana, J. Chem. Technol. Biotechnol., 2013, 88, 1551–1560 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |