
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKey Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited†
Marian C.
Bryan
 a,
Peter J.
Dunn‡
b,
David
Entwistle
c,
Fabrice
Gallou
d,
Stefan G.
Koenig
e,
John D.
Hayler
*f,
Matthew R.
Hickey
*g,
Shaun
Hughes
h,
Michael E.
Kopach
i,
Gerard
Moine‡
j,
Paul
Richardson
k,
Frank
Roschangar
l,
Alan
Steven
h and
Franz J.
Weiberth
m
a,
Peter J.
Dunn‡
b,
David
Entwistle
c,
Fabrice
Gallou
d,
Stefan G.
Koenig
e,
John D.
Hayler
*f,
Matthew R.
Hickey
*g,
Shaun
Hughes
h,
Michael E.
Kopach
i,
Gerard
Moine‡
j,
Paul
Richardson
k,
Frank
Roschangar
l,
Alan
Steven
h and
Franz J.
Weiberth
m
aGenentech, Inc, A Member of the Roche Group, 1 DNA Way, South San Francisco, California 94080, USA
bPfizer Global Supply, Ramsgate Road, Sandwich, UK
cCodexis, Inc., 200 Penobscot Drive, Redwood City, 94063, USA
dChemical & Analytical Development, Novartis Pharma AG, 4002 Basel, Switzerland
eSmall Molecule Process Chemistry, Genentech, Inc, A Member of the Roche Group, 1 DNA Way, South San Francisco, California 94080, USA
fAPI Chemistry, GlaxoSmithKline, Stevenage, Hertfordshire, SG1 2NY, UK. E-mail: John.Hayler@gsk.com
gBristol-Myers Squibb Co., One Squibb Drive, New Brunswick, New Jersey 08903, USA. E-mail: Matthew.Hickey@bms.com
hAstraZeneca, Macclesfield, SK10 2NA, UK
iSmall Molecule Design and Development, Eli Lilly and Company, Indianapolis, Indiana 46285, USA
jProcess Research and Synthesis, F. Hoffmann-La Roche Ltd., Grenzacherstrasse 124, CH-4070 Basel, Switzerland
kPfizer – Medicine Design, 10770 Science Center Drive, La Jolla, California 92121, USA
lChemical Development, Boehringer Ingelheim Pharmaceuticals, Ridgefield, Connecticut 06877, USA
mSynthesis Development, Sanofi US R&D, Waltham, Massachusetts 02451, USA
First published on 2nd November 2018
Abstract
In 2007 the members of the ACS Green Chemistry Institute® Pharmaceutical Roundtable assembled a list of key green chemistry research areas to both identify transformations that would benefit from improvements in process greenness and to encourage academic research to this end. The list provided the topics for a review of the literature, published in 2007 in the journal Green Chemistry. As part of the 10th anniversary of the founding of the Roundtable in 2015, the membership revisited the list. This article discusses the selection of the revised list, the updated 10 key green chemistry research areas, 2 solvent themes and the emerging area of medium molecules that resulted from the exercise.
Introduction
The ACS Green Chemistry Institute® Pharmaceutical Roundtable (GCIPR) was developed in 2005 to encourage the integration of green chemistry and green engineering into the pharmaceutical industry. One of the strategic priorities of the Roundtable is to inform and influence the research agenda and an early step was the publication of a paper on key green chemistry research areas (KRA) from a pharmaceutical manufacturers’ perspective.1 The publication was followed up with the ongoing GCIPR Research Grant Programme. For the 10th Anniversary of GCIPR, we decided to revisit the original KRA, the organization having 15 members† compared to six in 2006. The original six-member companies all participated in the exercise, although some company names have changed because of changes within the pharmaceutical industry. This paper discusses the selection and updated list of key green chemistry research areas, solvent themes and a brief commentary on the emerging area of medium molecules.Selection of the long list and vote
The original 2006 long list was augmented with areas of potential interest compiled by the UK pre-competitive research group, and suggestions from member companies. Each member company was asked to select their top 10 research areas and top 2 solvent themes, keeping in line with the selection made in 2006. The distinction between “current issues” and desired, enabling, transformations, the “aspirational reactions”, in 2006 was not made this time around. The long list and number of votes received is shown in the ESI.† The 10 Key Green Chemistry Research Areas and two Solvent Themes are shown below.Key Green Chemistry Research Areas
1. Development of effective and versatile methodology utilizing cheap/sustainable metals.*2. General methods for catalytic/sustainable (direct) amide or peptide formation.
3. Aliphatic and aromatic C–H activation, using green oxidants and giving predictable site selectivities.
4. Amide reductions avoiding LiAlH4 and diborane.
5. Direct substitution of alcohols.
6. Catalyst immobilization without significant loss in kinetics.*
7. Asymmetric hydrogenation of unfunctionalized olefins/enamines/imines.
8. Improved fluorination/trifluoromethoxylation.
9. Wittig chemistry without Ph3PO.‡
10. Alternatives for oxidations, C–O or C–N redox processes.
Solvent Themes
1. Viable replacements for polar aprotic solvents.2. Viable replacements for halogenated solvents.‡
* New research area, ‡ new research area, but on the 2006 long list.
Evolution, not revolution
It is interesting to make a comparison of the key research areas with the previous list published in 2007.1 Seven research areas have broadly survived, although in some cases have been more tightly defined, they are summarized in Table 1. Of note is the progression of the C–H activation research area from an “aspirational reaction” to a much more concise definition of the challenges in a rapidly expanding field.| 2006 research area | “Equivalent” research area |
|---|---|
| Amide Formation avoiding reagents with poor atom economy | General methods for catalytic/sustainable (direct) amide or peptide formation |
| OH activation for nucleophilic substitution | Direct substitution of alcohols |
| Reduction of amides without hydride reagents | Amide reductions avoiding LiAlH4 and diborane |
| Asymmetric hydrogenation of unfunctionalized olefins/enamines/imines | Asymmetric hydrogenation of unfunctionalized olefins/enamines/imines |
| New greener fluorination methods | Improved methods for fluorination and trifluoromethoxylation |
| C–H activation of aromatics | Aliphatic and aromatic C–H activation using green oxidants and giving predictable site selectivities |
| Replacements for dipolar aprotic solvents | Viable replacements for dipolar aprotic solvents |
Five areas on the list in 2006 failed to make the new list and these areas plus some explanation are given in Table 2.
| Research area | Reason for not being selected |
|---|---|
| Aldehyde or ketone + NH3 + “X” gives chiral amine | Substantial progress since 2006. Problem essentially solved |
| Solvent-less reactor cleaning | No application for research funding in this area received since 2007 |
| N-Centred chemistry avoiding azides, hydrazine etc. | Dropped down the vote. Just made the cut in 2006 and missed the cut by 1 vote |
| Oxidation/epoxidation methods without the use of chlorinated solvents | Broadened into two areas “Viable replacements for halogenated solvents” and “Alternatives for oxidations, C–O or C–N redox processes” |
| Safer and more environmentally friendly Mitsunobu reactions | Not offered as a choice, incorporated into direct substitution of alcohols |
The most satisfying omission is the first entry in Table 2. The 2006 research area was in the “aspirational” group, defined as “aldehyde or ketone plus ammonia plus “X” gives a chiral amine” where X (especially for the case of the ketone) was the source of the chirality. This problem has essentially been solved by the significant growth in transaminase enzymes over the past decade. These enzymes have become “X” in the equation with low cost amines such as diisopropylamine providing the source of ammonia. There are increasing numbers of both R-selective and S-selective transaminases which are commercially available and can be used in high throughput screening, many of the enzymes are process chemistry friendly and have been evolved using directed evolution.2 Examples of the use of such transaminases by process groups are the viable process to manufacture sitagliptin published by Merck3 and the Pfizer synthesis of a smoothened receptor inhibitor.4
Somewhat less satisfying is the omission of solvent-less reactor cleaning from the new list. As low volume specialty medicines replace high volume block-buster drugs (at least for the innovator companies) the percentage of solvent used in cleaning rather than in processing must be increasing. However, during the period 2006–2016, the Pharmaceutical Roundtable did not receive a single application for a research grant in this area. Hence it was the lack of interest from the scientific community rather than the lack of importance of this topic which caused it to drop from the list.
Interest in N-centred chemistry remained fairly constant. In 2006 this research area just made the cut whereas in the new list it “missed the cut” by a single vote. The other two topics not being reselected per se were due to changes in the description of both research areas as described in Table 2.
Of the five new areas, the two most popular were the development of effective and versatile methodology using cheap/sustainable metals (11 votes) and catalyst immobilization without significant loss of kinetics (8 votes) reflecting increasing awareness of elemental sustainability.5 Wittig chemistry (9) and halogenated solvents (2) were on the 2006 long-list.
Inclusion of “Preparation of non-small molecule APIs e.g. Oligonucleotides, antibody/drug conjugates, proteins” on the long list reflects the emergence of “medium molecule pharmaceuticals” over the last decade, it missed the cut by one vote and a discussion of this important and growing area is included below.
Key Green Chemistry Research Areas
Development of effective and versatile methodology utilizing cheap/sustainable metals
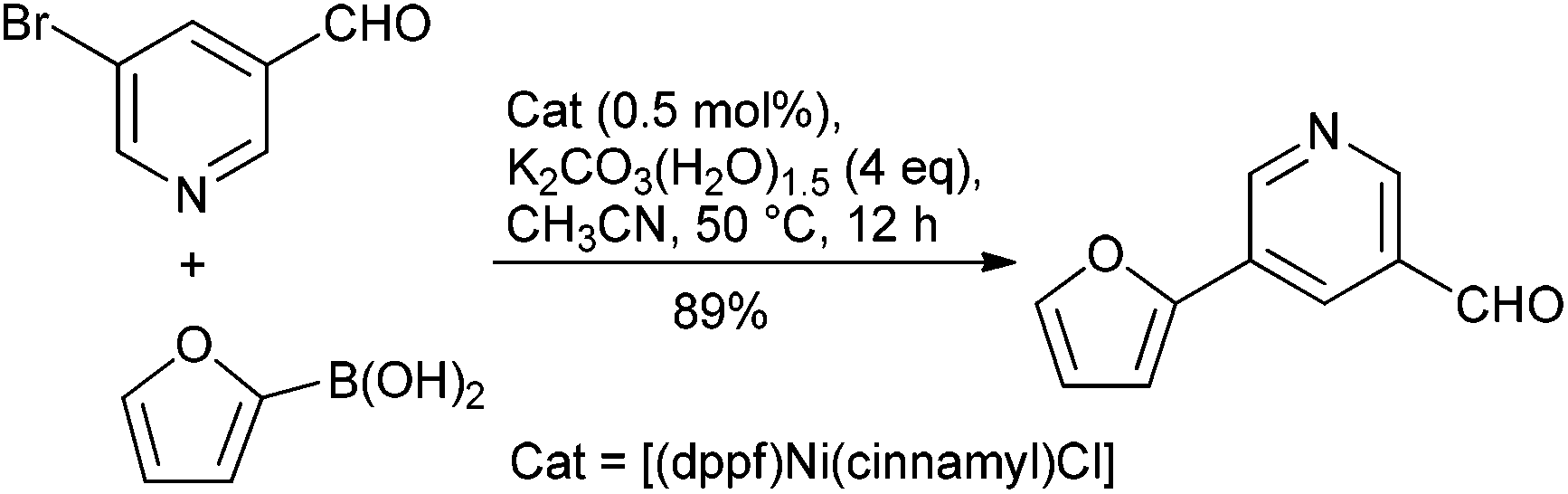
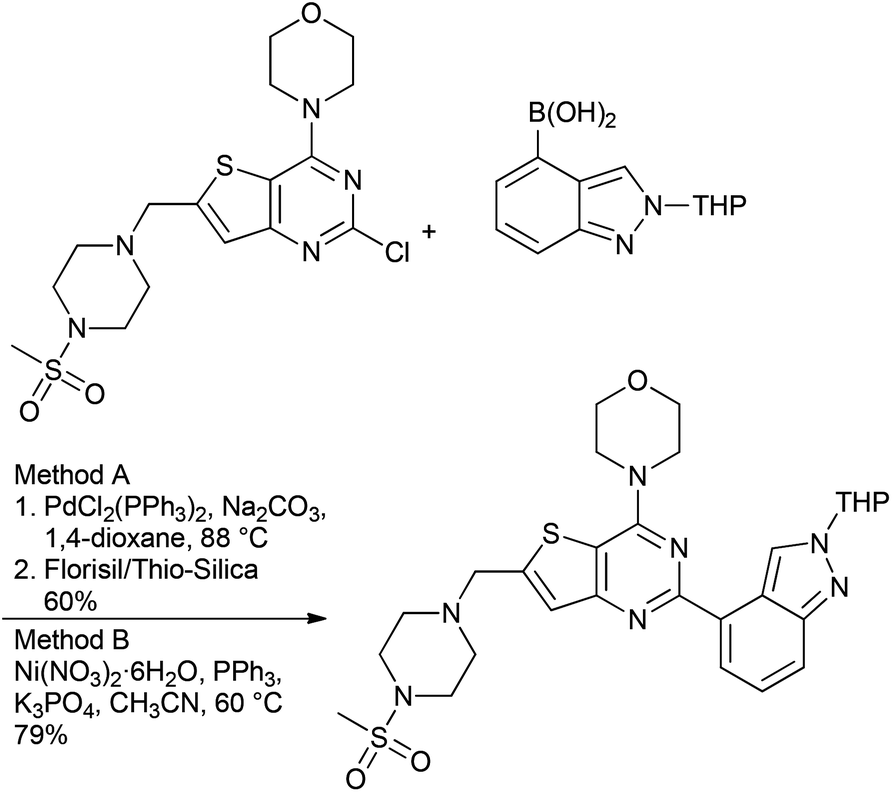
Catalysis is at the core of sustainable chemistry. Transition metal-catalysed cross-coupling reactions are widely used in the pharmaceutical industry in both medicinal chemistry and drug manufacturing.6 Many catalytic processes, however, rely on elements that are expensive, in short or fluctuating supply, potentially toxic or have a significant environmental footprint associated with their extraction and isolation.7 Although the use of palladium catalysis is most common,8 approaches involving non-precious metal catalysts might provide a complementary approach. Nickel, copper and iron catalysts are particularly appealing for several reasons: (a) Cost: Pd is much more expensive than Ni and without any comparison with Fe. (b) Toxicity: the limits for residual trace metals in orally administered drugs are more generous for Ni and Fe, compared to Pd (30 ppm Ni; 1300 ppm Fe; 10 ppm Pd).9 (c) Substrate scope: although Pd catalysis is versatile, new methods and catalyst systems may enable the coupling of important and challenging substrates, such as heterocycles and sterically-encumbered substrates. (d) New reactivity: Ni and Fe catalysts have proven uniquely effective in allowing for unconventional electrophilic coupling partners to be used in C–C and C–N bond forming reaction (e.g., pivalates, carbamates, sulfamates). (e) Green Chemistry opportunities: with growing concerns regarding the environment and sustainability, it is imperative to develop ‘greener’ methods which should translate to a dramatic decrease in the environmental impact. In this respect, the development of cobalt catalysis is a subject of interest owing to its greater earth abundance than Pd. However, this advantage is offset by the current short supply due to its use in batteries as well as the tighter ICH guidelines placed upon this metal because of its greater toxicity.9 In this respect, moving away from transition metals like Pd to non-precious metal catalysts like Ni and Fe combined with the fundamental concepts of green chemistry could bring potential utility from an industrial point of view (Table 3).Of the potential alternatives, studies into utilizing nickel in cross-couplings are perhaps the most advanced. The ability of nickel to mediate such reactions has been long established, and applications of nickel in homogeneous catalysis have been reviewed.14 However, couplings disclosed using nickel catalysts to date have suffered from high catalyst loadings (3–10%), and of more concern from a pharmaceutical perspective, limitations in terms of scope particularly with reactions leading to biologically relevant hetero-biaryl products. However, several promising systems have been developed with Ge and Hartwig reporting a single-component catalyst system, which enables such couplings under mild conditions using low loadings of the catalyst system (0.5 mol%) (Scheme 1).15 With the view to using more cost effective nickel-based catalysts that enable these reactions to take place under more environmentally friendly conditions, Garg and co-workers have reported on the use of the commercially available Ni(Cl)2(PCy3)2 as a pre-catalyst to mediate the Suzuki-coupling.16 The model reaction between a naphthyl sulfamate and phenylboronic acid using potassium phosphate as the base with 5 mol% catalyst was examined in >30 solvents to potentially identify greener alternatives, with over half the solvents providing a quantitative yield, and tert-amyl alcohol and 2-MeTHF being selected for further studies. Furthermore, Abe and co-workers have developed a NiCl2/morpholine based system for the coupling of a wide range of aryl and heteroaryl chlorides with aryl- and alkenyl-boronic acids.17 Although relatively high Ni loading is required (10 mol%), the amounts of residual Ni in the final products were shown to be <9.7 ppm.
 | ||
| Scheme 1 Single component Ni-based system. | ||
To illustrate the industrial applicability of this methodology, Tian and co-workers at Genentech provided a direct comparison of both a Ni and Pd-mediated Suzuki reaction for the scale-up of the Pi3K inhibitor, GDC-0941.18 In addition to a 19% improvement in yield compared to the optimized Pd-catalysed protocol, they also demonstrated that the Ni-catalyst could be easily removed via an aqueous ammonia wash whereas the Pd-methodology required the utilization of scavenger resins. The Ni-reaction was successfully performed to provide 54 kg of material (Scheme 2).
 | ||
| Scheme 2 Ni-Catalysed Suzuki-coupling for GDC-0941. | ||
For other base metals, examples of iron,19 cobalt,20 and copper-mediated21 coupling reactions have been reported, though these areas of research remain in their infancy. In some cases, there are concerns regarding potential trace contaminants in the metals under evaluation being the actual active catalyst,22 and several studies have demonstrated that mechanistically, coupling reactions mediated by base metal catalysts may be more complex than they initially appear.23 With this in mind, for the foreseeable future, developments in base-metal catalysis should occur in parallel with the increased efficiency in both use and recovery (such as catalyst immobilization) in the area of precious metal catalysis.
General methods for catalytic/sustainable (direct) amide or peptide formation
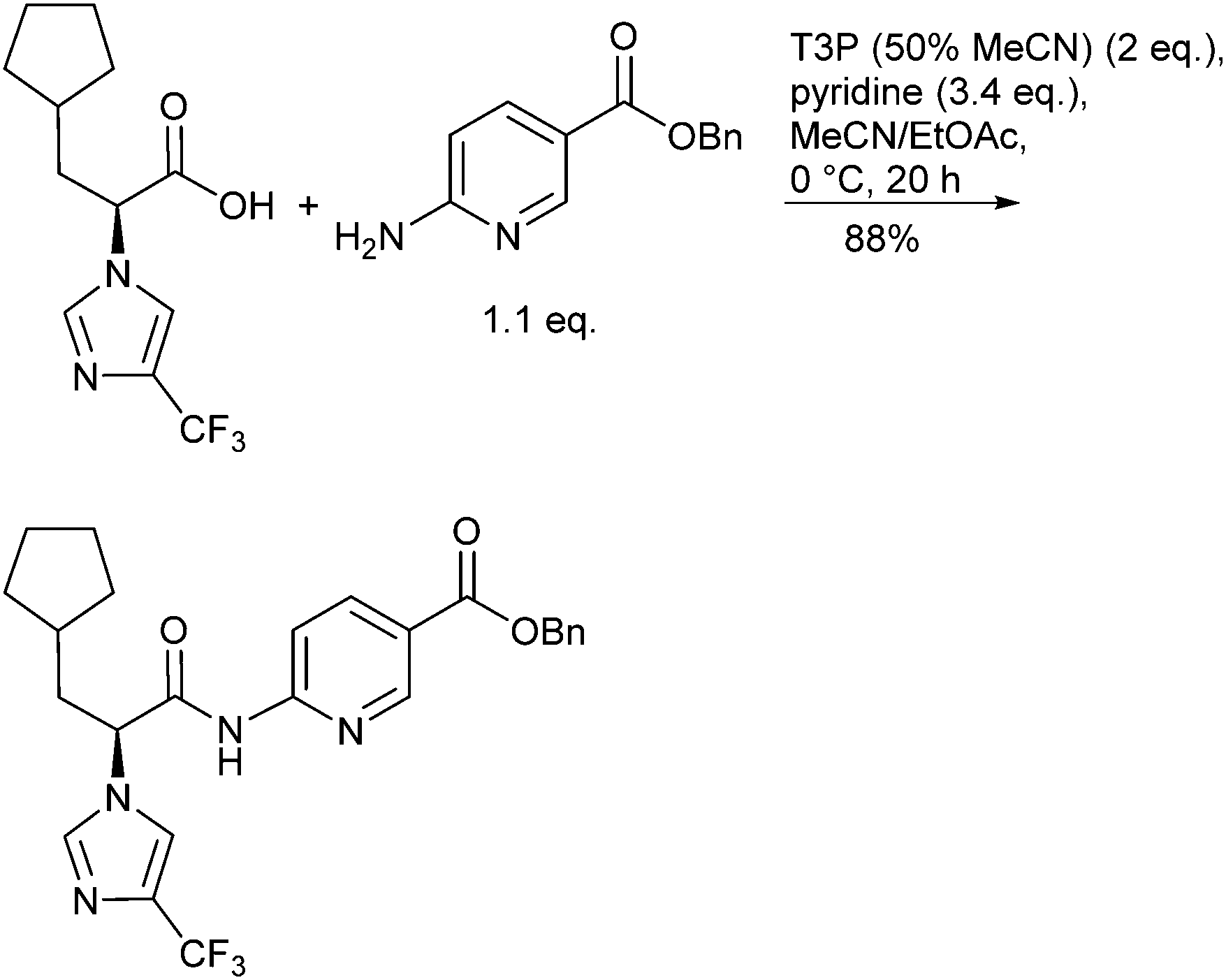
The green attributes desirable for efficiently forming amide bonds include performing these transformations catalytically or with improved atom efficiency, using more environmentally friendly solvents, and robustness on scale including reducing waste streams and simplifying/intensifying processes. The frequency and diversity of review articles reflect both the importance and the variety of synthetic methods available for generating amide bonds. For example, Allen and Williams24 reviewed metal-catalysed approaches to amides from acids, esters, aldehydes, alcohols, nitriles and oximes, along with aminocarbonylation methodology and the N-arylation of amides. Taylor and Bull25 surveyed the N-acylation of amines by common protocols including acid halides, mixed anhydrides, carbodiimide-mediated couplings, macrolactamization and N-acylation as a means for kinetic resolution, to name just a few. Valeur and Bradley26 reviewed pros and cons of coupling reagents, including polymer supported and other immobilized reagents, aimed at providing chemists criteria for selecting reagents to carry out their desired amidations. Adolfsson et al.27 reviewed catalytic methods for direct coupling of non-activated carboxylic acids with amines. Joullié and Lassen28 reviewed the evolution of amide bond formation with emphasis on peptide-bond formation. Lukasik and Wagner-Wysiecka29 discussed amide-bond formation using microwave techniques. Pattabiraman and Bode30 summarized recent novel approaches to forming amides, including the use of amine surrogates such as isonitriles, oxidative amidation of amines, transamidation processes and chemoselective ligations as applied to the synthesis of proteins. The Green Chemistry Institute (GCI) Pharmaceutical Roundtable periodically publish articles of interest that highlight green chemistry and green engineering principles including examples pertaining to amide bond formation.31 Many amide forming methodologies utilize unfavourable polar aprotic solvents; Lipshutz has challenged organic chemists to “Make the Switch”32 from chemistry that is highly dependent on organic solvents to a more sustainable one that is based on water as the reaction medium including forming amide bonds under aqueous micellar conditions.33 Watson et al.34 conducted a systematic screening of preferred solvents for amide bond formation involving HATU, COMU, DIC/HOBt, pyBOP and T3P protocols with a variety of carboxylic acids and amines. Dimethyl carbonate (DMC), EtOAc and 2-MeTHF were found to be practical replacements for the more commonly used but less environmentally friendly solvents DCM and DMF.Examples of efficient amidations performed on medium and multi-kilogram scale have been described. Dunetz35et al. reported on a screening of a variety of reagents for the coupling of a racemization-prone acid substrate with a poorly nucleophilic aminonicotinate (Scheme 3). Best success was achieved using T3P (n-propanephosphonic acid anhydride, 2 equiv.), pyridine as base and stirring for 20 h at 0 °C in ACN/EtOAc. Isolation was facilitated because the product precipitated as the reaction proceeded and the by-products from T3P were water soluble easing separation, although the aqueous waste stream would require treatment to remove phosphorus residues before discharge to avoid eutrophication. Thus, after quenching with 0.5 M aq. HCl, aging for 16 h at 20 °C and filtration, 34 kg of the chiral amide was obtained in 88% yield and 99% ee. Weiberth et al.36 have demonstrated direct amidation of an unactivated methyl ester with a benzylamine on pilot-plant scale using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as the catalyst.37 The optimum reaction, identified after a DoE study, employed a slight excess of the ester (1.06 equiv. relative to amine) with 0.28 equiv. of TBD. Dunetz et al.38 have recently thoroughly surveyed amide coupling reagents employed in the large-scale syntheses of pharmaceuticals. The most commonly used reagents for acid activation were found to be EDC, SOCl2, CDI, and (COCl)2, followed by PivCl, IBCF, T3P, and DCC.
 | ||
| Scheme 3 | ||
Ritter transformations39 convert nitriles and carbocation precursors (often alcohols or alkenes) to amides. Although useful, the reactions generate large amounts of acid waste under standard conditions. Anxionnat et al. describe a Ritter reaction catalysed by FeCl3·6H2O to prepare amides from a variety of substituted benzylic alcohols and nitriles and t-butyl acetate and nitriles in 40–96% yield.40 A microwave-assisted, Ca(II)-catalysed Ritter reaction under solvent-free conditions (but in presence of 2 equiv. water and using 2 equiv. nitrile) has also been reported.41 Ritter reactions have been performed in flow mode.42
Nature has been using enzymes to create amide bonds efficiently and sustainably for eons.43 Although biocatalytic processes are used on commercial scale to produce some specialty amides from nitriles with nitrile hydratases, penicillins and cephalosporins with penicillin G and cephalosporin acylases and from esters with lipases, including nicotinamide, acrylamide, and 5-cycanovaleramide to name a few,44,45 industrial application has generally been lacking.45 This is due in part because specialized technologies are required to develop scalable biocatalytic processes. An enzyme is selective and interacts with a substrate 3-dimensionally to align its active site with the reactive site of the substrate. This orientation allows an enzymatic transformation to occur with a specific substrate, but is restrictive for substrates that might bind and align less precisely. Thus, identifying or developing a suitable enzyme for a substrate could be a daunting task, especially for organizations that have limited expertise and resources in biocatalysis. Fortunately, there are contract research organizations that specialize in biocatalysis. These CROs have high-throughput screening libraries of naturally-occurring and mutant enzymes, specialize in molecular engineering technologies for modeling, modification, and directed evolution of engineered enzymes to optimize biocatalytic performance with a substrate,46 and have facilities to manufacture enzymes on an industrial scale. It can be anticipated that the future for the industrial preparation of amides using biocatalysts is bright as the culture for greener chemistry grows and as the access to, and speed of, bioengineering of enzymes increases, making routine application during route and process selection feasible.
In summary, amide bond formation is one of the most important and frequently performed transformations in organic synthesis. Progress is being made towards making this transformation more scalable and efficient, most notably via catalytic methods that can be expected to more frequently replace stoichiometric protocols in due time upon further development and demonstration of scalability. In addition, from a more global perspective, the increasing variety and pool of new synthetic methods allowing wider tolerability of functional groups in substrates together with advancements in biocatalysis and enzyme engineering provide diversity in strategies to concisely and efficiently assemble target molecules containing amide bonds.
Aliphatic and aromatic C–H activation, using green oxidants and giving predictable site selectivities
Transition metal-catalysed selective C–H functionalization is the conversion of an inert C(sp2/sp3)–H into a C–R bond, with R being any functional group. It represents an opportunity for green pharmaceutical manufacturing as it constitutes an ideal transformation in terms of atom economy and waste minimization when compared to classic catalytic cross-coupling reactions. Metal-free catalytic C–H bond activation has also been described.47 Overall, the methodology avoids the long and tedious functional group manipulations that often include lengthy and wasteful protection–deprotection sequences. Thus, it is not surprising that C–H activation strategies have received growing attention from the chemistry community over the past decade with more than 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 total publications.
000 total publications.
Mechanistically, C–H functionalization can proceed through manipulation of an aromatic or aliphatic C–H bond via (1) radical rebound mechanism (H atom abstraction followed by radical functionalization), (2) direct C–H insertion by means of metal carbenes and nitrenes, or most commonly (3) organometallic C–H activation and a C–M intermediate by way of s-bond metathesis, concerted metalation deprotonation, and oxidative addition. The C–M intermediate is typically treated with an oxidant to form the C-FG product via reductive elimination, and commonly used metals include Pd, Rh, Ir, Pt, Ru, Ni, Fe, Cu, and Co.
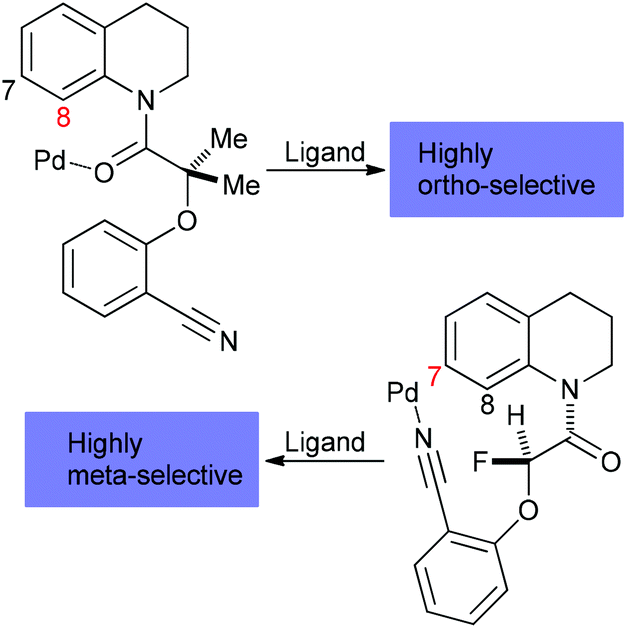
Currently, we face substantial hurdles with organometallic C–H bond functionalization reactions. First, besides high activation energy requirements for C–H bond breakage, it is difficult to control the regioselectivity due to the omnipresence of C–H moieties in organic molecules. In response to solving this problem, the concept of proximate cleavable metal-directing groups was introduced48 and elaborated49 to allow for high ortho-selectivity. Alternatively, removable, modifiable or traceless directing groups derived from carbonyls, amines, nitriles, and phenols have been employed.50 Expanding the scope to distal C–H activation, ligand-enabled methods to achieve aromatic meta C–H selectivity were developed,51 which did not require traceless directing groups and allowed for remote activation up to 11 bonds away from the directing group in the meta-position of a N-heterocycle via a “Pd crane” tactic (Scheme 4).52 Regioselective aromatic para C–H activation has not yet been attained, but aliphatic gamma, delta, and epsilon-C(sp3)–H functionalization could be accomplished through directed radical H abstraction.53
 | ||
| Scheme 4 | ||
A second main challenge of organometallic C–H bond activation lies within the catalytic cycle and its redox versatility, resulting in the general requirement for stoichiometric oxidants (e.g., MnO2, hypervalent iodine reagents), which in the case of common external additives render the transformations less attractive in terms of green chemistry, unless air can be used.54 Use of arguably less green hydroxylamine derivatives as both reactant and internal oxidant can obviate the need for external oxidants.55 Redox-neutral C–H activation strategies, in which the metal stays in the same oxidation state throughout, do alleviate this drawback.56 A milestone was nevertheless published in 2014 by Hartwig.57 In this seminal work, Hartwig developed a highly selective C–H iridium-mediated borylation of various heteroarenes, such as pyridines, pyrimidines, imidazoles, thiazoles, benzimidazoles, azaindoles etc., thus set for immediate functionalization via standard cross-coupling methodologies for example, (Scheme 5). Distal control is enabled owing to the rapidly occurring Heteroatom-H borylation that creates an unfavourable steric environment near the formed complex. Rules for predicting the regioselectivity of borylation are described. This methodology has proven to be extremely powerful and already found applications within the pharmaceutical industry.58 This work was followed by further extension.59
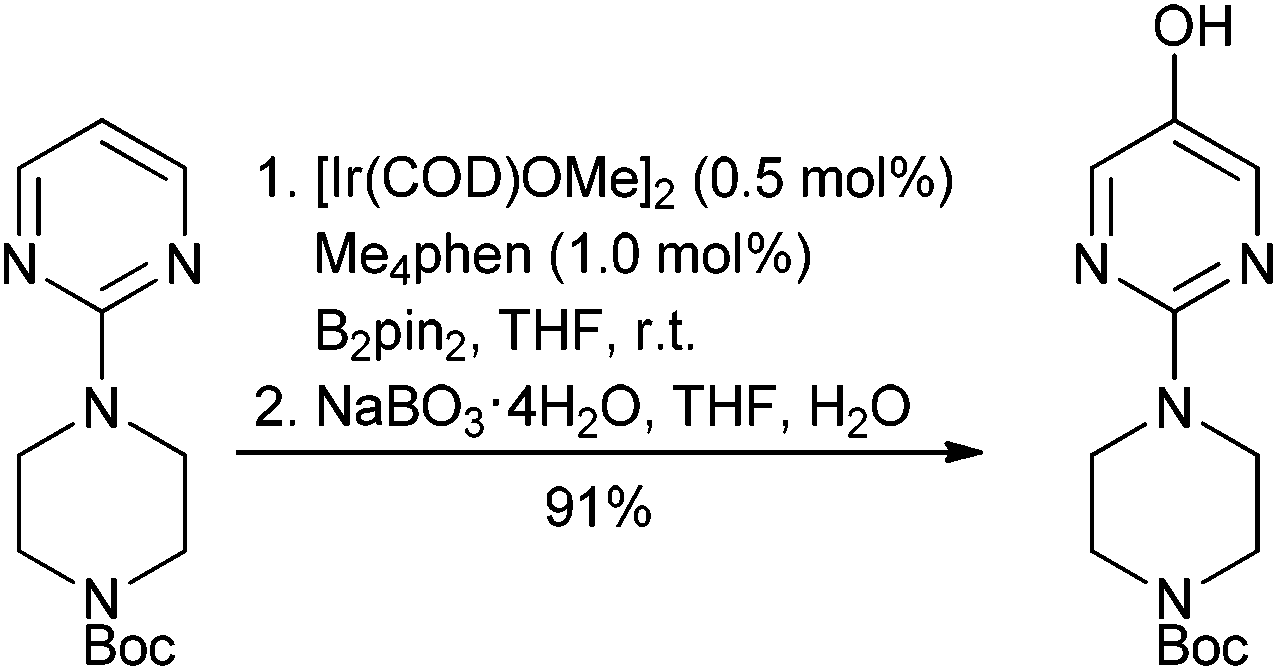 | ||
| Scheme 5 | ||
The same investigator reported a remarkable rhodium-catalysed intermolecular C–H silylation of arenes with high steric regiocontrol. The catalytic intermolecular C–H silylation occurs on unactivated arenes with very high regioselectivity through steric effects of substituents meta to a potential site of reactivity, and from the steric properties of both the rhodium catalyst and the silane.60
The enantioselectivity of C–H activation chemistry is a third key challenge. While progress has been made in terms of reactivity and enantioselectivity,61 substrate scope remains limited.
In summary, despite the recent advances, C–H functionalization is still limited in scope, selectivity, and its requirement for external oxidants. Collaborative efforts between industry, academia, and government, such as the ‘Center for Selective C–H Functionalization’ (CCHF),62 will play a key role in advancing the science and rendering C–H activation universally useful and practical.
Amide reductions avoiding LiAlH4 and diborane
In the 2007 perspective, the ‘reduction of amides without hydride reagents’ was ranked the third most desirable area for research,1 and a previous pharmaceutical industry transformation survey revealed that reduction of an amide to amine was reported as only 0.6% of the total reaction count.63 The scope of this research area has changed slightly to ‘amide reductions avoiding LiAlH4 and diborane’ and this category ranked fourth in the areas of interest. For the established reagents, such as LiAlH4, diborane complexes, DIBAL and Red-Al the issues of safety, atom efficiency and waste disposal are still major concerns and barriers to adoption.64 Given the wealth of amide syntheses and ubiquity of amines in drug substances, efficient and sustainable amide reduction is still very much an underused methodology in the pharmaceutical industry.Of the 534 citations of the 2007 perspective found at the time of writing this article, only 9 addressed amide reduction in any form. This however underestimates the activity in the area as a separate literature search revealed 766 tertiary amide reductions (across 142 articles) not using LiAlH4 or borane complexes from 2007 to March 2016.
The subtle change in scope from the 2007 perspective's ‘reduction of amides without hydride reagents’ to the current ‘amide reductions avoiding LiAlH4 and diborane’ introduces the use of less reactive hydrides such as the silanes and siloxanes. In fact, the majority of the relevant 766 tertiary amide reductions from 2007 to 2016 were silane/siloxane mediated (645 reactions over 110 articles). Initial reports focused on ruthenium based catalysts but there have been significant advances made in the catalytic reductive hydrosilylation of amide bonds, especially with non-precious Lewis acid catalysts based on iron,65 zinc,66,67 molybdenum,68 indium69 and non-metals such as boron.70 It should be noted that a specific potential safety concern exists with triethylsiloxane and Lewis acid catalysts, the potential for pyrophoric silane generation,71 but safer alternative silanes, siloxanes and disiloxanes have been reported such as methyldiethoxysilane,66 1,1,3,3-tetramethyldisioxane72 and polymethylhydrosiloxane (PMHS).67 One drawback of the disiloxanes is the potential to form intractable silicone gels as by-products making large scale application and clean up impractical. Table 4 gives a summary of the top 8 silanes and siloxanes used from 2007 onwards and leads to the conclusion that hydrosilylation, no matter how compelling the most recent developments are, will still generate silicon based waste which on a mass/mass basis could be worse than that created during the quenching of LiAlH4 based reactions and future research into improved atom efficiency or silane recycling would be welcomed.
| Silane | PHMS | PhSiH3 | Ph2SiH2/D2 | (Me2SiH)2O |
|---|---|---|---|---|
| Reactions | 122 | 116 | 95/5 | 44 |
| (articles) | (12) | (16) | (39) | (5) |
| Silane | Me2SiHPh | (EtO)3SiH | (iPr2Si)2NH | Et3SiH |
|---|---|---|---|---|
| Reactions | 41 | 33 | 25 | 22 |
| (articles) | (6) | (7) | (3) | (2) |
The natural progression to minimize the waste is to move to the more atom economical strategy of hydrogenation.64 Of the 766 in scope amide reduction reactions, 111 were hydrogenation based (over 26 articles) when monoselective imide reductions were removed. The majority of these were heterogeneous reactions that required high temperatures and pressures and are in general intolerant of a wide range of active functionality.73 Homogeneous catalysis has improved and for example ruthenium triphos complexes with acid additives have been successfully deployed with some improvements in substrate scope but still with high temperatures of 150 °C or more.74 More recently Yb(OTf)3·H2O has been used as a replacement for Brønsted acid additives which gave rise to a much wider substrate scope, but still under rather harsh conditions.75
Biocatalytic approaches for amide reduction have not noticeably progressed in the period between these two perspectives. The whole cell Clostridium sporogenes transformation76 reported in the 2007 perspective has been cited only 8 times since 2007, mainly in review, and no new articles for bioreduction of amides were found. As such in vitro biotransformation remains essentially an unexplored field.
In summary, there is still a considerable amount of development to be made in alternative methods before the use of stoichiometric hydride reagents can be fully laid to rest. Hydrosilylation approaches are relatively mild and have improved substrate scope, but are still inefficient in terms of atom economy and waste generation. Hydrogenation approaches have also greatly improved substrate scope compared to the state of the art in 2007, but the operating conditions are still very harsh potentially requiring specialized manufacturing plant.
Direct substitution of alcohols
The direct substitution of alcohols is an attractive prospect with the ideal process displacing a hydroxyl group resulting in water as the sole by-product. Such an approach offers increased efficiency and atom economy over the alternative stoichiometric activation and displacement protocol, and avoids the use of potentially genotoxic alkyl halides and sulfonate esters in alkylation reactions. Unfortunately, there are intrinsic thermodynamic and kinetic barriers to direct displacement rendering the methodology complex and therefore a robust general method is lacking. A survey carried out in 2005 of large scale GMP reactions found that 2% of all transformations were the conversion of alcohols to halides or sulfonate esters i.e. activation for SN2 displacement.77 Direct substitution can be broadly considered to follow three pathways: classical SN1 and SN2 reactions and redox hydrogen transfer approaches resulting in a net substitution. Good progress has been made in all three areas over the past decade.The scope of new methodologies for the SN1 reaction pathway are diverse with many examples of Brønsted and Lewis acid catalysed reactions resulting in direct displacement of allylic, benzylic and propargylic alcohols.78,79 The disadvantages of these methods are that in their majority they are substrate specific in regards to either the alcohol or the nucleophilic species. The use of Brønsted acid catalysed intramolecular SN2 substitution of enantio-enriched alcohols has been shown to proceed with some transfer of chirality.80 Catalyst or activator free direct alcohol substitution of aromatic allylic alcohol substrates with a wide range of amine and carbon nucleophiles has been demonstrated to take place in fluorinated alcohol solvents.81
Catalytic hydrogen auto-transfer, or hydrogen borrowing, where an alcohol is dehydrogenated, the resulting aldehyde or ketone condensed with an amine82 or carbon nucleophile83 and the unsaturated product reduced by the catalyst hydride, was remerging as an approach when the 2007 perspective was prepared.84 It has been applied to both primary and secondary alcohols, providing a complementary pathway to SN1 and SN2 reactions. Advances have been made with homogeneous, especially based on iridium and ruthenium, and heterogeneous catalysts with numerous metals.85 The use of homogeneous earth abundant metal catalysts (iron,86 cobalt87 and manganese88) is emerging. In general, the heterogeneous and earth abundant catalysts require more forcing conditions than their precious metal, homogeneous counterparts, as exemplified through the hydrogen borrowing synthesis of piribedil.89 Metal free, aldehyde catalysed hydrogen transfer reactions have been demonstrated.90
Organocatalytic nucleophilic substitution of alcohols has been demonstrated with two complementary approaches, initially for deoxychlorination by treatment with oxalyl chloride and either catalytic triphenylphosphine oxide (reacting via a chlorophosphonium intermediate)91 or with substituted cyclopropenones.92 Both approaches achieve nucleophilic substitution of secondary alcohols with inversion93 complementing the Mitsunobu reaction, an important sub-class of alcohol activation approaches that was highlighted as a separate research area in 2007. Progress has been made to improve the Mitsunobu reaction, with alternative reagents for both triphenylphosphine and diethyl azodicarboxylate (DEAD). Triphenyl bis(2,2,2-trifluoroethoxy)phosphorane, prepared from triphenylphosphine oxide enables a redox free Mitsunobu reaction avoiding the use of diazobased oxidants and with no net phosphorus waste as the triphenylphosphine oxide produced can be recovered from the reaction.94 The reagent was shown to be superior to conventional Mitsunobu conditions for the synthesis of a key intermediate in the synthesis of a GPR119 agonist.95 Hirose et al. demonstrate an iron phthalocyanine redox catalysed approach using air as the co-oxidant making the Mitsunobu reaction catalytic with respect to the azodicarboxylate oxidant. The DEAD reagent being replaced with phenyldiazocarboxylate that more readily undergoes oxidation by iron.96 Although the triphenylphosphine component remains stoichiometric, the improvements are important when considering the toxicity and processing risks associated with the alternative hydrazine by-product. Phospholane (1) has been used as a pre-catalyst making the Mitsunobu reaction catalytic in phosphine. Phenylsilane was used to regenerate the phosphine and 1.1 equiv. diisopropylazo-dicarboxylate (DIAD) used as the coupling partner. Furthermore, combination of this approach with a modification of the catalytic azo-carboxylate protocol described above enabled demonstration of a Mitsunobu reaction that is catalytic in both reagents, requiring a slight excess of phenylsilane.97 (Scheme 6) The reaction was run in boiling THF under an oxygen enriched atmosphere, which will require careful engineering controls to run safely on scale.
 | ||
| Scheme 6 | ||
While the Mitsunobu reaction and its variations generally give clean inversion, achieving enantioselective SN2 displacements; asymmetric SN1 and hydrogen transfer reactions remain a challenge. Enantioselective substitution of allylic alcohols, through iridium-catalysed amination and alkynylation has been demonstrated on a gram scale and effort was made during development with a view for industrial operation.98 Enantioselective, intermolecular, Brønsted acid catalysed, direct allylic amination has been demonstrated, the approach is based on the concept of chiral ion-pair catalysis and relies on the assumed formation of a contact ion-pair with a chiral phosphoramide, although the only applied to highly π-active aromatic allylic alcohols.99 The hydrogen borrowing mechanism removes chirality in a secondary alcohol substrate during the oxidation step, resulting a racemic product after reduction, however, asymmetric variants of the reaction are emerging with the use of a ruthenium catalyst and a chiral Josiphos ligand in the enantioselective preparation of β-aminoalcohols from racemic diols, via an asymmetric hydrogenation of an intermediate ketone.100 The use of Ellman's chiral sulfinamide auxiliary enabled ruthenium catalysed preparation of enantiomerically enhanced amines from racemic secondary alcohols101 and use of chiral phosphoric acids as an additive in combination with an iridium catalyst enabled preparation of chiral amines.102
Given the amount of progress made in all alcohol activation approaches it is surprising to find few published reports of scale-up of direct substitution approaches towards the preparation of pharmaceutically relevant substrates. Work carried out by AMRI to produce ICA-17043 exemplifies an InCl3 catalysed cyanation on a highly π-active tertiary alcohol conducted on approximately 100 kg scale103 (Scheme 7).
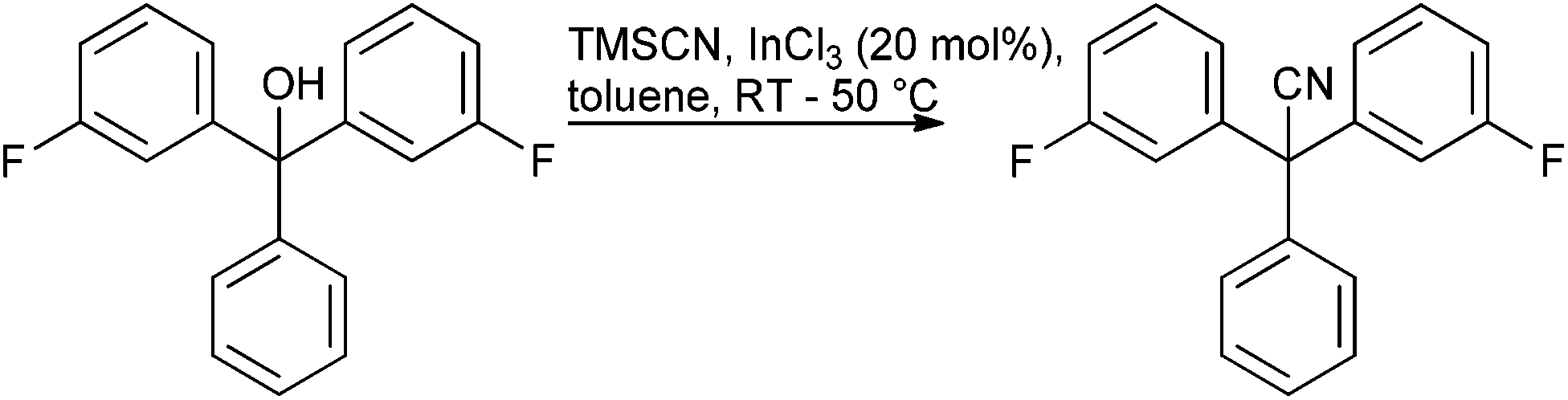 | ||
| Scheme 7 | ||
Berliner et al. at Pfizer utilized the hydrogen borrowing strategy to replace a sequential Swern oxidation and reductive amination procedure in the synthesis of 2 on 2.4 kg scale, an intermediate to a GlyT1 inhibitor104 (Scheme 8).
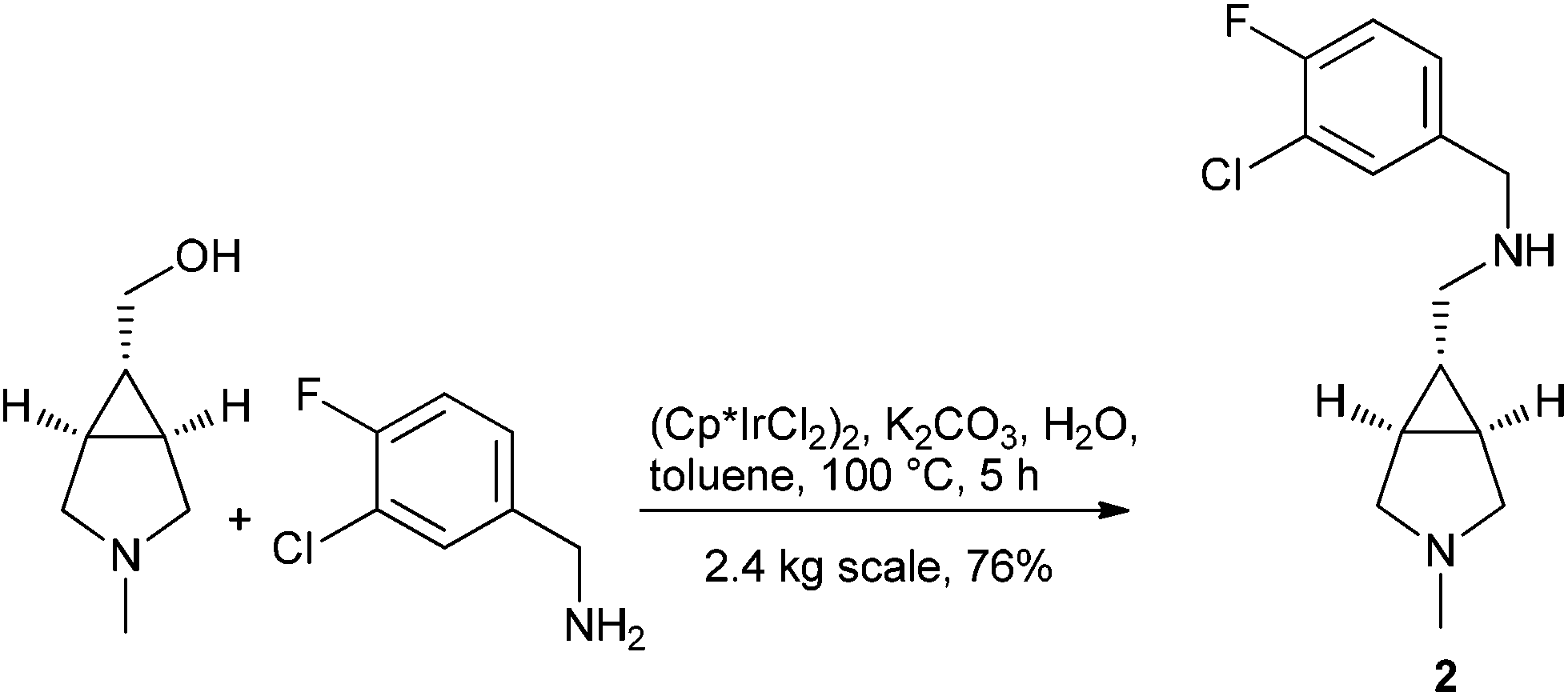 | ||
| Scheme 8 | ||
Leonard et al. have published an evaluation of the application of the borrowing hydrogen approach to pharmaceutical targets at AstraZeneca, discussing both successful application and the current limitations of the methodology. The authors’ highlight the often structural incompatibility of heteroatom-rich, complex polyfunctional pharmaceutical intermediates with transition metal catalysts and the need for more active and tolerant catalysts, capable of operating at low loadings in a broad range of solvents.105 Their comments confirm that despite substantial progress in developing chemo-catalytic approaches for the direct substitution of alcohols, the challenges of application to the synthesis of pharmaceutical intermediates provide numerous opportunities for continued research in this area.
The application of biocatalysis offers a metal free, stereoselective, approach to borrowing hydrogen through the combination of an alcohol dehydrogenase (ADH) to oxidise the alcohol and an amine dehydrogenase (AmDH), to convert the resulting carbonyl intermediate to an amine in a single cascade process. The aminolysis of primary and secondary alcohols to their corresponding amines has been reported with (R)-amines obtained from racemic secondary alcohols using a pair of complementary ADHs.106 The use of a non-selective ADH in the oxidation step, although with reduced substrate tolerance, was reported around the same time107 and a non-selective ADH with broader substrate scope was subsequently reported.108 The approach has been extended to the alkylation of amines to afford (R)-secondary amines using a reductive aminase (RedAm) in the final step (Scheme 9).109
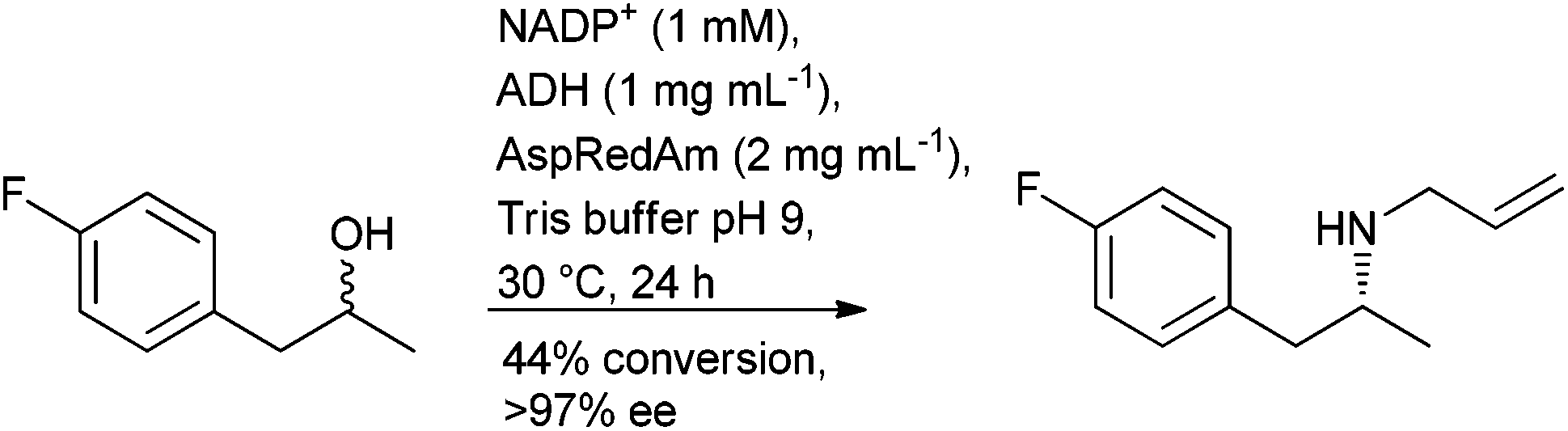 | ||
| Scheme 9 | ||
With the methods reported to-date affording the (R)-amine, and the ongoing progress in enzyme evolution, this could prove to be a fruitful area of research in the future.
Catalyst immobilization without significant loss in kinetics
Catalyst immobilization defined as “the transformation of a homogeneous catalyst into a heterogeneous one, which is able to be separated from the reaction mixture and preferably be reused for multiple times” continues to represent an intense area of academic interest.110 However, aside from the well-established supported catalysts utilized in hydrogenation (Pd/C, Rh/Al2O3etc.),111 and the emerging area of heterogeneously-catalysed alcohol oxidation,112 there has not been significant uptake within the pharmaceutical industry.113 At first inspection, this might seem surprising as this technology apparently offers numerous advantages from a sustainability standpoint in terms of the potential for recycling, easy separation of the catalyst thus avoiding contamination of the product, as well as enhanced stability. Given also the expense and diminishing supplies associated with precious metal catalysts, as well as the high environmental impact and cost associated with the preparation of many custom ligands, cost savings through catalyst recycling could be envisioned. Finally, the ability to incorporate supported catalysts into a flow system provides an outlet to not only broaden the scope of this technology,114 but also enhance the rate of such transformations through the reaction being influenced by a super-stoichiometric amount of catalyst at any one time.115In a critical overview, Hübner et al. address many of the perceived advantages of catalyst immobilization to provide a rationale for why immobilized transition metal complexes are not more extensively utilized in industry.116 Reduced cost may be somewhat of a misnomer as added costs incurred in the immobilization process are often ignored. Secondly, active homogeneous reaction catalysts in themselves are often unstable, and as such immobilizing them or the pre-catalysts from which they originate is redundant unless immobilization affords increased stability. With regard to recycling, the rate of reaction often falls with each subsequent recycle indicating some degree of catalyst deactivation. In the pharmaceutical industry, reaction parameters such as time, yield, and purity may require tight control, and given this, reuse of a catalyst can present a risk of producing an out-of-specification batch. In terms of catalyst deactivation, metal leaching can occur over time, presenting a risk of product contamination. With regard to the commonly employed Pd-catalysed cross-couplings,117 it is important to bear in mind that palladium-complexes are very easily transformed into catalytically active nanoparticles thus making the subsequent immobilization a very challenging endeavour.118
The major issues with catalyst immobilization are reduced TON (turnover number), and TOF (turnover frequency), which are observed in the majority of cases in comparison to the corresponding homogeneous systems.119 A case study on the asymmetric hydrogenation of an imine to produce the herbicide, (S)-metolachlor evaluated both homogenous and heterogeneous catalyst systems derived from a range of different immobilization techniques (covalent attachment, adsorbed, entrapped, extractable), and concluded that the latter systems were both more complex than their homogeneous counterparts.120 In most cases, the TON observed for the immobilized systems was significantly lower than the homogeneous case, and although the system immobilized on silica gel showed a comparable TON, the reaction rate (TOF) was four-fold slower, and the covalent attachment to the silica support both time-consuming and costly (Scheme 10).
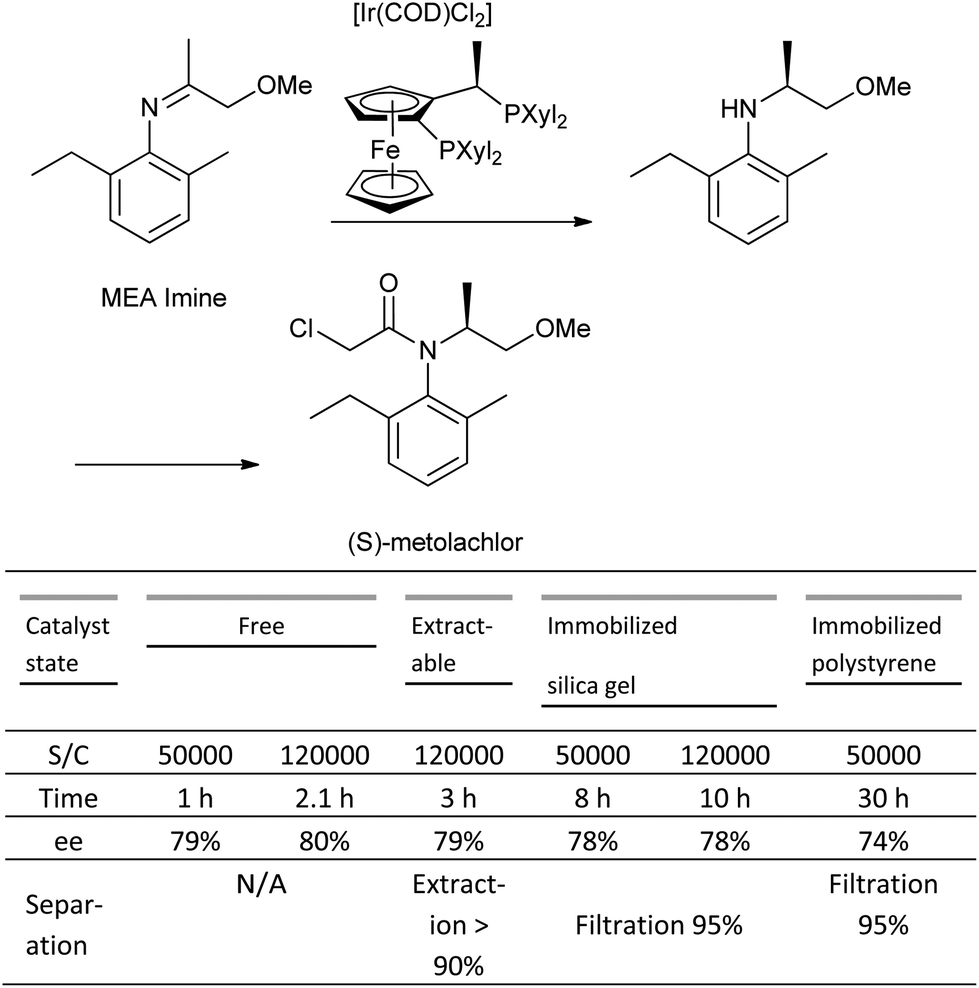 | ||
| Scheme 10 Comparison of immobilized Josiphos-based catalyst systems in the Ir-catalysed hydrogenation of MEA imine. | ||
Clearly, the immobilization of catalysts remains an exciting area particularly with the growth of interest in continuous manufacturing. In addition, there have been reported catalyst systems in which the immobilized system exhibits equal or superior reactivity and/or offers enhanced stability to the homogeneous equivalent.121 It is important to focus on these systems, which offer enhanced productivity, and learn what the beneficial effects the solid supports, linkers etc. have on stabilizing the active catalyst, kinetics, and mass transport as opposed to recycling.122 With these learnings in hand, one should be able to more efficiently predict whether immobilization is a strategy that will be beneficial for a particular system, and more efficiently synthesize the desired catalyst.
Asymmetric hydrogenation of unfunctionalized olefins/enamines/imines
The transition metal-catalysed asymmetric hydrogenation of unsaturated compounds, one of the most atom economical of chemical transformations in the chemist's toolbox, dominates the practice of asymmetric chemocatalysis in the commercial arena. What follows is a brief review of progress made since early 2007 where the functionality being hydrogenated is an olefin, enamine or imine devoid of potentially redundant directing functionality, such as the acetyl of an N-acetyl dehydroamino acid, whose coordination to a chiral catalyst could bias the delivery of hydrogen. The hydrogenation of such substrates is challenging because differentiation between prostereotopic faces relies on different interactions between the catalyst and olefin substituents which can appear to be similar in size.The asymmetric hydrogenation of minimally functionalized olefins has been subject to several reviews by the groups of Diéguez and Andersson.123–125 Since the 2007 publication, advances have centred on chiral iridium catalysts. Rhodium and ruthenium complexes continue to provide only low levels of enantioselectivity despite being highly selective for more functionalized alkenes. Useful levels of selectivity have been observed when the iridium bears a P,N ligand consisting of a phosphine, phosphinite or phosphite moiety and an oxazolidine, oxazole, thiazole or pyridine. This is exemplified with the use of chiral phosphite-oxazoline ligands for the hydrogenation of 1,1-disubstituted olefins126,127 (Scheme 11).
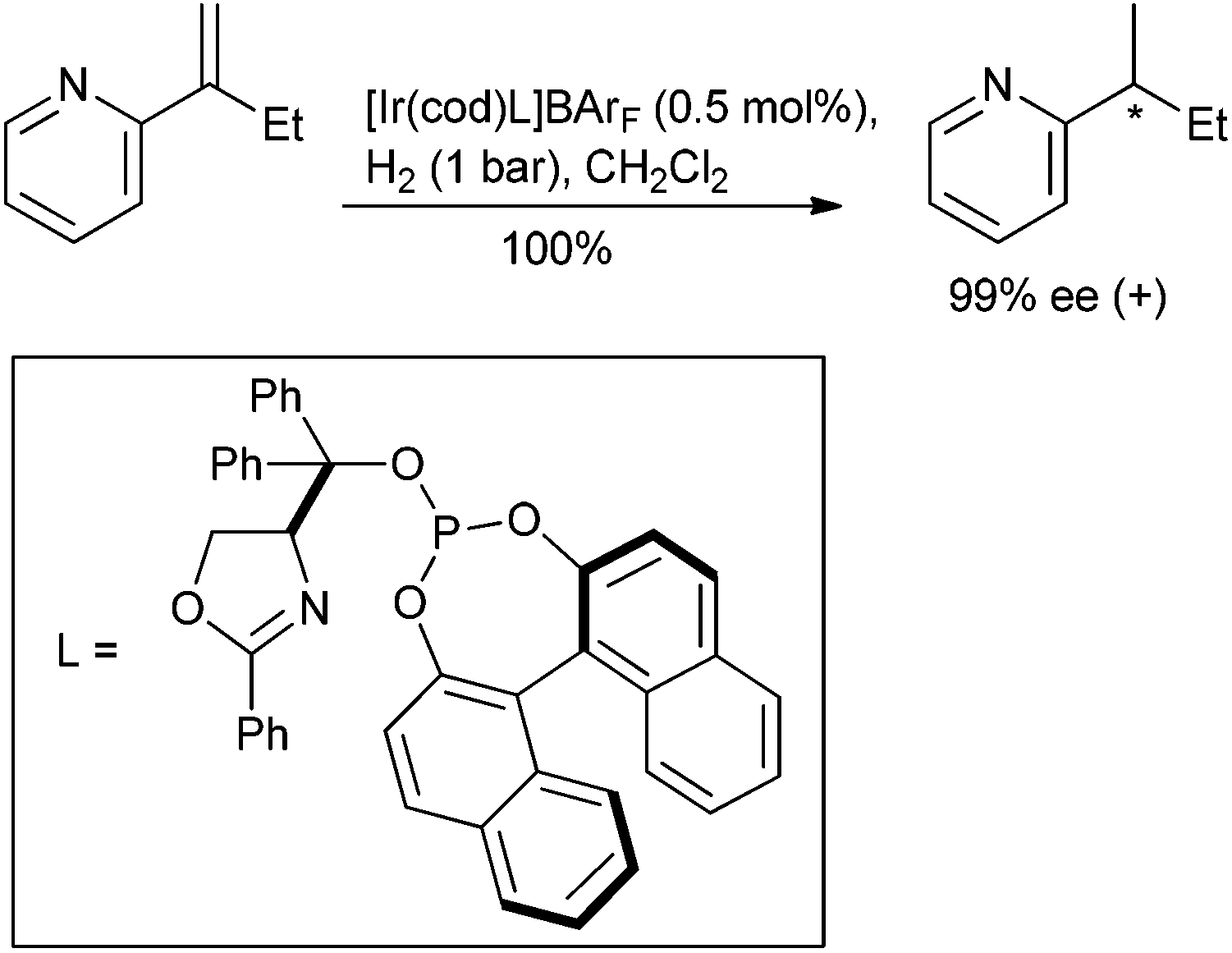 | ||
| Scheme 11 | ||
An innovation in the field surrounds the use of propylene carbonate as a solvent.128 In addition to being a ‘green’ solvent, its use is reported to slow the isomerisation of a 1,1-disubstituted olefin to an internal position, a pathway which typically erodes enantiomeric excesses. Its immiscibility with apolar solvents, also opens the door to catalyst recycling protocols where the product preferentially partitions into a hydrocarbon solvent.
Unsurprisingly, there are parallels between recent advances in the hydrogenation of both imines and enamines that are lacking in coordinating substituents. In both cases, configurational instability can lead to ambiguous interactions between the catalyst and substrate, lowering stereoselectivities. Given the structural similarity of the substrates and amine products, it is unsurprising that the latter can poison hydrogenation catalysts. Overcoming this problem using an acid additive, which protonates the basic nitrogen atom, has gained traction as a solution in this regard.129
The emergence of phosphoramidites as useful ligands for asymmetric hydrogenation can be attributed to their highly modular synthesis, air stability and comparative lower cost when compared to most bidentate ligands. When complexed to iridium, their utility in the current context is demonstrated by the asymmetric reduction of benzophenone imines that are unsubstituted on nitrogen,130 (Scheme 12) and cyclic N,N-dialkylenamines to cyclic tertiary amines.131 Again, as part of iridium complexes, phosphine-oxazoline (PHOX) derivatives have also emerged as a privileged ligand set for the reduction of minimally functionalized ketimines, as well as α-arylethenamines where the nitrogen only bears alkyl substituents.132 In an example of asymmetric induction from a chiral counterion, iridium(III) complexes derived from a monosulfonylated 1,2-diphenylethane-1,2-diamine and a chiral binaphthyl-based phosphoric acid have proven to be very effective catalysts for the hydrogenation of acyclic imines, including those derived from dialkyl ketones.133
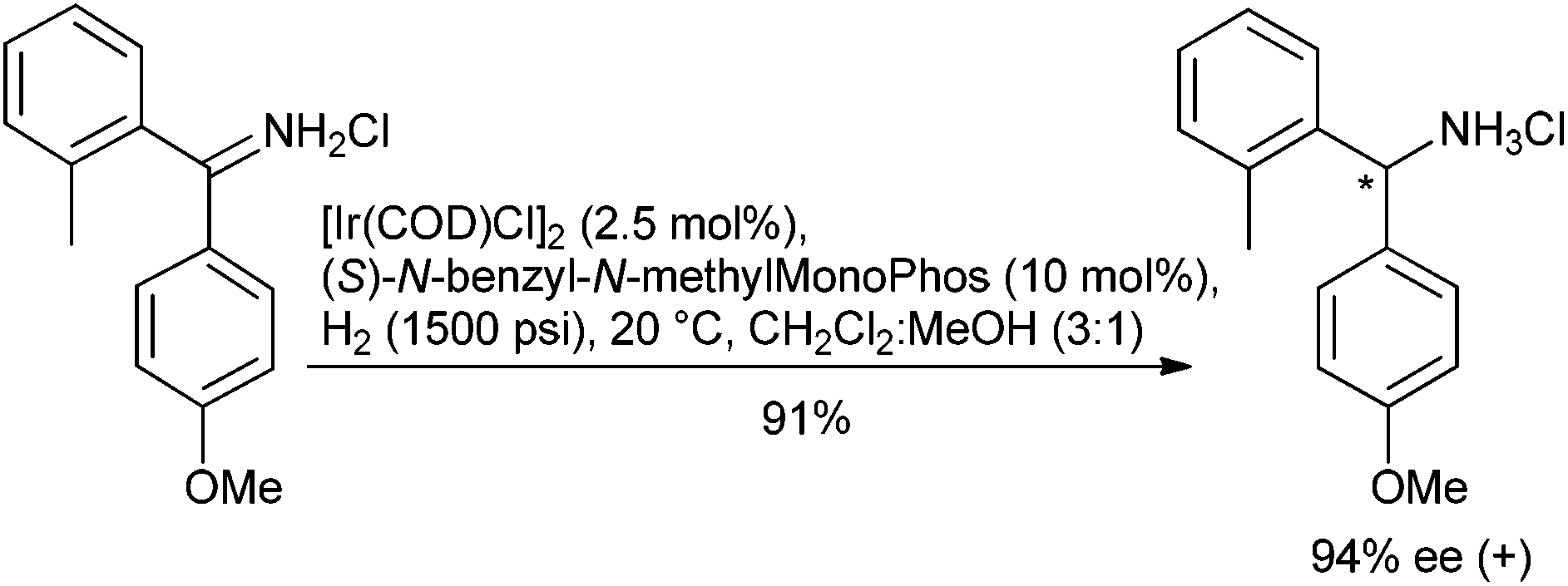 | ||
| Scheme 12 | ||
Progress has been made with ruthenium pre-catalysts, for example, the direct reductive amination of β-ketoesters using a Ru–DM-SEGPHOS complex in the presence of an ammonium salt has been used to furnish the corresponding β-amino ester with high enantioselectivities,134 obviating the need to start from an isolated β-enamine ester.135 Starting from a β-ketoamide precursor has also allowed the chemo-catalytic introduction of the stereogenic centre of sitagliptin to be streamlined such that it mimics the preferred biotransformation approach3 to this API136 (Scheme 13).
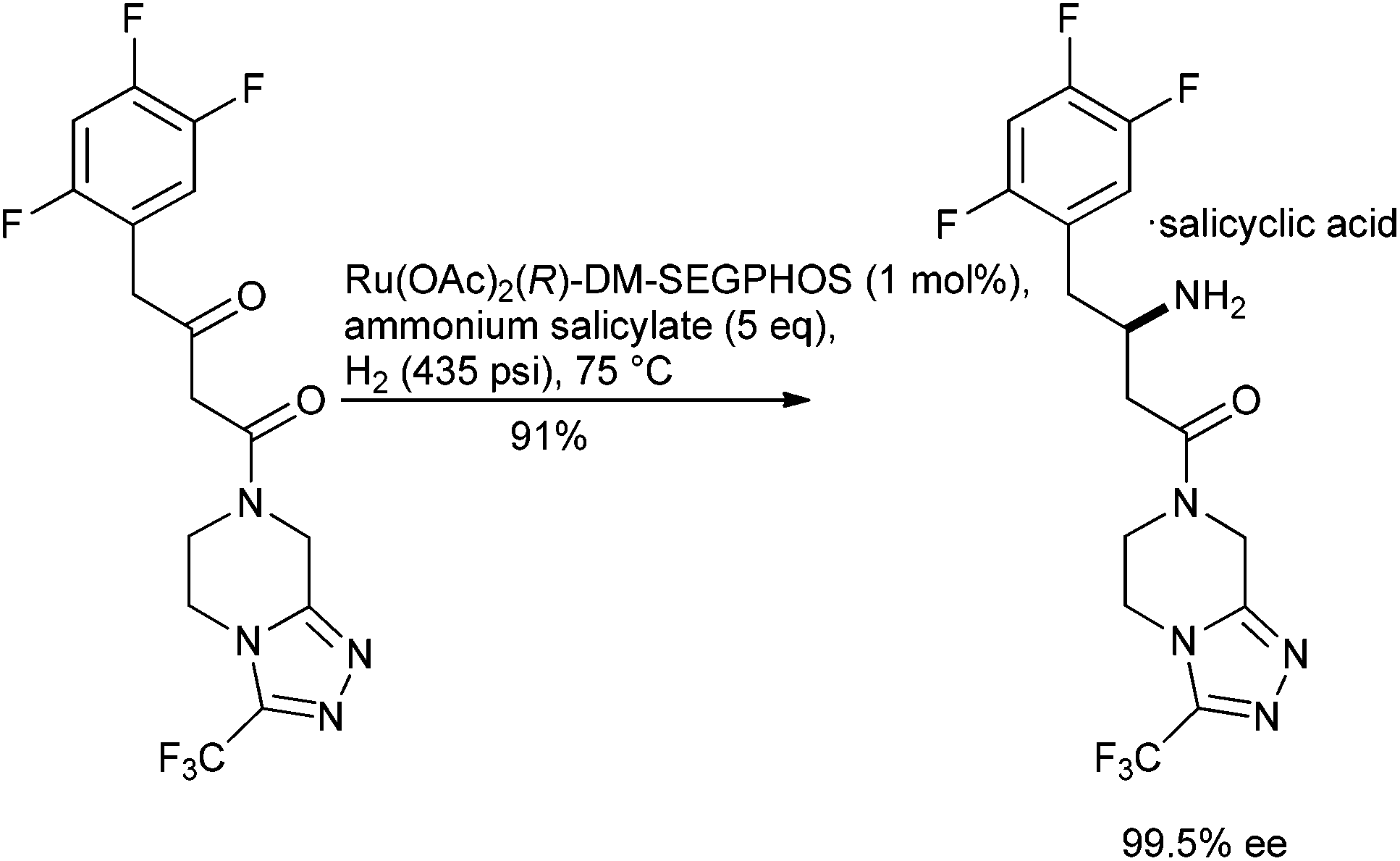 | ||
| Scheme 13 Preparation of sitagliptin. | ||
An emerging theme in the asymmetric hydrogenation of minimally functionalized olefins, imines and enamines is the development of catalysts based on first row transition elements. The cost, comparative abundance in the Earth's Crust and the effect of geopolitical influences on the availability of their heavier congeners (rhodium, ruthenium and iridium in particular), makes such catalysts an attractive proposition. First row transition elements also offer new opportunities for the effective transmission of chirality from the metal complex to challenging substrate classes due to their distinct electronic structures. Developments in this area are exemplified by the use of a C1-bis(imino)pyridine cobalt complex for the hydrogenation of trisubstituted alkenes.137 The explosion of interest in the application of frustrated Lewis pairs over the last decade has started to extend to the asymmetric hydrogenation of ketimines,138 an approach which is completely metal-free.
Despite being a mature technology, its atom economy has ensured the asymmetric hydrogenation of minimally functionalized olefins, imines and enamines remains a relevant technology into which research continues to be invested. Key challenges that remain include the development of single catalytic systems that tolerate diverse substituents, the hydrogenation of hindered tetra-substituted olefins, and the continued development of catalysts based on first row transition metals with low toxicity.
Improved fluorination/trifluoromethoxylation
The use of fluorine in the design of small molecule APIs as an isostere to modify physical properties, metabolism, membrane permeability and pharmacokinetic properties is a consistent tool for the discovery scientist139 with 32% of the small molecule APIs approved by FDA between 2014 & 2016 containing at least one fluorine atom.140 In a review of the chemistry to prepare the 40 fluorine containing small molecule APIs launched between 2001 & 2011; 30 of the APIs either contained an aryl fluoride or aryl trifluoromethyl group and two contained both functionalities. The remaining compounds contained fluoroalkyl chains and there was one fluorinated steroid.141 The fluorine was introduced through purchase of simple, predominately fluoro-aryl or trifluoromethyl-aryl, starting materials for 37 of the molecules. This synthetic strategy broadly reflects the green chemistry challenges of fluorination methodologies up to the turn of the century. Industrial manufacture of aryl fluorides is typically by halogen exchange (Halex) or the Balz–Schiemann process comprising diazotization followed by reaction of the diazonium salt with BF4− or anhydrous HF. Manufacture of –CF3 is by halogen exchange of –CCl3 or fluorination of –CO2H using anhydrous HF.142 The severe reaction conditions of these approaches are frequently incompatible with the structural complexity of small molecule APIs. In addition, anhydrous HF requires specialist handling and manufacturing facilities that are not usually available in the multi-purpose pilot and production plants typically used by the pharmaceutical industry.Fluorine, the simplest and most atom economical electrophilic reagent, also requires specialist equipment for use, although the hazards of handling fluorine gas have been to some degree mitigated through dilution with 90% of nitrogen, and employing a flow chemistry reactor as demonstrated in the single step synthesis of the antifungal medicine flucytosine (Scheme 14).143
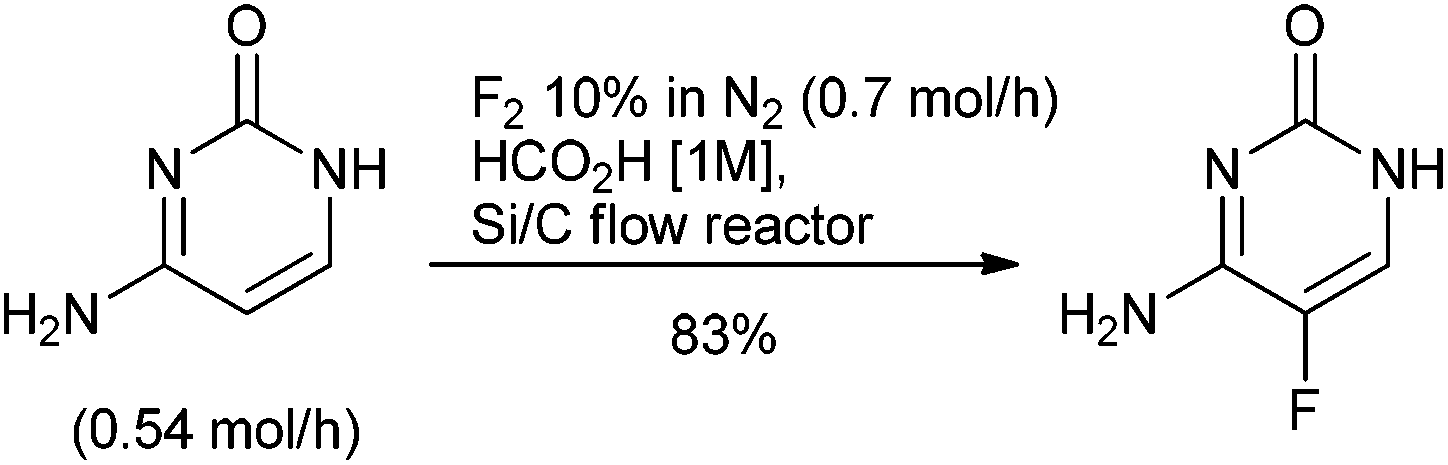 | ||
| Scheme 14 Synthesis of flucytosine using fluorine gas. | ||
In the 2007 publication, “green fluorination” was selected as an “aspirational” reaction, highlighting the need for improved: “catalysts for increasing the nucleophilicity of F−, milder conditions for conducting fluorine exchange reactions (ArCl to ArF), and safer and more economical sources of electrophilic fluorine”.1 In the subsequent decade, whilst not directly addressing all of these needs, there has been a large increase in the number of reagents and approaches towards fluorination offering greater flexibility to the medicinal and development chemist for the strategic introduction of fluorine under milder conditions.144
A good example is the development of complementary nucleophilic deoxyfluorinating reagents, showing improved stability and selectivity over time with a trend emerging towards safer, milder and therefore more selective reagents for fluorination.145 In this respect, the initial wave of aminosulfur trifluorides reagents, such as DAST, DeoxoFluor and MOST were introduced to replace SF4. However, use of the reagents presented their own challenges, for example, the original introduction of the gem-difluoromethyl functionality in maraviroc used diethylamino sulfur trifluoride (DAST). DAST is unstable at elevated temperature, making it unsuitable for scale-up146 and production of the intermediate was outsourced to a specialist company who developed alternative sulfur trifluoride reagents.147 The drive for safer reagents led to the emergence of reagents such as Xtalfluor-E and -M,148 Fluolead,149 TFFH,150 PhenoFluor,151 PyFluor152 and AlkylFluor153 (Fig. 1). Despite the improved safety profiles, and the ability to utilize these reagents for the late-stage functionalization of complex organic molecules, there remain areas for improvement as drawbacks still exist such as the need to use additional promoters and the frequent use of unfavourable solvents in these reactions, the restricted availability of some of these compounds prohibiting use on scale, as well as issues with elimination in numerous cases. In addition, development of suitable reagents, which can be extended to the introduction of 18F for PET-labeling (positron emission tomography) are a priority.
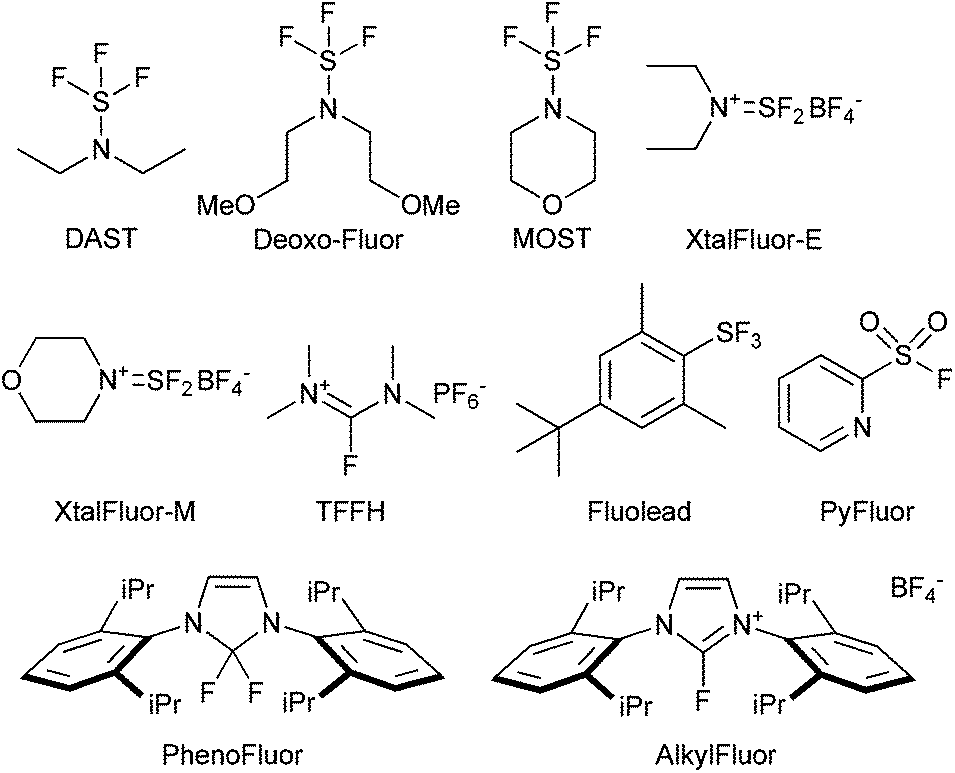 | ||
| Fig. 1 The evolution of reagents for deoxyfluorination, see ESI.† | ||
The high heat of hydration (approx. 120 kcal mol−1) of F− makes it a very poor nucleophile for direct halogen exchange unless dehydrated. Several reports have emerged providing methods to prepare either anhydrous TBAF154 or TMAF155 to facilitate aryl fluoride synthesis. In addition, two approaches show progress in this area: Kim et al. found TBAF(t-BuOH)4 to be an anhydrous reagent for halogen exchange of alkyl bromides or sulfonate esters.156 Whilst Gouverneur et al. applied silver catalysis for halogen exchange of bromide in aryl bromodifluoromethyl ethers or thio ethers and chloride in aryl chloro fluoromethoxy ethers with 18F−.157 A range of diverse mild approaches to arylfluorination have also been reported enabling the use of a variety of starting materials such as arylboronic acids with traditional electrophilic fluorination reagents.158 The development of sterically encumbered ligands to facilitate the reductive elimination of C–F has also led to the evolution of transition metal catalysed arylfluorination approaches.159 A driver for development of a number of the approaches above is the use of 18F in PET (two of the FDA approvals in 2014–2016 were PET ligands). Another approach in this developing field is the application of the fluorinase enzyme providing the mildest of reaction conditions.160
Free radical fluorination has emerged as a mild alternative approach, for example Baran's zinc fluorinated-methylsulfinates for di- and trifluoromethylation of heterocycles.161 Photo-induced fluorination further broadening the scope of alkyl fluorination and aryl trifluoromethylation. For example, Xia et al. achieve light induced, biarylketone catalysed selective benzylic C–H mono-fluorination (9-fluoreneone) or gem-difluorination (xanthone) using Selectfluor as the fluorine source.162 Beatty et al. use blue LEDs and a ruthenium catalyst to generate the trifluoromethyl radical from trifluoroacetic anhydride to achieve aryl and heteroaryl trifluoromethylation.163 The zinc reagent [(DMPU)2Zn(CF2H)2] has been used for the copper catalysed difluoromethylation of aryl and heteroaryl iodides at 60 °C164 or for room temperature, nickel catalysed difluoromethylation of aryl and heteroaryl iodides, aryl bromides and triflates.165 While geminal difluoroolefins have been prepared via carbene insertion of :CF2.166 Enantioselective fluorination, fluoroalkylation and trifluoromethylthiolation, in particular using organocatalysis enables the preparation of chiral fluorine containing molecules.167
The increased interest in structural diversity of fluorine containing functional groups is reflected in the expansion of the title of this key research area to include trifluoromethoxylation. Direct trifluoromethoxylation of simple arenes has been demonstrated with trifluoromethyl hypofluorite under free radical conditions,168 however, CF3OF is a toxic gas. Alternatively, O-trifluoromethylation of phenols has been achieved in a stepwise approach with alkylation using sodium bromodifluoroacetate followed by silver catalysed decarboxylative fluorination with SelectFluor II.169 Liu et al. achieve a silver mediated, oxidative trifluoromethylation using Ruppert's reagent, albeit the reaction requires many additives in high stoichiometry.170
In conclusion, substantial progress has been made to meet the goals outlined in the 2007 publication, if not by directly addressing the issues, through providing viable alternative approaches. Further opportunities exist to develop reagents and methodologies with improved atom economy and reaction mass efficiency, as well as the use of less problematic solvents.
Wittig chemistry without Ph3PO
The Wittig reaction has been a mainstay in the synthesis of olefins because of its wide applicability and functional group tolerance. Though it is an atom uneconomical process, E- or Z-olefins can be generated from triphenylphosphorous ylides with aldehydes and ketones, depending on the stabilizing substituents on the ylide.171 Variations have been developed in which the phosphorous reagent can be a phosphonate ester (Horner–Wadsworth–Emmons, HWE) to give E-olefins.172 Simple trialkylphosphine ylides have also been described. These accepted variations will not be discussed since the by-products are more readily removed post-reaction. Here, the focus will be improvements on the traditional Wittig reaction to minimize the impact of triphenylphosphine oxide (TPPO) waste.In the discovery space, where ease of throughput with multiple analogues is imperative, triphenylphosphine-functionalized solid supports can be used effectively for Wittig reactions (also as a reagent replacement for Mitsunobu, Appel halogenation of alcohols, reductive work-up of alkene ozonolysis and tetrazole formation).173 The supports are available in bulk and have good filtration properties being fibre-based resins.174,175 The reaction work-up is simplified to avoid chromatography (silica, solvent volumes) since the resultant phosphine oxide can be removed by simple filtration. This is in stark contrast to the often-tedious process involving multiple columns utilized to remove the TPPO and purify the desired product, generating large amounts of silica gel and solvent waste.176
Further along in development, where solid-supported reagents are more critically evaluated by process mass intensity (PMI) calculations, innovative organocatalytic Wittig-type reactions could find greater application.177 A general analysis of different catalytic phosphorus-based reactions (Mitsunobu, Appel, Wittig, etc.) was conducted with processes assigned into two categories: redox-shuttled or redox-neutral, depending on the oxidation state of the phosphorus reagent (Scheme 15). Recent publications in the catalytic Wittig reaction have fallen into the redox-shuttled process, including work with the novel phospholane oxide pre-catalysts from O'Brien and Werner (3, 4, and 5, respectively, Fig. 2).178
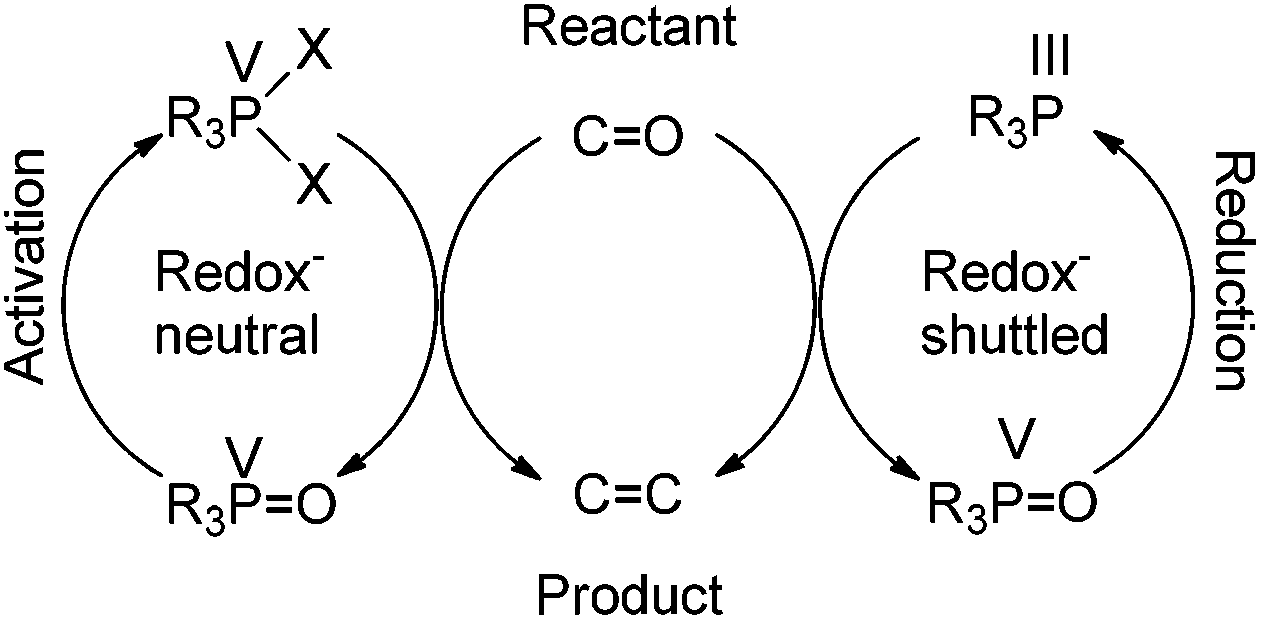 | ||
| Scheme 15 General approaches to phosphorus catalysis. | ||
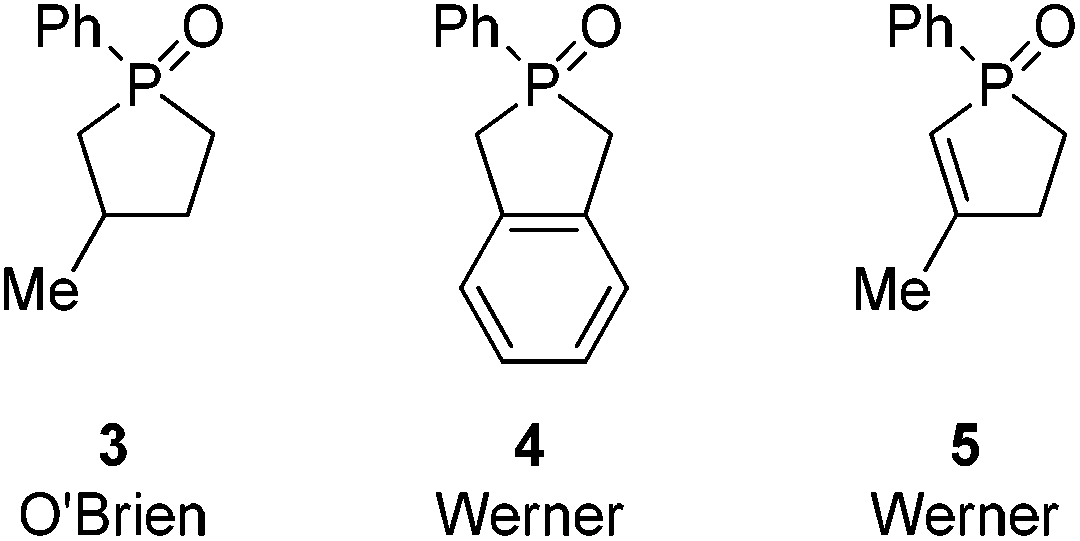 | ||
| Fig. 2 Novel Wittig pre-catalysts. | ||
The reports have shown that molecular phosphines can be used in a catalytic manner by selective reduction of the phosphine oxide by-product to the active phosphine, allowing catalytic cycle turnover. The compatibility of the reducing agents with the reaction conditions is critical. Reagents for phosphine oxide reduction have been reported ranging from harsh oxalyl chloride and trichloromethyl chloroformate (TCF), which cannot be readily applied to the catalytic Wittig reaction, to milder alkyl silanes and alkoxysilanes, including the very cheap and benign poly(methylhydrosiloxane) (PMHS).179
An all-important question about the true green benefit regarding total waste and energy consumption created by addition of the reducing agent and the reduction process has been addressed in a life cycle analysis (LCA).180 This report suggests that the catalytic Wittig reaction retains its favourable contributions even after considering CED (cumulative energy demand) and GHG (greenhouse gas emissions, an environmental metric). Thus, the positive impact of catalytic Wittig transformations in pharmaceutical syntheses could be large and measurable.
The original series of reports discuss a microwave-assisted catalytic Wittig reaction178f as well as an asymmetric catalytic olefination to desymmetrize a prochiral ketone.178g To further highlight the opportunities for catalytic Wittig reactions, a publication demonstrated that an iron hydride hydrosilylation catalyst could be repositioned in the presence of catalytic phosphine to achieve in situ phosphine oxide reduction.181 Key to this dual iron/phosphine catalysis was inclusion of a solvent that was unfavourable to the competitive carbonyl hydrosilylation, allowing the catalytic Wittig conditions to be applied to aromatic and aliphatic aldehydes.
Lastly, in situ recycling of the TPPO waste to perform other key reactions has recently gained attention in the literature. Examples include using the generated TPPO as a co-catalyst in aldol reactions and conjugate reductions.182 In these reports, the TPPO is used to catalyse the second transformation across a variety of substrates and can further incorporate an additional step such as furan formation.183 This strategy is finding application in the Mitsunobu reaction97 and should also be applicable during ozonolysis quenching, where TPPO waste is generated.
Recent improvements in the utilization of TPPO and derivatives via creation of novel phospholane oxides have reinvigorated interest in the catalytic Wittig olefination, for both medicinal and process chemistry applications. The present limitations are that relatively high catalyst loadings are still required, and non-stabilized ylides and Z-olefin products are not fully available. Further development of these catalytic reactions or leveraging phosphine oxide by-products in a subsequent step (in situ recycling) should support utilization of the powerful Wittig olefination and help the pharmaceutical industry continue to become a greener enterprise.
Alternatives for oxidations, C–O or C–N redox processes
In the 2007 Perspective, discussion of oxidation reactions was largely focused ‘oxidation and epoxidation without the use of halogenated solvents’. This perspective has shifted slightly, and now focuses on ‘Greener alternatives for oxidations and C–O or C–N redox processes’. While the use of halogenated solvents still represents a major concern to improve process greenness, this topic is discussed below and is no longer specific to oxidation reactions. Thus, oxidation and C–O or C–N redox processes will focus primarily on manipulation of the oxidation states of carbon–nitrogen and carbon–oxygen bonds.Many of these transformations use molecular oxygen (air) as the oxidant. Diligent safety evaluations should be conducted prior to implementing these procedures, as running reactions with flammable solvents poses a potential safety risk.
Research in photocatalytic oxidation has grown substantially over the past years, logging nearly 107 articles in SciFinder (May 2016), up from around 40 in 2007. These approaches are attractive as they can run using molecular oxygen or air as the terminal oxidant, often use visible light or sunlight as the photochemical activator and can be conducted in pure aqueous media, alleviating the major safety concerns regarding running oxidation reactions in flammable solvents. Additionally, the catalysts are typically supported on solid support, nanoparticles184 or as quantum dots – doped with the appropriate metal to promote the transformation.185 While these materials can be somewhat laborious to prepare, the possibility of repeated recovery and reuse minimizes the impact. To date, oxidations of alcohols to aldehydes, ketone and esters has been demonstrated. Limitations exist, especially with the demonstrated scope of the reactions. Additional application of these promising technologies will be required to promote widespread use within the pharmaceutical industry.
Another transformation of interest is the oxidation of amines to imines. This has been accomplished using catalytic systems involving alloxan as a co-catalyst, with either a synthetic Flavin186 or Cu(I) salt187 as the catalyst. In the case of the reaction using CuCl as the catalyst, air was the terminal oxidant and the reaction was run in acetonitrile. In both cases the reactions were run at ambient temperature and pressure and assay yields were often greater than 70%. In most cases, these transformations produce highly symmetric imine products; however, in the case of the Cu(I) system, asymmetric imines were generated by reaction between a benzylic amine and aniline, albeit in lower yield and selectivity. While the solvents employed are not ideal from a green chemistry standpoint, alterations would necessarily be made when used on the more complex substrates employed in the pharmaceutical industry.
Given the safety concerns regarding the use of air as a terminal oxidant in a reaction mediated in flammable or combustible solvents, alternative oxidants that produce benign by-products are highly sought after, principle among these is hydrogen peroxide. A competent oxidant, this reagent produces water as the by-product making it one of the most attractive from this perspective. When used in aqueous combination with 0.2 mol% CuSO4·5H2O and benzylic or primary alkyl amines, pseudo symmetric imines are produced in moderate to high yield.188
Another approach to viable aerobic oxidation in flammable solvents can be achieved through the use of reactions in flow reactors.189 When considering aerobic oxidation, a primary concern is the control of the heat generated during the reaction, as aerobic oxidations are highly exothermic. Use of flow provides a minimal volume for reaction and a larger surface to volume ratio than batch processing, which allows for more efficient heat management, and avoids a headspace. With the flexibility to control both flow rate and channel length, product stability can be enhanced and the potential for run-away reaction lessened by reducing contact time of the product with the harsh reaction conditions within the flow reactor.
Viable replacements for polar aprotic solvents
The 2007 paper authoring team were ahead of the curve in identifying dipolar aprotic solvents as key solvents for replacement.1 That paper accurately describes the challenges of handing and disposing of these materials, however since then significant regulatory challenges have emerged in Europe. N-Methylpyrrolidinone (NMP), N,N-dimethylacetamide (DMAc) and N,N-dimethylformamide (DMF) have been nominated to the candidate list of substances of very high concern (SVHC) as part of the European Union REACH regulations because of their potential reproductive toxicity.190 These nominations were made in June & December 2011 and December 2012 respectively. At the time of writing it is not clear whether these materials will be restricted (under Annex XVII) or made subject to Authorisation (under Annex XIV) but either of these approaches will make the industrial use of NMP, DMAc or DMF more difficult in the future.191 A publication by Ashcroft et al.192 gives the results of a survey of the process chemistry journal (Org. Process Res. Dev.) for the years 1997–2012. This survey looks at the percentage of processes which use these materials as a function of time, analyses why the solvents are used and suggests some strategies for minimization. However, if further progress is to be made with this solvent class, new solvents are required which are non-toxic but have the desired polarity to dissolve polar materials. The following solvents (Fig. 3), not all of which are presently available at commercial volume, offer potential to replace the current REACH impacted dipolar aprotic solvents. It is the Roundtable's hope that future research and development with these alternatives and totally new solvents will stimulate the market.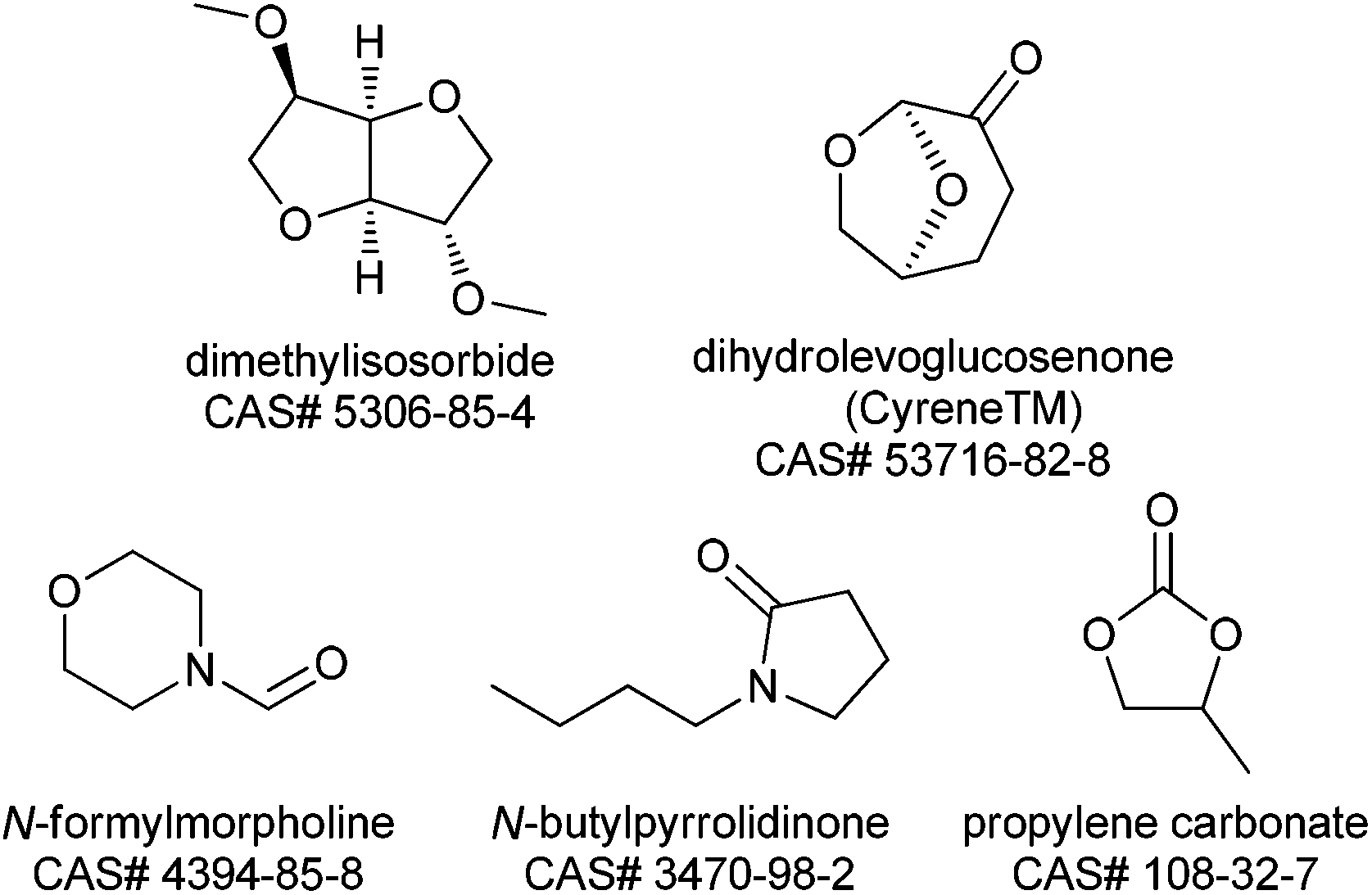 | ||
| Fig. 3 Potential dipolar aprotic solvent replacements. | ||
N-Formylmorpholine is a solid at room temperature (m.p. 20–23 °C), nevertheless experience within some Roundtable companies has shown it to have quite good solvating properties. There is some limited single dose toxicity data and skin and eye irritation data available (a summary of which can be found in Table 4 in the ESI†), however no reproductive toxicity studies have been reported. The Roundtable feels it is important to prioritize reproductive toxicity testing as part of the toxicity testing package of new polar aprotic solvents as this is the main issue with the existing ones. Currently the REACH registration process only requires reproductive/developmental toxicity testing where ≥10 tonnes per annum of a material is manufactured or imported.193 On the practical side the morpholine ring imparts high water solubility which can be useful in reaction work-up enabling use as a “drop in” replacement for the existing problematic solvents. Partition of high boiling polar aprotic solvents (N-formyl morpholine b.p. = 239 °C) is often the best way to remove the solvent from product. On the negative side multiple washes may be required to reduce the solvent to an acceptable level, increasing the PMI, a heavily contaminated aqueous waste stream is produced and recovery of the solvent by distillation requires high energy input.
Dimethylisosorbide is unique in that it is a chiral ethereal solvent derived from glucose.194 It has a very favourable toxicology profile and is used as a solubilizing agent for dermal administration of many pharmaceutical products. Dimethylisosorbide also has many non-pharmaceutical applications including sunscreen lotions, deodorants and cosmetics. It has found utility as a bio-sourced alternative in liquid detergent formulations and as a coalescent in water based paints. Dimethylisosorbide has a wide liquid range, (−50 to 234 °C), is stable to oxidation and is fully miscible with water, which potentially allows for facile removal from pharmaceutical processes (vide supra). Based on its full water miscibility, dimethylisosorbide may offer potential as a phase transfer catalyst. The polarity of dimethylisosorbide is less than DMF, DMAc and NMP, but higher than other solvent options such as ethylene glycol dimethyl ether, 2-methyltetrahydrofuran and cyclopentyl methyl ether.195 Dimethylisosorbide is a known solvent for benzoyl peroxide products and is reported to improve stability of the peroxide in aqueous media.196 Based on these findings, a logical extension may be direct preparation of useful peroxide intermediates in this solvent. In addition, dimethyl isosorbide has received some attention as potential solvent for solid phase peptide synthesis (SPPS), the most common approach to polypeptide synthesis.195 Despite the potential benefits, its use as a traditional pharmaceutical solvent for chemical reactions has been limited. One disadvantage is, like DMSO, its high skin permeability which is problematic when highly hazardous reagents are utilized. In addition, the viscosity of the solvent (∼5 MPa s) is also higher than most common organic solvents. A promising indicator for the future use of dimethylisosorbide is that it has recently been assessed in a solvent selection guide.197
Propylene carbonate is a valuable polar aprotic solvent which has low viscosity and high dielectric constant (64) which allows for excellent solvation properties. Propylene carbonate also has a very favourable toxicological profile as no significant toxic effects were observed in rats fed propylene carbonate, exposed to the vapour, or to the neat liquid.198 In addition, propylene carbonate is biodegradable and a green route of manufacture from propylene oxide and carbon dioxide is available.199 Propylene carbonate has found utility as a solvent for hydrogenation,200 palladium catalysed substitution,201 aldol,201 and Heck202 reactions. The main limitation with propylene carbonate is instability toward strong acid and bases resulting in CO2 liberation, the latter has limited its utility in SNAr and similar reaction classes.
Dihydrolevoglucosenone (Cyrene™) is a bio based solvent derived from cellulose.203 Cyrene is similar to NMP in Kamlet – Taft and Hansen solubility parameters.204 Initial use case studies reveal that Cyrene™ has given similar performance to NMP in an SNAr fluorination and in the Menschutkin reaction.204 Watson et al. also report utility of Cyrene in Sonogashira and Cacchi type annulation reactions. The authors also describe some limitations which include incompatibilities with strong acid and base.205 A preliminary Ames study has been reported without mutagenic effects.204
N-Butylpyrrolidinone (NBP) has emerged as a potential replacement for DMF and NMP. Hunt et al. reported that NBP performed well instead of NMP and DMF for Suzuki, Heck and other reactions where these solvents are normally in operation.206 Most notably NBP has been found to be non-reproductively toxic and non-mutagenic. While further studies are warranted, these initial results combined with the expected chemical inertness of NBP show promise.
Viable replacements for halogenated solvents
The survey from the CHEM21 project192 showed that the use of dichloromethane and 1,2-dichloroethane has remained consistent in process chemistry published over a 16-year period in the journal Organic Process Research and Development (see Table 5).207| 1997–2000 | 2000–2004 | 2005–2008 | 2009–2012 | |
|---|---|---|---|---|
| Dichloromethane | 44.7% | 41.5% | 50.0% | 46.1% |
| 1,2-Dichloroethane | 2.6% | 3.8% | 5.6% | 3.2% |
Halogenated solvents have seen significant industrial uses due to their strong solubilizing properties and low reactivity across broad reaction classes. In addition, low boiling points generally allow for facile removal from desired process streams. However, halogenated solvents are coming under increasing regulatory pressure around the globe. The first group of halogenated solvents were ozone depleting solvents such as tetrachloromethane which are severely limited by the Montreal Protocol. More recently, the use of 1,2-dichloroethane (DCE) in a manufacturing process performed in the EU became subject to Authorisation (sunset date November 2017) under the REACH legislation. This means that 1,2-dichloroethane can no longer be used in the EU unless a special authorisation has been obtained from the European Chemical Agency.208 Finally, the use of dichloromethane is restricted under the European Solvent Emissions Directive209 where emissions even at Pilot Plant scale are limited to 100 g per hour and special abatement is required to meet these tight limits. Due to its volatility, dichloromethane enters the atmosphere where it acts as a greenhouse gas and has been has been reported to IPCC to have 8.7 times more heat absorptive capacity than carbon dioxide. In addition, dichloromethane has an NTP listing as a probable carcinogen.210 Based on these considerations greener alternatives to halogenated solvents is a high roundtable priority. However, finding drop in replacements for dichloromethane and DCE is even more challenging than for dipolar aprotic solvents.
Future opportunities: greener peptide and oligonucleotide syntheses
The Roundtable started in 2005 with a focus on small molecule chemistry but has steadily evolved its mission to address the broader needs and interests of the industry. This includes large molecules which are the primary focus of the Roundtable greener biologics team.211 In fact, some Roundtable companies have evolved over the past decade from originally being small molecule focused to now primarily large molecule based organizations. In addition, an area of emerging importance is the concept of “medium molecules” which include polypeptide and oligonucleotide products. These medium molecule products can be roughly defined as API consisting of MW between 1000–5000. Many peptide and oligonucleotide products fit this definition and are an area of significant growth within the biopharmaceutical industry. For example, there are now approximately 140 peptide based active pharmaceutical ingredients (APIs) in clinical trials and more than 400 in pre-clinical development.212 However, the current state of the art in peptide and oligonucleotide syntheses utilize primarily legacy technologies, with little focus on green chemistry and engineering. Waste generated from current peptide and oligonucleotide processes is typically 3000–15000 kg per kg API (10–50-mer products) with multiple usages of highly hazardous reagents and solvents.213 A majority of the waste composition is DMF and NMP which is used in solid phase peptide synthesis (SPPS) and oligonucleotide synthesis. Many of the greener alternative solvents to traditional polar aprotics should find utility in this space, but will require significant efforts as a majority of these processes are operated by manufacturers using decades old practices where green chemistry has not been an area of focus. Contributing to the poor environmental profile is the pervasive and extensive use of chromatography to produce peptide and oligonucleotide products with required quality attributes. In 2016 the ACS Pharmaceutical Roundtable identified the need for greener syntheses of peptide and oligonucleotide products as an unmet need and a new Roundtable team formed with 100% participation from member companies to address these important areas.
Conclusion
The retention of ten of the original research areas and solvent themes in one way or another in the revised list is not very surprising as small molecule APIs, although changing, are mostly nitrogen containing molecules that present similar synthetic challenges. However, as discussed in this perspective, while considerable progress has been made to develop new methodologies to address the issues associated with those transformations from a sustainability perspective, there remains room for improvement. There are some common themes across many of the research areas, notably, the positive impact and potential of biocatalysis and chemo-catalysis to improve existing methodology or provide alternate solutions to the challenges, e.g., the preparation of chiral amines. As well as the goals to identify alternative catalysts with improved reactivity, recovery efficiency and, where appropriate, using more abundant metals. Almost all chemistry to prepare small molecule APIs is conducted in organic solvents where two common themes can be seen across the literature. Chemistry using air or oxygen as the terminal oxidant is frequently conducted above the flashpoint of the reaction solvent, presenting a significant hazard for scale-up. Interesting methodology across all chemistries is frequently run in solvents with undesirable environmental, health or safety profiles and in many cases at risk of future legislative control, often without any screening of alternative solvents reported. With the availability of equipment for small scale parallel experimentation, routine solvent screening for alternatives to undesirable solvents is encouraged. The publication of this perspective is to recognize the significant progress made in the decade since the original publication and to stimulate interest in continued research and development to improve the sustainability of those methodologies that are core to the discovery and development of new medicines.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank John Leonard (formerly AstraZeneca) and Alan Pettman (formerly Pfizer) for their assistance with the preparation of the long list through provision of the UK pre-competitive group214 list. Colleagues at our member companies who participated in the vote. Julie Manley (Guiding Green) for her help coordinating the vote across the member companies. Rakeshwar Bandichhor (Dr Reddy's), Louis Diorazio (AstraZeneca), Michael Justus (Johnson and Johnson), Ingrid Mergelsberg (Merck) and John Tucker (Amgen) for coordinating the vote at their respective companies.This manuscript was prepared by the authors on behalf of the American Chemical Society Green Chemistry Institute® Pharmaceutical Roundtable.215 The ACS GCI is a not-for-profit organization whose mission is to catalyze and enable the implementation of green and sustainable chemistry throughout the global chemistry enterprise. The ACS GCI Pharmaceutical Roundtable is composed of pharmaceutical and biotechnology companies and was established to encourage innovation while catalyzing the integration of green chemistry and green engineering in the pharmaceutical industry. The activities of the Roundtable reflect its members' shared belief that the pursuit of green chemistry and engineering is imperative for business and environmental sustainability.
References
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. S. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- W. P. Stremmer, Nature, 1994, 370, 389–391 CrossRef CAS PubMed; M. T. Reetz, L.-W. Wang and M. Bocola, Angew. Chem., Int. Ed., 2006, 45, 1236 CrossRef PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305 CrossRef CAS PubMed.
- Z. Peng, J. W. Wong, E. C. Hansen, A. L. A. Puchlopek-Dermenci and H. J. Clarke, Org. Lett., 2014, 16, 860 CrossRef CAS PubMed.
- Element Recovery and Sustainability, ed. A. Hunt, Royal Society of Chemistry, Cambridge, U.K., 2013 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed; T. W. J. Cooper, I. B. Campbell and S. J. F. MacDonald, Angew. Chem., Int. Ed., 2010, 49, 8082 CrossRef PubMed.
- http://www.napalladium.com/palladium/supply-and-demand/default.aspx (accessed October 2017).
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed.
- https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3D/Q3D_Step_4.pdf (accessed October 2017).
- http://www.infomine.com/investment/metal-prices/ (accessed October 2017).
- ICH Q3D step 4: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3D/Q3D_Step_4.pdf, other element = elemental impurities for which PDEs have not been established due to their low inherent toxicity and/or differences in regional regulations are not addressed in the guideline. The EMEA Oral pde is 13000 μg per day: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003587.pdf (accessed October 2017).
- https://education.jlab.org/itselemental/index.html (accessed October 2017).
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- S. Ge and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 12837 CrossRef CAS PubMed.
- S. D. Ramgren, L. Hie, Y. He and N. K. Garg, Org. Lett., 2013, 15, 3950 CrossRef CAS PubMed.
- T. Abe, T. Mino, K. Watanabe and M. Sakamoto, Eur. J. Org. Chem., 2014, 6983 CrossRef CAS.
- Q. Tian, Z. Cheng, H. M. Yajima, S. J. Savage, K. L. Greene, T. Humphries, M. E. Reynolds, S. Babu, F. Gosselin, D. Askin, I. Kurimoto, N. Hirata, M. Iwasaki, Y. Shimasaki and T. Miki, Org. Process Res. Dev., 2013, 17, 97 CrossRef CAS.
- T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, I. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674 CrossRef CAS PubMed; J. Wen, S. Qin, L.-F. Ma, L. Dong, J. Zhang, S.-S. Liu, Y.-S. Duan, Y.-S. Chen, C.-W. Hu and X.-Q. Yu, Org. Lett., 2010, 12, 2694 CrossRef PubMed . and references therein.
- J. M. Neely, M. J. Bezdek and P. J. Chirik, ACS Cent. Sci., 2016, 2, 935 CrossRef CAS PubMed.
- S. K. Gurung, S. Thapa, A. Kafte, D. A. Dickie and R. Giri, Org. Lett., 2014, 16, 1264 CrossRef CAS PubMed.
- R. B. Bedford, M. Nakamura, N. J. Gower, M. F. Haddow, M. A. Hall, M. Huwe, T. Hashimoto and R. A. Okopie, Tetrahedron Lett., 2009, 50, 6110 CrossRef CAS.
- For studies on an Fe-catalyzed Kumada coupling, see: A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773 CrossRef CAS PubMed ; For a Ni-mediated reductive coupling, see: S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef PubMed.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC.
- J. E. Taylor and S. D. Bull, N-Acylation Reactions of Amines, in Comprehensive Organic Synthesis, ed. P. Knochel and G. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 6, p. 427 Search PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- H. Lundberg, F. Tinnus, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC.
- M. M. Joullié and K. M. Lassen, ARKIVOC, 2010, 189 Search PubMed.
- N. Lukasik and E. Wagner-Wysiecka, Curr. Org. Synth., 2014, 11, 592 Search PubMed.
- B. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef PubMed.
- For an example, see: M. C. Bryan, L. Diorazio, Z. Fei, K. Fraunhoffer, J. Hayler, M. Hickey, S. Hughes, L. Humphreys, P. Richardson, M. Schober, A. Steven, T. White, S. Wuyts and J. Yin, Org. Process Res. Dev., 2017, 21, 153 CrossRef CAS.
- B. H. Lipshutz, J. Org. Chem., 2017, 82, 2806 CrossRef CAS PubMed.
- C. M. Gabriel, M. Kenner, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968 CrossRef CAS PubMed.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596 RSC.
- J. R. Dunetz, M. A. Berliner, Y. Xiang, T. L. Houck, F. H. Salingue, W. Chao, C. Yuandong, W. Shenghua, Y. Huang, D. Farrand, S. J. Boucher, D. B. Damon, T. W. Makowski, M. T. Barrila, R. Chen and I. Martínez, Org. Process Res. Dev., 2012, 16, 1635 CrossRef CAS.
- F. J. Weiberth, Y. Yu, W. Subotkowski and C. Pemberton, Org. Process Res. Dev., 2012, 16, 1967 CrossRef CAS.
- C. Sabot, K. A. Kumar, S. Meunier and C. Mioskowski, Tetrahedron Lett., 2007, 48, 3863 CrossRef CAS.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS.
- For a review on Ritter-type reactions, see: R. Bishop, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 6, p. 239 Search PubMed.
- B. Anxionnat, A. Guérinot, S. Reymond and J. Cossy, Tetrahedron Lett., 2009, 50, 3470 CrossRef CAS.
- S. Yaragorla, G. Singh, P. L. Saini and M. K. Reddy, Tetrahedron Lett., 2014, 55, 4657 CrossRef CAS.
- For examples, see: L. Audiger, K. Watts, S. C. Elmore, R. I. Robinson and T. Wirth, ChemSusChem, 2012, 5, 257 CrossRef CAS PubMed.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32 CrossRef CAS PubMed.
- R. N. Patel, Chapter 11. Applications of Biocatalysis for Pharmaceuticals and Chemicals, in Organic Synthesis using Biocatalysis, ed. A. Goswami and J. D. Stewart, Elsevier, Boston, USA, 2016, p. 339 Search PubMed.
- B. M. Dorr and D. E. Fuerst, Curr. Opin. Chem. Biol., 2018, 43, 127 CrossRef CAS PubMed.
- See recent reviews: (a) S. Jemli, D. Ayadi-Zouari, H. B. Hlima and S. Bejar, Crit. Rev. Biotechnol., 2016, 36, 246 CrossRef CAS PubMed; (b) M. Kaushik, P. Sinha, P. Jaiswal, S. Mahendru, K. Roy and S. Kukreti, J. Mol. Recognit., 2016, 29, 499 CrossRef CAS PubMed; (c) H. Yang, J. Li, H.-D. Shin, G. Du, L. Liu and J. Chen, Appl. Microbiol. Biotechnol., 2014, 98, 23 CrossRef CAS PubMed.
- M.-A. Légaré, M.-A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef CAS PubMed; C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef PubMed; W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef PubMed.
- M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef PubMed; K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef PubMed; K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef PubMed.
- K. Yuan, J.-F. Soulé and H. Doucet, ACS Catal., 2015, 5, 978 CrossRef CAS; F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef PubMed; J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef PubMed.
- Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS PubMed; X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed.
- T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871 CrossRef CAS PubMed.
- L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344 CrossRef CAS PubMed.
- H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC.
- Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097 CrossRef CAS PubMed.
- M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal, S. W. Krska and W. Shane, Chem. Soc. Rev., 2017, 45, 546 RSC.
- B. Su, T.-G. Zhou, P.-L. Xu, Z.-J. Shi and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 7205 CrossRef CAS PubMed; J. F. Hartwig, Acc. Chem. Res., 2017, 50, 549 CrossRef PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed.
- G.-J. Chang, P. Chen, T.-Y. Sun, X. Zhang, J.-Q. Yu and Y.-D. Wu, Chem. – Eur. J., 2015, 21, 11180 CrossRef CAS PubMed; K. S. L. Chan, H.-Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042 CrossRef PubMed; K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef PubMed.
- http://www.nsf-cchf.com CrossRef CAS PubMed; H. M. L. Davies and D. Morton, Angew. Chem., Int. Ed., 2014, 53, 10256 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- D. L. Dodds and D. J. Cole-Hamilton, Catalytic Reduction of Amides Avoiding LiAlH4 or B2H6, in Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, John Wiley & Sons, Inc., Hoboken, New Jersey, 2013, DOI:10.1002/9781118354520.ch01s.
- For a review see: D. S. Merel, M. L. T. Do, S. Gaillard, P. Dupau and J.-L. Renaud, Coord. Chem. Rev., 2015, 288, 50 CrossRef CAS.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed and ref. 6 within.
- O. O. Kovalenko, A. Volkov, F. Tinnis and H. Adolfsson, Org. Lett., 2015, 17, 446 CrossRef CAS PubMed.
- For a recent example see: A. Volkov, F. Tinnis, T. Slagbrand, I. Pershagena and H. Adolfsson, Chem. Commun., 2014, 50, 14508 RSC.
- For a recent example see: Y. Ogiwara, T. Uchiyama and N. Sakai, Angew. Chem., Int. Ed., 2016, 55, 1864 CrossRef CAS PubMed.
- For a recent example see: R. C. Chadwick, V. Kardelis, P. Lim and A. Adronov, J. Org. Chem., 2014, 79, 7728 CrossRef CAS PubMed.
- (a) S. Buchwald, Chem. Eng. News, 1993, 71, 2 CrossRef CAS; (b) S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 4971 CrossRef CAS.
- (a) S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032–15040 CrossRef CAS PubMed and H. Nagashima, Synlett, 2015, 26, 866 Search PubMed; (b) O. O. Kovalenko, A. Volkov, F. Tinnis and H. Adolfsson, Org. Lett., 2015, 17, 446 CrossRef CAS PubMed.
- For an example of heterogeneous hydrogenation in flow see: J. Coetzee, H. G. Manyar, C. Hardacre and D. J. Cole-Hamilton, ChemCatChem, 2013, 5, 2843 CrossRef CAS.
- J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinsky, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem. – Eur. J., 2013, 19, 11039 CrossRef CAS PubMed; T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217 CrossRef PubMed.
- J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432 RSC.
- O. Dipeolu, J. Gardiner and G. Stephens, Biotechnol. Lett., 2005, 27, 1803 CrossRef CAS PubMed.
- R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253 CrossRef CAS.
- Bismuth: H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef CAS PubMed ; Dodecylbenzenesulfonic acid: S. Shirakawa and S. Kobayashi, Org. Lett., 2007, 9, 311 CrossRef PubMed ; Gold: T. Ohshima, Y. Nakahara, J. Ipposhi, Y. Miyamotob and K. Mashima, Chem. Commun., 2011, 47, 8322 RSC ; Iron: P. Trillo, A. Baeza and C. Nájera, Eur. J. Org. Chem., 2012, 2929 CrossRef ; Phosphotungstic acid: G.-W. Wang, Y.-B. Shen and X.-L. Wu, Eur. J. Org. Chem., 2008, 4999 CrossRef.
- For a comprehensive review see: M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 48, 935 CrossRef CAS.
- A. Bunrit, C. Dahlstrand, S. K. Olsson, P. Srifa, G. Huang, A. Orthaber, P. J. R. Sjöberg, S. Biswas, F. Himo and J. S. M. Samec, J. Am. Chem. Soc., 2015, 137, 4646 CrossRef CAS PubMed.
- 2,2,2-Trifluoroethanol: P. Trillo, A. Baeza and C. Nájera, J. Org. Chem., 2012, 77, 7344 CrossRef CAS PubMed ; 1,1,1,3,3,3-Hexafluoro-isopropanol: F.-Z. Zhang, Y. Tian, G.-X. Li and J. Qu, J. Org. Chem., 2015, 80, 1107 CrossRef PubMed.
- Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC.
- F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862 CrossRef CAS PubMed; Y. Obora, ACS Catal., 2014, 4, 3972 CrossRef.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611 RSC; Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667 CrossRef CAS.
- K.-I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412 RSC.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086 CrossRef PubMed; T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381 CrossRef; S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483 CrossRef PubMed.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046 CrossRef CAS PubMed; G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300 CrossRef PubMed.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. B. David, N. A. E. Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298 CrossRef CAS PubMed; M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Chem. – Eur. J., 2016, 22, 12316 CrossRef PubMed; M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543 CrossRef PubMed; S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darce and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967 CrossRef PubMed; A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, J. Catal., 2017, 347, 57 CrossRef.
- See Table 2 in the ESI.†.
- Q. Xu, Q. Li, X. Zhu and J. Chen, Adv. Synth. Catal., 2013, 355, 73 CrossRef CAS; Q. Xu, J. Chen and Q. Liu, Adv. Synth. Catal., 2013, 355, 697 CrossRef.
- R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis and A. M. Poulton, J. Org. Chem., 2011, 76, 6749 CrossRef CAS PubMed.
- C. M. Vanos and T. H. Lambert, Angew. Chem., Int. Ed., 2011, 50, 12222 CrossRef CAS PubMed.
- J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993 RSC.
- X. Tang, C. Chapman, M. Whiting and R. Denton, Chem. Commun., 2014, 50, 7340 RSC.
- T. Koshizawa, Y. Yamazaki, T. Ohgiya and K. Shibuya, Tetrahedron, 2015, 71, 3231 CrossRef CAS.
- D. Hirose, T. Taniguchi and H. Ishibashi, Angew. Chem., Int. Ed., 2013, 52, 4613 CrossRef CAS PubMed.
- J. A. Buonomo and C. C. Aldrich, Angew. Chem., Int. Ed., 2015, 54, 13041 CrossRef CAS PubMed.
- M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470 CrossRef CAS PubMed; J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 7532 CrossRef PubMed.
- M. Zhuang and H. Du, Org. Biomol. Chem., 2014, 12, 4590 RSC.
- A. E. Putra, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6146 CrossRef.
- N. J. Oldenhuis, V. M. Dong and Z. Guan, J. Am. Chem. Soc., 2014, 136, 12548 CrossRef CAS PubMed.
- Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 1399 CrossRef CAS PubMed; Z.-Q. Rong, Y. Zhang, R. H. B. Chua, H.-J. Pan and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4944 CrossRef PubMed.
- B. I. Mobele, S. Venkatraman, G. McNaughton-Smith, C. Gibb, L. G. Ulysse, C. A. Lindmark, S. Shaw, B. Marron, K. Spear and M. J. Suto, Org. Process Res. Dev., 2012, 16, 1385 CrossRef CAS.
- M. A. Berliner, S. P. A. Dubant, T. Makowski, K. Ng, B. Sitter, C. Wager and Y. Zhang, Org. Process Res. Dev., 2011, 15, 1052 CrossRef CAS.
- J. Leonard, A. J. Blacker, S. P. Marsden, M. F. Jones, K. R. Mulholland and R. Newton, Org. Process Res. Dev., 2015, 19, 1400 CrossRef CAS.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525 CrossRef CAS PubMed.
- F.-F. Chen, Y.-Y. Liu, G.-W. Zheng and J.-H. Xu, ChemCatChem, 2015, 7, 3838 CrossRef CAS.
- M. P. Thompson and N. J. Turner, ChemCatChem, 2017, 9, 3833 CrossRef CAS.
- S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491 CrossRef CAS PubMed.
- Definition taken from: Z. Wang, K. Ding and Y. Uozumi, An Overview of Heterogeneous Asymmetric Catalysis, in Handbook of Asymmetric Heterogenous Catalysis, ed. K. Ding and Y. Uozumi, Wiley-VCH Verlag GmbH & Co., 2008, pp. 1–24 CrossRef CAS PubMed; See also: N. End and K.-U. Schöning, Top. Curr. Chem., 2004, 242, 241 CrossRef CAS PubMed.
- R. Nerozzi, Platinum Metals Rev., 2012, 56, 236 CrossRef.
- R. Ciriminna, V. Pandarus, F. Beland, Y.-J. Xu and M. Pagliaro, Org. Process Res. Dev., 2015, 19, 1554 CrossRef CAS.
- This discussion does not include immobilized enzymes. For a review on the extensive use of immobilized enyzmes in industry, see: R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC.
- See for example: A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790 RSC.
- For an example of this effect, see: S. Ötvös and F. Fülöp, Catal. Sci. Technol., 2015, 5, 4926 RSC.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3 CrossRef.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- For a discussion on the ideal recoverable catalyst, see: J. A. Gladysz, Pure Appl. Chem., 2001, 73, 1319 Search PubMed.
- B. Pugin and H.-U. Blaser, Top. Catal., 2010, 53, 953 CrossRef CAS.
- See for example: S. Desikan and L. K. Doraiswamy, Chem. Eng. Sci., 2000, 55, 6119 CrossRef CAS; J. O. Krause, S. Lubbad, O. Nuyken and M. R. Buchmeiser, Adv. Synth. Catal., 2003, 345, 996 CrossRef; J. S. Kinksbury, S. B. Garber, J. M. Giftos, B. L. Gray, M. M. Okamoto, R. A. Farrer, J. T. Fourkas and A. H. Hoveyda, Angew. Chem., Int. Ed., 2001, 40, 4251 CrossRef; K. M. Chepiga, Y. Feng, N. A. Brunelli, C. W. Jones and H. M. L. Davies, Org. Lett., 2013, 15, 6136 CrossRef PubMed.
- S. Desikan and L. K. Doraiswamy, Ind. Eng. Chem. Res., 1995, 34, 3524 CrossRef CAS.
- O. Pàmies, P. G. Andersson and M. Diéguez, Chem. – Eur. J., 2010, 16, 14232 CrossRef PubMed.
- M. Diéguez, P. G. Andersson and O. Pàmies, Ir-catalyzed hydrogenation of minimally functionalized olefins using phosphite-nitrogen ligands, in Innovative Catalysis in Organic Synthesis, ed. P. G. Andersson, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 153–165 Search PubMed.
- C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346 CrossRef CAS PubMed.
- J. Mazuela, J. J. Verendel, M. Coll, B. Schaffner, A. Borner, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2009, 131, 12344 CrossRef CAS PubMed.
- J. Mazuela, P.-O. Norrby, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2011, 133, 13634 CrossRef CAS PubMed.
- J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verevkin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971 CrossRef CAS PubMed.
- Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060 CrossRef CAS PubMed.
- G. Hou, R. Tao, Y. Sun, X. Zhang and F. Gosselin, J. Am. Chem. Soc., 2010, 132, 2124 CrossRef CAS PubMed.
- G.-H. Hou, J.-H. Xie, P.-C. Yan and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366 CrossRef CAS PubMed.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2009, 15, 2266 CrossRef CAS PubMed.
- C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2008, 130, 14450 CrossRef CAS PubMed.
- K. Matsumura, X. Zhang, K. Hori, T. Murayama, T. Ohmiya, H. Shimizu, T. Saito and N. Sayo, Org. Process Res. Dev., 2011, 15, 1130 CrossRef CAS.
- G. Hou, W. Li, M. Ma, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12844 CrossRef CAS PubMed.
- D. Steinhuebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316 CrossRef CAS PubMed.
- M. R. Friedfeld, M. Shevlin, G. W. Margulieux, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3314 CrossRef CAS PubMed.
- D. Chen, Y. Wangand and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475 CrossRef CAS PubMed.
- See for example: E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; C. D. Murphy and G. Sandford, Expert Opin. Drug Metab. Toxicol., 2015, 11, 589 CrossRef PubMed.
- “New Drugs at FDA: CDER's New Molecular Entities and New Therapeutic Biological Products”: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm (accessed December 2017).
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- Fluorinated aliphatic compounds and Fluorinated aromatic compounds, monographs, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Sons, Inc., 2014, Online ISBN: 9780471238966 Search PubMed; Fluorine compounds, organic, monograph, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley and Sons, Inc., 2014, Online ISBN: 9783527306732 Search PubMed.
- A. Harsanyi, A. Conte, L. Pichon, A. Rabion, S. Grenier and G. Sandford, Org. Process Res. Dev., 2017, 21, 273 CrossRef CAS.
- A dedicated issue of Chemical Reviews on fluorine chemistry was published in 2015 providing comprehensive state of the art reviews of a range of approaches: V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563 CrossRef CAS PubMed.
- See table 3 in ESI.†.
- Bretherick's Handbook of Reactive Chemical Hazards, ed. P. G. Urben, Elsevier, 7th edn, 2007, vol. 1–2, p. 641, Online ISBN: 978-0-0805-2340-8, entry 1678 Search PubMed.
- S. Haycock-Lewandowski, A. Wilder and J. Ahman, Org. Process Res. Dev., 2008, 12, 1094 CrossRef CAS.
- A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401 CrossRef PubMed.
- T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199 CrossRef CAS PubMed.
- G. Bellavance, P. Dube and B. Nguyen, Synlett, 2012, 569 CAS.
- F. Sladojevich, S. I. Arlow, P. Tang and T. Ritter, J. Am. Chem. Soc., 2013, 135, 2470 CrossRef CAS PubMed.
- M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571 CrossRef CAS PubMed.
- N. W. Goldberg, X. Shen, J. Li and T. Ritter, Org. Lett., 2016, 18, 6102 CrossRef CAS PubMed.
- H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050 CrossRef CAS PubMed.
- S. D. Schimler, S. J. Ryan, D. C. Bland, J. E. Anderson and M. S. Sanford, J. Org. Chem., 2015, 80, 12137 CrossRef CAS PubMed.
- D. W. Kim, H.-J. Jeong, S. T. Lim and M.-H. Sohn, Angew. Chem., Int. Ed., 2008, 47, 8404 CrossRef CAS PubMed.
- T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. L. Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991 CrossRef CAS PubMed.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed; T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804 Search PubMed.
- C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS PubMed; A. C. Sather and S. L. Buchwald, Acc. Chem. Res., 2016, 49, 2146 CrossRef PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634 CrossRef PubMed.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- H. Serizawa, K. Ishii, K. Aikawa and K. Mikami, Org. Lett., 2016, 18, 3686 CrossRef CAS PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536 CrossRef CAS PubMed.
- M. Hu, C. Ni, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496 CrossRef CAS PubMed; Z. Zhang, W. Yu, C. Wu, C. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 273 CrossRef PubMed.
- J.-H. Lin and J.-C. Xiao, Tetrahedron Lett., 2014, 55, 6147 CrossRef CAS; X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef PubMed.
- F. Venturini, W. Navarrini, A. Famulari, M. Sansotera, P. Dardani and V. Tortelli, J. Fluorine Chem., 2012, 140, 43 CrossRef CAS.
- M. Zhou, C. Ni, Z. He and J. Hu, Org. Lett., 2016, 18, 3754 CrossRef CAS PubMed.
- J.-B. Liu, C. Chen, L. Chu, Z.-H. Chen, X.-H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2015, 54, 11839 CrossRef CAS PubMed.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS.
- Name Reactions for Homologations, ed. J. J. Li, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2009(Pt. 1), p. 420 Search PubMed.
- (a) J. Westman, Org. Lett., 2001, 3, 3745 CrossRef CAS PubMed; (b) G. L. Thomas, C. Böhner, M. Ladlow and D. R. Spring, Tetrahedron, 2005, 61, 12153 CrossRef CAS.
- https://www.acs.org/content/acs/en/greenchemistry/research-innovation/tools-for-green-chemistry/medchem-tips-and-tricks.html (accessed 2017).
- Supported arsine and telluride reagents have been described for Wittig-type reactions but will not be covered here: Arsenic: P. Wang, C.-R. Liu, X.-L. Sun, S.-S. Chen, J.-F. Li, Z. Xie and Y. Tang, Chem. Commun., 2012, 48, 290 RSC ; Tellurium: Z.-Z. Huang, S. Ye, W. Xia and Y. Tang, Chem. Commun., 2001, 1384 RSC.
- N. Shimojuh, Y. Imura, K. Moriyama and H. Togo, Tetrahedron, 2011, 67, 951 CrossRef CAS.
- Catalytic Wittig-type reactions were first described with trialkylarsine and dialkyltelluride-based reagents nearly thirty years ago, however, these variants present obvious health concerns and will not be covered here. Arsenic: L. Shi, W. Wang, Y. Wang and Y. Huang, J. Org. Chem., 1989, 54, 2027 CrossRef CAS ; Tellurium: X. Huang, L. Xie and H. Wu, Tetrahedron Lett., 1987, 28, 801 CrossRef.
- (a) C. J. O'Brien, J. L. Tellez, Z. S. Nixon, L. J. Kang, A. L. Carter, S. R. Kunkel, K. C. Przeworski and G. A. Chass, Angew. Chem., Int. Ed., 2009, 48, 6836 CrossRef PubMed; (b) C. J. O'Brien, F. Lavigne, E. E. Coyle, A. J. Holohan and B. J. Doonan, Chem. – Eur. J., 2013, 19, 5854 CrossRef PubMed; (c) C. J. O'Brien, Z. S. Nixon, A. J. Holohan, S. R. Kunkel, J. L. Tellez, B. J. Doonan, E. E. Coyle, F. Lavigne, L. J. Kang and K. C. Przeworski, Chem. – Eur. J., 2013, 19, 15281 CrossRef PubMed; (d) E. E. Coyle, B. J. Doonan, A. J. Holohan, K. A. Walsh, F. Lavigne, E. H. Krenske and C. J. O'Brien, Angew. Chem., Int. Ed., 2014, 53, 12907 CrossRef CAS PubMed; (e) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2014, 6630 CrossRef CAS; (f) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2014, 6873 CrossRef CAS; (g) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2015, 3286 CrossRef CAS; (h) M.-L. Schirmer, S. Adomeit and T. Werner, Org. Lett., 2015, 17, 3078 CrossRef CAS PubMed; (i) M.-L. Schirmer, S. Adomeit, A. Spannenberg and T. Werner, Chem. – Eur. J., 2016, 22, 2458 CrossRef CAS PubMed.
- H. A. van Kalkeren, F. L. van Delft and F. P. J. T. Rutjes, ChemSusChem, 2013, 6, 1615 CrossRef CAS PubMed.
- H. A. van Kalkeren, A. L. Blom, F. P. J. T. Rutjes and M. A. J. Huijbregts, Green Chem., 2013, 15, 1255 RSC.
- S. Rommel, C. Belger, J.-M. Begouin and B. Plietker, ChemCatChem, 2015, 7, 1292 CrossRef CAS.
- (a) J. Lu and P. H. Toy, Chem. – Asian J., 2011, 6, 2251 CrossRef CAS PubMed; (b) L. Chen, Y. Du, X.-P. Zeng, T.-D. Shi, F. Zhou and J. Zhou, Org. Lett., 2015, 17, 1557 CrossRef CAS PubMed.
- J.-J. Cao, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4976 CrossRef CAS PubMed.
- (a) A. Mondal, D. Mukherjee, B. Adhikary and M. A. Ahmed, J. Nanopart. Res., 2016, 18, 117 CrossRef; (b) C. Santra, A. Aurouxand and B. Chowdhury, RSC Adv., 2016, 6, 45330 RSC.
- M. Mahyari, Y. Bide and J. N. Gavgani, Appl. Catal., A, 2016, 517, 100 CrossRef CAS.
- A. T. Murray, M. J. H. Dowley, F. Pradaux-Caggiano, A. Baldansuren, A. J. Fielding, F. Tuna, C. H. Hendon, A. Walsh, G. C. Lloyd-Jones, M. P. John and D. R. Carbery, Angew. Chem., Int. Ed., 2015, 54, 8997 CrossRef CAS PubMed.
- A. T. Murray, R. King, J. V. G. Donnelly, M. J. H. Dowley, F. Tuna, D. Sells, M. P. John and D. R. Carbery, ChemCatChem, 2016, 8, 510 CrossRef CAS.
- K. Marui, A. Nomoto, H. Akashi and A. Ogawa, Synthesis, 2016, 48, 31 CAS.
- A. Gavriilidis, A. Constantinou, K. Hellgardt, K.-K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595 RSC.
- Regulation (EC) 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH); Candidate List of Substances of Very High Concern for Authorization, http://echa.europa.eu/candidate-list-table. (accessed December 2017).
- For a good discussion of the potential impact of REACH on dipolar aprotic solvents and an explanation of the technical terms Restriction and Authorisation see: L. Berkamp and N. Herbatschek, Rev. Eur. Community Int. Env. Law, 2014, 23, 221 CrossRef.
- C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740 CrossRef CAS.
- For an introduction to the REACH registration process and testing requirements see: https://echa.europa.eu/regulations/reach/registration (accessed June 2018).
- P. Tundo, A. Arico, G. Gauthier, L. Rossi, A. Rosamilia, H. S. Bevinakatti, R. L. Sievert and C. P. Newman, ChemSusChem, 2010, 3, 566 CrossRef CAS PubMed.
- S. Lawrenson, M. North, F. Peogneguy and A. Routledge, Green Chem., 2017, 19, 952 RSC.
- (a) R. L. De Villez, US Patent, 4923900, 1990 Search PubMed; (b) R. L. De Villez, US Patent, 5086075, 1992 Search PubMed.
- C. M. Adler, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC.
- K. R. Stone, Environmental Profile for Propylene Carbonate, U.S. Environmental Protection Agency, 1998 Search PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966 RSC.
- J. Bayardon, J. Holz, B. Schaeffer, V. Andrushko, S. Verevkin, A. Preetz and A. Boerner, Angew. Chem., Int. Ed., 2007, 46, 5971 CrossRef CAS PubMed.
- B. Schaeffner, J. Holz, S. Verevkin and A. Boerner, ChemSusChem, 2008, 1, 249 CrossRef CAS PubMed.
- H. L. Parker, J. Sherrad, A. J. Hoat and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739 CrossRef CAS.
- K. Koseki, T. Ebata, H. Kawakami, H. Matushita, K. Itoh and Y. Naoi, US Pat, 5112994, 1992 Search PubMed.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, R. C. McElroy, T. J. Farmer, T. Duncan, W. Raverts, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650 RSC.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005 CrossRef CAS PubMed.
- J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmer and A. J. Hunt, Green Chem., 2016, 18, 3990 RSC.
- The 50.0% figure for dichloromethane for years 2005–2008 does not mean that 50.0 of steps use dichloromethane but rather that a synthesis will be made up of several steps and in 50% of these syntheses at least one step will use dichloromethane.
- For an up to date list of chemicals which are subject to authorisation under REACH legislation see the following link. https://echa.europa.eu/information-on-chemicals/registered-substances (accessed December 2017).
- VOC Solvent Emissions Directive 199/13/EC (11th March 1999).
- P. M. Schlosser, A. S. Bale, C. F. Gibbons, A. Wilkins and G. S. Cooper, Environ. Health Perspect., 2015, 123, 114 CrossRef CAS PubMed.
- K. Brown, K. Budzinski, S. Ho, J. Millard, J. Johnson, P. Dahlin, B. Owen and R. Kottmeier, Toward Sustainable Engineering Practices in Biologics Manufacturing, Bioprocess International, Dec. 18th, 2015 Search PubMed.
- J. Thundimadathil, Specialty Chemicals Magazine, 2013, p. 26 Search PubMed.
- S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685 RSC.
- Now ChemSInC: https://www.linkedin.com/pulse/introducing-chemsinc-chemical-synthesis-industries-john-leonard (accessed December 2017).
- https://www.acs.org/content/acs/en/greenchemistry/industry-business/pharmaceutical.html .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc01276h |
| ‡ Retired. |
| This journal is © The Royal Society of Chemistry 2018 |