
Unveiling the role of choline chloride in furfural synthesis from highly concentrated feeds of xylose†
S.
Jiang,  a
C.
Verrier,
b
M.
Ahmar,
b
C.
Ma,
c
Y.
Queneau,
b
M.
Pera-Titus,
c
F.
Jérôme
a
and
K.
De Oliveira Vigier
*a
a
C.
Verrier,
b
M.
Ahmar,
b
C.
Ma,
c
Y.
Queneau,
b
M.
Pera-Titus,
c
F.
Jérôme
a
and
K.
De Oliveira Vigier
*a
a
C.
Verrier,
b
M.
Ahmar,
b
C.
Ma,
c
Y.
Queneau,
b
M.
Pera-Titus,
c
F.
Jérôme
a
and
K.
De Oliveira Vigier
*a
Abstract
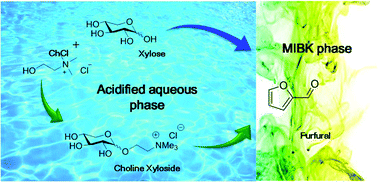
Furfural is a biomass derived compound used for the synthesis of fuels and chemicals. Herein we show that choline chloride allows the conversion of highly concentrated feeds of xylose (up to 50 wt%) to furfural (up to 75%) and that it can be recycled. Such a beneficial effect was observed from the formation of a choline xyloside intermediate exhibiting higher reactivity than xylose.
 Please wait while we load your content...
Please wait while we load your content...

