
A call to (green) arms: a rallying cry for green chemistry and engineering for CO2 capture, utilisation and storage
 *a
and
David J.
Heldebrant
*b
*a
and
David J.
Heldebrant
*b
Abstract
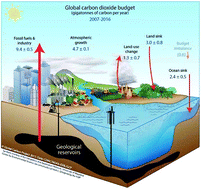
Chemists, engineers, scientists, lend us your ears… Carbon capture, utilisation, and storage (CCUS) is among the largest challenges on the horizon and we need your help. In this perspective, we focus on identifying the critical research needs to make CCUS a reality, with an emphasis on how the principles of green chemistry (GC) and green engineering can be used to help address this challenge. We identify areas where GC principles can readily improve the energy or atom efficiency of processes or reduce the environmental impact. Conversely, we also identify dilemmas where the research needs may be at odds with GC principles, and present potential paths forward to minimise the environmental impacts of chemicals and processes needed for CCUS. We also walk a different path from conventional perspectives in that we postulate and introduce potential innovative research directions and concepts (some not yet experimentally validated) in order to foster innovation, or at least stoke conversation and question why certain approaches have not yet been attempted. With elements of historical context, technological innovation, critical thinking, and some humour, we issue a call to arms and hope you may join us in this fight.
- This article is part of the themed collections: Needs of Green Chemistry and 2018 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

