
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of polysaccharide-based copolymers†
F.
Grimaud
 a,
P.
Faucard
b,
L.
Tarquis
a,
S.
Pizzut-Serin
a,
P.
Roblin
c,
S.
Morel
a,
S.
Le Gall
b,
X.
Falourd
b,
A.
Rolland-Sabaté
d,
D.
Lourdin
b,
C.
Moulis
a,
M.
Remaud-Siméon
a and
G.
Potocki-Veronese
*a
a,
P.
Faucard
b,
L.
Tarquis
a,
S.
Pizzut-Serin
a,
P.
Roblin
c,
S.
Morel
a,
S.
Le Gall
b,
X.
Falourd
b,
A.
Rolland-Sabaté
d,
D.
Lourdin
b,
C.
Moulis
a,
M.
Remaud-Siméon
a and
G.
Potocki-Veronese
*a
aLISBP, CNRS, INRA, INSAT, Université de Toulouse, F-31400 Toulouse, France. E-mail: veronese@insa-toulouse.fr; Fax: +33 (0)561 559 400
bUR1268 Biopolymères Interactions Assemblages, INRA, F-44300 Nantes, France
cUniversité de Toulouse, LGC UMR 5503 (CNRS/UPS/INPT), 118 route de Narbonne, 31062 Toulouse, France
dUMR408 Sécurité et Qualité des Produits d'Origine Végétale, INRA, Université Avignon, F-84000 Avignon, France
First published on 7th August 2018
Abstract
The design of enzymatic routes for the production of biosourced copolymers represents an attractive alternative to chemical synthesis from fossil carbon. In this paper, we explore the potential of glycosynthesizing enzymes to produce novel block copolymers composed of various covalently-linked α-glucans with contrasting structures and physicochemical properties. To this end, various glucansucrases able to synthesize α-glucans with different types of α-osidic bonds from sucrose were tested for their ability to elongate oligosaccharide and polysaccharide acceptors with different structures from the native polymer synthesized by each enzyme. We showed that two enzymes – namely, the alternansucrase from Leuconostoc mesenteroides NRRL B-1355 (specific for α(1 → 6)/α(1 → 3)-linked alternan synthesis) and the dextransucrase DSR-MΔ1 from Leuconostoc citreum NRRL B-1299 (specific for α(1 → 6)-linked dextran formation) – were able to elongate α(1 → 4)-linked amylose and α(1 → 6)/α(1 → 3)-linked alternan respectively. Carrying out stepwise acceptor reactions, and after optimization of the acceptor size and donor/acceptor ratio, two types of diblock copolymers were synthesized – a dextran-b-alternan and an alternan-b-amylose – as well as the triblock copolymer dextran-b-alternan-b-amylose. Their structural characterization, performed by combining chromatographic, NMR and permethylation analyses, showed that the copolymer polymerization degree ranged from 29 to 170, which is the highest degree of polymerization ever reported for an enzymatically synthesized polysaccharide-based copolymer. The addition of dextran and alternan blocks to amylose resulted in conformational modifications and related flexibility changes, as demonstrated by small angle X-ray scattering.
1. Introduction
Due to their carbon neutrality and renewability, biosourced polymers are seen as an attractive alternative to synthetic polymers derived from fossil carbon when it comes to producing a wide range of materials. However, the intrinsic properties of natural polymers, for example in terms of rigidity and solubility, are often insufficient to meet functionality requirements for most non-food uses. In this context, the design of block copolymers is a promising strategy. Indeed, the physicochemical properties of polymers directly derive from their molecular structure and supramolecular organization, which vary considerably based on the nature of the covalently assembled building blocks that they are constructed from. Until now, the design of polysaccharide-based copolymers was mainly focused on the combination of cellulose, amylose, dextran, alginate or chitosan building blocks with synthetic blocks for obtaining hybrid materials used as surfactants, chromatography supports, or controlled release systems.1–5 Very few studies have looked at the synthesis of copolymers formed solely of blocks of different polysaccharides.6,7 Moreover, in most of these cases, the grafting of polysaccharides (or polysaccharide primers) onto synthetic or polysaccharidic blocks was achieved by chemical reaction8 or polymerization from chemically modified monomers.9 The in vivo or in vitro enzymatic synthesis of biosourced copolymers has also scarcely been investigated. Yadav et al.10 demonstrated the potential of using a glycosyltransferase for the microbial production of modified bacterial cellulose (MBC) with increased biodegradability properties. In this case, the MBC was a statistical copolymer rather than a block copolymer, since the glucosyl (Glc) and N-acetyl-glucosaminyl (GlcNAc) units were incorporated on a random basis by cellulose synthase from Gluconacetobacter xylinus, which uses both UDP-Glc and UDP-GlcNAc as the glycosyl donor. Using the same principle, another type of glycan-synthesizing enzyme, α(1 → 4) glucan phosphorylase (SP, EC 2.4.1.1) from Aquifex aeolicus VF5, belonging to the GT35 family of carbohydrate active enzymes (CAZy11), was tested for the in vitro enzymatic synthesis of hybrid polysaccharides.12–15 The substrate promiscuity of this enzyme towards glycosyl donors (α-D-Glc-1-phosphate, α-D-GlcNAc-1-phosphate and α-D-Man-1-phosphate) was exploited to produce non-natural polysaccharides such as hetero-mannosides and aminopolysaccharides.13–15 The three sugar-phosphate donors were used in the presence of a maltotriose acceptor to obtain different copolymers with maltotriose at the reducing end, on which were grafted α(1 → 4)-mannan, α(1 → 4)-linked glucosaminoglucans or heteromannans (composed of either Glc and GlcNAc units, or Glc and Man units). Nevertheless, the degree of polymerization of the products was limited to 30 residues,14 with a very short block of only three glucosyl residues at the reducing end. Moreover, the impact of the Glc/GlcN or Glc/Man composition on the polymer chain conformation, and therefore its physicochemical properties, was not investigated.Retaining transglucosylases, which catalyse polymerization reactions from non-nucleotide sugars, represent another category of glycan-synthesising enzymes that could be of interest in the production of block copolymers. Some α-transglucosylases, such as dextran dextrinases16–18 (DDase; EC2.4.1.2; CAZy family GH15), 4,3-α-glucanotransferase19 (4,3-α-GTase; E.C. 2.4.1.-; CAZy family GH70) and 4,6-α-glucanotransferase20,21 (4,6-α-GTase; E.C. 2.4.1.-; CAZy family GH70), have the ability to cleave the α(1 → 4)-linkages of maltooligosaccharides (MOSs) and successively transfer the glucosyl units from one MOS chain to an acceptor molecule. If the acceptor molecule is another MOS chain, α(1 → 6) (DDase and 4,6-α-glucanotransferase)16–19 or α(1 → 3) (4,3-α-glucanotransferase) linkages can be formed.19 Besides, the 4,6-α-GTase from Lactobacillus reuteri 121 has been used to produce isomalto/malto-polysaccharides (IMMPs) with a DP of up to 30.22 These products contain one or more α(1 → 4)-linked glucosyl units at the reducing end, and at least one α(1 → 6) linkage at the non-reducing end. However, there are other α-transglucosylases, such as glucansucrases (GSs) from the GH13 and GH70 families, which also appear to be very interesting candidates for producing block copolymers. GSs use sucrose, a low cost agroresource, as the glucosyl donor, and catalyse the formation of homopolymers of α-D-glucosyl units. Depending on their characteristics, a wide variety of α-glucans with different molar masses, as well as with different distributions, numbers or types of glucosidic linkage (namely, α(1 → 2), α(1 → 3), α(1 → 4) and/or α(1 → 6)), can be obtained.23–25
Of the GSs, dextransucrases (DS; E.C. 2.4.1.5; CAZy family GH70) catalyse the synthesis of dextrans that are α-glucans of molar masses ranging from 104 to 109 g mol−1 with at least 50% α(1 → 6) linkages and various branching points. Recently, a recombinant dextransucrase from Leuconostoc citreum NRRL B-1299 (DSR-M) with a rather unusual specificity was characterized. Unlike other dextransucrases, which usually produce very high molar mass polymers, this enzyme produces only low molar mass linear α(1 → 6)-linked dextran.26,27 Alternansucrase (ASR; E.C. 2.4.1.140; CAZy family GH70) specificity is also remarkable due to the mixture of α(1 → 6) and α(1 → 3) linkages found in the main polymer chain. The first ASR to be characterized, isolated from L. mesenteroides NRRL B-1355, produces a high molar mass alternan,28 4 × 107 g mol−1 in size,29 containing 58% α(1 → 6) and 42% α(1 → 3) linkages28 mainly alternated. This alternan displays interesting properties such as a high solubility in water, low viscosity and resistance to dextranase degradation.30 A third example is amylosucrases (AS; E.C. 2.4.1.4; CAZy family GH13), which catalyse the synthesis of an insoluble amylose-like polysaccharide containing exclusively α(1 → 4)-linked glucosyl residues from sucrose,31 the size of which does not exceed 2 × 104 g mol−1.32 The potential of amylosucrases has also been extensively investigated in terms of the synthesis of hyperbranched α-glucans and glycodendrimers through elongation of native glycogen chains.33,34 In addition to polymer formation, GSs can also transfer the glucosyl unit of sucrose to a large variety of carbohydrate acceptors, including mono-, di- and trisaccharides such as galactose, xylose, maltose, isomaltose, mannose, maltotriose, panose, cellobiose, nigerose, lactose, raffinose and melibiose, as well as aglycon molecules such as polyols or flavonoids.25,35–39 However, there are very few publications describing the glucosylation using GSs of oligosaccharides with a DP higher than 3. The first example of this involved the strict elongation of maltooligosaccharides (MOSs) with a DP between 2 and 8 by the GTF-I and GTF-S enzymes from S. mutans 6715![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 and the dextransucrase from L. mesenteroides NRRL B-512FM.41 Only up to six glucosyl units could be added to the acceptor with a DP of 8. Although the physicochemical properties of these oligomers were not characterized, these findings reveal that GSs can be used for gluco-co-oligomer formation. In this study, we have gone one step further and explored various enzymatic routes for the production of gluco-copolymers composed of di- and tri-α-glucan blocks that differ in terms of their linkage types and physical properties. To this end, various combinations of glucansucrases and acceptor blocks were screened, and the synthesis conditions optimized. The new block copolymers that were produced were then characterized in detail in order to determine their structure and conformation in solution.
40 and the dextransucrase from L. mesenteroides NRRL B-512FM.41 Only up to six glucosyl units could be added to the acceptor with a DP of 8. Although the physicochemical properties of these oligomers were not characterized, these findings reveal that GSs can be used for gluco-co-oligomer formation. In this study, we have gone one step further and explored various enzymatic routes for the production of gluco-copolymers composed of di- and tri-α-glucan blocks that differ in terms of their linkage types and physical properties. To this end, various combinations of glucansucrases and acceptor blocks were screened, and the synthesis conditions optimized. The new block copolymers that were produced were then characterized in detail in order to determine their structure and conformation in solution.
2. Experimental
Enzyme sources
The amylosucrase gene from Deinoccocus geothermalis DSM11300 (DgAS) (accession number ABF44874) was amplified by means of PCR from a pGST-DgAS plasmid template42 using the two following primers: 5′-CACCATGCTGAAAGACGTGCTCACTTCTG-3′ as the forward primer and 5′-TGCTGGAGCCTCCCCGGCGGTCAGC-3′ as the reverse primer. The addition of a CACC sequence to the 5′-forward primer allowed the gene to be correctly inserted into the pENTR/D-TOPO® vector (Life Technologies). The recombinant gene was then transferred to the pET-53-DEST (Novagen) destination vector using the Gateway® LR Clonase® II enzyme mix (Life Technologies). Recombinant clones were selected on Luria–Bertani (LB) agar plates supplemented with 100 μg mL−1 of ampicillin. Plasmids were extracted using the Sigma-Aldrich GenElute HP Plasmid Miniprep kit, and verified using restriction analyses and sequencing (GATC Biotech). DgAS was produced as a (His)6-DgAS fusion protein by the recombinant E. coli BL21 star strain carrying the pET-53-DEST-DgAS plasmid, after 24 hours in a ZYM 5052 medium at 28 °C. The alternansucrase from Leuconostoc mesenteroides NRRL B-1355 (ASR) was produced as a (His)6-ASR fusion protein by the recombinant E. coli BL21 strain carrying the pBad asr C-APY-del Δthio plasmid.28 The dextransucrase from Leuconostoc citreum NRRL B-1299 (DSR-M Δ1) was produced as a (His)6-DSR-M Δ1 fusion protein by the recombinant E. coli BL21 star strain carrying the pET-53-DEST-dsrMΔ1 plasmid.27 (His)6-DgAS, (His)6-ASR and (His)6-DSR-M Δ1 were further purified by means of affinity chromatography using ProBond nickel-charged resin (Invitrogen), following the manufacturer's instructions, as described elsewhere.27,42 In order to make the reading of this study easier, (His)6-DgAS, (His)6-ASR and (His)6-DSR-M Δ1 will be referred to hereafter as DgAS, ASR and DSR-M respectively.Enzyme activity assays
One unit of glucansucrase corresponds to the amount of enzyme that catalyses the production of one μmol of fructose per minute under the assay conditions. Assays involving ASR and DSR-M were performed at 30 °C in a 50 mM sodium acetate buffer at pH 5.75 with 100 g L−1 sucrose, while assays involving amylosucrase (DgAS) were performed at 50 °C in a 50 mM Tris-HCl buffer at pH 7.0 with 100 g L−1 sucrose. The concentration of the fructose released during the reactions was determined using the dinitrosalycilic (DNS) acid method,43 using fructose as the standard.Production of acceptors
Maltooligosaccharides and the amylose block were synthesized in vitro from sucrose and maltose using the purified recombinant DgAS, under the same conditions as those described for NpAS.44 The synthesis reaction was performed at 50 °C for a period of 24 hours in a 50 mM Tris-HCl buffer at pH 7.0, from 600 mM sucrose as the glucosyl donor and 100 mM maltose as the acceptor, using 1 U mL−1DgAS. Oligoalternans and the low molar mass (LMM) alternan block were synthesized in vitro using the purified ASR.28 This synthesis reaction was performed at 30 °C for a period of 24 hours in a 50 mM sodium acetate buffer at pH 5.75, from 1 M sucrose as the sole substrate, using 1 U mL−1 ASR. As described elsewhere,28 under these conditions, ASR synthesizes three α-glucan populations: the HMM alternan (around 107 g mol−1), the LMM alternan, and oligoalternans. Isomaltooligosaccharide (dextran 1500 g mol−1) was purchased from Sigma-Aldrich. In order to prepare the blocks that would serve as acceptors for elongation, maltooligosaccharides, short amylose, isomaltooligosaccharides, oligoalternans, and LMM alternans were subsequently fractionated by means of preparative size-exclusion chromatography (SEC) using a 3L column of Biogel P6DG (Biorad). Sugars were eluted with water at 60 °C using a flow rate of 10 mL min−1. High-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) was used to determine the DP distribution of each fraction.Oligosaccharide elongation and copolymer synthesis
Reactions were performed at 30 °C, in a 50 mM sodium acetate buffer at pH 5.75 containing 1 U mL−1 of purified ASR or DSR-M, with acceptor concentrations between 10 and 50 g L−1 and sucrose concentrations between 50 and 150 g L−1. The reaction media were stirred at 30 °C for 24 hours to allow total sucrose consumption. After 24 hours, the reaction media were heated for five minutes at 95 °C to stop the reaction. Two controls were included in the experiment. The first relates to the enzyme with sucrose and the second relates to the enzyme in the presence of the acceptor. The second control was used to determine whether the enzyme was able to modify the acceptor in the absence of sucrose. After a reaction period of 24 hours, the acceptor modification (i.e. elongation) was checked using HPAEC-PAD.Copolymer purification
The reaction products obtained from sucrose and the amylose block acceptor using ASR were purified by means of preparative SEC, as described in the previous section. The reaction products obtained from sucrose and the alternan block acceptor using DSR-M were purified by means of preparative SEC using a HiPrep 26/60 column of Sephacryl S200 HR (GE Healthcare). Sugars were eluted at 25 °C in water using a flow rate of 1.5 mL min−1. The reaction products obtained from sucrose and the alternan-b-amylose acceptor using DSR-M were purified using a semi-preparative 9 × 250 mm Dionex Carbopac PA100 column. A gradient of sodium acetate in 150 mM NaOH was applied at a flow rate of 2.5 mL min−1 as follows: 0–10 min, 0–200 mM; 10–15 min, 200–200 mM; 15–30 min, 200–300 mM. Carbohydrate detection was performed in the same way as analytic HPAEC-PAD (see below). After HPAEC-PAD separation, the samples were desalted online using a carbohydrate membrane desalter (CMD; Dionex) according to the manufacturer's recommendations. The resulting purified polymer fractions were freeze-dried.α-Glucan structure analysis
The percentage of the alternan-b-amylose copolymer made up of the amylose block was determined as follows: % of the amylose block = % of α(1 → 4) linkages in alternan-b-amylose. Similarly, the percentage of the dextran-b-alternan copolymer made up of the alternan block was determined using the following formula: % of the alternan block = (% of α(1 → 3) linkages in alternan-b-dextran)/(% of α(1 → 3) linkages in the alternan block) × 100. With regard to the triblock dextran-b-alternan-b-amylose, the following formulae were used: % of amylose in the triblock = % of α(1 → 4) linkages in the dextran-b-alternan-b-amylose; % of the alternan block in the triblock = [(% of α(1 → 3) linkages in dextran-b-alternan-b-amylose)/(% of α(1 → 3) in alternan-b-amylose)] × [(% of α(1 → 3) in alternan-b-amylose + % of α(1 → 6) in alternan-b-amylose)]/100; % dextran block in the triblock = 100 − % alternan block − % amylose block.
The proportion of each type of osidic linkage was calculated as follows:
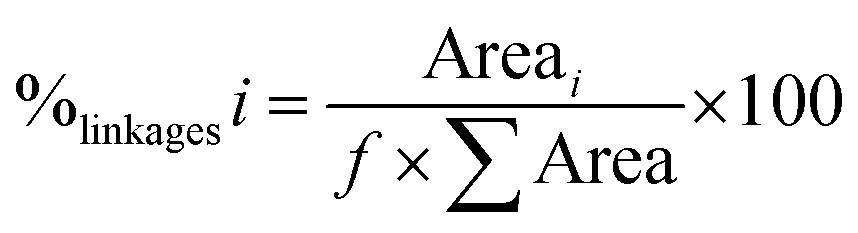 sinθ/λ, and the scattering angle was taken to be 2θ. The samples were circulated in a thermostated quartz capillary with a diameter of 1.5 mm and a wall thickness of 10 μm, which had been placed in a vacuum chamber.
sinθ/λ, and the scattering angle was taken to be 2θ. The samples were circulated in a thermostated quartz capillary with a diameter of 1.5 mm and a wall thickness of 10 μm, which had been placed in a vacuum chamber.
In order to obtain the oligosaccharide form factor, concentrated solutions of amylose, alternan-b-amylose and dextran-b-alternan-b-amylose (50 μL at 50 mg mL−1) were injected into a size exclusion column (BIOSEC, Agilent) using an Agilent high-performance liquid chromatography (HPLC) system, and eluted directly into the SAXS flow-through capillary cell at a flow rate of 200 μL min−1.
SAXS data were collected online for the duration of the elution time, with a time frame of 1 s, and a dead time between frames of 0.5 s. For each sample, the frames corresponding to the main elution peak were checked for the stability of the associated radius of gyration and global curve shape, and the resulting selection of curves was averaged, as described elsewhere.49 Selected frames corresponding to the main elution peak were averaged using FOXTROT, a custom homemade application. A large number of frames were collected during the first minutes of elution, and these were averaged to account for buffer scattering, which was subsequently subtracted from the signal during the elution of the polymer. Data reduction to absolute units, frame averaging, and subtraction were carried out using FOXTROT.
3. Results and discussion
Three different GSs, specific for the synthesis of α-glucans of various structures and properties, were tested for copolymer synthesis: the DSR-M dextransucrase from Leuconostoc citreum NRRL B-1299, the ASR alternansucrase from L. mesenteroides NRRL B-1355 and the DgAS amylosucrase from Deinoccocus geothermalis DSM11300. The approach consisted of using one GS to elongate the α-glucans produced by the other two. In order to select the best GS/α-glucan combination, we first tested the ability of each enzyme to glucosylate short oligosaccharides, whose structure differed from that of their natural acceptors: α(1 → 6)-linked (IMOS), α(1 → 6)/α(1 → 3)-linked (OAL) and α(1 → 4)-linked (MOS) oligosaccharides for DSR-M, ASR and DgAS respectively. Six acceptor reactions were carried out and the reaction products were analysed by means of HPAEC-PAD. A given enzyme was considered to be able to glucosylate a given acceptor if acceptor conversion into new products was observed, which was determined by the presence of new peaks in the HPAEC chromatogram. As can be seen from Table 1, only the three couples DSR-M/OAL, ASR/MOS and ASR/IMOS produced positive results. The acceptor reaction conditions were then optimized by varying the acceptor concentration from 10 to 50 g L−1, and the sucrose concentration from 50 to 150 g L−1. The best acceptor glucosylation was always obtained using an acceptor/donor mass ratio of 0.15. These conditions were selected for the next experiments. Almost total acceptor consumption, alongside the appearance of new peaks in the HPAEC-PAD chromatograms (Fig. 1), proved that covalent grafting had occurred. Similar results were obtained for the three couples being tested, providing the first piece of evidence to suggest that some GSs are able to glucosylate α-linked glucooligosaccharide acceptors with DPs > 8 and structures different from those of the enzyme's natural acceptor.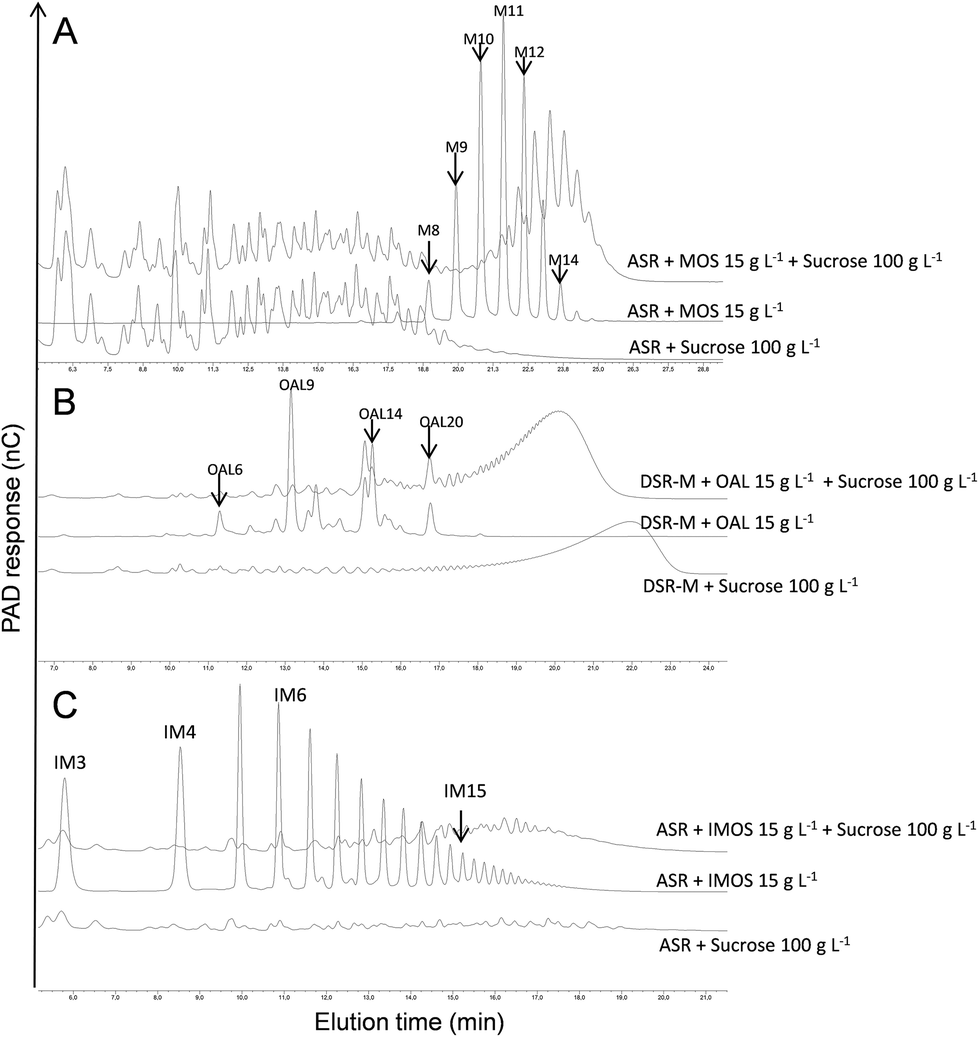 | ||
| Fig. 1 HPAEC-PAD chromatograms of the acceptor reaction media involving the following couples: ASR/maltooligosaccharides (A), DSR-M/oligo-alternan (B), and ASR/isomaltooligosaccharides (C). Two controls are also presented below the acceptor reaction products, these being (i) the enzyme with sucrose alone, and (ii) the enzyme with the acceptor alone. Only chromatograms corresponding to the final reaction times are reported here. For the control ‘enzyme + acceptor’, it was verified that the chromatograms obtained at the initial and final reaction times were identical. | ||
| Enzyme | Product from sucrose only | Acceptorsa | ||
|---|---|---|---|---|
| MOSb DP 8–14 | OALc DP 6–20 | IMOSd DP 1–25 | ||
| a The reactions resulting in the elongation of the acceptors are indicated by the symbol “+”. b Maltooligosaccharides (MOS) with 100% α(1 → 4) linkages. c Oligo-alternan (OAL) with 63% α(1 → 6) and 37% α(1 → 3) linkages. d Isomaltooligosaccharides (IMOS) with 95% α(1 → 6) and 5% α(1 → 3) linkages. Linkage percentages determined by 1H proton NMR. | ||||
| ASR | Alternan (58% of α(1 → 6) and 42% of α(1 → 3)) | + | n.d. | + |
| DSR-M | Dextran (100% of α(1 → 6)) | − | + | n.d. |
| DgAS | Amylose (100% of α(1 → 4)) | n.d. | − | − |
Only MOSs of up to DP 8 were indeed shown to be glucosylated with GTF-I and GTF-S from S. mutans 6715,40 and the dextransucrase DSR-S from L. mesenteroides NRRL-B-512FM.41 Subsequently, with a view to synthesizing copolymers, and not just oligosaccharides with new structures, the potential of DSR-M and ASR to act on α-glucans with higher DPs was investigated. Accordingly, α-glucan acceptors of intermediate molar mass were prepared that could be easily separated from the natural polymers synthesized by the glucansucrases selected to perform the elongation. At this stage, and in order to limit the number of combinations, we selected the following couples: DSR-M/OAL and ASR/MOS.
Amylose block synthesis and elongation using ASR
DgAS was used to produce α(1 → 4)-glucan fractions with suitable DPs, which would be tested as primers for copolymer synthesis with ASR. The synthetic reaction was performed using a sucrose donor/maltose acceptor ratio of 6. The resulting amylose-like products with DPs between 14 and 23 were fractionated by means of SEC. This fraction (DP 14–23) was further incubated with ASR and 100 g L−1 sucrose.The HPAEC-PAD analysis of the reaction products showed that the initial α(1 → 4)-glucan chains were used as acceptors, leading to the formation of new compounds with higher retention times, as revealed by the clear displacement of the peaks corresponding to the initial acceptor (Fig. 2A). This population is, in theory, composed of an α(1 → 3)/α(1 → 6)-glucan block at the non-reducing end, grafted to the non-reducing end of the α(1 → 4)-glucan block acceptor. It was thus considered to be an alternan-b-amylose copolymer. To further characterize the polymer, the diblock was separated by means of SEC from the fructose side-product and the natural α-glucans produced by ASR (OAL). The purity of the fraction was evaluated by means of HPAEC-PAD, which revealed the absence of contamination from the natural ASR products (Fig. 2A). The various α-glucosidic linkages comprising the pure diblock were determined by means of 1H NMR in order to evaluate the relative proportion of each glucan block (Fig. 2B and Table 2). In the alternan-b-amylose copolymer, α(1 → 4) linkages made up 69%, indicating that the alternan block made up 31%. As the average DP of the initial amylose fraction was 20 (determined by HPAEC-PAD analysis), it was estimated that the amylose chains were grafted with oligoalternans with a DP of 9, assuming that all the chains were homogeneously elongated.
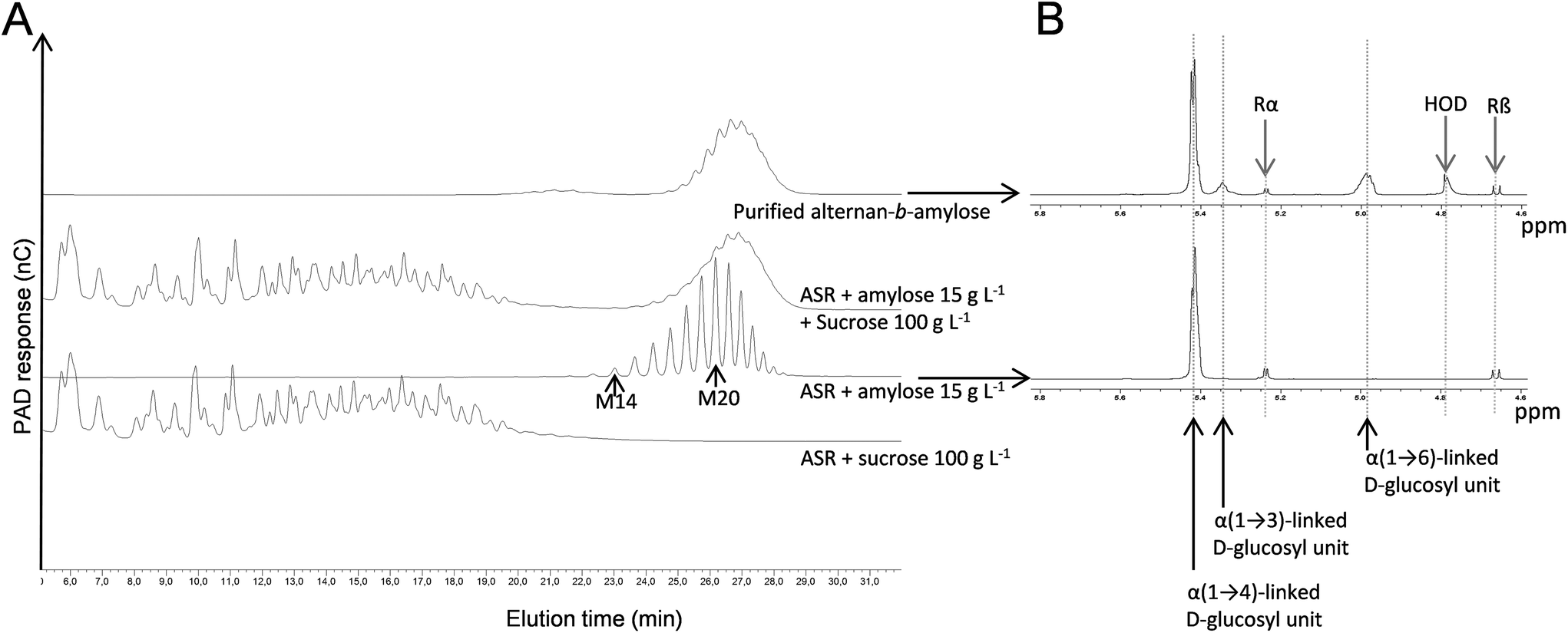 | ||
| Fig. 2 HPAEC-PAD chromatograms of the copolymer synthesis reaction medium involving ASR and amylose (A), and 1H NMR spectra of alternan-b-amylose and amylose (B). The chemical shifts at 5.45 ppm, 5.35 and 5 ppm were assigned to α(1 → 4), α(1 → 3) and α(1 → 6) linkages respectively. Rα and Rβ correspond to the anomeric signals of the reducing (→4)-D-Glcp units. | ||
| Samples | Enzymes | Synthesis conditions | % of α-glucosidic linkages | Average DP | Estimated synthesis yields (%) | Production yields (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Donor | Acceptor | α(1 → 6) | α(1 → 3) | α(1 → 4) | |||||
| Alternan | ASR | Sucrose | — | 62.9 | 37.1 | 0 | 33 | ||
| Dextran-b-alternan | DSR-M | Sucrose | Alternan | 91.0 | 9.0 | 0 | 171 | 44.3 | 8.7 |
| Amylose | DgAS | Sucrose | Maltose | 0 | 0 | 100 | 20 | ||
| Alternan-b-amylose | ASR | Sucrose | Amylose | 21.3 | 9.9 | 68.8 | 29 | 35.8 | 11.7 |
| Dextran-b-alternan-b-amylose | DSR-M | Sucrose | Alternan-b-amylose | 61.3 | 4.5 | 34.2 | 59 | 45.2 | 12.9 |
The structure of the copolymer was also analysed by means of permethylation. The relative proportions of each type of permethylated glucosyl units are reported in Table 3. The presence of 3 substituted, 6 substituted and 3,6 di-substituted glucosyl units confirms that the amylose chains were grafted with glucosyl units linked in α(1 → 6), α(1 → 3) and branching points. This is in accordance with the structure proposed for alternan46 and what has been elsewhere deduced from the analysis of the ASR products obtained from sucrose and maltose.51
| Glct | (→3)-Glcp-1→ | (→4)-Glcp-1→ | (→6)-Glcp-1→ | (→3,6)-Glcp-1→ | |
|---|---|---|---|---|---|
| % | % | % | % | % | |
| Alternan | 8.8 | 21.6 | 0 | 62.3 | 6.9 |
| Dextran-b-alternan | 2.5 | 5.1 | 0 | 88.9 | 3.1 |
| amylose | 6.6 | 0 | 93.4 | 0 | 0 |
| Alternan-b-amylose | 5.4 | 5.2 | 58 | 28.4 | 1.9 |
| Dextran-b-alternan-b-amylose | 2.7 | 1.8 | 33.5 | 60.1 | 1.1 |
Alternan block synthesis and elongation using DSR-M
At the same time, ASR was also used to synthesize LMM alternans directly from sucrose. After synthesis and fractionation by means of preparative SEC, a fraction with an average molar mass of 5388 g mol−1 (estimated from the HPSEC data with dextran calibration) was selected for extension by DSR-M (Fig. 3A). An HPAEC-PAD analysis of the reaction products revealed the presence of two α-glucan populations, one corresponding to the polymer synthesized by DSR-M from sucrose only, and a second that had been eluted later than the initial alternan acceptor (Fig. 3A).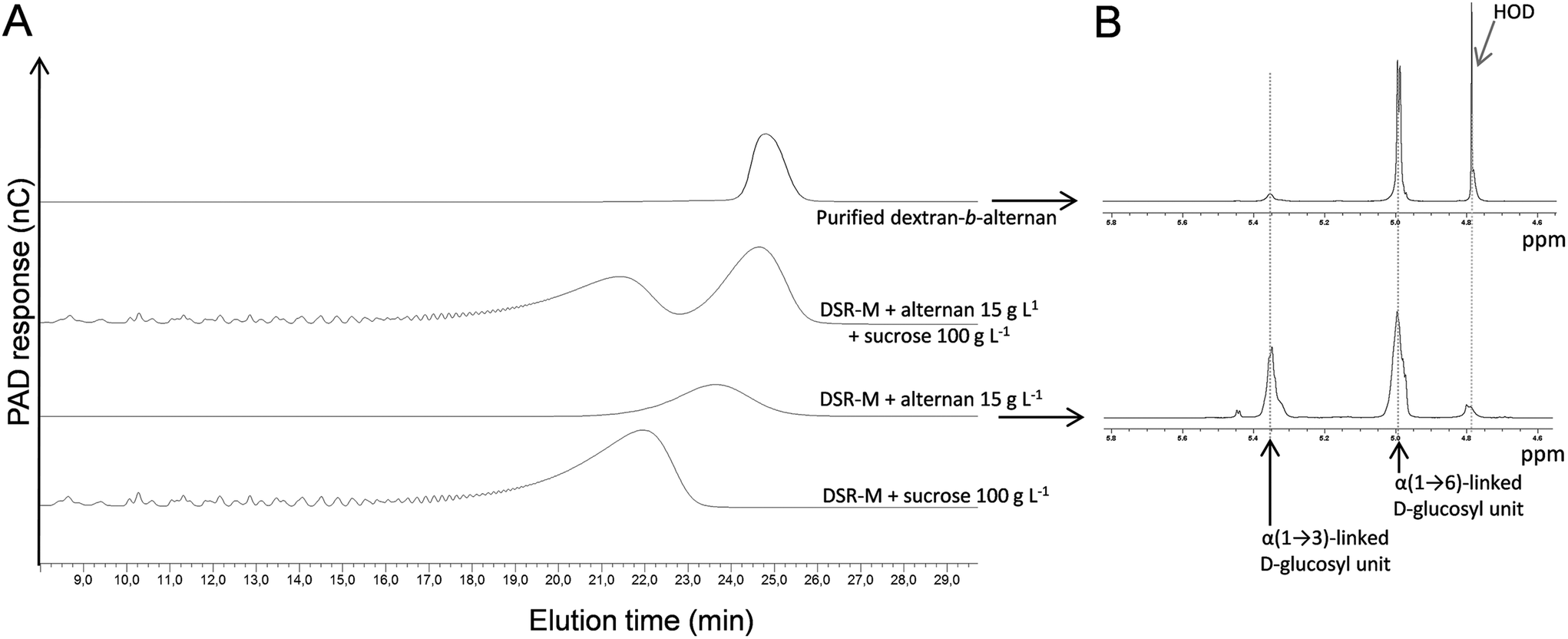 | ||
| Fig. 3 HPAEC-PAD and 1H NMR analysis of the copolymer synthesis reaction media involving DSR-M and alternan: (A) HPAEC-PAD chromatograms and (B) 1H NMR spectra of dextran-b-alternan and alternan. The chemical shifts at 5.35 and 5 ppm were assigned to the α(1 → 3) and α(1 → 6) linkages respectively. | ||
These results indicate that the alternan block was elongated by DSR-M, resulting in a diblock referred to here as a dextran-b-alternan copolymer, since DSR-M is specific for linear α(1 → 6) glucan formation. The diblock was isolated by means of preparative SEC (Fig. 3A) and analysed by means of 1H NMR and permethylation (Tables 2 and 3).
According to the 1H NMR analysis, the number of α(1 → 3) linkages (including the α(1 → 3,6) branching points) decreased from 37% in the alternan acceptor to 9% in the diblock, indicating that the alternan and dextran blocks represented 24% and 76% of the diblock respectively. Methylation data confirmed the increased proportion of α(1 → 6) linkages in the diblock and the decrease of α(1 → 3) linkages and branching points. Finally, from the HPSEC-MALLS analysis of the diblock, a weight-average molar mass of 27700 g mol−1 (DP171) was determined. Since the molar mass of the alternan block was 5388 g mol−1 (DP of 33), it was calculated that the alternan and dextran blocks made up 19% and 81% of the diblock respectively. These values do not significantly differ from those obtained from the 1H NMR analysis (24% and 76% respectively). Based on these results, it would appear that the DP of the alternan block in the diblock ranged from 31 to 36 and that of the dextran block from 135 to 140.
Production, purification and analysis of a triblock copolymer
As it had been proven that DSR-M was able to glucosylate LMM alternan, an attempt was made to glucosylate the alternan-b-amylose diblock in order to synthesize a triblock. The purified alternan-b-amylose copolymer was incubated with DSR-M and sucrose.After the reaction, it was observed by means of HPAEC-PAD that the concentration of the alternan-b-amylose copolymer had decreased, indicating a covalent modification of the diblock copolymer structure (Fig. 4). Surprisingly, the elution time of the modified copolymer in the HPAEC chromatogram was lower than that of the acceptor. We can exclude that DSR-M caused hydrolysis of the diblock, since no modification of the diblock was observed when the enzyme was incubated with the diblock alone. A more plausible explanation is that the decrease in the retention time was due to the higher number of α(1 → 6)-linked glucosyl units in the triblock, introduced by DSR-M.
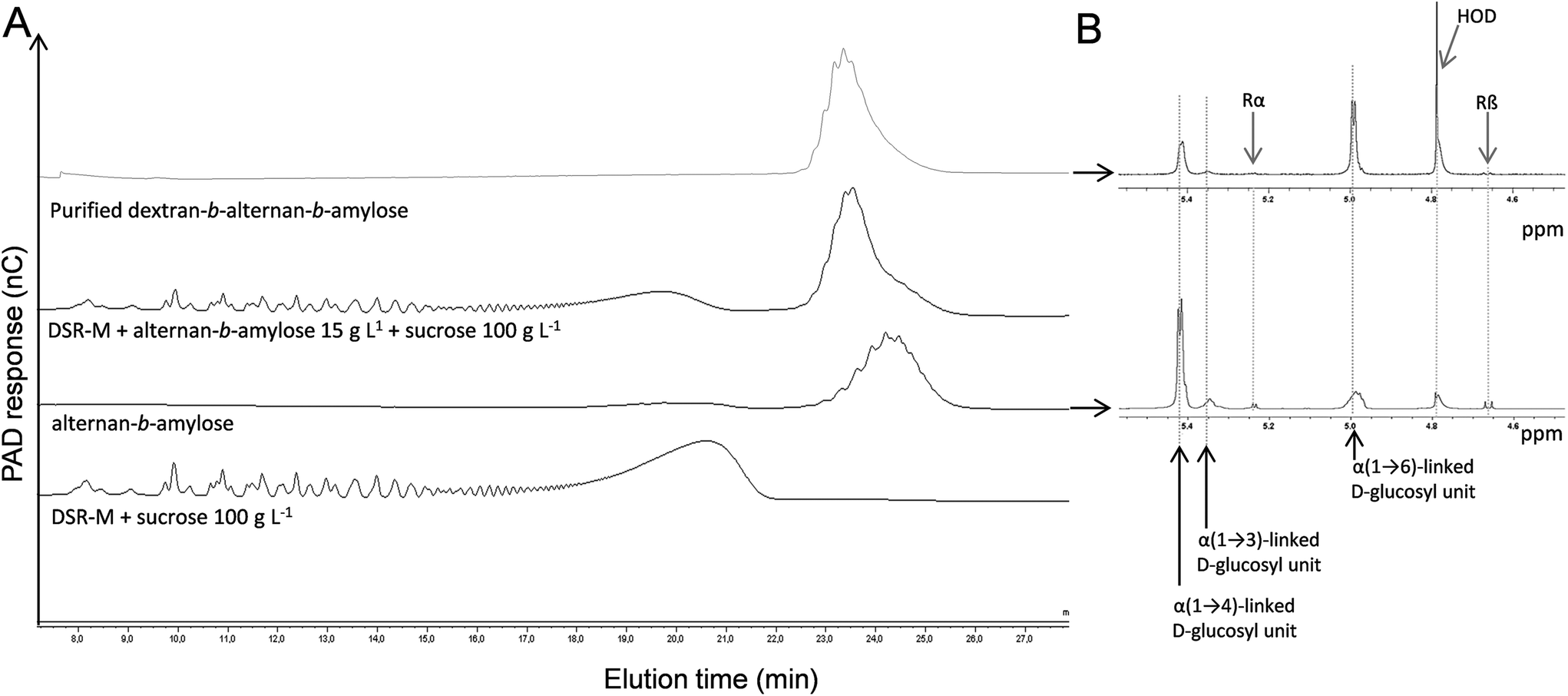 | ||
| Fig. 4 HPAEC-PAD chromatograms of the purified copolymer obtained from the DSR-M/alternan-b-amylose pair (A), and 1H NMR spectra of dextran-b-alternan-b-amylose and alternan-b-amylose (B). The chemical shifts at 5.45 ppm, 5.35 and 5 ppm were assigned to α(1 → 4), α(1 → 3) and α(1 → 6) linkages respectively. Rα and Rβ correspond to the anomeric signals of the reducing -(1 → 4)-D-Glcp units. | ||
As shown in Fig. 5, HPAEC-PAD elution times indeed depend on a product's linkage composition, α(1 → 4)-linked oligosaccharides being eluted much later than those that are α(1 → 6)-linked. In order to investigate the osidic linkage composition of the triblock copolymer, the product was isolated by means of preparative HPAEC-PAD. The product purity was then verified by means of analytical HPAEC-PAD, and the linkage pattern was analysed using 1H NMR (Fig. 4B and Table 2) and permethylation (Table 3). The results show that the proportion of α(1 → 6) linkages increased in this purified fraction, whereas α(1 → 3) and α(1 → 4) linkages decreased, while maintaining an almost constant α(1 → 3)/α(1 → 4) linkage ratio. This is again consistent with the elongation of the diblock by DSR-M, which is specific for the formation of α(1 → 6) linkages. Low numbers of α(1 → 3,6)-linked glucosyl units in the triblock were also detected, indicating the presence of branched molecules (Table 3).
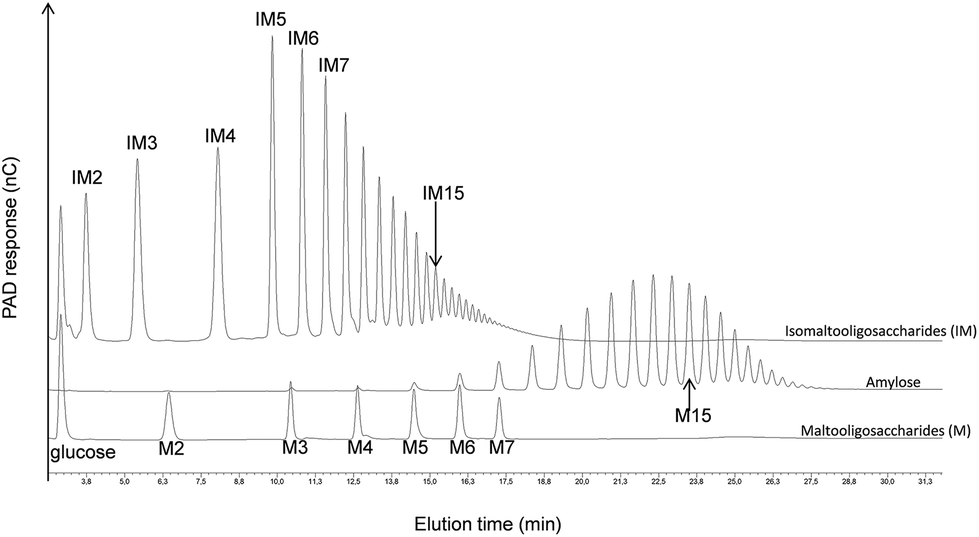 | ||
| Fig. 5 HPAEC-PAD chromatograms showing the difference in retention times between α(1 → 4)-glucans (maltooligosaccharides and amylose) and α(1 → 6)-glucans (isomaltooligosaccharides). | ||
NMR analysis indicated that the proportion of α-(1 → 4) linkages in the triblock was 34%. It was thus deduced that the amylose block represented 34% of the triblock. Since the alternan block comprised 31% of the alternan-b-amylose copolymer, the proportions of the alternan and dextran blocks in the triblock were 14% and 52% respectively.
As the average DP of the initial alternan-b-amylose copolymer was 29, we can deduce that approximately 30 α-(1 → 6)-linked glucosyl units were grafted to the alternan-b-amylose. Thus, the dextran-b-alternan-b-amylose copolymer should contain 59 glucosyl residues (30 in the dextran block, 9 in the alternan and 20 in the amylose block) with a calculated molar mass of 9558 g mol−1. These values are average values, as it was assumed that all the chains were elongated to the same extent. Altogether, these results indicate that a triblock copolymer could be obtained by following a stepwise process using the DgAS, ASR and DSR-M enzymes. Examples of structural arrangements of this triblock copolymer are presented in Fig. 6.
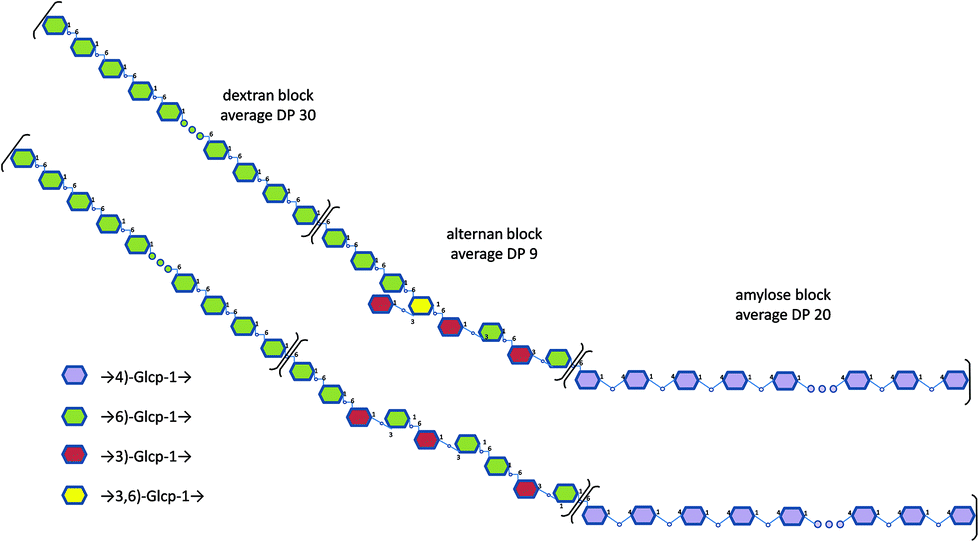 | ||
| Fig. 6 Examples of structural models proposed for the triblock copolymer dextran-b-alternan-b-amylose. | ||
The specific locations of the osidic linkages, especially in the alternan block, are putative as several structures could correspond to our data.
Copolymer conformation in solution
SAXS was used to analyse the conformation of the initial amylose block, as well as of the alternan-b-amylose and dextran-b-alternan-b-amylose copolymers. The SAXS curves obtained for each polymer are shown in Fig. 7. LogI(q) versus log(q) was used in order to highlight the region of small q angles where the Guinier approximation and different power law regimes can be determined. The radius of gyration (Rg) values obtained for the amylose block, alternan-b-amylose diblock and dextran-b-alternan-b-amylose triblock were 15.1, 17.1 and 30.0 Å, respectively. It can therefore be deduced that the addition of each block caused an increase in the copolymer Rg. The SAXS curves with a form factor corresponding to a worm-like chain were then fitted using SASfit.52 The worm-like model was adjusted, taking the contour length, persistence length and polymer radius as the fitting parameters.
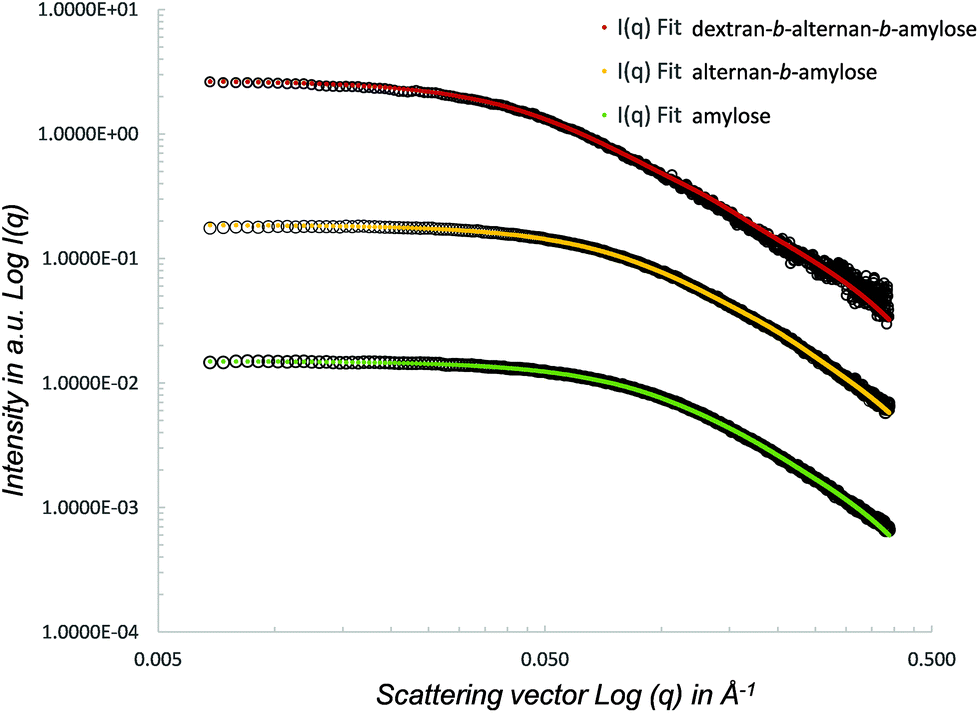 | ||
| Fig. 7 SAXS curves of the purified triblock copolymer and its diblock and monoblock components. | ||
A worm-like conformation is usually used to describe the low DP polysaccharide conformation in solution, and has been previously used to investigate the conformational tracking of amylose synthesized by amylosucrase from sucrose as the sole substrate.53 Here again, it was observed that the worm-like model fits very well with the data collected from the single, di- and triblocks. The contour length (RL) of the fitted worm-like chain ranged from 85.5 to 271.0 Å (Table 4). This model was also used to estimate the polymers’ DPs using a contour length (RL)/monomer size (radius) ratio. The calculated DPs were 20, 30 and 59 for amylose, alternan-b-amylose and dextran-b-alternan-b-amylose, respectively. These values are very close to those deduced from the HPAEC-PAD and 1H NMR analyses. The flexibility of the products in solution was assessed based on the persistence length (RLL) factor values. The addition of the alternan block to the amylose block increased the flexibility, as revealed by the RRL shift from 20.7 to 15.4 Å. With regard to dextran-b-alternan-b-amylose, the fitted values obtained from the worm-like model differed slightly from the experimental data for q values higher than 0.1 Å. In this specific case, the RLL value must therefore be taken with caution. Nevertheless, the logI(q) versus log(q) slope is lower for the experimental values than for the worm-like model, indicating that the addition of the dextran block increased the flexibility of the diblock copolymer. It has been previously demonstrated by means of theoretical and experimental analyses that polysaccharides (amylose, dextran, mutan and cellulose) with the same monosaccharide composition but different linkage types present different behaviours in solution.54–56 Using 13C NMR relaxation experiments, Tylianakis and co-workers also showed that dextran flexibility was higher than that of amylose.57 The higher flexibility of α(1 → 6)-linked chains is due to the presence of an extra bond in the inter-residue linkages, which grants a higher degree of freedom.58,59 This is very consistent with our SAXS analysis results, as each copolymer block differs from the others in terms of its linkage type, which in turn affects its conformation and flexibility. The contrasting properties of each block would be expected to positively impact the applicative potential of the copolymers synthesized in this study. Due to its helical conformation, amylose could certainly prove to be of great interest in drug and flavour complexation. However, as a result of the weak solubility of amylose preparations in water, these kinds of applications would require amylose functionalization, such as hydroxypropylation, in order to increase the water solubility of the inclusion complexes.60,61 In this study, we demonstrated that the covalent grafting of alternan and dextran blocks to amylose increases the flexibility. Such copolymer structures could offer advantages with respect to the encapsulation of bioactive molecules, since the amylose block could serve as the complexing agent, while the alternan and dextran blocks could be used to increase the water solubility, due to their greater flexibility.
| Amylose | Alternan-b-amylose | Dextran-b-alternan-b-amylose | |
|---|---|---|---|
| Contour length RL in Å | 85.5 | 131.3 | 271.0 |
| Persistence length (RLL) in Å | 20.7 | 15.4 | 21.5 |
| Radius (R) in Å | 4.7 | 4.3 | 4.8 |
| Gyration radius (Rg) in Å | 15.1 | 17.1 | 30.0 |
| Maximal distance (Dmax) in Å | 70.0 | 80.0 | 120.0 |
4. Conclusions
To our knowledge, this study represents the first successful enzymatic synthesis of biosourced block copolymers from sucrose by means of a stepwise approach. Previous studies involved a glycosyltransferase,10,40 a glycoside-phosphorylase,12–15 or transglycosidases,22,41 and produced either statistical copolymers or co-oligomers (of a total size of less than 30 glycosyl units) rather than block copolymers. Here, we demonstrated that the selected glucansucrases (DSR-M and ASR) transfer α-glucosyl residues to acceptor polymers (i) with a conformation and linkage composition different from that of their natural acceptors, and (ii) a size of up to 30 glucosyl residues, producing copolymers comprising up to 171 residues. The original synthesis method designed for this study successfully resulted, for the first time, in several diblock polysaccharides – dextran-b-alternan with a molar mass of 27700 g mol−1 and alternan-b-amylose with a molar mass of 4700 g mol−1 – and one triblock of dextran-b-alternan-b-amylose with a molar mass of approximately 9500 g mol−1. Whether all GH70 enzymes are able to perform such reactions remains to be established. It was noted that ASR elongated amylose, whereas DSR-M did not, revealing that both the enzyme's specificity and its acceptor's site topology are important. With respect to the objective of producing HMM copolymers on a large scale, the problem appears to be more complex, as the purification techniques currently available do not allow HMM polymer acceptors to be easily separated from either the HMM polymers produced by acceptor elongation or those naturally produced by many glucansucrases. Fractionation based on polymer solubility differences could be a solution, but this remains tricky. Moreover, enzyme engineering may be required in some cases to improve the accommodation of long polymer chains differing in terms of their structure from those of the natural polymer. Nevertheless, the conformational analysis of the new copolymers synthesized here showed that covalent grafting of alternan and dextran to amylose increases the polymer flexibility. While such properties are likely to generate interest in terms of how these copolymers might be used to encapsulate various molecules of interest, such as aromas or drugs, their potential for such uses will have to be assessed. Scaling up this fully green production process to the dozen gram level will allow us to obtain more information on the structure/property relationship of these copolymers, as well as their potential applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The HPLC analyses were performed using the equipment at the ICEO facility, which is dedicated to the screening, discovery and characterization of new enzymes. ICEO, part of the Integrated Screening Platform of Toulouse (PICT, IBiSA), is supported by grants from the Région Midi-Pyrénées, the European Regional Development Fund and the Institut National de la Recherche Agronomique (INRA). The permethylation analyses were performed with the equipments of the BIBS platform of Nantes (UR1268 BIA, IBiSA, Phenome-Emphasis-FR (grant number ANR-11-INBS-0012)). We are grateful to MetaSys, the Metabolomics and Fluxomics Center at the Laboratory for the Engineering of Biological Systems and Processes (Toulouse, France) for the NMR experiments. We would like to cordially thank Marion Claverie, Nelly Monties, Gianluca Cioci, Karine Cahier and Brigitte Laillet for their technical support. This work was financed by the Agence Nationale de la Recherche (grant number ANR 14-CE27-0011-02).References
- K. Akiyoshi, M. Kohara, K. Ito, S. Kitamura and J. Sunamoto, Macromol. Rapid Commun., 1999, 20, 112–115 CrossRef.
- N. Enomoto, S. Furukawa, Y. Ogasawara, H. Akano, Y. Kawamura, E. Yashima and Y. Okamoto, Anal. Chem., 1996, 68, 2798–2804 CrossRef PubMed.
- C. Lemarchand, P. Couvreur, M. Besnard, D. Costantini and R. Gref, Pharm. Res., 2003, 20, 1284–1292 CrossRef.
- K. Loos and A. H. E. Muller, Biomacromolecules, 2002, 3, 368–373 CrossRef PubMed.
- C. Schatz and S. Lecommandoux, Macromol. Rapid Commun., 2010, 31, 1664–1684 CrossRef PubMed.
- Y.-C. Lin, F.-j. Tan, K. G. Marra, S.-S. Jan and D.-C. Liu, Acta Biomater., 2009, 5, 2591–2600 CrossRef PubMed.
- M. Rinaudo, Carbohydr. Polym., 2011, 83, 1338–1344 CrossRef.
- D. Hatanaka, Y. Takemoto, K. Yamamoto and J.-I. Kadokawa, Journal, 2014, 2, 34–44 Search PubMed.
- S. Kobayashi, Proc. Jpn. Acad., Ser. B, 2007, 83, 215–247 CrossRef PubMed.
- V. Yadav, B. J. Paniliatis, H. Shi, K. Lee, P. Cebe and D. L. Kaplan, Appl. Environ. Microbiol., 2010, 76, 6257–6265 CrossRef PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef PubMed.
- J. Kadokawa, R. Shimohigoshi, K. Yamashita and K. Yamamoto, Org. Biomol. Chem., 2015, 13, 4336–4343 RSC.
- R. Baba, K. Yamamoto and J.-i. Kadokawa, Carbohydr. Polym., 2016, 151, 1034–1039 CrossRef PubMed.
- K. Yamashita, K. Yamamoto and J.-i. Kadokawa, Biomacromolecules, 2015, 16, 3989–3994 CrossRef PubMed.
- R. Shimohigoshi, Y. Takemoto, K. Yamamoto and J. Kadokawa, Chem. Lett., 2013, 42, 822–824 CrossRef.
- E. J. Hehre and M. H. With the assistance of Doris, J. Biol. Chem., 1951, 192, 161–174 Search PubMed.
- X. Z. Mao, S. Wang, F. F. Kan, D. Z. Wei and F. L. Li, Appl. Biochem. Biotechnol., 2012, 168, 1256–1264 CrossRef PubMed.
- M. Naessens, A. Cerdobbel, W. Soetaert and E. J. Vandamme, J. Ind. Microbiol. Biotechnol., 2005, 32, 323–334 CrossRef PubMed.
- J. Gangoiti, S. S. van Leeuwen, G. J. Gerwig, S. Duboux, C. Vafiadi, T. Pijning and L. Dijkhuizen, Sci. Rep., 2017, 7, 39761 CrossRef PubMed.
- H. Leemhuis, W. P. Dijkman, J. M. Dobruchowska, T. Pijning, P. Grijpstra, S. Kralj, J. P. Kamerling and L. Dijkhuizen, Appl. Microbiol. Biotechnol., 2013, 97, 181–193 CrossRef PubMed.
- X. F. Meng, J. Gangoiti, Y. X. Bai, T. Pijning, S. S. Van Leeuwen and L. Dijkhuizen, Cell. Mol. Life Sci., 2016, 73, 2681–2706 CrossRef PubMed.
- H. Leemhuis, J. M. Dobruchowska, M. Ebbelaar, F. Faber, P. L. Buwalda, M. van der Maarel, J. P. Kamerling and L. Dijkhuizen, J. Agric. Food Chem., 2014, 62, 12034–12044 CrossRef PubMed.
- A. Jeanes, W. C. Haynes, C. A. Wilham, J. C. Rankin, E. H. Melvin, M. J. Austin, J. E. Cluskey, B. E. Fisher, H. M. Tsuchiya and C. E. Rist, J. Am. Chem. Soc., 1954, 76, 5041–5052 CrossRef.
- I. Andre, G. Potocki-Veronese, S. Morel, P. Monsan and M. Remaud-Simeon, Carbohydrates in Sustainable Development I: Renewable Resources for Chemistry and Biotechnology, 2010, vol. 294, pp. 25–48 Search PubMed.
- C. Moulis, I. Andre and M. Remaud-Simeon, Cell. Mol. Life Sci., 2016, 73, 2661–2679 CrossRef PubMed.
- D. Passerini, M. Vuillemin, L. Ufarté, S. Morel, V. Loux, C. Fontagné-Faucher, P. Monsan, M. Remaud-Siméon and C. Moulis, FEBS J., 2015, 282, 2115–2130 CrossRef PubMed.
- M. Claverie, G. Cioci, M. Vuillemin, N. Monties, P. Roblin, G. Lippens, M. Remaud-Simeon and C. Moulis, ACS Catal., 2017, 7, 7106–7119 CrossRef.
- G. Joucla, S. Pizzut, P. Monsan and M. Remaud-Simeon, FEBS Lett., 2006, 580, 763–768 CrossRef PubMed.
- S. L. Isenberg, A. K. Brewer, G. L. Côté and A. M. Striegel, Biomacromolecules, 2010, 11, 2505–2511 CrossRef PubMed.
- T. D. Leathers, M. S. Nunnally and G. L. Côté, Biotechnol. Lett., 2009, 31, 289–293 CrossRef PubMed.
- G. P. De Montalk, M. Remaud-Simeon, R. M. Willemot, P. Sarcabal, V. Planchot and P. Monsan, FEBS Lett., 2000, 471, 219–223 CrossRef.
- G. Potocki-Veronese, J. L. Putaux, D. Dupeyre, C. Albenne, M. Remaud-Simeon, P. Monsan and A. Buleon, Biomacromolecules, 2005, 6, 1000–1011 CrossRef PubMed.
- F. Grimaud, C. Lancelon-Pin, A. Rolland-Sabate, X. Roussel, S. Laguerre, A. Vikso-Nielsen, J. L. Putaux, S. Guilois, A. Buleon, C. D'Hulst and G. Potocki-Veronese, Biomacromolecules, 2013, 14, 438–447 CrossRef PubMed.
- J. L. Putaux, G. Potocki-Veronese, M. Remaud-Simeon and A. Buleon, Biomacromolecules, 2006, 7, 1720–1728 CrossRef PubMed.
- G. L. Cote and B. Y. Tao, Glycoconjugate J., 1990, 7, 145–162 CrossRef.
- M. A. A. Morales, M. Remaud-Simeon, R. M. Willemot, M. R. Vignon and P. Monsan, Carbohydr. Res., 2001, 331, 403–411 CrossRef.
- D. Daude, E. Champion, S. Morel, D. Guieysse, M. Remaud-Simeon and I. Andre, ChemCatChem, 2013, 5, 2288–2295 CrossRef.
- P. Monsan, M. Remaud-Simeon and I. Andre, Curr. Opin. Microbiol., 2010, 13, 293–300 CrossRef PubMed.
- J. F. Robyt and S. H. Eklund, Carbohydr. Res., 1983, 121, 279–286 CrossRef PubMed.
- D. Fu and J. F. Robyt, Carbohydr. Res., 1991, 217, 201–211 CrossRef PubMed.
- D. Fu and J. F. Robyt, Arch. Biochem. Biophys., 1990, 283, 379–387 CrossRef PubMed.
- S. Emond, S. Mondeil, K. Jaziri, I. Andre, P. Monsan, M. Remaud-Simeon and G. Potocki-Veronese, FEMS Microbiol. Lett., 2008, 285, 25–32 CrossRef PubMed.
- J. B. Sumner and S. F. Howell, J. Biol. Chem., 1935, 108, 51–54 Search PubMed.
- D. Popov, A. Buléon, M. Burghammer, H. Chanzy, N. Montesanti, J. L. Putaux, G. Potocki-Véronèse and C. Riekel, Macromolecules, 2009, 42, 1167–1174 CrossRef.
- P. Faucard, F. Grimaud, D. Lourdin, J.-E. Maigret, C. Moulis, M. Remaud-Siméon, J.-L. Putaux, G. Potocki-Véronèse and A. Rolland-Sabaté, Carbohydr. Polym., 2018, 181, 337–344 CrossRef PubMed.
- F. K. Seymour, R. D. Knapp, E. C. M. Chen, S. H. Bishop and A. Jeanes, Carbohydr. Res., 1979, 74, 41–62 CrossRef.
- K. R. Anumula and P. B. Taylor, Anal. Biochem., 1992, 203, 101–108 CrossRef PubMed.
- D. P. Sweet, R. H. Shapiro and P. Albersheim, Carbohydr. Res., 1975, 40, 217–225 CrossRef.
- G. David and J. Perez, J. Appl. Crystallogr., 2009, 42, 892–900 CrossRef.
- L. S. Sciarini, A. Rolland-Sabate, S. Guilois, P. Decaen, E. Leroy and P. Le Bail, Green Chem., 2015, 17, 291–299 RSC.
- G. L. Côté and S. Sheng, Carbohydr. Res., 2006, 341, 2066–2072 CrossRef PubMed.
- I. Breßler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 CrossRef PubMed.
- P. Roblin, G. Potocki-Véronèse, D. Guieysse, F. Guerin, M. A. V. Axelos, J. Perez and A. Buleon, Biomacromolecules, 2013, 14, 232–239 CrossRef PubMed.
- B. A. Burton and D. A. Brant, Biopolymers, 1983, 22, 1769–1792 CrossRef.
- H. Elmgren, Carbohydr. Res., 1987, 160, 227–241 CrossRef.
- S. Cui and Q. Wang, in Food Carbohydrates, CRC Press, 2005 Search PubMed.
- M. Tylianakis, A. Spyros, P. Dais, F. R. Taravel and A. Perico, Carbohydr. Res., 1999, 315, 16–34 CrossRef.
- I. M. Neelov, D. B. Adolf, T. C. B. McLeish and E. Paci, Biophys. J., 2006, 91, 3579–3588 CrossRef PubMed.
- G. Lee, W. Nowak, J. Jaroniec, Q. Zhang and P. E. Marszalek, Biophys. J., 2004, 87, 1456–1465 CrossRef PubMed.
- G. Wulff, A. Steinert and O. Höller, Carbohydr. Res., 1998, 307, 19–31 CrossRef.
- G. Wulff, G. Avgenaki and M. S. P. Guzmann, J. Cereal Sci., 2005, 41, 239–249 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Copolymer production yields and copolymer synthesis yields. See DOI: 10.1039/c8gc01251b |
| This journal is © The Royal Society of Chemistry 2018 |