
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal azide–alkene cycloaddition reactions: straightforward multi-gram access to Δ2-1,2,3-triazolines in deep eutectic solvents†
Filip
Sebest
 a,
Luis
Casarrubios
ab,
Henry S.
Rzepa
a,
Andrew J. P.
White
a and
Silvia
Díez-González
*a
a,
Luis
Casarrubios
ab,
Henry S.
Rzepa
a,
Andrew J. P.
White
a and
Silvia
Díez-González
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: s.diez-gonzalez@imperial.ac.uk
bDepartamento de Química Organica I, Facultad de Ciencias Químicas, Universidad Complutense and Centro de Innovación en Química Avanzada (ORFEO–CINQA), 28040 Madrid, Spain
First published on 7th August 2018
Abstract
The multi-gram synthesis of a wide range of 1,2,3-triazolines via azide–alkene cycloaddition reactions in a Deep Eutectic Solvent (DES) is reported. The role of DES in this transformation as well as the origin of the full product distribution was studied with an experimental/computational-DFT approach.
Introduction
1,3-Dipolar cycloadditions, also known as Huisgen reactions, are convergent and atom-economical reactions that offer a convenient synthetic route for five-membered heterocyclic molecules.1 To date, few reactions can match dipolar cycloadditions in the variety of bonds that can be involved and the increase of molecular complexity in the obtained products. Hence, the extensive research coverage received by these reactions over the years is more than justified and has notably allowed uncovering alternative catalytic routes leading to broader and more selective reactions, including enantioselective ones. A particularly high-profile example of this is the copper-mediated azide–alkyne cycloaddition reaction,2 and this transformation has become the emblem of Click chemistry,3 a concept coined by Sharpless in 2001 and closely related to that of Green chemistry.While the use of metals (mainly copper, but also ruthenium, iridium, or silver)4 has been instrumental for the study and application of triazoles, the preparation of 1,2,3-triazolines from azides and alkenes remains limited to highly activated alkenes, such as strained and electron-rich alkenes,5 and therefore out of the scope of Click chemistry. Simple alkenes do not react with azides or react very slowly and reaction times of several days or even months are not overly uncommon.6 Even when a triazoline can be formed, it often rearranges/further reacts and other heterocycles (such as triazoles,7 pyrazolines,8 isoindoles,9 amidines,10 pyrrol- and indol-izidines11) are isolated instead. In consequence, the promise of 1,2,3-triazolines as versatile synthons12 and biologically active compounds remains unfulfilled.13
A thermal azide–alkene cycloaddition is expected to deliver two regioisomeric triazolines, either 1,4- or 1,5-disubstituted, and the product distribution is mainly dictated by electronic factors.14 Thermodynamically, dipolar cycloadditions leading to either triazoles or triazolines are virtually identical. However, while triazoles are aromatic and highly stable, triazolines are typically more reactive than the starting materials due to the numerous decomposition pathways available to them.15 The most commonly encountered products are then imines (often hydrolysed into amines and carbonyl derivatives) and aziridines. The formation of aziridines upon nitrogen extrusion from the corresponding triazolines could be a simple alternative to metal-mediated aziridination reactions with azides as nitrene source.16 However, the formation of aziridines from the corresponding triazolines depends strongly on the position and electronic nature of the substituents on the five-membered ring,12 which has led to apparently conflicting reports over the years.17,18 Unlike the reaction with alkynes, to the best of our knowledge no metal-mediated version of the azide–alkene cycloaddition has been reported and therefore the scope of this reaction remains severely limited.
On the other hand, it was recognised early on that the reaction media could play a key role in numerous non-catalysed dipolar cycloaddition reactions.19 As part of our interest in developing user-friendly and greener methodologies, we turned our attention to Deep Eutectic Solvents (DESs). DESs are combinations of two or three inexpensive, non-toxic and safe components that produce a non-flammable mixture with a much lower melting point than any of the individual components. These mixtures, which are liquids at the reaction temperature, are held together via hydrogen bonding interactions. The interesting physico-chemical properties of DESs are often compared to those of ionic liquids, with the additional advantages of lower costs and much higher user- and environment-friendliness (i.e. water tolerance).20 The benefits of using DESs instead of ionic liquids or standard organic solvents are obviously dependent on the actual DES components. Those made of urea, glycerol, water or choline chloride are particularly promising in Green chemistry.
The applications of DESs are steadily spreading in separation, materials, organic and organometallic chemistry.21 In particular, DESs have been applied to several cycloaddition reactions such as Lewis acid-catalysed Diels–Alder reactions,22 formation of pyrrolidines from the corresponding azomethine ylides,23 and isoxazoles from nitrile oxides.24 From azides, either tetrazoles25 or triazoles26 have been prepared in DES. In many of these examples the beneficial effect of DES as reaction media was rationalised either through the Lewis acidity of one of the components, or the formation of hydrogen bonds between either of the components and the cycloaddition partners.
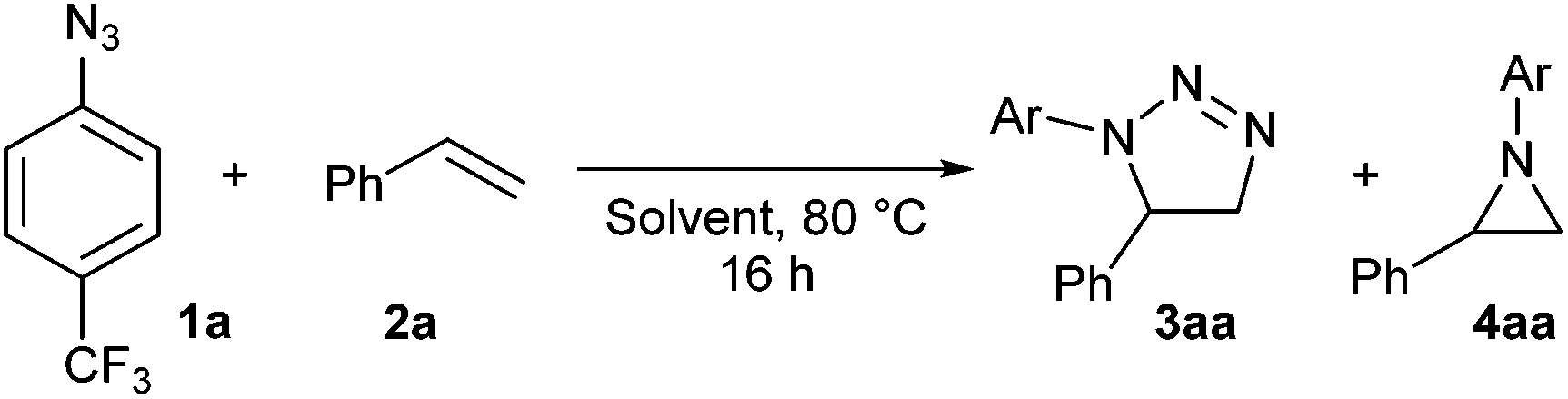
Herein we report straightforward azide–alkene cycloaddition reactions in Deep Eutectic Solvents to yield 1,2,3-triazoline cycloadducts, or in some cases aziridines, where organic solvents or ionic liquids only led to disappointing results. Preliminary studies to ascertain the role of DES in the azide–alkene cycloaddition reaction and the origin of the aziridine products are also presented.
Results and discussion
Optimisation of the reaction conditions
4-Trifluoromethylphenyl azide and styrene were chosen as model substrates and they were reacted at 80 °C overnight (Table 1). Mixtures of 1,5-disubstituted triazoline 3aa and aziridine 4aa were obtained in most reactions. A number of known decomposition products of triazolines –an imine, aniline or acetophenone (Table 1)– were evidenced by NMR in many of these reactions but only as traces. Disappointing NMR yields were obtained in all tested organic solvents independently of their characteristics due to severe decomposition of cycloadducts and/or the starting azide (up to 80% of the total mass, Table 1, entries 1–10), which made it virtually impossible to isolate any reaction product. In stark contrast with these results, a clean reaction was obtained when the model substrates were heated in a mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 of choline chloride and urea (DES, Table 1, entry 12).
:2 of choline chloride and urea (DES, Table 1, entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | 1a (%) | 3aa (%) | 4aa (%) |
a Reaction conditions: Azide 1a (1 mmol), styrene 2a (1.3 equiv.) in 2 mL of solvent.
b Observed trace decomposition products: 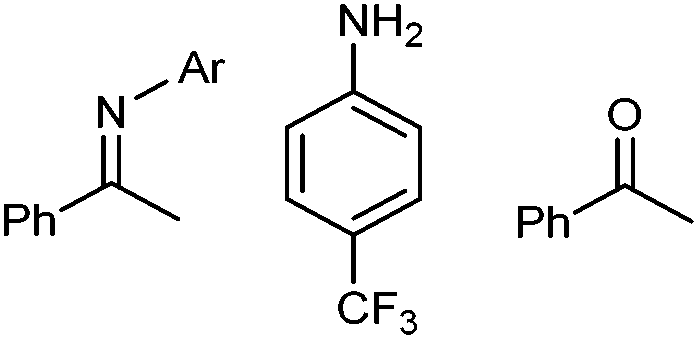 .
c
1H NMR yields/recovery are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. .
c
1H NMR yields/recovery are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard.
|
||||
| 1 | Toluene | 25 | 5 | 13 |
| 2 | MeCN | 23 | <5 | 5 |
| 3 | CHCl3 | 19 | <5 | 7 |
| 4 | 1,4-Dioxane | 30 | 18 | 14 |
| 5 | THF | 14 | 30 | 13 |
| 6 | TBME | 37 | 11 | 10 |
| 7 | Diglyme | 0 | 10 | 11 |
| 8 | EtOAc | 38 | <5 | 7 |
| 9 | EtOH | 22 | 5 | <5 |
| 10 | DMSO | 16 | <5 | 0 |
| 11 | Water | 12 | 0 | 0 |
| 12 | DES | 7 | 55 | 18 |
It is important to note that both reaction products are stable under the reaction conditions and they were recovered quantitatively when heated separately at 80 °C in DES for 16 h. However, partial decomposition of triazoline 3aa was observed when heated in standard organic solvents (i.e. DMSO, 1,4-dioxane) at the same temperature (vide infra). We believe that this increased stability of triazolines in DES is key to explaining the success of these reactions.
Capitalising on this promising result, the optimisation studies then focused on cycloaddition reactions in deep eutectic solvents and different reaction times and temperatures were screened. At 80 °C no further conversion was observed when the reaction mixture was heated for 24 h instead of 16 h. Comparable conversions into 3aa and 4aa were observed at 90 °C after 8 h, but again the reaction did not proceed further after longer heating periods. Higher reaction temperatures led to significant formation of decomposition products.
Next, a number of commonly encountered DESs were tested (Table 2). In all cases, the starting azide was fully converted when 2 equivalents of styrene were employed and overall similar reaction outcomes were obtained with most of the tested DESs. The original choice, choline chloride/urea (1:2), led to the highest conversion into the desired triazoline 3aa. When choline iodide was used instead as the Lewis base partner, significant formation of 4-trifluoromethylaniline (∼10%) was observed and the resulting DES was only liquid above 70 °C, which made its manipulation cumbersome. Other DESs remained valuable options, except when a more acidic partner such as glycolic acid was used as one of the components and only a complex mixture of decomposition products was obtained (Table 2, entry 6).27 A similar outcome was observed when a popular ionic liquid, BMIM·PF6 (BMIM = N-butyl-N′-methylimidazolium), was used as solvent (Table 2, entry 7).
|
|
|||
|---|---|---|---|
| Entry | Solvent | 3aa (%) | 4aa (%) |
| a Reaction conditions: Azide 1a (1 mmol), styrene 2a (2 equiv.) in 2 mL of solvent. b 1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. | |||
| 1 | Choline chloride/urea (1:2) |
67 | 22 |
| 2 | Choline iodide/urea (1:2) |
60 | 11 |
| 3 | Choline chloride/ethylene glycol (1:2) |
61 | 13 |
| 4 | Choline chloride/water (1:2) |
61 | 18 |
| 5 | Choline chloride/glycerol (1:2) |
61 | 12 |
| 6 | Choline chloride/glycolic acid (1:2) |
Decomposition | |
| 7 | BMIM·PF6 | Decomposition | |
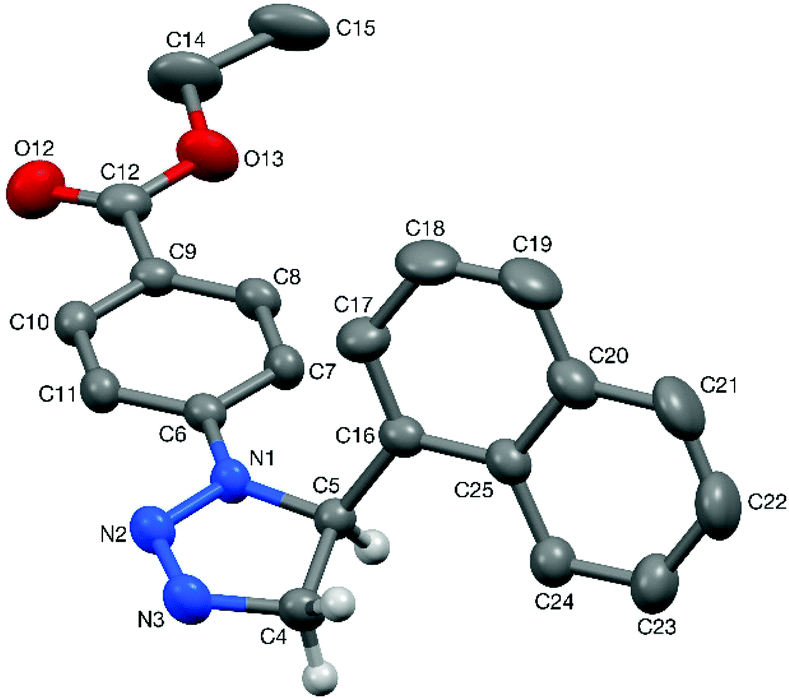
After this screening, the term DES in this article will refer solely to a 1:2 mixture of choline chloride and urea.28 To date, only a handful of 1,2,3-triazolines have been structurally characterised,29 and therefore single crystals were grown for 3aa and 4aa to confirm the assigned structures (Fig. 1 and 2). Crystals of 3aa contained two independent molecules and in both of them the triazoline ring has a small envelope deformation with C5 lying ca. 0.20 Å out of the {N1,N2,N3,C4} plane in molecule 3aa-A (0.18 Å in 3aa-B), which atoms are coplanar to better than 0.01 Å in both instances. In each case, the 4-CF3-phenyl group is almost co-planar to the {N1,N2,N3,C4} plane, while the phenyl ring on C5 is oriented such that its plane approaches eclipsed with respect to the N1–C5 bond, with the N1–C5–C16–C21 torsion angles being ca. 28° in both molecules. Aziridine scaffolds are far more common, and in the case of 4aa, the torsion angle between the two substituents is 138° (Fig. 2).
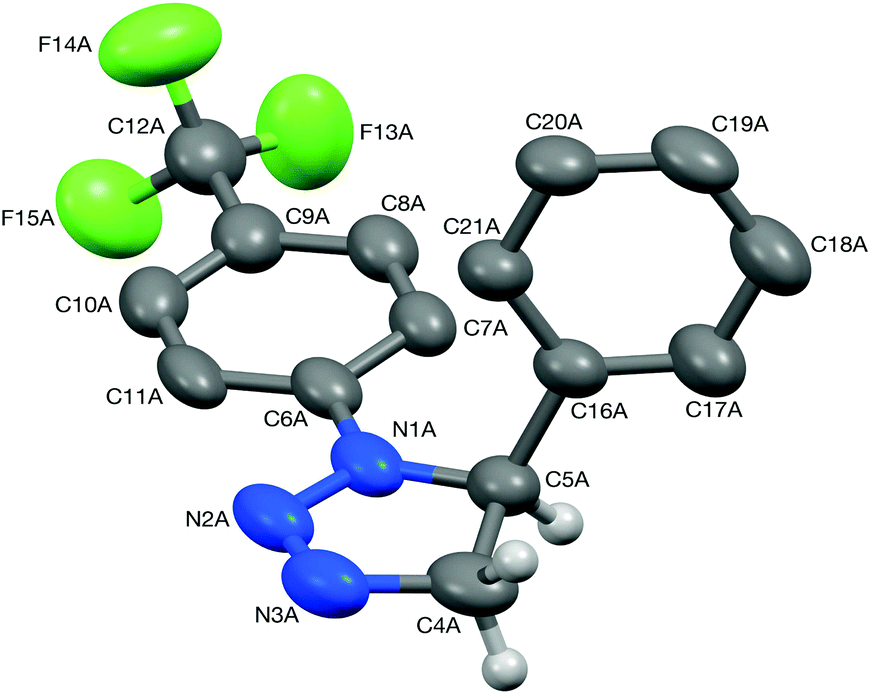 | ||
| Fig. 1 Structure of one (3aa-A) of the two independent molecules present in the crystal of triazoline 3aa (50% probability ellipsoids). Most hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°): N1A–N2A 1.380(6), N2A–N3A 1.241(8), N3A–C4A 1.471(8), C4A–C5A 1.538(8), N1A–C5A 1.462(7); N3A–N2A–N1A 112.2(5), C4A–C5A–C16A 113.2(4). | ||
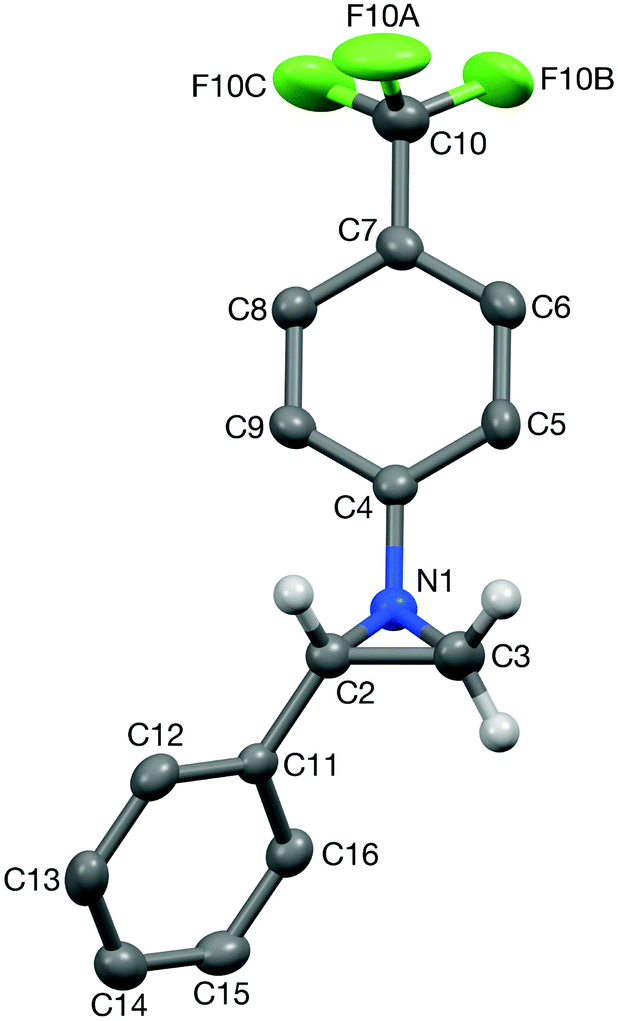 | ||
| Fig. 2 Structure of aziridine 4aa (50% probability ellipsoids). Most hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°): N1–C2 1.477(4), N1–C3 1.451(5), C2–C3 1.484(5); C3–N1–C2 60.9(2), C4–N1–C2 18.3(3). | ||
Scope of the reaction
With a set of optimised conditions in hand, we explored the scope of the reaction with a range of aryl/heteroaryl azides and styrene (Table 3). In all cases, the desired 1,5-disubstituted triazolines were the major product in these reactions and the 1H NMR conversions for the corresponding aziridines ranged between 6 and 36%. Proportionally, the highest formation of aziridines was observed when using azides bearing electron-donating substituent(s) (Table 3, entries 12–15). Reactions with heteroaryl azides as cycloaddition partners (Table 3, entries 16–18) were carried at lower temperatures (60–70 °C) as substantial decomposition of the reaction products was observed at 80 °C.|
|
||||
|---|---|---|---|---|
| Entry | R |
3:4b (%) |
3 (%) | 4 (%) |
| a Reaction conditions: Azide 1X, styrene 2a (2 equiv.) in DES (0.5 M). b 1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. c Isolated yields. d Reaction time = 8 h. e Reaction carried out at 70 °C for 24 h. f Reaction carried out at 60 °C for 32 h. | ||||
| 1 | 4-CF3 (1a) | 72:24 |
58 | 18 |
| 2d | 4-NO2 (1b) | 68:6 |
67 | — |
| 3 | 4-CN (1c) | 71:14 |
57 | 4 |
| 4 | 4-COOEt (1d) | 63:24 |
57 | 11 |
| 5 | 4-I (1e) | 60:27 |
54 | 20 |
| 6 | 4-Br (1f) | 71:31 |
56 | 24 |
| 7 | 4-Cl (1g) | 70:30 |
58 | 21 |
| 8 | 4-F (1h) | 66:29 |
56 | 15 |
| 9 | H (1i) | 53:30 |
54 | 23 |
| 10 | 3-CF3 (1j) | 76:21 |
76 | — |
| 11 | 3,5-(CF3)2 (1k) | 76:10 |
68 | — |
| 12 | 4-Me (1l) | 54:35 |
48 | 28 |
| 13 | 4-iPr (1m) | 52:33 |
46 | 27 |
| 14 | 4-OMe (1n) | 56:36 |
51 | 29 |
| 15 | 2,4,6-(Me)3 (1o) | 57:29 |
56 | 11 |
| 16e |
|
69:15 |
62 | 14 |
| 17 |
|
35:6 |
— | — |
| 18f |
|
43:11 |
33 | — |
Even if the main purpose of this work was the preparation of 1,2,3-triazolines, many of the formed aziridines 4 were also isolated and fully characterised due to the inherent importance of this heterocyclic motif, both in chemistry and biology.30 Traces of aniline derivatives were observed in many of these reactions, but such a decomposition product was only significant when a nitro substituent was present in the starting azide (Table 3, entry 2, 23% by 1H NMR). On the other hand, imine formation was only competitive in the reaction of heteroaryl azide 1r (Table 3, entry 18). Indeed, whereas triazolines bearing ortho-electron-donor groups in their 1-aryl substituents were found relatively stable, this was not the case when the ortho substituent was an electron-withdrawing one. For example, in the reaction with 2-trifluoromethylphenyl azide, only traces of the desired triazoline were observed together with 30% of aziridine, 18% of imine and 6% of aniline. In the case of 2-nitrophenyl azide, only formation of imine (18%) and aniline (16%) was evidenced by 1H NMR.
While the optimisation studies were carried out using 1 mmol of aryl azide, the reactions could be easily scaled up and the scope of the cycloaddition was explored using 3 to 5 g of azide (15–27 mmol). This led to the multigram isolation of 1,2,3-triazolines, typically by simple precipitation and recrystallisation from the reaction mixture, which minimises the need for volatile organic compounds. Column chromatography was still necessary in some cases, as for the isolation of oily triazolines and aziridines, since the latter remained in the filtrate during the first recrystallization step. While triazoline 3aa and aziridine 4aa could be separated on silica, significant decomposition (∼40% of the formed materials) was observed as expected for products sensitive to acidic conditions. This could be easily avoided using basified silica (triethylamine), instead. No decomposition was observed with cellulose either, although the separation of 3aa and 4aa was poor and required several columns to obtain an acceptable purity. When basified alumina or florisil were used as stationary phase only triazoline 3aa was recovered, albeit contaminated with some decomposition by-products. Finally, purification on neutral alumina resulted in the substantial decomposition of both products. Overall, basified silica led to the best separation of the formed reaction products while minimising their decomposition during purification.
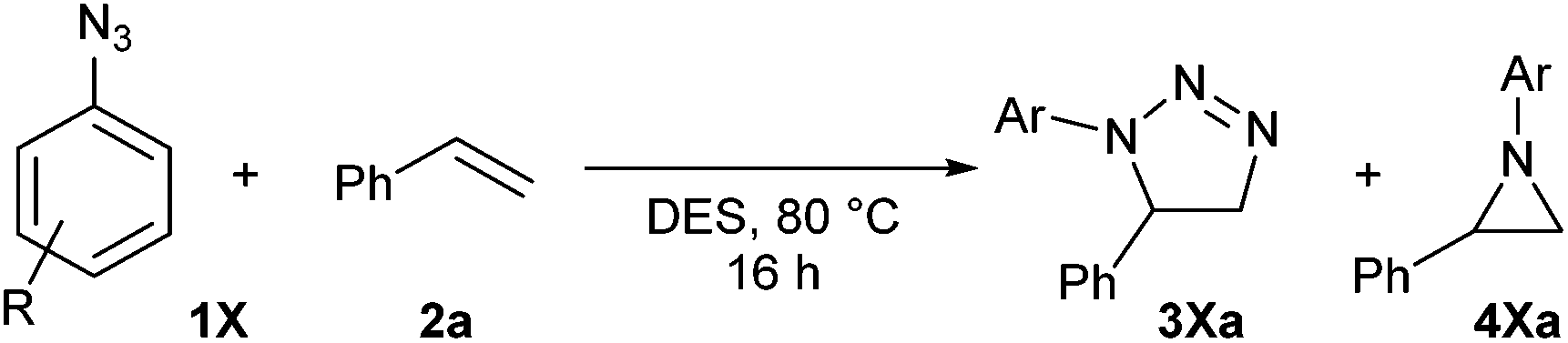
Benzylic azides were also screened in DES (Table 4), although lower conversions into cycloadducts were observed in these reactions, particularly when no electron-withdrawing groups were present. Longer reactions times could be used in order to maximise azide conversion, but higher reaction temperatures only led to significant decomposition of the reaction mixture. The reaction of bis(azide) 1v led to the formation of four different products, with that bearing a triazoline and an aziridine ring being the major product (34va, Table 4, entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Azide |
3:4b (%) |
3 (%) | 4 (%) | |
a Reaction conditions: Azide 1X, styrene 2a (2 equiv. per azide group) in DES (0.5 M).
b
1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard.
c Isolated yields.
d Reaction carried out at 85 °C.
e Reaction time = 24 h.
f Four reaction products could be isolated in this reaction: .
g Reaction carried out at 70 °C. .
g Reaction carried out at 70 °C.
|
|||||
| 1d |
|
1s | 36:34 |
28 | 29 |
| 2d,e |
|
1t | 50:30 |
42 | 22 |
| 3 |
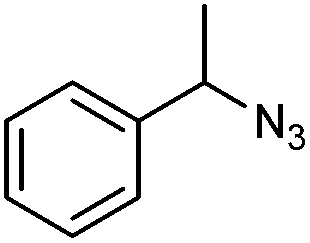
|
1u | 18:8 |
— | — |
| 4e,f |
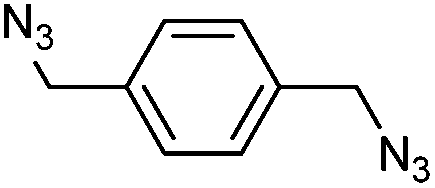
|
1v | 9:24:42:15 |
6 (31va) | 1 (44va) |
| 20 (33va) | |||||
| 29 (34va) | |||||
| 5g |
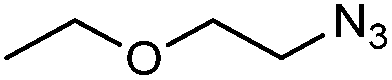
|
1w | n.d. | 16 | 5 |
The reaction of 1-azido-2-ethoxyethane with styrene has been reported to yield a mixture of regioisomeric triazolines after heating neat at 50 °C for 30 days.31 However, when we tried to reproduce this transformation, we obtained a mixture of 1,5-disubstituted triazoline and aziridine (49% and 35% 1H NMR yield, respectively). This reaction could also be performed in DES, albeit low conversions were obtained after 16 h of stirring (Table 4, entry 5). Only starting materials were recovered from the reactions with other aliphatic azides (i.e. decyl azide and adamantyl azide) at 80 °C. When the reaction temperature was raised to 90 °C, 8% of the desired triazoline was observed by 1H NMR with decyl azide, but higher temperatures only led to severe decomposition of the reaction mixture.
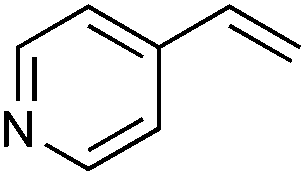
Next, the generality of the reaction in terms of alkene substitution was studied (Table 5). Two other styrene derivatives were screened bearing either an electron-withdrawing or an electron-donating group at the para position (Table 5, entries 1 and 2). While only formation of triazoline 3dc was observed in the reaction with 4-methoxystyrene, a lower ratio of triazoline formation was observed with the 4-chloro analogue. A similar triazoline to aziridine ratio was also obtained with a naphthyl derivative (Table 5, entry 3). The structure of triazoline 3dd was confirmed by X-ray crystallography (Fig. 3) because several broad signals were observed in its 1H NMR spectrum, which in our experience is unusual for this family of compounds. Interestingly, the reaction of 4-vinylpyridine afforded the corresponding aziridine 4de as the major reaction product (Table 5, entry 4). This was also the only product that could be isolated analytically pure from this reaction.
 | ||
| Fig. 3 Structure of triazoline 3dd (50% probability ellipsoids). Most hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°): N1–N2 1.3724(18), N2–N3 1.2515(19), N3–C4 1.477(2), C4–C5 1.542(2), N1–C5 1.4681(19); N3–N2–N1 111.97(14), C4A–C5A–C16A 113.99(12). | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alkene |
3:4b (%) |
3 (%) | 4 (%) | |
| a Reaction conditions: Azide 1d, alkene 2X (2 equiv.) in DES (0.5 M). b 1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. c Isolated yields. d Reaction carried out at 85 °C. e Reaction carried out at 75 °C. f Reaction carried out at 65 °C. g Reaction carried out at 60 °C. | |||||
| 1 |
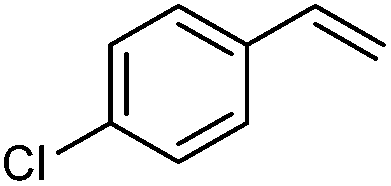
|
2b | 53:31 |
39 | — |
| 2 |
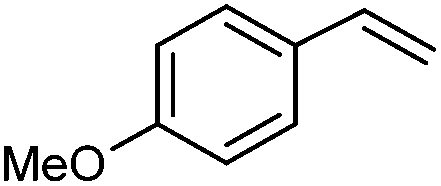
|
2c | 86:0 |
70 | — |
| 3 |
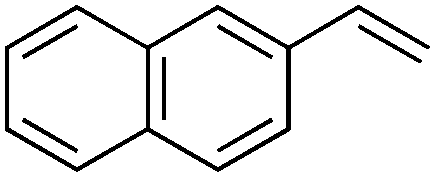
|
2d | 56:25 |
42 | 14 |
| 4 |
|
2e | 31:40 |
— | 31 |
| 5 |
|
2f | 44:16 |
41 | 3 |
| 6 |
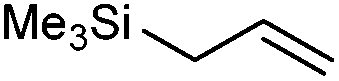
|
2g | 67:<5 |
62 | — |
| 7 |
|
2h | 5:30 |
— | 16 |
| 8d |
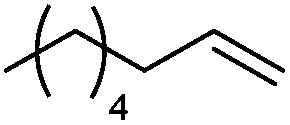
|
2i | 52:15 |
43 | 26 |
| 9e |
|
2j | 74:0 |
66 | — |
| 10f |

|
2k | 90:0 |
85 | — |
| 11g |
|
2l | 56:0 |
37 | — |
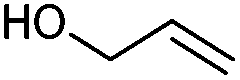
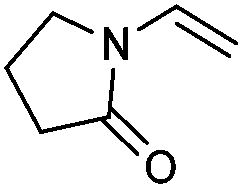
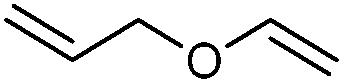
We also found that the allylic substituent has a profound impact on the cycloaddition outcome. Indeed, the reaction of allyltrimethylsilane delivered triazoline 3dg in good yields and only traces of the corresponding aziridine. However, allylic alcohol led to the formation of 30% of aziridine 4dh and only 5% of triazoline (Table 5, entries 6 and 7). Significant aniline formation (∼25% 1H NMR yield), indicative of product decomposition, was observed in all reactions with allylic derivatives. Even an alkene as non-activated as 1-octene could be successfully used under our reaction conditions (Table 5, entry 8). As expected,5 electron-richer alkenes led to completely selective reactions and no aziridine formation was observed (Table 5, entries 9 and 10). These alkenes were particularly reactive cycloaddition partners and triazoline 3dk could be prepared at 65 °C in high yields. This accrued reactivity was exploited with allyl vinyl ether (Table 5, entry 11), which delivered 5-(allyloxy)triazoline 3dl with only traces of the triazoline issue of the reaction of the allyl moiety. Nevertheless, the presence of an allyl group remained problematic and around 30% of the overall mass could not be accounted for in this case.
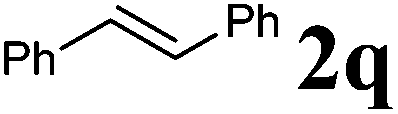
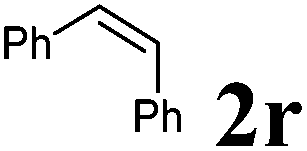
Next, the effect of introducing additional substituents on the alkene partner was explored. In general, longer reaction times and/or higher reaction temperatures were required to overcome the increased steric bulk in these reactions (Table 6). 1,1-Disubstituted alkenes led to the exclusive formation of the corresponding triazolines with the notable exception of 1,1-diphenylethene (Table 6, entries 1–3). Norbornene yielded triazoline 3dp quantitatively at room temperature (Table 6, entry 4), as expected for a strained substrate.5a In stark contrast, unstrained 1,2-disubstituted alkenes led to the stereospecific formation of the corresponding aziridines (trans-aziridines from E-alkenes and cis-aziridines from Z-alkenes). The crystal structure of 4dq confirmed the stereospecificity of the aziridine formation,32 which is in agreement with the expected configurational stability of singlet biradicals (vide infra).33
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alkene | Conditions |
3:4b (%) |
3 (%) | 4 (%) |
| a Reaction conditions: Azide 1d, alkene 2X (2 equiv.) in DES (0.5 M). b 1H NMR yields are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. c Isolated yields. | |||||
| 1 |
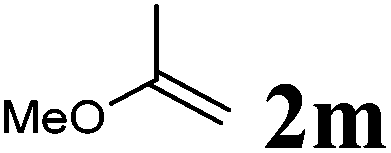
|
75 °C, 96 h | 92:0 |
87 | — |
| 2 |
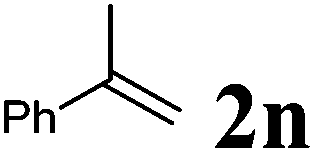
|
90 °C, 16 h | 66:0 |
51 | — |
| 3 |
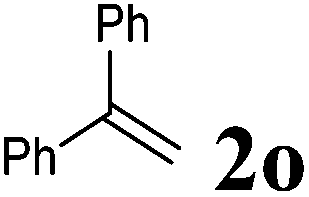
|
100 °C, 72 h | 30:17 |
24 | 11 |
| 4 |
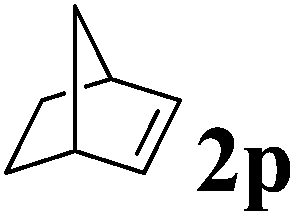
|
RT, 16 h | 100:0 |
99 | — |
| 5 |
|
100 °C, 48 h | 0:60 |
— | 53 |
| 6 |
|
110 °C, 16 h | 0:42 |
— | 36 |
| 7 |
|
90 °C, 32 h | 0:26 |
— | 21 |
| 8 |
|
95 °C, 32 h | 0:26 |
— | 21 |
Aziridines 4dq–4dt were accompanied by decomposition products, which suggests that triazolines bearing substituents on the 4-position are considerably less stable and supports the notion of aziridines being formed from 1,4-disubstituted triazolines, as discussed below.
From these results, it is clear that the product distribution is most strongly influenced by the alkene substitution, while the nature of the azide has a crucial effect on the overall conversion into cyclised products.
Finally, we screened a 1,2-diene under our reaction conditions, cyclohexylallene (Scheme 1). In this case, no aziridine formation was observed and instead two conjugated cycloadducts were obtained (together with aniline and other unidentified decomposition products), triazoline 3du and triazole 5du, issue of a double bond migration in 3du. The observed regioselectivity is in accordance with the few reports available on intermolecular azide–allene cycloadditions.34
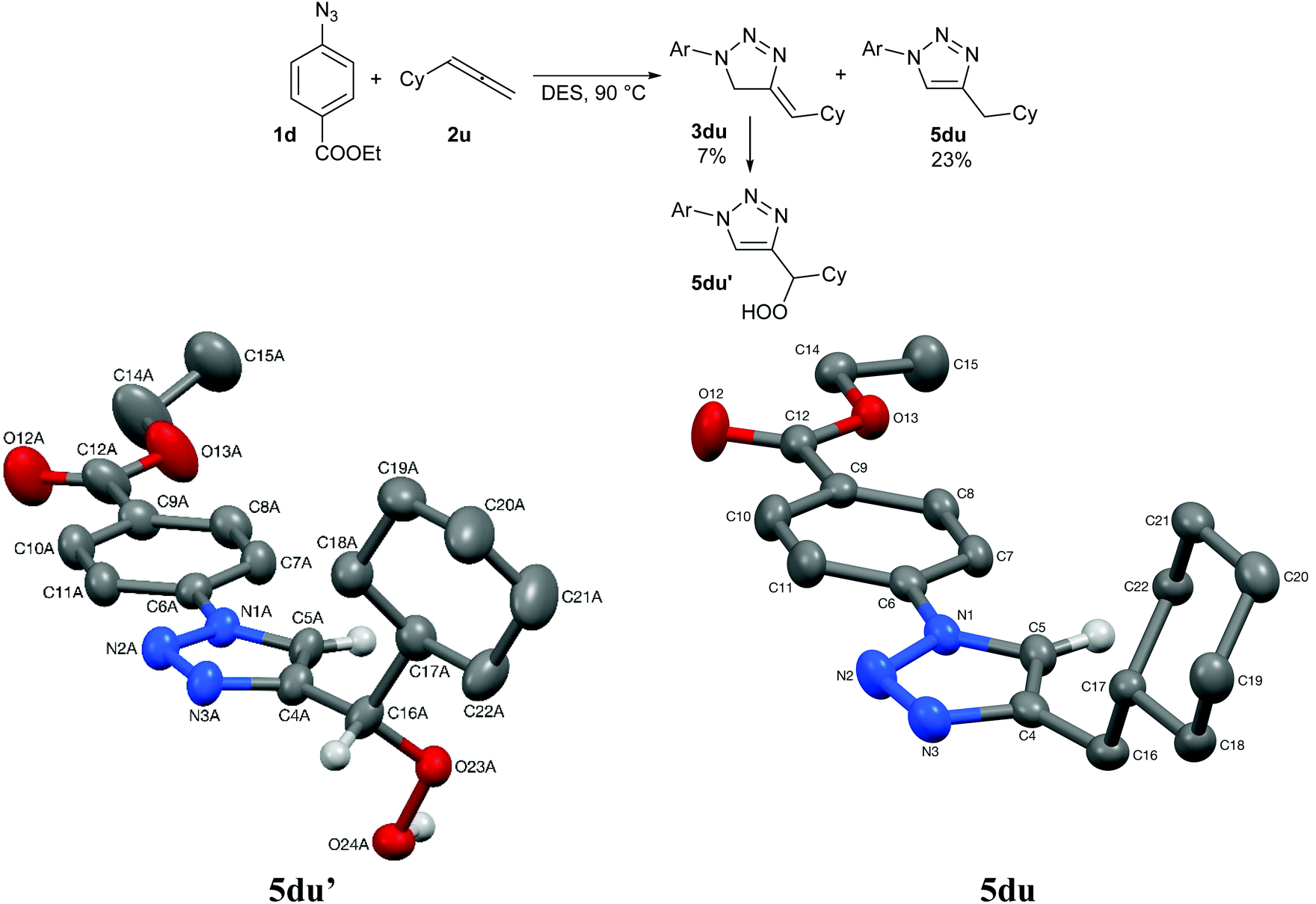 | ||
| Scheme 1 Intermolecular azide–allene cycloaddition reaction in DES. Structures of one (5du′-A) of the two independent molecules present in the crystal of triazoles 5du′ (left) and triazole 5du (right) are 50% probability ellipsoids. Most hydrogens are omitted for clarity. | ||
No conversion of triazoline 3du into triazole 5du was observed during its isolation/purification. However, attempts to grow crystals of 3du from a hot solution in methanol led to the characterisation of peroxide 5du′ instead. While triazole 5du′ was not present in the reaction mixture, traces of it could already be evidenced in the 1H NMR of triazoline 3du after a pentane/diethyl ether wash, which shows its high reactivity.
Mechanistic studies and origin of the aziridines
(A) The DES effect: As mentioned in the introduction, no metal-based catalysts have been reported for azide–alkene cycloaddition reactions. We could only find two reports evoking the use of organocatalysts in this reaction. Garcia-Garibay and co-workers reported the use of dimethylurea as catalyst in the cycloaddition reaction of aryl azides and electron-poor 1,2-disubstituted alkenes.35 Alternatively, tetramethylammonium hydrogen sulfate was used to mediate the cycloaddition of enones to isolate triazoles after in situ oxidation of the intermediate triazoline.36 When we subjected one of our reactions to these reported conditions, only disappointing results were obtained (Table 7, entries 1–3). Little, if any, cycloadducts were obtained, accompanied by significant decomposition of the reaction mixture as observed by 1H NMR.|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions (R = –COOEt) | 1d (%) | 3da (%) | 4da (%) | ArNH 2 (%) |
| 1 | Dimethylurea (10 mol%), toluene, 80 °C, 16 h | 12 | 23 | 12 | — |
| 2 | [NBu4][HSO4] (20 mol%), toluene, 80 °C, 16 h | 58 | — | — | 7 |
| 3 | [NBu4][HSO4] (20 mol%), DMF, 80 °C, 16 h | 42 | 9 | 5 | 10 |
| 4 | Standard conditions: DES, 80 °C, 16 h | 5 | 63 | 24 | 7 |
| Entry | Conditions (R = –CF3) | 1a (%) | 3aa (%) | 4aa (%) | ArNH 2 (%) |
|---|---|---|---|---|---|
| a 1H NMR yields/recoveries are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. | |||||
| 5 | Urea (15 mol%), THF/water, 80 °C, 16 h | 20 | 19 | 11 | 6 |
| 6 | ChCl (15 mol%), THF/water, 80 °C, 16 h | 16 | 22 | 10 | 6 |
| 7 | Urea (30 mol%), ChCl (15 mol%), THF/water, 80 °C, 16 h | 0 | 42 | 16 | 5 |
| 8 | Standard conditions: DES, 80 °C, 16 h | <5 | 72 | 24 | <5 |
Furthermore, in previous examples of dipolar cycloadditions in DES, an organocatalytic effect is often suggested through H-bonding interactions between the cycloaddition partners and DES components.23,24,26b While such interactions might be established with any organic azide through its internal nitrogen atom, most of the alkenes screened in this work are not susceptible to forming hydrogen bonds. We nevertheless tested each of the DES components as organocatalysts for cycloaddition. In these tests, THF/water mixtures and vigorous stirring were required in order to ensure homogeneous solutions. Virtually identical results were obtained with either 15 mol% of choline chloride or urea (Table 7, entries 5 and 6) and even if the conversion of triazoline 3aa remained modest it should be noted that only decomposition was obtained when the cycloaddition was carried out in THF/water in the absence of an additive. Using both DES components in sub-stochiometric quantities further improved the results, but none of these conditions were competitive when compared to the developed reactions in DES, which led to the highest and cleanest conversions (Table 7, entries 4 and 8).
Overall, it appears plausible that the observed “DES effect” in the azide–alkene reaction is mainly due to the increased thermal stability of the formed cycloadducts when compared to standard organic solvents. However, an organocatalytic effect of one or both DES components cannot be completely ruled out at this time.
(B) Formation and evolution of triazolines: It is well established that the cycloaddition of aryl azides and simple alkenes proceeds through an asynchronous concerted mechanism.37 In comparison, the factors determining the final reaction product distribution are less studied.35,38 Accordingly, we next carried out a computational mechanistic study to rationalise the obtained experimental results and focused our efforts on 1,2,3-triazolines as the origin of the isolated aziridines. The formation of the three-membered heterocycles via a nitrene–alkene cycloaddition was not considered since formation of nitrenes from the reported aryl azides is highly unlikely at the reaction temperatures employed.39 Indeed, azide 1d was quantitatively recovered after heating it in DES at 80 °C for 24 h or at 90 °C for 8 h. Hence, the formation of aziridines 4 from transient 1,4-disubstituted triazolines seems to be the most likely option. However, we could not prepare such a triazoline even when following literature procedures (vide supra, Table 4, entry 5). On the other hand, we observed that triazoline 3aa was stable under our reaction conditions (Table 8, entry 1) and only minor decomposition was observed in other solvents such as DMSO and dioxane (Table 8, entries 2 and 3). At 100 °C, only 58% of the starting triazoline was recovered but the main decomposition product was the corresponding aniline and only small amounts of aziridine were formed in comparison (Table 8, entry 4). No triazoline was recovered at 100 °C in either DMSO or dioxane, with imine 6aa being the major thermolysis product in the latter case (Table 8, entries 5 and 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions | 3aa (%) | 4aa (%) | 6aa (%) | 7a (%) | ArNH 2 (%) |
| a 1H NMR yields/recoveries are the average of two independent experiments and were determined with respect to 1,3,5-trimethoxybenzene as internal standard. | ||||||
| 1 | DES, 80 °C, 16 h | >95 | — | — | — | — |
| 2 | DMSO, 80 °C, 16 h | 89 | — | 12 | — | — |
| 3 | 1,4-Dioxane, 80 °C, 16 h | 93 | — | 5 | — | — |
| 4 | DES, 100 °C, 16 h | 58 | 6 | 7 | 7 | 29 |
| 5 | DMSO, 100 °C, 16 h | Complete decomposition | ||||
| 6 | 1,4-Dioxane, 100 °C, 16 h | — | 9 | 30 | <5 | <5 |
Calculations40 were performed at the B3LYP41+GD3BJ42/6-311++G(d,p)43 (triple-ζ) SCRF continuum solvation level, using the CPCM44 model and N-methylformamide mixture level parameters (ε = 181.56) to simulate the high polarity of the eutectic solvent used experimentally. Calculations using the alternative ωB97XD45 functional and/or the Def2-QZVPP46 quadruple-ζ basis set are reported for selected systems. All transition states were characterised by normal coordinate analysis, revealing one imaginary mode corresponding to the intended reaction. IRC47 calculations on these transition states confirmed the identity and synchronicity of the reactions. Unless stated otherwise, all energy values are free energies ΔG‡353 (kcal mol−1) for a standard state of 1 atm (0.041 M), normalized to a relative free energy of 0 kcal mol−1 for the reactant pre-complex. Full coordinates for all the stationary points, together with IRC animations and other data are available via a FAIR data repository.48
The reaction between azide 1d and styrene was chosen for the computational study. In this reaction, small quantities of imine 6da and the corresponding aniline were also formed as minor by-products (Scheme 2). An overview of the located stationary points and transition states and their corresponding energies is shown in Scheme 3.
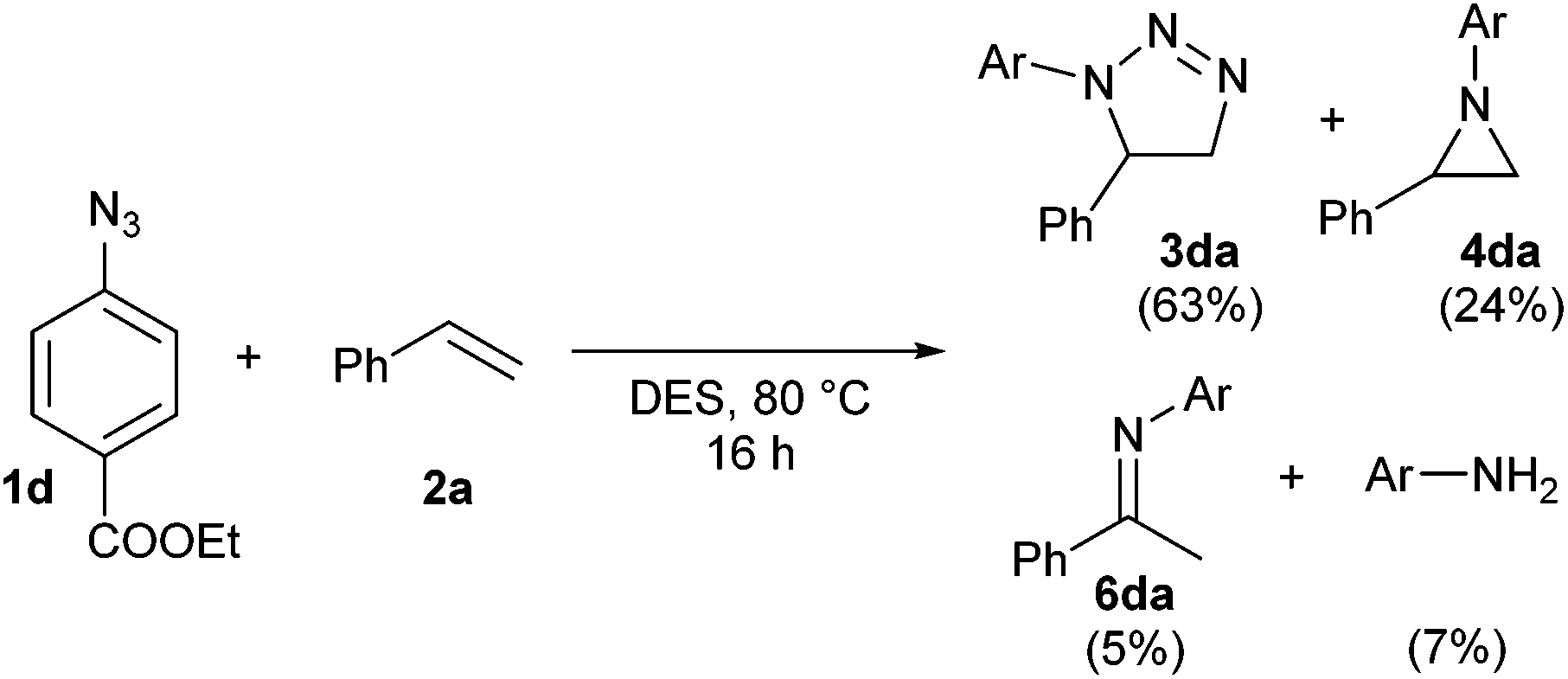 | ||
| Scheme 2 Complete product distribution in the model reaction. | ||
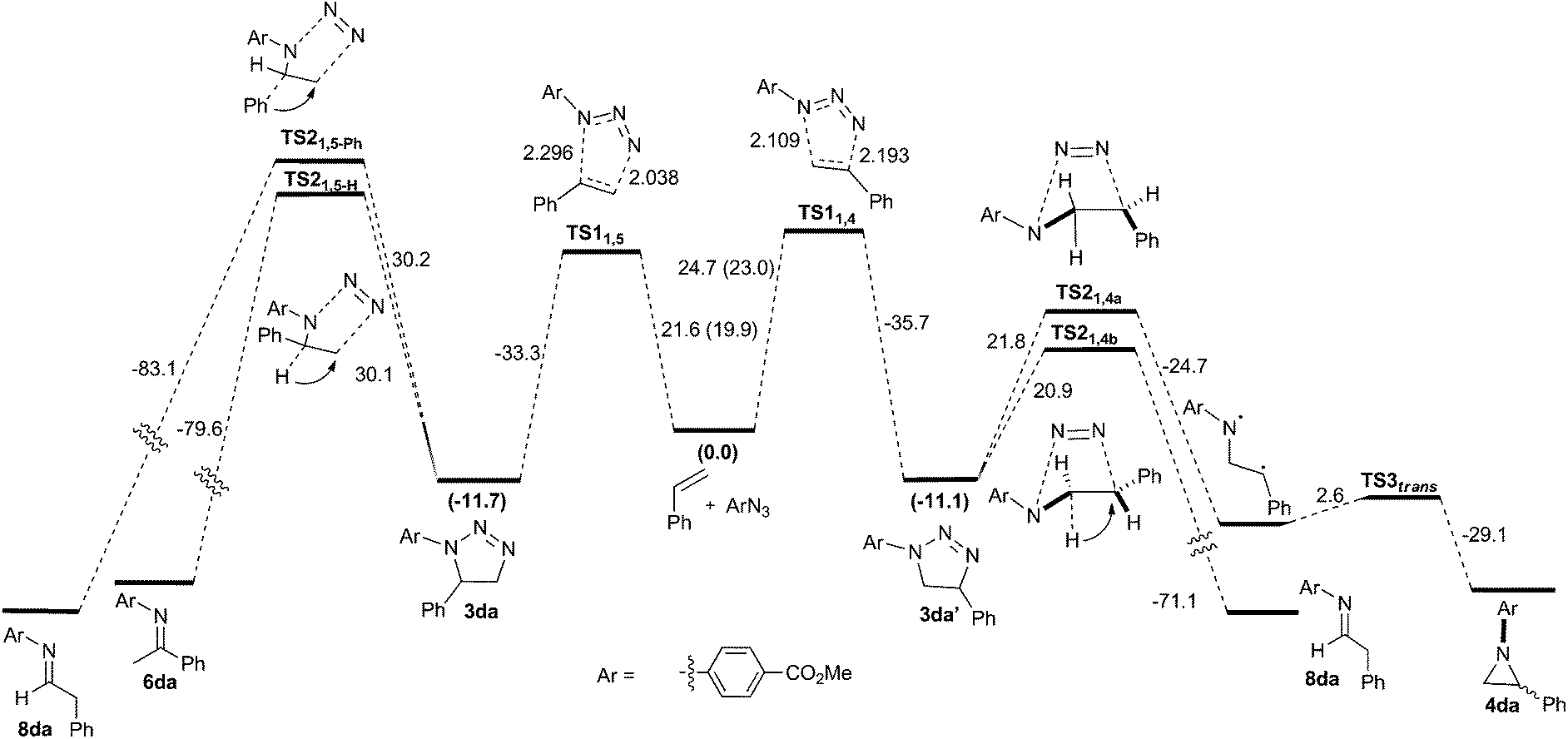 | ||
| Scheme 3 Complete reaction profile with ΔG‡353 calculated at the B3LYP+GD3BJ/6-311++G(d,p)/SCRF level. | ||
At the B3LYP+GD3BJ/6-311++G(d,p)/SCRF level, two concerted asynchronous cycloaddition transition states resulting in the formation of regioisomeric triazolines 3da and 3da′ were located as TS11,5 and TS11,4, respectively (Scheme 3). The basis set effect (BSE) on the difference in the activation free energies ΔΔG‡353 for 6-311++G(d,p) vs. Def-QZVPP was found to be insignificant, whilst the effect on the barrier is to increase it by ∼2 kcal mol−1. The regioselectivity ΔΔG‡353 was more sensitive to the DFT functional, ranging from 1.3 for the B3LYP method with no dispersion included, to 2.2 for the ωB97XD method which includes a built-in second generation D2 dispersion correction and 2.8 kcal mol−1 using B3LYP with explicit inclusion of the third generation GD3BJ correction (Table 9). These differences qualitatively match the experimentally observed regioselectivity favouring the formation of 3da as the major product (57% isolated yield). The reaction is predicted exoenergic by >10 kcal mol−1 but only if dispersion is included in the model, since this affects both the interaction energies and the entropies, both of which propagate to the free energies.
| Method | TS11,4 | TS11,5 | ΔΔG‡353 | ΔΔG353c |
|---|---|---|---|---|
| a Bimolecular energetic barriers ΔG‡353 are reduced by 1.7 kcal mol−1 when corrected for the 0.5 M experimental concentration. b Def2-QZVPP basis set. c ΔΔG353 product1,4, product1,5. Full FAIR data is available via a data repository at DOI: 10.14469/hpc/4325. | ||||
| B3LYP/6-311++G(d,p) | 30.8 | 29.5 | 1.27 (0.95)b | −4.9, −4.1 |
| B3LYP+GD3BJ/6-311++G(d,p) | 24.7(26.7)b | 21.6(24.0)b | 3.08 (2.75)b | −11.1, −11.7 |
| ωB97XD/6-311++G(d,p) | 29.0 | 26.7 | 2.29 (2.22)b | −17.5, −18.3 |
To understand the origin of the minor products shown in Scheme 2, we next modelled the nitrogen extrusion processes. That from 1,5-disubstituted triazoline was investigated just at the B3LYP+GD3BJ/6-311++G(d,p) level, the reference (resting) state now being the product of the first reaction 3da (Table 10). No transition state for aziridine formation could be located from 3da. Instead, it was computed to evolve into two different imines 6da(H) and 8da(Ph)viaTS21,5-H and TS21,5-Ph, a process that also involves the migration of either a H or Ph group to the emerging primary cation via relatively high energy barriers (Table 10 and Fig. 4). The presence of a dipolar “hidden intermediate”49 is shown by the large increase in dipole moment in the region of the transition state (Fig. 4b). As previously stated, prolonged heating at 80 °C of a pure sample of a 1,5-disubstituted triazoline led to the corresponding imine as the only decomposition product (see Table 8). Partial hydrolysis of this imine during work-up would explain the detection of anilines in the reaction crudes. Competitive TS21,5-Ph would produce aldimine 8da, a more unstable product that would completely hydrolyse into aniline and an aliphatic aldehyde, which in turn is likely to oligomerise under the reaction conditions. The overall reactions are now strongly exoenergic.
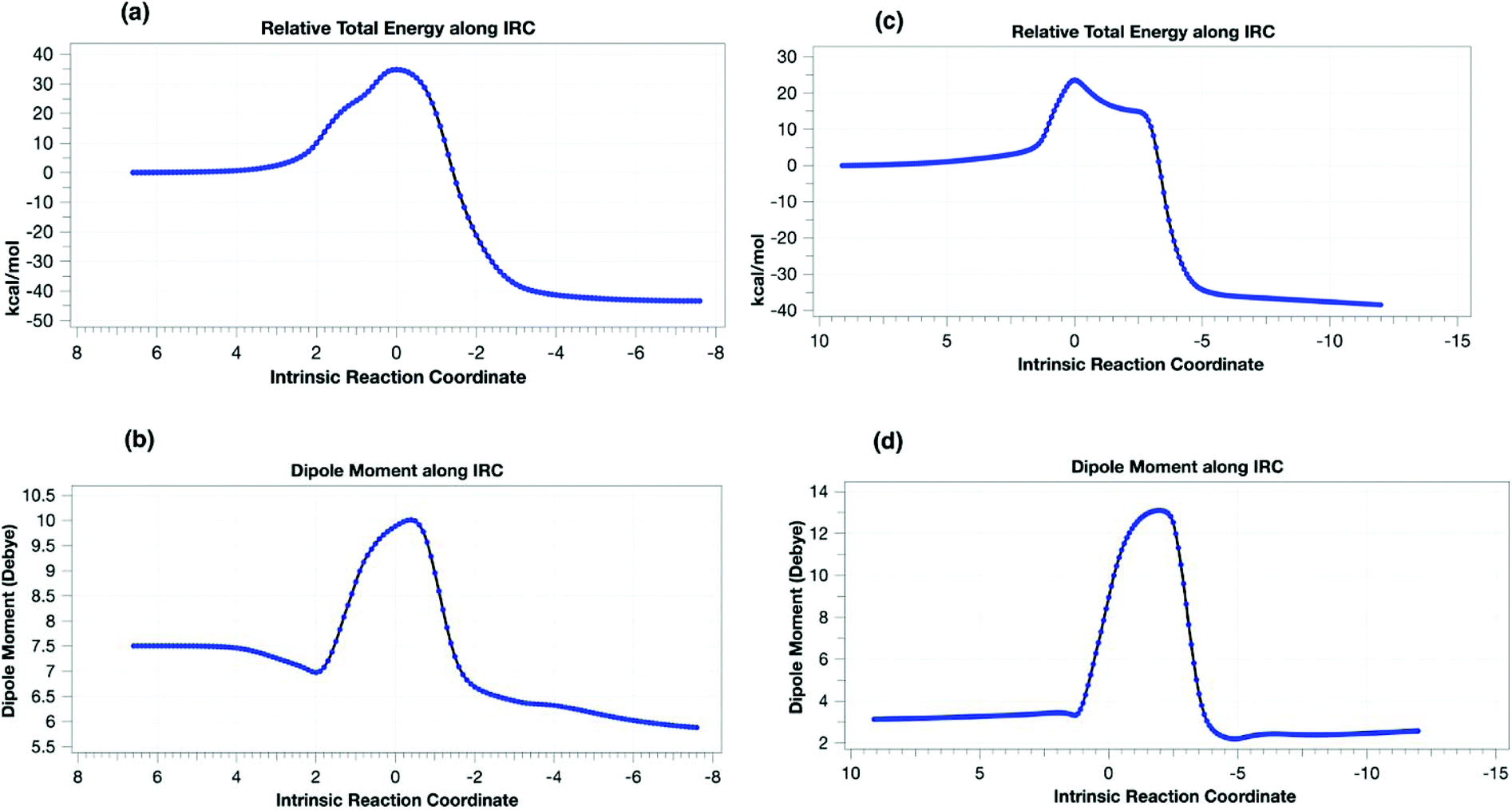 | ||
| Fig. 4 Computed properties along an intrinsic reaction coordinate (IRC) for (a) energy profile for TS21,5-H, (b) dipole moment response for TS21,5-H, (c) energy profile for TS21,4-b and (d) dipole moment response for TS21,4-b. | ||
| Method | ΔG‡353 | ΔΔG353b |
|---|---|---|
| a At the B3LYP+GD3BJ/6-311++G(d,p) level, relative to 3da or 3da′ in kcal mol−1. b Overall reaction free energies. c Energy of intermediate biradical. Full FAIR data is available via a data repository at DOI: 10.14469/hpc/4326. | ||
| TS21,5-H | 30.1 | −53.0 |
| TS21,5-Ph | 30.2 | −49.4 |
| TS21,4a | 21.8 | −42.4 (−13.9)c |
| TS21,4b | 20.9 | −61.3 |
Two diastereomeric transition states (TS21,4a and TS21,4b) were located for the loss of dinitrogen from 1,4-disubstituted triazoline 3da′. The activation free energies are now low enough to explain the absence of reactant 3da′ in the reaction products (Table 10). The IRC for TS21,4a revealed the formation of a singlet dipolar biradical followed by a relatively low energy (e.g. ΔG‡353trans = 2.6 kcal mol−1) for electrocyclic ring closure to the corresponding aziridines cis-4da and trans-4da. In contrast, TS21,4b was found to evolve into imine 8da upon a migration of a hydrogen atom (Fig. 4), which would be expected to easily hydrolyse into the corresponding aniline. The transition state again has transient cationic character (Fig. 4c) as revealed by the dipole moment response along the IRC.
Overall, our calculations are in good agreement with the experimental results. A thermal dipolar cycloaddition step leads to the formation of two regioisomeric triazolines. Whereas the 1,5-disubstituted triazoline 3da would be kinetically relatively stable at the reaction temperature and render only small quantities of imine by-products at most, the 1,4-disubstituted regioisomer 3da′ readily reacts to form either the corresponding aziridine or imines with dinitrogen evolution. We have experimentally shown that the product distribution is also highly dependent on the aryl alkene and azide substitutions. This is consistent with the insight revealed by the calculations for nitrogen extrusion in particular, which show significant dipolar/ionic character localised in the region of the transition state for this step. It has indeed been established that variation in the substituents may indeed induce dramatic changes in reaction mechanisms.50 A full study of the influence of such substituents upon the reactions described here will be reported separately.
Conclusions
A range of di- and tri-substituted Δ2-1,2,3-triazolines have been prepared and fully characterised. The studied azide–alkene cycloadditions in DES typically led to mixtures of 1,2,3-triazolines and aziridines, although the overall product distributions were largely influenced by the alkene substitution. The benefits of DES in this context are twofold. First, the DES employed herein is non-flammable and it is composed of two safe and inexpensive components, urea and choline chloride. Secondly, much cleaner reaction crudes were obtained in DES when compared to standard organic solvents, and typically several grams of the desired triazolines could then be isolated analytically pure after a simple recrystallisation step.Furthermore, a mechanism for the reaction between aromatic azides and olefins to yield triazolines and/or aziridines has been proposed. All our calculations have shown that this interesting process can proceed through two divergent pathways, cycloaddition followed by simple loss of dinitrogen to a biradical intermediate and then cyclization to the corresponding aziridines or loss of dinitrogen together with concomitant group migration to form imines. The process seems to be highly dependent on the nature of both the starting azide and the alkene and might also be dynamically controlled. Further studies on DES applications and dynamic calculations are underway in our laboratories.
Experimental section
General procedure for the dipolar cycloadditions
Azide (1.0 equiv.) and alkene (2.0 equiv.) were stirred in deep eutectic solvent (choline chloride/urea = 1:2; 0.5 M) at 80 °C for 16 h, unless stated otherwise. The reaction mixture was cooled down to room temperature, diluted with water (2 mL per mmol of azide) and pentane/Et2O (3:1; 1 mL per mmol of azide) and stored at −18 °C overnight. If triazoline precipitated overnight, this was triturated at room temperature, collected by filtration, washed three times with ice cold pentane/Et2O and recrystallised from boiling ethanol if necessary. Occasionally, the washings and/or filtrate from this recrystallisation were further purified by column chromatography as described next, either to improve the triazoline recovery, or to isolate the corresponding aziridine. If no solid precipitated at −18 °C, the mixture was extracted with EtOAc, the combined organic phases were washed with water and brine, dried over MgSO4, filtered, concentrated under reduced pressure and purified by column chromatography on basified silica (eluent: petroleum ether/EtOAc 0 → 25% gradient basified with 2% v/v NEt3 unless stated otherwise; reaction crude was dry-loaded onto stationary phase). Occasionally, the product recovered after column chromatography required washing with ice-cold pentane/Et2O (1:1).
3aa: Following the general procedure from 1-azido-4-trifluoromethylbenzene (5.00 g, 26.7 mmol) and styrene (6.18 mL, 53.4 mmol), the title compound was isolated as a white solid (3.68 g after recrystallisation, 0.82 g after column: 58% overall). From this reaction, aziridine 4aa was also isolated as a white solid (1.28 g, 18%) after column chromatography and recrystallisation from pentane at −18 °C.
 :20); IR: νmax 1612 (m), 1523 (m), 1502 (m), 1458, 1431, 1365 (m), 1316 (m), 1193, 1153 (m), 1111 (s), 1067 (s), 1010 (m), 990 (m), 963 (m), 926 (m), 841 (m), 820 (s), 751 (s), 698 (s) cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.45 (d, J = 8.5 Hz, 2H, H2), 7.35–7.24 (m, 5H, HAr), 7.15 (d, J = 8.5 Hz, 2H, H3), 4.94 (dd, J = 12.5; 8.0 Hz, 1H, H6), 4.88 (dd, J = 17.5; 12.5 Hz, 1H, H5), 4.37 (dd, J = 17.5; 8.0 Hz, 1H, H5); 13C NMR (CDCl3, 101 MHz): δ 142.9 (C4), 139.5 (C7), 129.5 (C9), 128.4 (C10), 126.5 (q, J = 3 Hz, C2), 125.9 (C8), 124.3 (q, J = 269 Hz, C11), 124.0 (q, J = 33 Hz, C1), 114.4 (C3), 76.1 (C5), 57.4 (C6); 19F NMR (CDCl3, 377 MHz): δ −61.9 (s); HRMS (ES+) calculated for C15H13N3F3 292.1062, found 292.1058 ([M + H]+); elemental analysis calculated for C15H12N3F3: C, 61.85; H, 4.15; N, 14.43, found: C, 61.95; H, 4.27; N, 14.27.
:20); IR: νmax 1612 (m), 1523 (m), 1502 (m), 1458, 1431, 1365 (m), 1316 (m), 1193, 1153 (m), 1111 (s), 1067 (s), 1010 (m), 990 (m), 963 (m), 926 (m), 841 (m), 820 (s), 751 (s), 698 (s) cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.45 (d, J = 8.5 Hz, 2H, H2), 7.35–7.24 (m, 5H, HAr), 7.15 (d, J = 8.5 Hz, 2H, H3), 4.94 (dd, J = 12.5; 8.0 Hz, 1H, H6), 4.88 (dd, J = 17.5; 12.5 Hz, 1H, H5), 4.37 (dd, J = 17.5; 8.0 Hz, 1H, H5); 13C NMR (CDCl3, 101 MHz): δ 142.9 (C4), 139.5 (C7), 129.5 (C9), 128.4 (C10), 126.5 (q, J = 3 Hz, C2), 125.9 (C8), 124.3 (q, J = 269 Hz, C11), 124.0 (q, J = 33 Hz, C1), 114.4 (C3), 76.1 (C5), 57.4 (C6); 19F NMR (CDCl3, 377 MHz): δ −61.9 (s); HRMS (ES+) calculated for C15H13N3F3 292.1062, found 292.1058 ([M + H]+); elemental analysis calculated for C15H12N3F3: C, 61.85; H, 4.15; N, 14.43, found: C, 61.95; H, 4.27; N, 14.27.
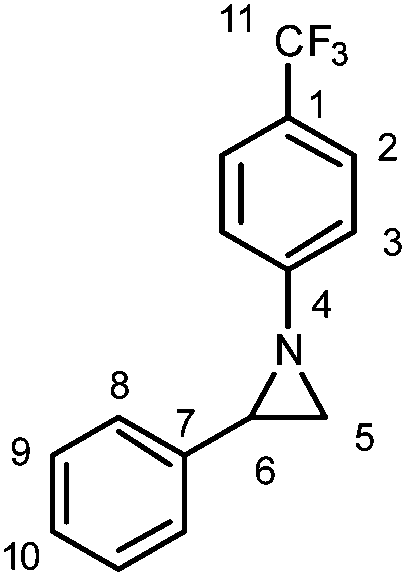 :20); IR: νmax 3053, 1614 (m), 1516, 1464, 1418, 1389, 1327 (s), 1279 (s), 1185, 1163 (m), 1127 (m), 1101 (s), 1076 (m), 1065 (s), 1010 (m), 981 (m), 914, 867, 843 (s), 790, 767 (m), 758 (s), 714 (s), 698 (s) cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.50 (d, J = 8.5 Hz, 2H, H2), 7.38–7.36 (m, 4H, HAr), 7.34–7.29 (m, 1H, HAr), 7.11 (d, J = 8.5 Hz, 2H, H3), 3.16 (dd, J = 6.5; 3.5 Hz, 1H, H6), 2.49 (dd, J = 6.5; 1.0 Hz, 1H, H5), 2.47 (dd, J = 3.5; 1.0 Hz, 1H, H5); 13C NMR (CDCl3, 101 MHz) δ 157.6 (C4), 138.6 (C7), 128.6 (C9), 127.7 (C10), 126.3 (q, J = 3 Hz, C2), 126.2 (C8), 124.6 (q, J = 32 Hz, C1), 124.4 (q, J = 269 Hz, C11), 120.7 (C3), 41.7 (C6), 37.7 (C5); 19F NMR (CDCl3, 377 MHz): δ −61.8 (s); HRMS (ES+) calculated for C15H13NF3 264.1000, found 264.0988 ([M + H]+); elemental analysis calculated for C15H12NF3: C, 68.44; H, 4.59; N, 5.32, found: C, 68.39; H, 4.69; N, 5.29.
:20); IR: νmax 3053, 1614 (m), 1516, 1464, 1418, 1389, 1327 (s), 1279 (s), 1185, 1163 (m), 1127 (m), 1101 (s), 1076 (m), 1065 (s), 1010 (m), 981 (m), 914, 867, 843 (s), 790, 767 (m), 758 (s), 714 (s), 698 (s) cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.50 (d, J = 8.5 Hz, 2H, H2), 7.38–7.36 (m, 4H, HAr), 7.34–7.29 (m, 1H, HAr), 7.11 (d, J = 8.5 Hz, 2H, H3), 3.16 (dd, J = 6.5; 3.5 Hz, 1H, H6), 2.49 (dd, J = 6.5; 1.0 Hz, 1H, H5), 2.47 (dd, J = 3.5; 1.0 Hz, 1H, H5); 13C NMR (CDCl3, 101 MHz) δ 157.6 (C4), 138.6 (C7), 128.6 (C9), 127.7 (C10), 126.3 (q, J = 3 Hz, C2), 126.2 (C8), 124.6 (q, J = 32 Hz, C1), 124.4 (q, J = 269 Hz, C11), 120.7 (C3), 41.7 (C6), 37.7 (C5); 19F NMR (CDCl3, 377 MHz): δ −61.8 (s); HRMS (ES+) calculated for C15H13NF3 264.1000, found 264.0988 ([M + H]+); elemental analysis calculated for C15H12NF3: C, 68.44; H, 4.59; N, 5.32, found: C, 68.39; H, 4.69; N, 5.29.
Conflicts of interest
There are no conflict to declare.Acknowledgements
This research was financially supported by Imperial College London and the EPSRC (DTP studentship to F. S. and EP/K030760). L. C. gratefully acknowledges financial support of the Spanish government for a grant in the 2017 Salvador de Madariaga program.Notes and references
- (a) 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley-Interscience, New York, 1984, vol. 1 and 2 Search PubMed; (b) R. Huisgen, Proc. Chem. Soc., 1961, 357–369 Search PubMed.
- For selected, complementary reviews, see: (a) C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686–701 CrossRef PubMed; (b) S. Chassaing, V. Bénéteau and P. Pale, Catal. Sci. Technol., 2016, 6, 923–957 RSC; (c) E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC; (d) S. Díez-González, Catal. Sci. Technol., 2011, 1, 166–178 RSC; (e) Special issue on Click chemistry, Chem. Soc. Rev., 2010, 39, 1221–1408; ; (f) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef PubMed.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef.
- (a) P. Scheiner, J. H. Schomaker, S. Deming, W. J. Libbey and G. P. Nowack, J. Am. Chem. Soc., 1965, 87, 306–311 CrossRef; (b) R. Huisgen, L. Möbius and G. Szeimies, Chem. Ber., 1965, 98, 1153–1158 CrossRef; (c) M. E. Munk and Y. K. Kim, J. Am. Chem. Soc., 1964, 86, 2213–2217 CrossRef; (d) Y. Nomura, Y. Takeuchi, S. Tomoda and M. M. Ito, Bull. Chem. Soc. Jpn., 1981, 54, 261–266 CrossRef; (e) P. K. Kabada, J. Org. Chem., 1992, 57, 3075–3078 CrossRef; (f) X. Zhang, Q. Zhang, Y. Wu, C. Feng, C. Xie, X. Fan and P. Li, Macromol. Rapid Commun., 2016, 37, 1311–1317 CrossRef PubMed; (g) T. Slagbrand, A. Volkov, P. Trillo, F. Tinnis and H. Adolfsson, ACS Catal., 2017, 7, 1771–1775 CrossRef.
- (a) P. Scheiner, Tetrahedron, 1967, 24, 349–356 CrossRef; (b) P. K. Kadaba and S. B. Edelstein, J. Org. Chem., 1990, 55, 5891–5894 CrossRef.
- For selected examples, see: (a) D. R. Roque, J. L. Neill, J. W. Antoon and E. P. Stevens, Synthesis, 2005, 2497–2502 Search PubMed; (b) S. S. van Berkel, A. J. Dirks, M. F. Debets, F. L. van Delft, J. J. L. M. Cornelissen, R. J. M. Nolte and F. P. J. T. Rutjes, ChemBioChem, 2007, 8, 1504–1508 CrossRef PubMed; (c) D. Gangaprasad, J. P. Raj, T. Kiranmye, R. Sasikala, K. Karthikeyan, S. K. Rani and J. Elangovan, Tetrahedron Lett., 2016, 57, 3105–3108 CrossRef.
- (a) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1966, 99, 475–490 CrossRef; (b) J. S. Yadav, B. V. Subba Reddy and V. Geetha, Synlett, 2002, 513–515 CrossRef.
- B. Wei-Qiang and S. Chiba, Org. Lett., 2009, 11, 729–732 CrossRef PubMed.
- S. Xie, S. A. Lopez, O. Ramström, M. Yan and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2958–2966 CrossRef PubMed.
- W. H. Pearson, S. C. Bergmeier, S. Degan, K.-C. Lin, Y.-F. Poon, J. M. Scheryantz and J. P. Williams, J. Org. Chem., 1990, 55, 5719–5738 CrossRef.
- J. Bourgois, M. Bourgois and F. Texier, Bull. Soc. Chim. Fr., 1978, 9–10, 485–527 Search PubMed.
- P. K. Kadaba, Curr. Med. Chem., 2003, 10, 2081–2108 CrossRef PubMed.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley-Interscience, New York, 1984, vol. 1, pp. 1–176 Search PubMed.
- (a) P. K. Kadaba, B. Stanovnik and M. Tišler, Adv. Heterocycl. Chem., 1984, 37, 217–361 CrossRef; (b) P. Scheiner, in Selective Organic Transformations, ed. B. S. Thyagarajan, Wiley-Interscience, New York, 1970, vol. 1, pp. 327–362 Search PubMed.
- (a) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 5538–5540 CrossRef PubMed; (b) D. M. Jenkins, Synlett, 2012, 1267–1270 CrossRef.
- For examples of aziridines isolated from the corresponding fused Δ2-triazolines, see: (a) F. D. Chattaway and G. D. Parkes, J. Chem. Soc., 1925, 127, 1307–1311 RSC; (b) A. Mustafa, S. M. A. D. Zayed and S. Khattab, J. Am. Chem. Soc., 1956, 78, 145–149 CrossRef; (c) P. Scheiner, J. Org. Chem., 1965, 30, 7–10 CrossRef PubMed; (d) P. Scheiner, J. Am. Chem. Soc., 1968, 90, 988–992 CrossRef; (e) D. Belei, E. Bicu, P. G. Jones and M. L. Birsa, J. Heterocycl. Chem., 2011, 48, 129–134 CrossRef.
- For examples that showed no aziridine formation from the corresponding 1,2,3-triazolines, see: (a) D. Pocar, P. Trimarco and G. Bombieri, J. Heterocycl. Chem., 1998, 35, 687–692 CrossRef; (b) S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, P. Macchi, N. Casati and S. Cenini, Organometallics, 2005, 24, 4710–4713 CrossRef.
- P. K. Kadaba, Synthesis, 1973, 71–84 CrossRef.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- (a) D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Partor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612–632 CrossRef; (b) J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147–5157 CrossRef.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Green Chem., 2002, 4, 24–26 RSC.
- R. Singh and A. Singh, J. Iran. Chem. Soc., 2017, 14, 1119–1129 CrossRef.
- J. M. Pérez and D. J. Ramón, ACS Sustainable Chem. Eng., 2015, 3, 2343–2349 CrossRef.
- S. A. Padvi and D. S. Dalal, Synth. Commun., 2017, 47, 779–797 CrossRef.
- (a) F. Ilgen and B. König, Green Chem., 2009, 11, 848–854 RSC; (b) M. A. P. Martins, G. C. Paveglio, L. V. Rodrigues, C. P. Frizzo, N. Zantta and H. G. Bonacorso, New J. Chem., 2016, 40, 5989–5992 RSC.
- 1,2,3-Triazolines are notoriously acid sensitive, although synthetically useful reactions have been developed under controlled conditions, see: (a) D. S. Reddy, W. R. Judd and J. Aubé, Org. Lett., 2003, 5, 3899–3902 CrossRef PubMed; (b) K. B. Hong, M. G. Donahue and J. N. Johnston, J. Am. Chem. Soc., 2008, 130, 2323–2328 CrossRef PubMed; (c) T. L. Troyer, H. Mucalski, K. B. Hong and J. N. Johnston, Org. Lett., 2011, 13, 1790–1792 CrossRef PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- (a) A. Gieren, Chem. Ber., 1973, 106, 288–311 CrossRef; (b) K. Kaas, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1458–1463 CrossRef; (c) S. Murata, Y. Mori, Y. Satoh, R. Yoshidome and H. Tomioka, Chem. Lett., 1999, 28, 597–598 CrossRef; (d) G. Mlostoń, K. Urbaniak, A. Linden and H. Heimgartner, Helv. Chim. Acta, 2002, 85, 2644–2656 CrossRef . See also ref. 18b.
- (a) S. Stanković, M. D′hooghe, S. Catak, H. Eum, M. Waroquier, V. Van Speybroeck, N. De Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643–665 RSC; (b) C. Botuha, F. Chemla, F. Ferreira and A. Perez-Luna, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and K. S. Chattopadhyay, John Wiley & Sons, 2011, pp. 3–39 Search PubMed; (c) G. S. Singh, M. D′hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080–2135 CrossRef PubMed.
- L. A. Lanovaya, M. F. Mishchenko and E. D. Korniets, Chem. Heterocycl. Compd., 1987, 23, 660–664 CrossRef.
- See ESI† for full crystallographic details.
- P. Scheiner, J. Am. Chem. Soc., 1966, 88, 4759–4760 CrossRef.
- (a) R. F. Bleinholder and H. Shechter, J. Am. Chem. Soc., 1968, 90, 2131–2137 CrossRef; (b) P. Battioni, L. Vo-Quang and Y. Vo-Quang, Bull. Soc. Chim. Fr., 1978, Part II, 415–427 Search PubMed; (c) D. K. Wedegaertner, R. K. Kattal, I. Harrison and S. K. Cristie, J. Org. Chem., 1991, 56, 4463–4467 CrossRef.
- T. S. Chung, S. A. Lopez, K. N. Houk and M. A. Garcia-Garibay, Org. Lett., 2015, 17, 4568–4571 CrossRef PubMed.
- (a) N. Singh, S. K. Pandey and R. P. Tripathi, Carbohydr. Res., 2010, 345, 1641–1648 CrossRef PubMed. See also; (b) J. Zhang and C.-W. T. Chang, J. Org. Chem., 2009, 74, 685–695 CrossRef PubMed.
- (a) R. Sustmann, Tetrahedron Lett., 1971, 12, 2717–2720 CrossRef; (b) K. N. Houk, Acc. Chem. Res., 1975, 8, 361–369 CrossRef; (c) A. K. Chandra, T. Uchimaru and M. T. Nguyen, J. Chem. Soc., Perkin Trans. 2, 1999, 2117–2121 RSC; (d) J.-C. Fan, J. Liang, Y. Wang and Z.-C. Shang, J. Mol. Struct.: THEOCHEM, 2007, 821, 145–152 CrossRef; (e) G. O. Jones and K. N. Houk, J. Org. Chem., 2008, 73, 1333–1342 CrossRef PubMed; (f) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef PubMed; (g) B. Braida, C. Walter, B. Engels and P. C. Hiberty, J. Am. Chem. Soc., 2010, 132, 7631–7637 CrossRef PubMed; (h) S. A. Lopez, M. E. Munk and K. N. Houk, J. Org. Chem., 2013, 78, 1576–1582 CrossRef PubMed.
- (a) C. Silva López, O. Nieto Faza, K. S. Feldman, M. R. Iyer and D. K. Hester II, J. Am. Chem. Soc., 2007, 129, 7638–7646 CrossRef PubMed; (b) A. Contini and E. Erba, RSC Adv., 2012, 2, 10652–10660 RSC.
- (a) P. A. S. Smith and J. H. Hall, J. Am. Chem. Soc., 1962, 84, 480–485 CrossRef; (b) L. Horner and A. Christmann, Angew. Chem., Int. Ed. Engl., 1963, 2, 599–607 CrossRef.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (c) A. D. Becke, Phys. Rev., 1998, A38, 3098–3100 Search PubMed; (d) P. J. Stevens, F. J. Devlin, C. F. Chablowski and M. J. Frish, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- (a) W. J. Hehre, L. Random, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Phys. Chem., 1972, 56, 2257–2261 CrossRef; (c) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2008, 8, 1057–1065 RSC.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef; (b) H. P. Hratchian and H. B. Schlegel, in Theory and Applications of Computational Chemistry: The First 40 Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. Scuseria, Elsevier, Amsterdam, 2005, pp. 195–249 Search PubMed.
- F. Sebest, L. Casarrubios, H. S. Rzepa, A. J. P. White and S. Díez-González, Imperial College Research Services Data Repository, 2018, DOI:10.14469/hpc/4062 and sub-collections therein.
- (a) D. Cremer and E. Kraka, Acc. Chem. Res., 2010, 43, 591–601 CrossRef PubMed; (b) H. S. Rzepa and C. Wentrup, J. Org. Chem., 2013, 78, 7565–7574 CrossRef PubMed.
- C. D.-T. Nielsen, W. J. Mooij, D. Sale, H. S. Rzepa, J. Burés and A. C. Spivey, submitted for publication.
Footnote |
| † Electronic supplementary information (ESI) available: FAIR data for NMR spectra, computational and crystallographic data, see ref. 48. CCDC 1843142–1843147. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8gc01797b |
| This journal is © The Royal Society of Chemistry 2018 |