
Identification of a new natural gastric lipase inhibitor from star anise†
Jannet
Kamoun
abc,
Renaud
Rahier‡
 c,
Mohamed
Sellami
a,
Imed
Koubaa
d,
Pascal
Mansuelle
e,
Régine
Lebrun
e,
Alexandra
Berlioz-Barbier
c,
Michele
Fiore
c,
Karine
Alvarez
f,
Abdelkarim
Abousalham
c,
Frédéric
Carrière
b and
Ahmed
Aloulou‡
*a
c,
Mohamed
Sellami
a,
Imed
Koubaa
d,
Pascal
Mansuelle
e,
Régine
Lebrun
e,
Alexandra
Berlioz-Barbier
c,
Michele
Fiore
c,
Karine
Alvarez
f,
Abdelkarim
Abousalham
c,
Frédéric
Carrière
b and
Ahmed
Aloulou‡
*a
aUniversity of Sfax, National School of Engineering of Sfax, Laboratory of Biochemistry and Enzymatic Engineering of Lipases, Sfax, Tunisia. E-mail: aloulou@uic.edu
bAix-Marseille University, CNRS, Bioénergétique et Ingénierie des Protéines, UMR 7281, Marseille, France
cUniv Lyon, Université Lyon 1, CNRS, Institut de Chimie et de Biochimie Moléculaires et Supramoléculaires, UMR 5246, Métabolisme, Enzymes et Mécanismes Moléculaires, Villeurbanne, France
dUniversity of Sfax, Faculty of Science of Sfax, Laboratory of Chemistry of Natural Products, Sfax, Tunisia
eAix-Marseille University, CNRS, Institut de Microbiologie de la Méditerranée, Plateforme Protéomique, Fédération de Recherche 3479, Marseille, France
fAix-Marseille University, CNRS, Architecture et Fonction des Macromolécules Biologiques, UMR 7257, Marseille, France
First published on 24th November 2018
Abstract
The identification and isolation of bioactive compounds are of great interest in the drug delivery field, despite being a difficult task. We describe here an innovative strategy for the identification of a new gastric lipase inhibitor from star anise for the treatment of obesity. After plant screening assays for gastric lipase inhibition, star anise was selected and investigated by bioactivity guided fractionation. MALDI-TOF mass spectrometry and peptide mass fingerprinting allowed the detection of an inhibitor covalently bound to the catalytic serine of gastric lipase. A mass-directed screening approach using UPLC-HRMS and accurate mass determination searching identified the flavonoid myricitrin-5-methyl ether (M5ME) as a lipase inhibitor. The inhibitory activity was rationalized based on molecular docking, showing that M5ME is susceptible to nucleophilic attack by gastric lipase. Overall, our data suggest that M5ME may be considered as a potential candidate for future application as a gastric lipase inhibitor for the treatment of obesity.
1. Introduction
Gastric and pancreatic lipases play a key role in the gastrointestinal digestion of dietary fat suggesting that they may represent a drug target for the treatment of obesity.1,2 Gastric lipase is the key enzyme for the gastrointestinal digestion of dietary triglycerides.3 Lipid digestion is initiated in the stomach by acid stable gastric lipase and continues in the duodenum through the synergistic actions of gastric lipase and colipase-dependent pancreatic lipase, leading to the formation of absorbable monoglycerides and free fatty acids (FFAs).4 Inhibiting these lipases to reduce fat absorption is the main pharmacological approach adopted for the treatment of obesity.5 Orlistat, a hydrogenated derivative of lipstatin produced by Streptomyces toxytricini, is the only FDA-approved anti-obesity drug that specifically targets digestive lipases.6 This drug is an active site-directed inhibitor that reacts with the nucleophilic serine residue of the catalytic triad of digestive lipases within the gastrointestinal tract.7 However, orlistat, which is considered relatively too expensive to afford, is also known to inhibit various other enzymes and could lead to undesirable side effects, which might affect its current clinical use.8 Therefore, looking for new cost-effective digestive lipase inhibitors is highly demanded nowadays.Much interest has been focussed on plant polyphenols that have been reported to play a significant role in the prevention of obesity and becoming overweight.9,10 More precisely, polyphenol-rich extracts from plant sources have been reported to inhibit pancreatic lipase in vitro.11–14
Bioactivity guided fractionation is a useful approach for the chemical and biological screening of complex plant extracts.15,16 Mass spectrometry (MS)-based affinity approaches have emerged as effective tools in small-molecule drug discovery. In these approaches the mass spectrometer can be exploited to rapidly screen drug candidates for specific interactions with targets of interest.17 Coupling bioaffinity selection to MS has been developed in the screening of natural product extracts18 and synthetic combinatorial libraries.19 Accordingly, direct and indirect bioaffinity screening approaches have been used. In the indirect screening approach, the complex protein–ligand is dissociated prior to the MS analysis, allowing thus the identification of the target ligand, whereas in the direct screening approach, the MS analysis can be directly performed on the whole complex.20 With the availability of MS technologies, the direct bioaffinity screening approach could be a potential way to facilitate tentative identification of the relevant compound within simplified natural mixtures.
UPLC-HRMS is a modern powerful tool to identify known and unknown compounds in a complex mixture. The use of UPLC-HRMS in combination with a QTOF mass spectrometer for target and non-target screening purposes has gained popularity in recent years.21 Indeed, UPLC provides fast and high-resolution separation, which increases LC-MS sensitivity and minimizes matrix interference arising from minimal simple preparation. Coupling of the UPLC system with QTOF-MS thus provides deep analysis of the sample composition.22
To identify a natural gastric lipase inhibitor from plants, we proposed herein a new strategy based on bioactivity guided fractionation along with direct bioaffinity screening using MALDI-TOF-MS and peptide mass fingerprinting (PMF), non-target screening using UPLC-HRMS, and in silico molecular docking.
2. Materials and methods
2.1. Preparation of plant crude extracts for the screening study
Dried plant species, purchased from local markets, were cleaned and finely powdered using a grinder. All powdered samples were separately extracted by the maceration technique using hexane, ethyl acetate, ethanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 w/v ratio). The different extracts were evaporated to dryness in vacuo yielding dried extracts. All extracts were dissolved in DMSO to obtain a final concentration of 100 mg mL−1 and were assayed for their anti-lipase potency.
:10 w/v ratio). The different extracts were evaporated to dryness in vacuo yielding dried extracts. All extracts were dissolved in DMSO to obtain a final concentration of 100 mg mL−1 and were assayed for their anti-lipase potency.
2.2. Bioactivity guided fractionation
The star anise crude phenolic extract (SACPE) was prepared from star anise powder via maceration in ethanol:water (7:3 v/v) (1:10 w/v ratio) for 24 hours. The solid–liquid mixture was filtered with fritted glass number 3. Then, the filtrate was evaporated to dryness in vacuo yielding dried SACPE.
Star anise powder was extracted with hexane (1:10 w/v) for 48 h to remove the lipoidal material.23 After filtration, the filtrate was evaporated under vacuum to yield the star anise hexanic extract. The residue was air-dried at room temperature overnight to obtain dried defatted star anise (DFSA). Thus, the star anise phenolic extract (SAPE) was prepared from DFSA via maceration in ethanol:water (7:3 v/v) (1:10 w/v ratio) for 24 h.
SAPE was successively fractionated using liquid–liquid partition and solid–liquid extraction to obtain star anise polyphenol-rich fractions (SAPRFs). In the first step, liquid–liquid partitioning of SAPE with ethyl acetate gives two fractions SAPRF1 and SAPRF2. These fractions were evaporated under vacuum (45 °C), lyophilized and subjected to the inhibition test against DGL activity. SAPE, SAPRF1 and SAPRF2 were then analyzed by TLC on aluminum sheets coated with 0.2 mm silica gel 60 to characterize their chemical profile. The migration was performed with a mixture of chloroform:methanol (8:2 v/v). After that, the spots were revealed with iodine vapor. In a second step, SAPRF2 was re-suspended in ethyl acetate for solid–liquid extraction. After 3 hours of maceration, the mixture was filtered. Similarly, this second step led to two fractions, SAPRF3 and SAPRF4, which were dried under vacuum. Subsequently, SAPRF2 and the fractions produced (SAPRF3 and SAPRF4) were subjected to the DGL inhibition test and TLC analysis under the same experimental conditions of the first step. SAPRF4 was thereby selected for further investigations. A flow chart for the preparation of various fractions from star anise powder is given in Fig. 1.
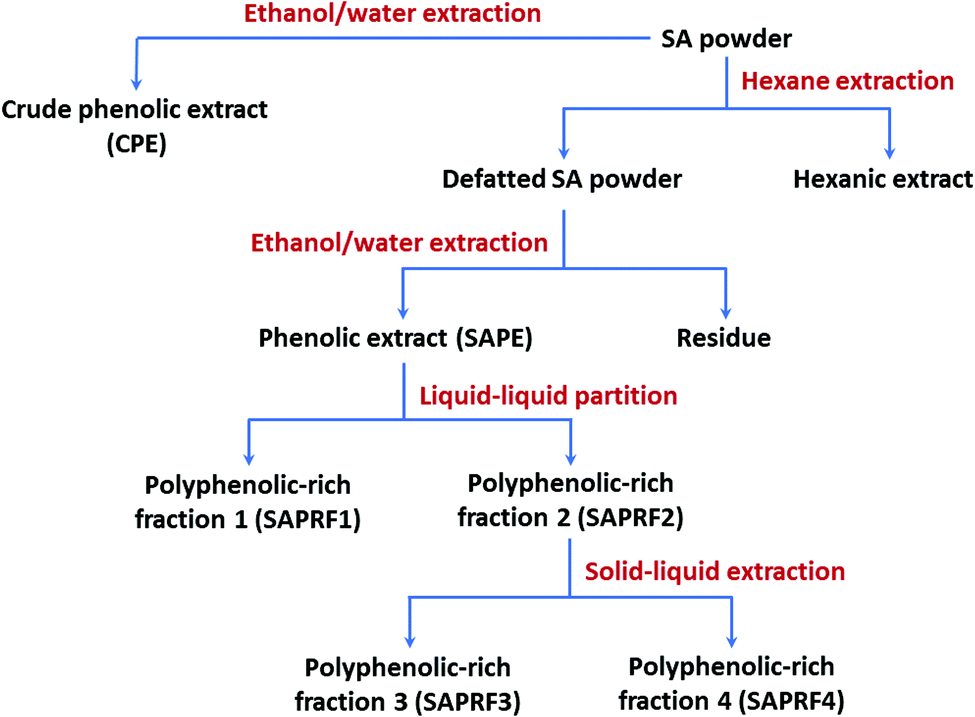 | ||
| Fig. 1 Bioactivity-guided fractionation scheme of star anise. | ||
2.3. Lipase activity measurements using the pH-stat technique
Recombinant DGL,24 HPL,25 and LipY26 were produced and purified as described previously. Pancreatic colipase was purified from lipid-free porcine pancreatic powder.27 Lipases were stored in 150 mM NaCl buffered solutions (pH 6 for DGL, pH 7 for HPL, and pH 8 for LipY).Enzymatic activity was assayed at 37 °C by measuring the amount of FFA released from a mechanically stirred tributyrin emulsion, using 0.1 M NaOH with a pH-stat (Metrohm 718 STAT Titrino, Switzerland) adjusted to a fixed end point.28 Emulsions were formed by mixing 0.5 mL tributyrin with 14.5 mL buffer solution. The activities of DGL were determined using the following assay solution: 150 mM NaCl, 2 mM sodium taurodeoxycholate (NaTDC), and 2 mM bovine serum albumin at pH 5.5.24 In the case of HPL, the activities were determined using the standard assay solution: 0.3 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM CaCl2, and 4 mM NaTDC, in the presence of 5 molar excess of colipase.25 With LipY, the assay solution was 2.5 mM Tris-HCl (pH 7.5), 300 mM NaCl and 3 mM NaTDC.26 Under the above assay conditions, enzymatic activities were expressed as international units: 1 U = 1 μmol FFA released per minute. The specific activities of DGL, HPL, and LipY, expressed in U per mg of pure enzyme, were found to be 340 ± 21, 8021 ± 79 and 129 ± 2 U mg−1, respectively.
2.4. Assay for anti-lipase activity
The lipase-inhibitor pre-incubation method was used to test, in an aqueous medium and in the absence of a substrate, the possible direct reactions between lipases and inhibitors.5 Lipase-inhibitor pre-incubations were performed at 25 °C, at different times and at various inhibitor extract amounts (aI), in the presence of 4 mM NaTDC.5 The final enzyme concentration in the incubation medium was 1 μM. The residual lipase activity was measured by the pH-stat. The amount of the inhibitor fraction (aI50) and the half-inactivation time (t½), corresponding to 50% of residual enzyme activity, were determined.In each case, control experiments were performed in the absence of the inhibitor fraction but with the same volume of DMSO. It is worth noting that DMSO at a final volume concentration less than 10% has no effect on the enzyme activity.
2.5. UPLC-HRMS experiment
The system used for UPLC-HRMS was an ultra-performance liquid chromatography Ultimate 3000 (Thermo Fisher Scientific, Villebon-sur-Yvette, France) coupled to a high-resolution hybrid quadrupole-time of flight mass spectrometer (Impact II, Bruker, Brême, Germany) equipped with an electrospray ionization source (ESI) (Bruker, Brême Germany). Instrument control and data collection were performed using Data Analysis 5.0 software.Chromatographic separation was performed on a polar C18 column (10 cm × 2.1 mm × 1.7 μm, Luna Omega, Phenomenex, France). The column oven temperature was set at 40 °C and an injection volume of 7 μL of the sample was loaded. The sample compounds were separated at a flow rate of 0.4 μL min−1 using (A) H2O (Milli-Q) with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. The analytes were separated using the following gradient: from 0% to 100% of B in 30 min and kept constant for 5 min. The column was re-equilibrated for 10 min at the initial composition of the gradient before runs.
The ESI interface was operated on the full scan mode (m/z 50–2000) in negative and positive ion modes. The parameters for the ESI ion source were as follows: the capillary voltage, 3.0 kV; the source temperature, 200 °C; the operating pressure of the nitrogen flow for the nebulizer gas, 45 Psi. Ultraviolet detectors measured the absorption over the range of 250 and 280 nm.
Before each acquisition batch, external calibration of the high-resolution mass spectrometer was performed with a sodium formate cluster solution. The calibration solution was injected at the beginning of each run. MS/MS analyses were carried out with the Data Dependent Acquisition mode for precursor ions with an intensity superior to 2000 counts using a stepping of the collision energy ramp.
Annotation of each signal was performed by interrogation of different databases (ChemSpider, MassBank, Drugbank, HMDB) using a home-made software providing annotation of LC-MS data according to parent mass accuracy (<5 ppm). After the generation of a short list of potential candidates for each signal, the correlation of isotope patterns according to putative atomic compositions was checked to reduce the list of putative annotation (mSigma < 30). The investigation of experimental MS/MS data using a web-based spectral database and/or an in silico fragmentation tool as MetFrag© was necessary to obtain structural information and annotate more precisely each compound.
2.6. MALDI-TOF mass spectrometry analysis
DGL was pre-incubated for 30 min at 25 °C with a large excess of SAPRF4 (aI = 30 μg) to abolish any residual lipolytic activity. A blank experiment was performed in the absence of SAPRF4. MALDI-TOF analysis of the entire non-inhibited or inhibited DGL was carried out with a Bruker Micoflex II mass spectrometer (Daltonik, Deutchland) using a saturated solution of α-cyano-4-hydroxycinnamic acid in acidified water (0.1% TFA) and acetonitrile (30:70 v/v).28 Mass spectra were acquired in the positive ion mode, using the Flex Analysis™ software program (Bruker, Daltonik, Deutchland). Protein identification was performed using the MASCOT™ version 2.2 search engine (Matrix Science, London, UK) and the NCBI protein database. Theoretical and experimental peptide masses were obtained using the BioTools™ software program (Bruker, Daltonik, Deutchland).
2.7. Peptide mass fingerprinting
Non-inhibited and inhibited DGL (with SAPRF4) were first separated by SDS-PAGE. The protein bands were then excised from the gel and subjected to the in-gel trypsin digestion procedure as previously described.28 For peptide identification, the in-gel digested peptides were analyzed by a proteomic approach including MALDI-TOF/MS and electrospray ionization (ESI) quadrupole time of flight (QTOF) MS/MS (Waters, Manchester) coupled with a nano flow UPLC nano Acquity (Waters, Manchester). For UPLC-ESI-QTOF analysis, the samples were dissolved in the loading buffer (3% acetonitrile/0.1% TFA in water) and desalted on a C18 nano trap (Symmerty C18, 180 μm × 2 cm, 5 μm, Waters) mounted on a 6-port valve, before on-line elution onto a C18 column (BEH 130 C18, 100 μm × 10 cm, 1.7 μm, Waters). Gradient elution was performed from 3% to 50% of mobile phase B (100% acetonitrile/0.1% formic acid) in A (0.1% formic acid in water) for 30 min. The column was rinsed for 6 min with 85% of B and then brought back in 1 min to the initial condition. Between each sample, a blank (injection of loading buffer only) was done using the same chromatographic method. The peptides were detected in the mass spectrometer in a positive ion mode using the MSE mode. Data acquisitions of the spectra were performed using the Micromass software Protein Lynx Global Server 2.5.2 (Waters).2.8. Molecular docking
In silico molecular docking of the M5ME inhibitor present in the active site of DGL (PDB entry codes: 1K8Q – 2.70 Å resolution24) was performed using the AutoDock Vina29 program of UCSF Chimera software.30 The grid box size was chosen to fit the whole active site cleft and to allow non-constructive binding positions. The inhibitor structure model was built using the Avogadro program.31 Binding modes were scored and ranked based on the most favorable energies.3. Results
3.1. Screening for the anti-lipase properties of crude medicinal plant extracts
Twenty extracts, prepared from five medicinal plants with anti-obesity potential (Illicium verum, Glycyrrhiza glabra, Salvia officinalis, Thymus vulgaris, and Rosmarinus officinalis),32 were screened for their inhibitory effect against DGL activity at an aI value of 1 mg. The ethanolic extract of the fruits of Illicium verum (star anise) was found to have the strongest inhibitory activity (81%) against DGL (Table S1†). While the ethanolic extract of Rosmarinus officinalis and the roots of Glycyrrhiza glabra showed a moderate inhibitory activity (43 and 44%, respectively) toward DGL, the remaining medicinal plant extracts displayed weak inhibitory activities. Star anise was thereby selected for further processing.3.2. Effect of defatting star anise on the anti-lipase activity
Since the star anise hexanic extract showed a weak inhibitory rate (18%, Table S1†) and a marked yield (around 7%, data not shown), we evaluated the anti-lipase activity of the phenolic extract from star anise before and after defatting. SACPE (non-defatted) and SAPE (defatted) samples were prepared as described in the Experimental section. Using an aI value of 1 mg of each extract, the lipase inhibitory activity of SAPE (100%) was higher than that of SACPE (89%).3.3. Bioactivity guided fractionation of SAPE
SAPE was fractionated using the scheme shown in Fig. 1. Two steps were used to fractionate SAPE. In the first step, the fractionation of SAPE led to SAPRF1 and SAPRF2, which were analyzed on TLC (Fig. S1A†). One can note that the spot intensities in SAPRF1 and SAPRF2 have increased compared to the initial extract (SAPE). The DGL activity inhibitory test was conducted on each of these two fractions. At an aI value of 500 μg of each fraction, SAPRF2 totally abolished DGL residual activity, while SAPRF1 led only to 25% residual DGL activity. Subsequently, as the SAPRF2 fraction was found to be more active, it was further subjected to a second fractionation step. Similarly, the latter led to SAPRF3 and SAPRF4 fractions (Fig. 1) which were subjected to the lipase activity inhibitory test and TLC analysis (Fig. S1B†). Unlike SAPRF3, SAPRF4 was found to display promising anti-lipase potency with 100% DGL inhibition rate at an aI value of 250 μg. TLC analysis showed that SAPRF3 is a moderately polar fraction, while SAPRF4 seems to have a strong polar character.3.4. Inhibitory effect of SAPRF4 against lipases of medical interest
The inhibitory activity of the SAPRF4 fraction was evaluated against two mammalian digestive lipases (DGL and HPL) and one microbial lipase belonging to the hormone-sensitive lipase (HSL) family (LipY).26 With each lipase, linear kinetics corresponding to the FFAs (μmol) released vs. time (min) was obtained in the presence and absence of the inhibitor fraction. A dose-dependent effect was observed upon increasing the amount of SAPRF4 on DGL, HPL and LipY (Fig. 2). At an aI value of only 30 μg, DGL activity was totally abolished after a 30 min incubation period (Fig. 2A), while HPL was strongly inactivated (96%) at an aI value of up to 1 mg (Fig. 2B). However, LipY activity was not completely abolished even at a high SAPRF4 amount, showing a residual activity of ∼20% (Fig. 2C). The aI50 values were found to be 7 μg, 82 μg and 5 μg for DGL, HPL and LipY, respectively (Table 1), indicating that SAPRF4 is a potent inhibitor toward both DGL and LipY.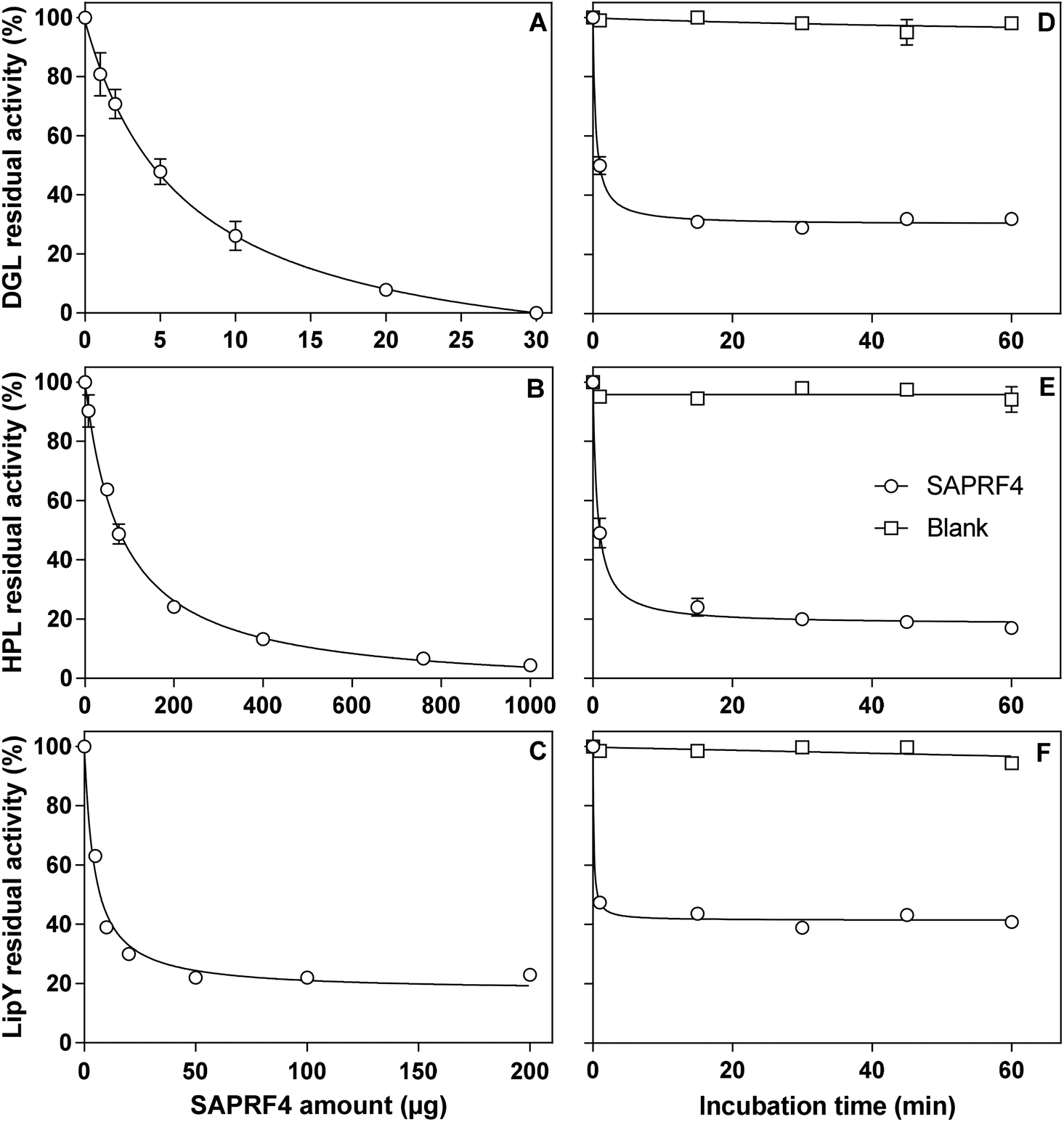 | ||
| Fig. 2 Evaluation of the inhibitory effect of the SAPRF4 fraction against DGL, HPL and LipY lipases. Each lipase was pre-incubated in the presence of various SAPRF4 amounts for 30 min at 25 °C and the residual activities of DGL (A), HPL (B) and LipY (C) were measured. Alternatively, the residual activities of DGL (D), HPL (E) and LipY (F) were measured as a function of the incubation time at a constant SAPRF4 amount. The aI value used in these latter experiments was chosen so that the average residual activities of DGL, HPL and LipY obtained after 30 min of incubation were in the 15–20% range. The results are expressed as mean values of at least three independent assays. | ||
| Lipases | a I50 (μg) | t ½ (min) |
|---|---|---|
| a SAPRF4 amount (aI) used for the determination of t½ was 10 μg, 200 μg and 10 μg for DGL, HPL and LipY, respectively. The results are expressed as mean values of at least three independent assays (CV % < 5.0%). | ||
| DGL | 7 | 0.4 |
| HPL | 82 | 0.6 |
| LipY | 5 | 0.1 |
The influence of the incubation time on the level of inhibition of DGL, HPL and LipY by the SAPRF4 fraction was further investigated (Fig. 2D–F). The residual activity of these lipases decreased rapidly and reached a plateau value after approximately 30 min of incubation. From these inhibition curves, the values of the half-inactivation times (t½) were then determined and found to be 0.4 min, 0.6 min and 0.1 min for DGL, HPL and LipY, respectively (Table 1). Such values reflect an extremely high rate of inhibition of these lipases by SAPRF4.
3.5. Analysis of the inhibitor-modified DGL complex with mass spectrometry
To investigate whether the inhibitory compound within SAPRF4 forms a covalent bond with the catalytic serine of DGL, MALDI-TOF-MS analysis was conducted on both non-inhibited and inhibited DGL (Fig. 3). A clear shift in the molecular mass of DGL was observed reflecting a covalent binding of the inhibitor to the lipase.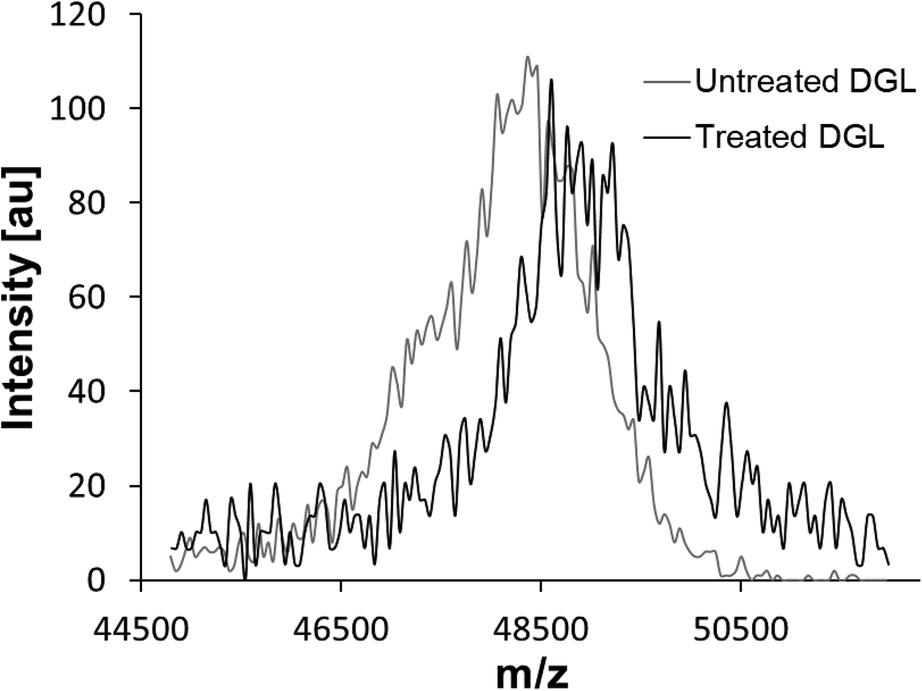 | ||
| Fig. 3 MALDI-TOF mass spectrometry analysis of untreated (grey spectrum) and treated (black spectrum) DGL. A 30 min incubation was performed at 25 °C in the presence or absence of a SAPRF4 amount of 30 μg. | ||
To deduce the exact molecular mass of the inhibitor contained in the SAPRF4 fraction, PMF analysis of non-inhibited and inhibited DGL was performed. The results showed that the peptide L147–K168 (LHYVGHSQGTTIGFIAFSTNPK) containing the catalytic S153 residue was detected at a molecular mass of 2376.05 Da with non-inhibited DGL (Fig. 4A). This catalytic peptide appeared concomitantly with a second peptide detected at a molar mass of around 2414.82 Da. By contrast, when DGL was incubated with SAPRF4, a mass increase of +489.52 Da was observed in the PMF spectrum for the catalytic peptide (Fig. 4B), while the peptide detected at a molar mass of around 2414.82 Da remained unchanged. This mass shift (489.52 Da) observed here is supposed to correspond to the molecular mass of the natural inhibitor bound to DGL.
 | ||
| Fig. 4 PMF spectra of untreated (A) and treated (B) DGL. A 30 min incubation was performed at 25 °C in the presence or absence of a SAPRF4 amount of 30 μg. Left panels, region of the unmodified isotopic peptide L147–K168 containing the catalytic Ser153 and detected at 2376.05 Da. This peak is overlaid with a second one corresponding to the peptide detected at 2414.82 Da. Right panels, region in which a new isotopic peptide was expected to occur, resulting from the covalent binding of the natural inhibitor of interest to the catalytic serine. Mass shift was calculated as the difference between the experimental m/z of the modified peptide and the theoretical m/z of the unmodified catalytic peptide. | ||
To tentatively identify the molecular formula and the structure of the phytochemical compound that binds covalently to DGL, we analyzed the SAPRF4 fraction by UPLC-HRMS. Seven major phenolic compounds have been identified (Fig. S2A and Table S2†). To empirically calculate their potential molecular formulas, various criteria have been used among them: the isotopic pattern, the fragmentation pattern and the high-resolution accurate mass (with error below 0.4 ppm at the time of matching the theoretical mass with the measured mass). Mass-directed identification showed that the natural DGL inhibitor compound within the SAPRF4 fraction might correspond to myricitrin-5-methyl ether (M5ME) with a molecular mass of 479.1184 Da and a molecular formula of C22H22O12 (Fig. 5).
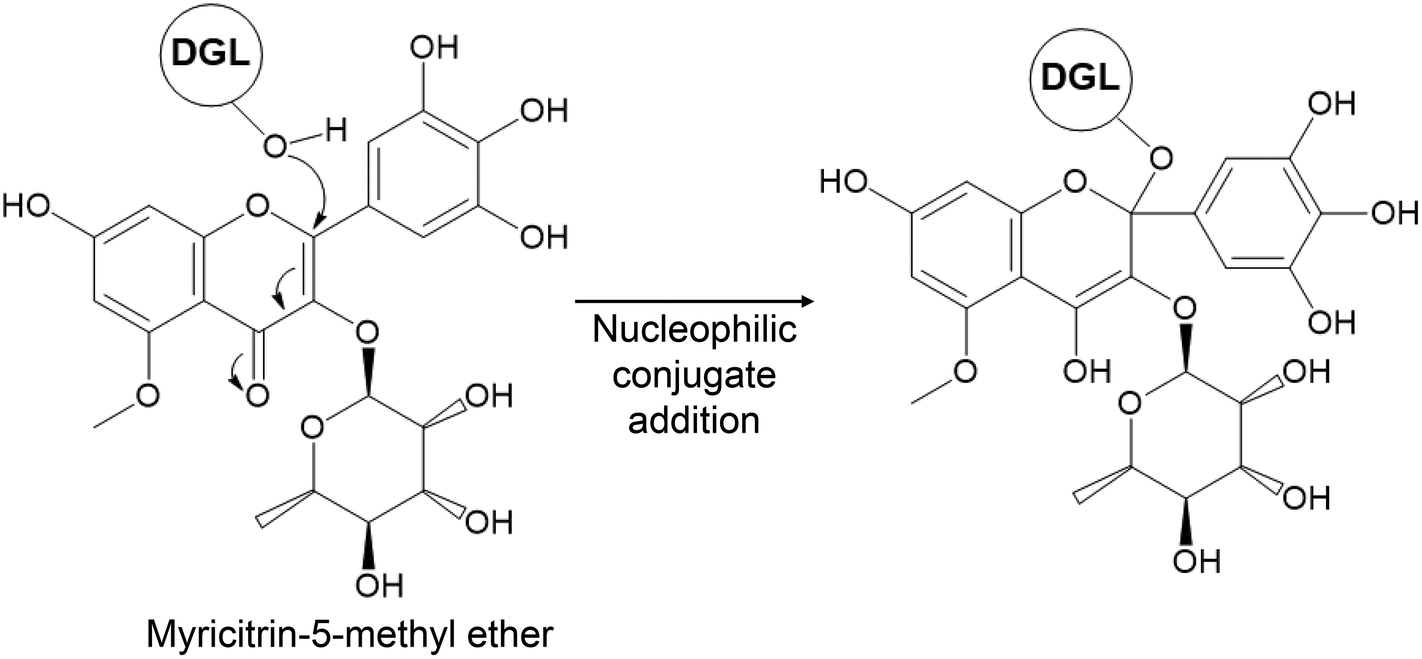 | ||
| Fig. 5 Chemical structure of the flavonoid myricitrin-5-methyl ether (M5ME) and proposed mechanism underlying DGL inhibition by M5ME. The nucleophilic attack by the hydroxyl group of the catalytic Ser153 residue on the inhibitor reactive site leads to a 1,4-nucleophilic addition on a double bond conjugated to a ketone and the formation of an enol function. | ||
3.6. Molecular docking
To shed light on DGL-M5ME binding interactions, in silico molecular docking was performed using the AutoDock Vina program. The open conformation of DGL, in complex with a phosphonate inhibitor covalently bound to its catalytic serine residue,24 was used as the reference model to improve the reliability of docking results. The lipase active site was found to be fully accessible thus enabling docking experiments. Automated docking resulted in several possible conformations of the M5ME inhibitor within the DGL catalytic pocket, with favorable binding energies ranging from −8.3 to −9.5 kcal mol−1 (Fig. S3A†). For subsequent analysis, we retained the best matching conformation that directly exposes the most reactive carbon atom of the M5ME inhibitor towards the catalytic serine, within a calculated distance of 3.8 Å which is sufficient to promote the nucleophilic attack (Fig. S3B†). In this configuration, we found that M5ME could fit properly into the DGL catalytic pocket (Fig. 6), which consists of a deep canyon of approximately 20 Å length, 7 Å width, and 20 Å depth.24 Moreover, like the phosphonate inhibitor, M5ME shows a relatively flat structure, with measured lengths of around 12 Å, widths of 5.5 Å, and depths of 9 Å (Fig. S3C†). For the sake of comparison, the previously reported phosphonate inhibitor only measures 6 Å in length, 2 Å in width and 10 Å in depth, and thus requires the presence of β-octyl glucoside or 1-amino-anthracene to completely fill the catalytic pocket.24 Interestingly, a more detailed analysis of the environment of the M5ME compound, bound to the catalytic serine, revealed a significant stabilizing effect of the DGL-M5ME tetrahedral intermediate by the neighboring oxyanion hole (Fig. 5). Indeed, an oxygen originating from the pyran ring of M5ME points towards the backbone nitrogen atoms of Leu67 and Gln154 residues within hydrogen bond distances of 2.37 Å and 2.08 Å, respectively.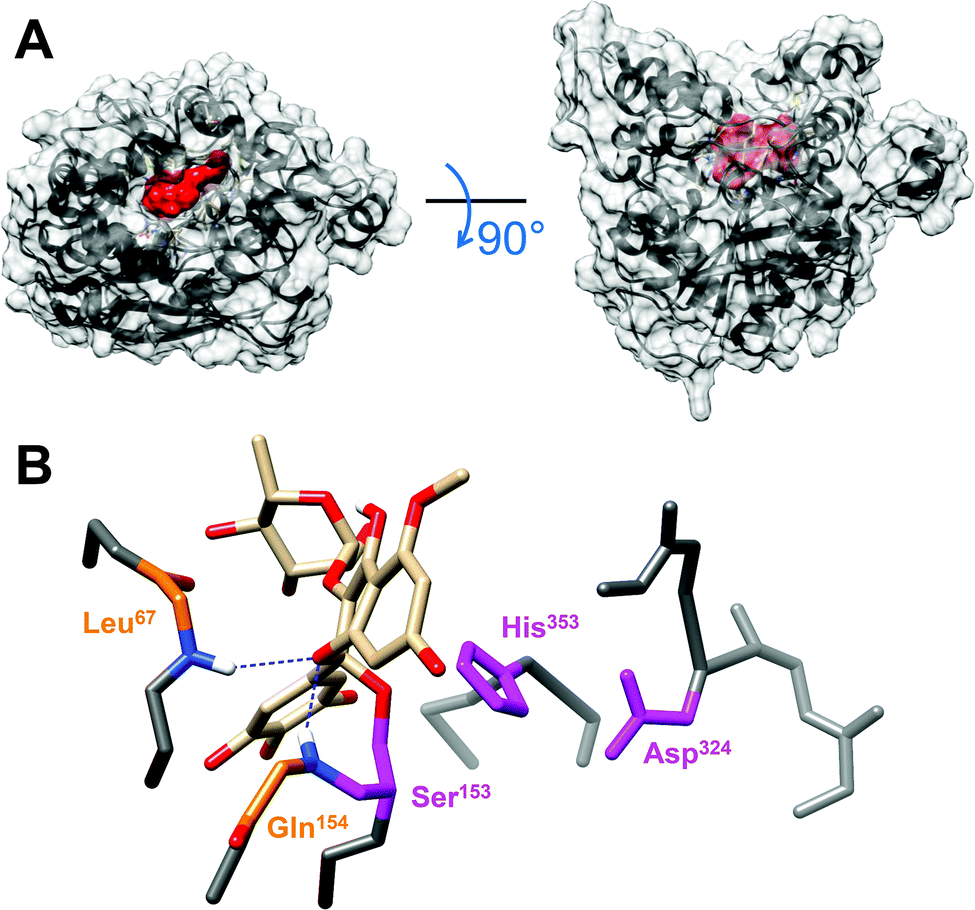 | ||
| Fig. 6 Visualization of the DGL-M5ME binding interaction by molecular docking. (A) Molecular surface representation of the DGL active site crevice with the bound M5ME. (B) View of the active site of DGL with the bound M5ME. The backbone nitrogen atoms of residues Leu67 and Gln154 are at a hydrogen bonding distance from an oxygen atom originating from the inhibitor. The catalytic residues Ser153, His353 and Asp324 are highlighted in pink and the inhibitor is represented in CPK coloring (oxygen, red). | ||
4. Discussion
Chinese star anise, the fruit of Illicium verum, is used in traditional medicine to treat stomachache, colic, vomiting, insomnia, skin inflammation, rheumatic pain, dyspepsia, facial paralysis, asthma, and bronchitis.33 A great deal of research effort is being devoted to testing the putative beneficial effects of star anise extracts. Modern pharmacology studies demonstrated that crude extracts and active compounds of star anise possess wide pharmacological actions.34 However, to the best of our knowledge, none has yet examined the effect of star anise extracts on digestive lipase activities.DGL provides a good model for human gastric lipases.24,35 A screening of a collection of 20 extracts from five medicinal plants using solvents with different polarities allowed the identification of an ethanolic extract from star anise, which efficiently inhibited gastric lipase (Table S1†). The inhibitory effect of the star anise ethanolic extract on DGL might be caused by the presence of phenolic compounds within the extract. It was reported that ethanol/water extracts of star anise contained high amounts of phenolic compounds.36 The use of water in combination with ethanol contributes to the establishment of a moderately polar medium that ensures the extraction of phenolic compounds.37 Plant phenolic compounds, such as flavones, flavanols, tannins and chalcones, have been reported as pancreatic lipase inhibitors,14 thus supporting the use of the star anise polyphenol-rich extract for further bioactivity guided fractionation (Fig. 1).
Our results showed that the removal of the lipoidal material from star anise before phenolic extraction enhanced the anti-lipase activity, which might be related to the fact that the concentration of phenolics in SACPE is lower than that in SAPE, suggesting thereby that the anti-lipase effect is related to star anise phenolic compounds. One of the hallmarks of bioactivity guided fractionation is the trend of increasing potency with increasing fractionation. Indeed, the aI value that totally abolishes gastric lipase activity moved from 1 mg SAPE to 30 μg SAPRF4 after two fractionation steps. According to TLC analysis, SAPRF4 produced by this fractionation approach is a less complex mixture than the initial SAPE (Fig. S1†). We therefore sought to evaluate SAPRF4 potency and selectivity against other lipases of medicinal interest.
Inhibiting lipases has potential applications in the field of medicine. The digestive lipase inhibitor orlistat used for the treatment of obesity has been shown to also inhibit HSL38 and microbial lipases.8,39 For the sake of comparison, SAPRF4 was found to inhibit efficiently DGL, HPL, and LipY (a microbial lipase belonging to the HSL family). The ability of SAPRF4 to inhibit the catalytic activity of these lipases was assessed in terms of aI50 and t½ values (Table 1). The inhibition of HSL could be a potential approach to reduce the levels of circulating FFAs linked to insulin resistance in obese patients.40 Consequently, SAPRF4 might be an effective candidate for the treatment of obesity and diabetes. Moreover, as LipY contributes to the growth and pathogenicity of M. tuberculosis,26 it would be a therapeutic target for SAPRF4 as an anti-microbial agent.
On the bioaffinity interaction level, inhibitors can bind either covalently or non-covalently with their biological target.16,41 MALDI-TOF mass spectrometry is an analytical instrument tool that can measure the protein-inhibitor complex mass when a specific covalent binding occurs, reflecting thereby an approximate molecular mass of the inhibitor.38 Direct MALDI-TOF mass spectrometry analysis of DGL totally inhibited by SAPRF4 showed the occurrence of an increase in the molecular masses of DGL, which is compatible with the formation of a covalent complex with the inhibitor. PMF analysis of a trypsin digest of DGL inhibited or not with SAPRF4 showed an increase in the molecular mass of the catalytic serine-containing peptide (Fig. 4) corresponding in size to the approximate molecular mass of the inhibitor. This process might involve a nucleophilic attack between the enzyme's catalytic serine and the inhibitor, leading to the formation of a covalent inhibitor-modified enzyme. Based on PMF analysis, it has been also shown previously that synthetic inhibitors, oxadiazolones28 and cyclophostin analogs,8,42 form a covalent bond with the catalytic serine residue of lipases. To the best of our knowledge, this is the first study that used covalent inhibitor-modified enzyme complex data to tentatively identify a lipase inhibitor in a natural extract.
After achieving the approximate molecular mass of the covalent lipase inhibitor, the identification of the compound of interest within SAPRF4 would be rather more straightforward. Thus, we applied UPLC-HRMS for mass-directed screening to tentatively identify the expected lipase inhibitor in SAPRF4. Using this non-target screening, it was possible to separate and detect seven chromatographic peaks containing high resolution MS information (Fig. S2†). The tentative identification of these peaks was performed according to their high-resolution accurate mass and isotopic patterns to empirically determine their molecular formulas. By inspecting all mass spectra around the m/z of 489.52 in the chromatogram of SAPRF4, one ion having an m/z of 479.1187 was found to fit with the PMF analysis data. This ion, assigned to the molecular formula C22H22O12, corresponds likely to the flavonoid myricitrin-5-methyl ether (Fig. 5) that has not been identified previously from star anise.
To gain insights into the lipase-inhibitor binding interactions at the molecular level, M5ME was docked into the active site of DGL (Fig. 6). The final selection of the M5ME “bioactive” conformation suggests that the inhibition mechanism involves a nucleophilic attack by the hydroxyl group of the catalytic Ser on a reactive carbon atom of the inhibitor, thus leading to the formation of a stoichiometric enzyme–inhibitor covalent complex (Fig. 5). Interestingly, the nucleophilic attack could be favored by some surrounding residues, including the backbone nitrogen atoms of Leu67 and Gln154 that belong to the previously described DGL oxyanion hole.24
Much more interest has been shifted on plant flavonoids for their possible anti-obesity effects. In vitro and in vivo studies provided evidence on the potential role that flavonoids play in the management of obesity.43 M5ME, a myricetin glycoside, was isolated from the flower of Rhododendron poukhanense as a potent antioxidant flavonoid. Although no lipase inhibiting activity was reported in the literature for M5ME, the structurally fairly similar quercetin and kaempferol were reported to be potent pancreatic lipase inhibitors.44 Moreover, myricetin was found to exert in vivo strong anti-obesity and anti-hyperlipidaemic activities by modulating the lipid metabolism.45
To our knowledge, this is the first report that tentatively identified a new natural gastric lipase inhibitor from a natural product. The combination of in silico molecular docking results with those of lipase inhibition and mass spectrometry provides scientific evidence for the potential medical use of the flavonoid myricitrin-5-methyl ether as a gastric lipase inhibitor for the treatment of obesity.
Abbreviations
| DGL | Dog gastric lipase |
| HPL | Human pancreatic lipase |
| LipY | Mycobacterial Rv3097c-encoded lipase |
| SA | Star anise |
| M5ME | Myricitrin-5-methyl ether |
| FFA | Free fatty acid |
| NaTDC | Sodium taurodeoxycholate |
| SAPRF | Star anise polyphenol-rich fraction |
| MALDI-TOF-MS | Matrix-assisted laser desorption ionization-time of flight mass spectrometry |
| PMF | Peptide mass fingerprinting |
| TLC | Thin layer chromatography |
| UPLC-HRMS | Ultra-performance liquid chromatography high resolution hybrid quadrupole-time of flight mass spectrometry |
Author contributions
J. K. carried out the experiments and performed the analyses. R. R., M. S., P. M., R. L., A. B.-B. and M. F. contributed to the experiment realization and analysis. I. K., K. A., A. A. and F. C. contributed to the experiment design and analysis. J. K. and A. A. conceived the experiments and wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Ministry of Higher Education and Scientific Research, Tunisia. Dr Stéphane Canaan and Dr Jean-François Cavalier (CNRS, Marseille) are acknowledged for their generous gift of LipY and for useful discussions.References
- A. Aloulou and F. Carriere, Gastric lipase: an extremophilic interfacial enzyme with medical applications, Cell. Mol. Life Sci., 2008, 65, 851–854 CrossRef CAS.
- A. Aloulou, D. Puccinelli, J. Sarles, R. Laugier, Y. Leblond and F. Carriere, In vitro comparative study of three pancreatic enzyme preparations: dissolution profiles, active enzyme release and acid stability, Aliment. Pharmacol. Ther., 2008, 27, 283–292 CrossRef CAS.
- F. Carriere, E. Rogalska, C. Cudrey, F. Ferrato, R. Laugier and R. Verger, In vivo and in vitro studies on the stereoselective hydrolysis of tri- and diglycerides by gastric and pancreatic lipases, Bioorg. Med. Chem., 1997, 5, 429–435 CrossRef CAS.
- F. Carriere, J. A. Barrowman, R. Verger and R. Laugier, Secretion and contribution to lipolysis of gastric and pancreatic lipases during a test meal in humans, Gastroenterology, 1993, 105, 876–888 CrossRef CAS.
- H. Lengsfeld, Physiology of Gastrointestinal Lipolysis and Therapeutical Use of Lipases and Digestive Lipase Inhibitors, Lipases Phospholipases Drug Dev., 2005, 195–223, DOI:10.1002/3527601910.ch10.
- H. L. Daneschvar, M. D. Aronson and G. W. Smetana, FDA-Approved Anti-Obesity Drugs in the United States, Am. J. Med., 2016, 129, 879 CrossRef PubMed e871–876.
- F. Carriere, C. Renou, S. Ransac, V. Lopez, J. De Caro, F. Ferrato, A. De Caro, A. Fleury, P. Sanwald-Ducray, H. Lengsfeld, C. Beglinger, P. Hadvary, R. Verger and R. Laugier, Inhibition of gastrointestinal lipolysis by Orlistat during digestion of test meals in healthy volunteers, Am. J. Physiol.: Gastrointest. Liver Physiol., 2001, 281, G16–G28 CrossRef CAS PubMed.
- V. Point, R. K. Malla, S. Diomande, B. P. Martin, V. Delorme, F. Carriere, S. Canaan, N. P. Rath, C. D. Spilling and J. F. Cavalier, Synthesis and kinetic evaluation of cyclophostin and cyclipostins phosphonate analogs as selective and potent inhibitors of microbial lipases, J. Med. Chem., 2012, 55, 10204–10219 CrossRef CAS PubMed.
- L. Titta, M. Trinei, M. Stendardo, I. Berniakovich, K. Petroni, C. Tonelli, P. Riso, M. Porrini, S. Minucci, P. G. Pelicci, P. Rapisarda, G. Reforgiato Recupero and M. Giorgio, Blood orange juice inhibits fat accumulation in mice, Int. J. Obes., 2010, 34, 578–588 CrossRef CAS.
- R. B. Birari, S. Gupta, C. G. Mohan and K. K. Bhutani, Antiobesity and lipid lowering effects of Glycyrrhiza chalcones: experimental and computational studies, Phytomedicine, 2011, 18, 795–801 CrossRef CAS.
- Y. Gu, W. J. Hurst, D. A. Stuart and J. D. Lambert, Inhibition of key digestive enzymes by cocoa extracts and procyanidins, J. Agric. Food Chem., 2011, 59, 5305–5311 CrossRef CAS PubMed.
- M. Yoshikawa, H. Shimoda, N. Nishida, M. Takada and H. Matsuda, Salacia reticulata and its polyphenolic constituents with lipase inhibitory and lipolytic activities have mild antiobesity effects in rats, J. Nutr., 2002, 132, 1819–1824 CrossRef CAS PubMed.
- N. Yuda, M. Tanaka, M. Suzuki, Y. Asano, H. Ochi and K. Iwatsuki, Polyphenols extracted from black tea (Camellia sinensis) residue by hot-compressed water and their inhibitory effect on pancreatic lipase in vitro, J. Food Sci., 2012, 77, H254–H261 CrossRef PubMed.
- R. B. Birari and K. K. Bhutani, Pancreatic lipase inhibitors from natural sources: unexplored potential, Drug Discovery Today, 2007, 12, 879–889 CrossRef CAS PubMed.
- F. E. Koehn and G. T. Carter, The evolving role of natural products in drug discovery, Nat. Rev. Drug Discovery, 2005, 4, 206–220 CrossRef CAS PubMed.
- M. G. Weller, A unifying review of bioassay-guided fractionation, effect-directed analysis and related techniques, Sensors, 2012, 12, 9181–9209 CrossRef CAS PubMed.
- S. A. Hofstadler and K. A. Sannes-Lowery, Applications of ESI-MS in drug discovery: interrogation of noncovalent complexes, Nat. Rev. Drug Discovery, 2006, 5, 585–595 CrossRef CAS PubMed.
- L. L. Cummins, S. Chen, L. B. Blyn, K. A. Sannes-Lowery, J. J. Drader, R. H. Griffey and S. A. Hofstadler, Multitarget affinity/specificity screening of natural products: finding and characterizing high-affinity ligands from complex mixtures by using high-performance mass spectrometry, J. Nat. Prod., 2003, 66, 1186–1190 CrossRef CAS PubMed.
- B. M. Johnson, D. Nikolic and R. B. van Breemen, Applications of pulsed ultrafiltration-mass spectrometry, Mass Spectrom. Rev., 2002, 21, 76–86 CrossRef CAS PubMed.
- H. Vu, N. B. Pham and R. J. Quinn, Direct screening of natural product extracts using mass spectrometry, J. Biomol. Screening, 2008, 13, 265–275 CrossRef CAS.
- D. C. Hilton, R. S. Jones and A. Sjodin, A method for rapid, non-targeted screening for environmental contaminants in household dust, J. Chromatogr. A, 2010, 1217, 6851–6856 CrossRef CAS PubMed.
- M. Ibáñez, J. V. Sancho, F. Hernández, D. McMillan and R. Rao, Rapid non-target screening of organic pollutants in water by ultraperformance liquid chromatography coupled to time-of-light mass spectrometry, TrAC, Trends Anal. Chem., 2008, 27, 481–489 CrossRef.
- M. A. Ramirez-Coronel, N. Marnet, V. S. Kolli, S. Roussos, S. Guyot and C. Augur, Characterization and estimation of proanthocyanidins and other phenolics in coffee pulp (Coffea arabica) by thiolysis-high-performance liquid chromatography, J. Agric. Food Chem., 2004, 52, 1344–1349 CrossRef CAS PubMed.
- A. Roussel, N. Miled, L. Berti-Dupuis, M. Riviere, S. Spinelli, P. Berna, V. Gruber, R. Verger and C. Cambillau, Crystal structure of the open form of dog gastric lipase in complex with a phosphonate inhibitor, J. Biol. Chem., 2002, 277, 2266–2274 CrossRef CAS PubMed.
- V. Belle, A. Fournel, M. Woudstra, S. Ranaldi, F. Prieri, V. Thome, J. Currault, R. Verger, B. Guigliarelli and F. Carriere, Probing the opening of the pancreatic lipase lid using site-directed spin labeling and EPR spectroscopy, Biochemistry, 2007, 46, 2205–2214 CrossRef CAS PubMed.
- P. Santucci, S. Diomande, I. Poncin, L. Alibaud, A. Viljoen, L. Kremer, C. de Chastellier and S. Canaan, Delineating the physiological roles of the PE and catalytic domain of LipY in lipid consumption in mycobacteria-infected foamy macrophages, Infect. Immun., 2018 DOI:10.1128/IAI.00394-18.
- C. Chapus, P. Desnuelle and E. Foglizzo, Stabilization of the C-terminal part of pig and horse colipase by carboxypeptidase and trypsin inhibitors, Eur. J. Biochem., 1981, 115, 99–105 CrossRef CAS.
- V. Point, K. V. Pavan Kumar, S. Marc, V. Delorme, G. Parsiegla, S. Amara, F. Carriere, G. Buono, F. Fotiadu, S. Canaan, J. Leclaire and J. F. Cavalier, Analysis of the discriminative inhibition of mammalian digestive lipases by 3-phenyl substituted 1,3,4-oxadiazol-2(3H)-ones, Eur. J. Med. Chem., 2012, 58, 452–463 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera–a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- J.-C. Charrié, B. Chastel, C. Cieur, P. Combe, M. Damak, K. Hedayat and C. Saigne-Soulard, Plantes médicinales - Phytothérapie clinique intégrative et médecine endobiogénique, Lavoisier S.A.S., 2017 Search PubMed.
- Y. Y. Sung, W. K. Yang, A. Y. Lee, D. S. Kim, K. J. Nho, Y. S. Kim and H. K. Kim, Topical application of an ethanol extract prepared from Illicium verum suppresses atopic dermatitis in NC/Nga mice, J. Ethnopharmacol., 2012, 144, 151–159 CrossRef CAS PubMed.
- G. W. Wang, W. T. Hu, B. K. Huang and L. P. Qin, Illicium verum: a review on its botany, traditional use, chemistry and pharmacology, J. Ethnopharmacol., 2011, 136, 10–20 CrossRef CAS.
- S. Fernandez, S. Chevrier, N. Ritter, B. Mahler, F. Demarne, F. Carriere and V. Jannin, In vitro gastrointestinal lipolysis of four formulations of piroxicam and cinnarizine with the self emulsifying excipients Labrasol and Gelucire 44/14, Pharm. Res., 2009, 26, 1901–1910 CrossRef CAS PubMed.
- A. Padmashree, N. Roopa, A. D. Semwal, G. K. Sharma, G. Agathian and A. S. Bawa, Star-anise (Illicium verum) and black caraway (Carum nigrum) as natural antioxidants, Food Chem., 2007, 104, 59–66 CrossRef CAS.
- C. Liyana-Pathirana and F. Shahidi, Optimization of extraction of phenolic compounds from wheat using response surface methodology, Food Chem., 2005, 93, 47–56 CrossRef CAS.
- Y. Ben Ali, H. Chahinian, S. Petry, G. Muller, R. Lebrun, R. Verger, F. Carriere, L. Mandrich, M. Rossi, G. Manco, L. Sarda and A. Abousalham, Use of an inhibitor to identify members of the hormone-sensitive lipase family, Biochemistry, 2006, 45, 14183–14191 CrossRef CAS PubMed.
- R. Dhouib, A. Ducret, P. Hubert, F. Carriere, S. Dukan and S. Canaan, Watching intracellular lipolysis in mycobacteria using time lapse fluorescence microscopy, Biochim. Biophys. Acta, 2011, 1811, 234–241 CrossRef CAS PubMed.
- G. Boden, Interaction between free fatty acids and glucose metabolism, Curr. Opin. Clin. Nutr. Metab. Care, 2002, 5, 545–549 CrossRef CAS.
- H. Gohlke and G. Klebe, Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors, Angew. Chem., Int. Ed., 2002, 41, 2644–2676 CrossRef CAS.
- V. Point, R. K. Malla, F. Carriere, S. Canaan, C. D. Spilling and J. F. Cavalier, Enantioselective inhibition of microbial lipolytic enzymes by nonracemic monocyclic enolphosphonate analogues of cyclophostin, J. Med. Chem., 2013, 56, 4393–4401 CrossRef CAS PubMed.
- N. Al Shukor, K. Raes, G. Smagghe and J. Van Camp, in Flavonoids and antioxidants, Studium Press LLC, New Delhi, India, 2016, vol. 40, pp. 496–514 Search PubMed.
- T. Sergent, J. Vanderstraeten, J. Winand, P. Beguin and Y.-J. Schneider, Phenolic compounds and plant extracts as potential natural anti-obesity substances, Food Chem., 2012, 135, 68–73 CrossRef CAS.
- C. J. Chang, T. F. Tzeng, S. S. Liou, Y. S. Chang and I. M. Liu, Myricetin Increases Hepatic Peroxisome Proliferator-Activated Receptor alpha Protein Expression and Decreases Plasma Lipids and Adiposity in Rats, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 787152 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fo02009d |
| ‡ Present address: University of Illinois at Chicago, Department of Chemistry, Chicago, Illinois 60607, USA. |
| This journal is © The Royal Society of Chemistry 2019 |