
From CO2 methanation to ambitious long-chain hydrocarbons: alternative fuels paving the path to sustainability
Filipe
Marques Mota
 and
Dong Ha
Kim
*
and
Dong Ha
Kim
*
Department of Chemistry and Nano Science, Ewha Womans University, 52 Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea. E-mail: dhkim@ewha.ac.kr
First published on 16th November 2018
Abstract
The clean and sustainable CO2 reutilization toward products of higher value is of great interest in a background of established environmental concerns and reducing the use of fossil fuels. As promising alternative fuels, hydrocarbons are more valuable than CO, alcohols or formate and can be directly used in existing infrastructures with high energy densities. The prominent development of catalysts capable of selectively converting CO2 into hydrocarbons, from methane to short olefins and long carbon-chains, has been reflected in an expanding volume of exploratory works, which suitably demand interpretive and continuous revision. In the past decades, conventional studies on the thermochemical conversion of CO2 have consistently unlocked meaningful pathways toward the synthesis of hydrocarbons covering a fairly wide range of molecular weights. Conversely, both electrochemically and photochemically driven reactions have only now started to unveil encouraging results, with an extensive number of critical citations outlining the continuous emergence of very recently published reports. In a field in need of urgent development, the authors provide, in a clear form, a detailed retrospective on benchmark catalysts, pioneering approaches and competitive developments in this subject, mechanistic difficulties, emerging stability issues, and reactor design, while highlighting the latest noteworthy reports. Most importantly, this review highlights the advances toward an increase in the hydrocarbon chain-length in the synthesis of highly competitive alternative fuels. Comparisons of valuable thermochemical, electrochemical and photochemically driven strategies in the conversion of CO2 to hydrocarbons are expected to serve as guidelines to disclose promising pathways in a field where mechanistic uncertainties remain a bottleneck for determining the product selectivity. The authors summarize leading and inquisitive perspectives with a focus on the viability and practicability of each approach at a larger scale, while tentatively paving the way to stimulate progress in this field.
 Filipe Marques Mota | Dr Filipe Marques Mota received his PhD degree in 2013 in Bioscience Engineering from K.U. Leuven (Belgium), in collaboration with IFP Energies Nouvelles (France) in the development of hydroconversion catalysts. He later worked as a research fellow at the Institute for Basic Science (Korea) in the synthesis and application of hierarchical nanostructured materials. He is currently a research professor in the Department of Chemistry and Nano Science at Ewha Womans University. His research interests focus on current challenges in the electrocatalytic and photocatalytic reutilization of CO2, and in the development of energy storage devices. |
 Dong Ha Kim | Prof. Dong Ha Kim received his PhD degree from the Department of Fiber and Polymer Science at Seoul National University (2000). He worked as a postdoctoral researcher at the University of Massachusetts at Amherst and at the Max Planck Institute for Polymer Research. He assumed a faculty position in the Department of Chemistry and Nano Science at Ewha Womans University (2006). His research interests include the development of hybrid nanostructures for energy storage and conversion, environmental remediation, memory devices, display devices, and theragnosis. He has authored 160 SCI publications and holds 31 Korean and 2 US patents. |
1. Introduction
In the past decades, environmental concerns and societal dependence on dwindling oil resources have provided the impetus for the development of viable alternatives to fossil fuels. With an ever-increasing global demand for energy that is predominantly produced worldwide from fossil fuels, new and effective technologies have been the focus of attention in the scientific community. The development and expansion of natural gas to liquid (GTL) and coal to liquid (CTL) technologies, and the significant progress being made in the production of biofuels from algae and woody biomass have been highlighted.1–4CO2 emissions that lead to increasing atmospheric concentrations are major bottlenecks in the cement, metal, bioethanol, oil refining and petrochemical industries, with fossil fuel combustion in power plants being the primary source of CO2 (Fig. 1). Current total global anthropogenic emissions amount to ca. 35.5 GtCO2 per year, reflecting an increase in the CO2 in the atmosphere from 278 ppm at the beginning of the industrial revolution to current values exceeding 400 ppm.5,6 With CO2 levels being widely accepted nowadays as one of the main reasons for global warming,7,8 reducing carbon emissions on an adequate timescale to hinder further risks of climate change demands an urgent increase in the use of renewable energy.9–11 Notwithstanding attempts to decarbonize the world economy, to date, the sum of constant non-carbon-emitting sources of energy remains scarce. Despite an increasing emphasis on renewable energy, the annual global investment in fossil energy has increased more than 2-fold over the period 2000–2013.12 In this context, carbon capture followed by strategic sequestration (e.g. geological or deep-sea storage and mineralization) or fixation by reaction have emerged as research topics of heightened concern.13–16 When put in perspective, under typical CO2 injection and storage conditions (10 MPa and 40 °C), the storage of the currently produced CO2 per day equals 1033 million barrels (MMbbl), which is 10-fold higher than the current daily global oil production rates (ca. 87–91 MMbbl).17 At the current rates of growth, this value is predicted to attain a 20-fold increase in 2050.18 These prospects underline the importance of discussing the premise of carbon reutilization. To date, the utilization of captured CO2 is bottlenecked by the purity and partial pressure of the gas. While higher partial pressures facilitate the capture and separation of CO2, power stations responsible for the highest CO2 emissions exhibit significantly lower gas partial pressures when compared with those originating from petrochemical plants. Alternatively, CO2 is much more abundantly present in ocean water than in air (up to 100 and 0.7 mg L−1, respectively), with seawater revealing a net gain of dissolved CO2 of 2 Gt per year.19–21
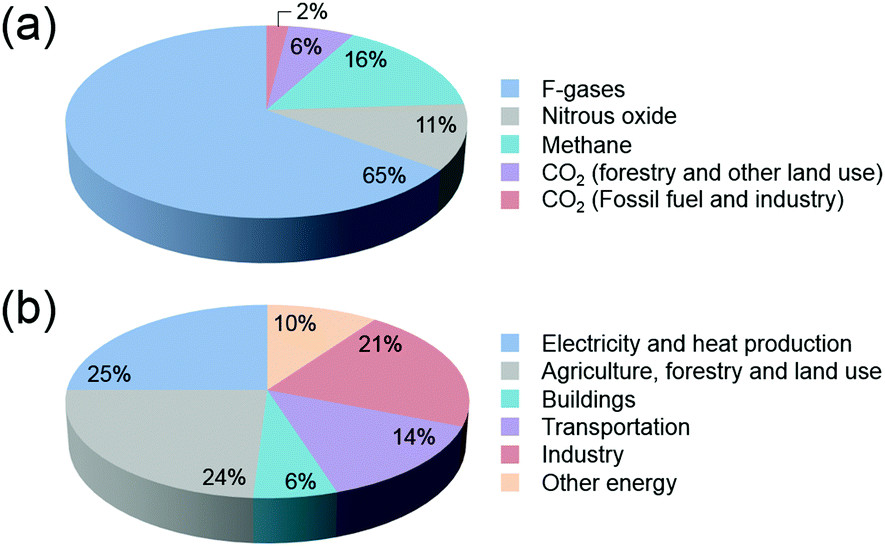 | ||
| Fig. 1 (a) Global greenhouse gas production by human activities; (b) categorization based on the corresponding economic sectors. Source: IPCC (2014) based on emission values in 2010.8 | ||
The focus on the reutilization of carbon dioxide dates from the 1850s when the chemical transformation of CO2 into chemicals, plastics, and fuels gained initial attention. Current examples of commercialized technologies include the synthesis of salicylic acid (1869),22 sodium carbonate via the Solvay process (1882),23 and urea production (1922).24 The current annual global CO2 utilization remains, however, on the order of 200 Mt, the equivalent of less than 0.6% of the total amount of anthropogenic emissions per year. In addition, with nearly 75% of the formed products, the incorporated CO2 is released once the products are used, therefore hindering the long-term sequestration of the reutilized CO2. CO2 capture, distillation, and the overall availability of large-volume sources with high purity reflect severe limitations in the economic viability of promising technologies; the proposals for the large-scale implementation of innovative platforms require the careful analysis of associated opportunity costs.17
In any catalytic reaction, product selectivity along with the activity and stability of a selected material define the promising character of a catalyst. Once CO2 is separated, the high stability of the molecule poses a challenge to its subsequent conversion. The chemically stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond possesses a dissociation energy of ca. 750 kJ mol−1, requiring an equally high energy input and catalytically active materials for CO2 reduction to occur. This high energetic demand emerges as a bottleneck in the large-scale implementation of the currently developed technologies for CO2 hydrogenation; e.g. to methanol and methane. Concerning product selectivity, several routes have been developed for the selective reutilization of CO2 toward a sustainable energy cycle for targeted fuels and chemicals, e.g. chemical reforming, photochemical, biological, mineralization and electrochemical methods. Recently, Li–CO2 electrochemistry has been proposed for simultaneous CO2 fixation and energy storage.25–27 Conversely, using CO2 as a building block to yield formic acid, carboxylic acids, urea, organic and cyclic carbamates, and organic, inorganic and cyclic carbonates has been widely reported, along with the synthesis of oxygenates, e.g., methanol and dimethyl ether (DME), as promising and easily stored and transported fuels in internal combustion engines (Fig. 2).28–36 Alternatively, the synthesis of CO2 is often directed toward the formation of syngas (CO and H2). The development of two-step cascade processes is a promising approach considering the viability of subsequent industrial platforms, e.g., biotechnological syngas fermentation technologies, Fischer–Tropsch (FT) synthesis to produce fuels and chemicals, or the synthesis of alcohols (in particular, methanol) or methane.37
O bond possesses a dissociation energy of ca. 750 kJ mol−1, requiring an equally high energy input and catalytically active materials for CO2 reduction to occur. This high energetic demand emerges as a bottleneck in the large-scale implementation of the currently developed technologies for CO2 hydrogenation; e.g. to methanol and methane. Concerning product selectivity, several routes have been developed for the selective reutilization of CO2 toward a sustainable energy cycle for targeted fuels and chemicals, e.g. chemical reforming, photochemical, biological, mineralization and electrochemical methods. Recently, Li–CO2 electrochemistry has been proposed for simultaneous CO2 fixation and energy storage.25–27 Conversely, using CO2 as a building block to yield formic acid, carboxylic acids, urea, organic and cyclic carbamates, and organic, inorganic and cyclic carbonates has been widely reported, along with the synthesis of oxygenates, e.g., methanol and dimethyl ether (DME), as promising and easily stored and transported fuels in internal combustion engines (Fig. 2).28–36 Alternatively, the synthesis of CO2 is often directed toward the formation of syngas (CO and H2). The development of two-step cascade processes is a promising approach considering the viability of subsequent industrial platforms, e.g., biotechnological syngas fermentation technologies, Fischer–Tropsch (FT) synthesis to produce fuels and chemicals, or the synthesis of alcohols (in particular, methanol) or methane.37
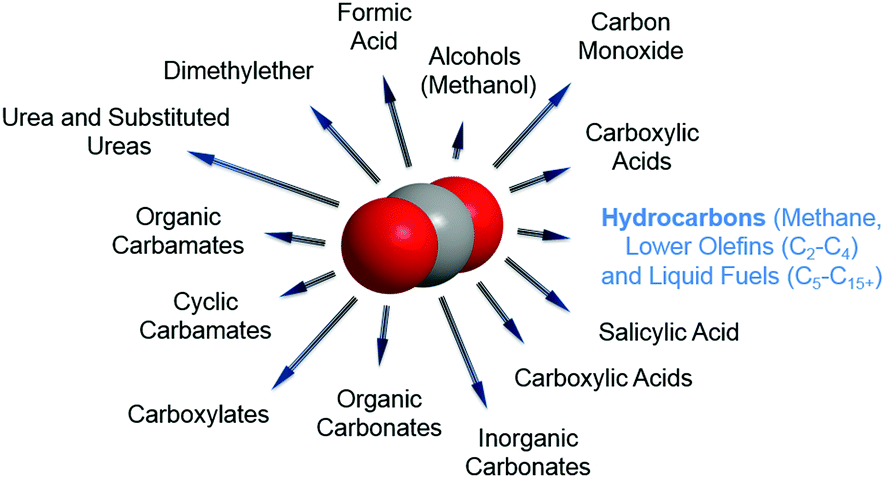 | ||
| Fig. 2 Representative spectrum of possible products from the reutilization of CO2. | ||
1.1. Scope of the review
Within the plethora of emerging products assessed in the reutilization of CO2, in this review, the authors provide a comprehensive dissection of reports of marked importance in the synthesis of hydrocarbons through thermochemical, electrochemical and photochemical approaches. The production of hydrocarbons reflects their role as the backbone of our energy infrastructure, with unparalleled energy density, and an emerging and valuable platform in ambitious space explorations. In addition to being a major component of natural and shale gas, relatively inexpensive and widely available, in the past decades, the production of methane (CH4) has emerged as a pathway to a potential future energy-carrier and an alternative to an envisioned H2 and methanol-driven sustainability. CH4 production from CO2 in the presence of H2 serves as an alternative to the well-known bottlenecks in the storage and transportation of hydrogen related to its low volume density. This shift toward enhanced CH4 production rates finds expected application within the current commercial infrastructure for electricity and heat generation as a substitute for gasoline, diesel or LPG in vehicles, and is appropriate in geographical regions where natural gas supplies are not abundant, and regulated CO2 emissions may demand alternative energy storage routes.Although synthesizing CH4 is the less energetically demanding reaction pathway, extending the hydrocarbon chain to either paraffins or olefins is preferable from an economic point of view. These can either be stored as an energy carrier or directly used as a petrochemical feedstock (C2–C4) or as liquid fuels (C5–C15+). Lower olefins (C2–C4) for instance, are conventionally synthesized by the thermal or catalytic cracking of naphtha or vacuum gas oil, or by the dehydrogenation of alkanes of equal carbon number. With extensive application in the chemical industry as building blocks in the synthesis of polymers, solvents, drugs, cosmetics, and detergents and an overall two-fold boost in the global production capacity over the past 15 years,38 the increasing demand for olefins from alternative feedstocks has been incited by both environmental and economic factors in recent years. The extensive market demand for olefins leading to the subsequent emission of ca. 1.2–1.8 tons of CO2 per ton of olefin produced further underlines the importance of the selective reutilization of CO2 to these products.
As the precursor to polyethylene, accounting for ca. 62% of the global ethylene consumption in 2016, C2H4 plays a major role in the plastic industry and benefits from an expanding global market and price point when compared to other C2 value-added products. In 2021, ethylene global supply is projected to surpass the 170 million metric tons mark, with an increasing non-steam cracker percentage of ca. 6% predicted for the same year.39 Taking into account the past market oscillations of the prices of conventional fossil fuels in recent years, the collected average prices still distinctly place ethylene (0.989 USD per kg) above the value-added products previously listed, e.g., formic acid (0.4–0.6 USD per kg) or acetic acid (0.6 USD per kg).40 Within the recently overviewed global market sizes, ethylene (155–248 billion USD) emerges two and three orders of magnitude above carbon monoxide (2.7–3.2 billion USD) and formic acid (0.62 billion USD), respectively.41
The synthesis of value-added transportation fuels accomplishing an idealized closed carbon cycle remains the most challenging to date. In the context of a depletion of fossil sources, the production of gasoline and diesel from CO2, containing typically C4–C12 and C12–C20, respectively, would represent a dramatic shift in sustainability. Despite the strong fluctuation in fuel prices in recent years, global consumptions continue to dramatically increase, reflecting the ever-increasing growth of global energy consumption. In particular, gasoline global market consumption has currently surpassed the annual 1000 million metric tons mark.42 The noteworthy dependence of fossil fuels aggravated by drastic fluctuations in the oil market further underlines the need for the development of alternative fuels from conventional non-fossil sources.
The above-discussed economic interest in a direct CO2RR to hydrocarbons demands the continuous updating of the current strategies for urgent development. Electrochemical and photochemical-based exploratory works, in particular, have reflected a dynamic development of promising approaches in recent years that deserve clear dissection in the delineation of impending strategic paths. Representative reviews available in the current literature tend, however, to cover the entire spectrum of formed products in the reutilization of CO2, failing to provide the necessary level of detail presented here. The authors will further focus exclusively on heterogeneous catalytic systems. The facilitated separation of catalysts and formed products and reactants is of prime concern in the design of commercially practical processes and is notably advantageous in comparison with homogeneous systems. The following sections provide a detailed retrospective on the benchmark and innovative catalytic systems in terms of achieved activity, selectivity and stability levels. In addition, discussions on the proposed mechanistic pathways and the current associated difficulties, pioneering alternative approaches, and competitive developments are reviewed herein, while highlighting the latest noteworthy reports.
2. Thermochemical CO2 reduction reaction (t-CO2RR)
The extensive background in t-CO2RR to hydrocarbons is a direct reflection of its ever-increasing attention in the community and of an accelerated development in light of analogous CO-based technologies. In addition, the thermochemically-driven reaction has a greater potential to be applied on a large scale as compared to the energetically promising alternative electrochemical and photochemical strategies in the production of transportation fuels. For the reasons above, extensive progress has been made in the production of C1 chemicals, e.g., CO, formic acid, and methanol. The reduction of the fully oxidized carbon to hydrocarbons requires the promotion of the kinetics of a reaction highly limited by the transfer of at least eight electrons. Although great progress has been attained in the selective synthesis of CH4, selectively producing long-chain hydrocarbons still faces a series of drawbacks and engineering bottlenecks. To date, the study of suitable catalysts essentially relies on single metal or alloy-based catalysts dispersed on metal oxides. Achieving progress in the development of CO2RR platforms relies on an in-depth knowledge of its underlying chemical mechanism, with this drawback being inspected below.2.1. Mechanistic difficulties
Assessing and eventually disentangling potential mechanistic pathways permits the optimization of the overall CO2 conversion rates through the minimization of relevant thermodynamic or kinetic barriers of the intermediate reactions, and allows an enhancement of the overall product selectivity. When tentatively reducing CO2 to the simplest hydrocarbon, CH4, in the presence of H2, the latter is easily dissociated over the ideally well-dispersed metallic phase. Subsequent spill-over of the resulting reactive atomic hydrogen species occurs on the support, where the hydrogenation of the involved carbon intermediates takes place. Mechanistically, the reduction of the CO2 molecule has been recognized to significantly depend on the selected catalyst, the choice of the support (cf. Section 2.2.3), operating conditions, and CO2 to H2 reactant ratios.43Chen and colleagues have recently summarized possible pathways for t-CO2RR to CH4 on metal/oxide catalysts; the adapted schematic diagram is depicted in Fig. 3.44 A possible direct hydrogenation of CO2 to synthetic natural gas claimed in earlier years is represented in Fig. 4.45 As evidenced in Fig. 3, both proposed pathways – the RWGS + CO-Hydro and formate-based – reflect an emerging parallelism in the synthesis of alcohols during the production of hydrocarbons, a key point in the development of selective catalysts. Reaction pathways involving the initial conversion of CO2 to CO, followed by the methanation of the adsorbed intermediates (CO*) have been widely argued. The formation of CO* is proposed to occur through the direct reverse water gas shift (RWGS) reaction pathway (Fig. 3), or following a direct C–O bond cleavage (Fig. 4). The formation of CO* is particularly emphasized over representative Ni/Al2O3 state-of-the-art catalysts in the selective CO2RR to CH4.43,46,47
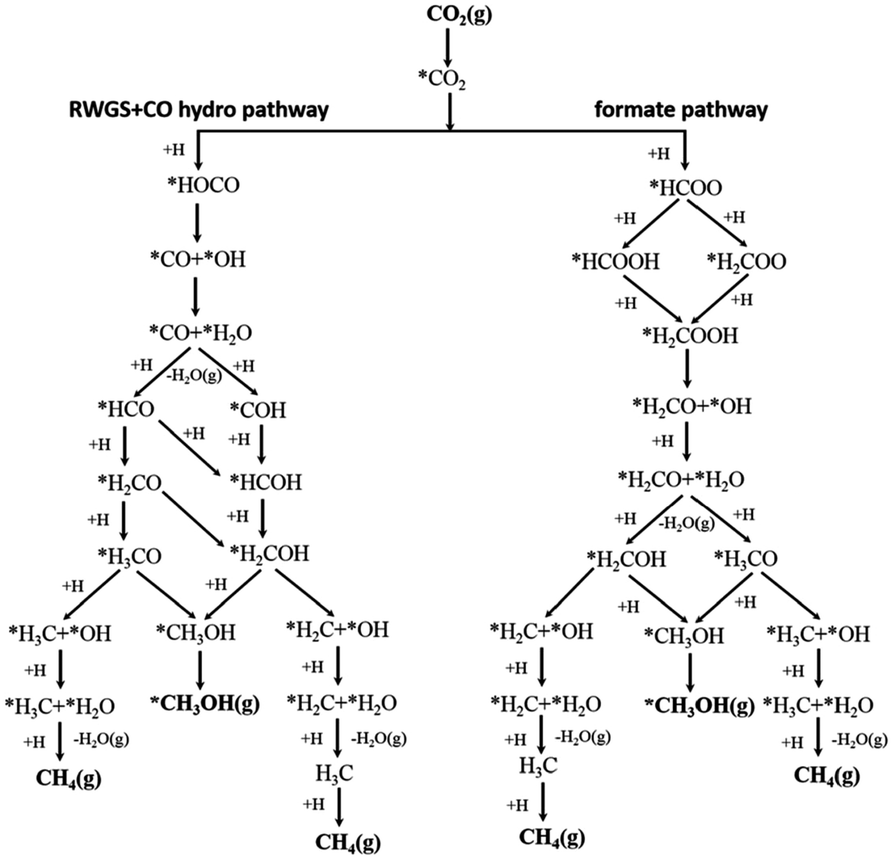 | ||
| Fig. 3 Schematic representation of the proposed RWGS + CO-hydro and formate mechanistic pathways in the selective t-CO2RR to methane. Adapted with permission from ref. 44. Copyright The Royal Society of Chemistry 2016. | ||
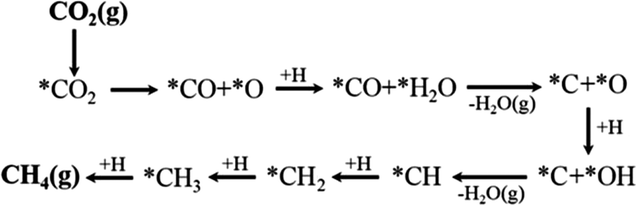 | ||
| Fig. 4 Proposed direct C–O bond cleavage pathway in CO2 methanation. | ||
The stability of the CO* intermediate determines whether the molecule will desorb or undergo further reduction. The reduction of CO* has been suggested to follow two distinct pathways: (1) the dissociation of CO* into carbon and oxygen species that are subsequently hydrogenated to CH4 and water, respectively; (2) the H2-assisted transformation of CO* to HCO* or COH* species.48 Despite the common formation of a CO* intermediate, higher CH4 selectivity and lower activation energies have been reported when the atmosphere is switched from CO to CO2, in accordance with the suggested CO* favored distribution during t-CO2RR. Equally under debate is the formation and decomposition of observed surface formate species (Fig. 3). For instance, over Ni/Y2O3, formate species were proposed to be a reaction intermediate leading to enhanced catalytic activity but Behm claimed that the detected formate only played a minor role in the reaction, acting as a spectator species.43,46
When extending the carbon-chain from CH4 to short olefins or products in the range of gasoline or even diesel, CO2 has been proposed to be first converted into a more reactive CO* intermediate via RWGS, followed by the modified Fischer–Tropsch (FT) reaction, which conventionally utilizes syngas (CO and H2) to generate long-chain hydrocarbons.49–52 The proposed mechanism for a CO2-based FT pathway is schematically simplified in Fig. 5. The conventional FT synthesis reaction utilizing syngas, first reported in the 20s by Hans Fischer and Franz Tropsch, applies iron and cobalt-based catalysts at high (HTFT) and low temperatures (LTFT), respectively. The properties of the resulting synthetic crudes (syncrudes) are significantly different depending on the type of temperature-based process, with three aspects being of importance to highlight (1) the probability of chain growth, (2) the degree of hydrogenation, and (3) secondary product formation.53 The probability of chain growth over the selected catalyst determines the carbon number distribution, often expressed by using the Anderson–Schultz–Flory (ASF) equation. HTFT syncrude is characterized by a light carbon number distribution, rich in olefins and oxygenates, with aliphatic hydrocarbons being predominantly linear. To a minor extent, naphthenes, mostly in the naphtha fraction, and aromatics, with increasing content as carbon number increases, are present. In contrast, ca. 50% of the hydrocarbon mass of LTFT syncrude is in the C22+ fraction, and becomes more paraffinic with increasing carbon number.54 Wax production is followed by selective hydrocracking to give the desired transportation fuels either in the naphtha or diesel ranges.
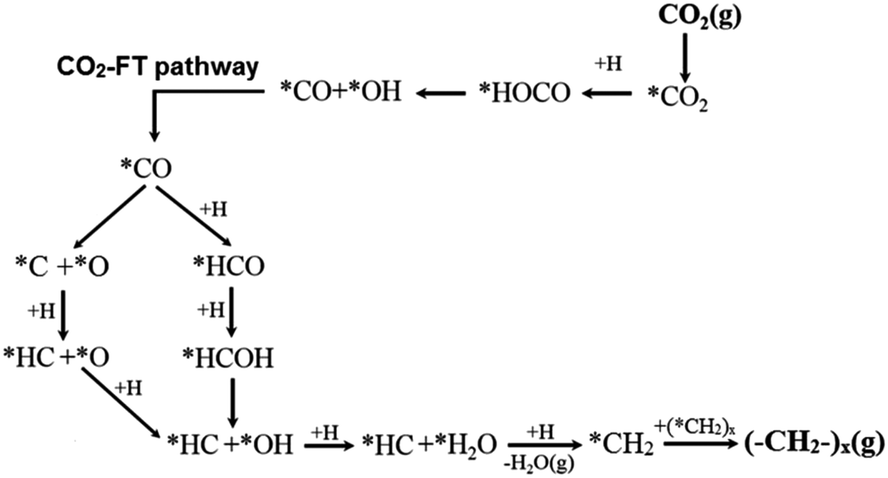 | ||
| Fig. 5 Schematic representation of the proposed CO2-based Fischer–Tropsch mechanistic pathway. Adapted with permission from ref. 44. Copyright The Royal Society of Chemistry 2016. | ||
In agreement with the proposed mechanism in Fig. 5, a stronger CO* binding energy is expected to favor the synthesis of hydrocarbons, while a weakly adsorbed CO* favors the RWGS reaction with CO evolution.55 The presumed parallelism between the CO2RR and the conventional CO-based FT synthesis, with the emergence of CO as an intermediate in the former process and presumably similar chain-growth pathways, have served as valuable guidance in continuous exploratory works. For these reasons, several comparative studies have assessed the applicability of state-of-the-art FT catalytic systems using CO2 as a feedstock. Leading groups have developed structure–function studies to enhance the selectivity of CO2RR using conventional FT catalysts to attain C5+ hydrocarbon yields comparable to those produced with CO as the feedstock.56 When switching the atmosphere from syngas to mixtures of CO2 and H2, state-of-the-art Fe-based CO2RR catalysts ideally hinder the formation of CH4 and favor the high olefinic nature of the yielded products (cf. Section 2.2.2).57–60 Directing the CO2-based Fischer–Tropsch mechanistic pathway toward the selective production of light olefins generally produced from fossil fuels including naphtha, gas oil, and light alkanes via catalytic cracking processes remains an alternative of interest as indicated in the introduction of this Review. Promoting the oligomerization of formed olefins to longer carbons is an alternative of equal importance, with the effect of residence time and co-feed being crucial to directing the readsorption of these products in the initiation of surface chains.61
An overall drawback in the direct application of conventional FT catalysts during t-CO2RR is, however, the need for an additional component able to direct the RWGS for initial CO production. Notwithstanding the thermodynamical viability of the combination of the two reactions, reaction conditions for CO2RR have been found to significantly differ from those of CO-based FT. Mechanistic difficulties have been highlighted in the past years, reflecting the prominent CO2 inertness during adsorption and reaction steps, and subsequent C–C coupling barriers. The above issues are further dissected in Section 2.2.2 in the application of suitable catalysts under CO2 and H2 mixtures.
2.2. Overview of catalysts
To date, extensive research has drawn critical conclusions regarding the activity and the selectivity of unsupported metal catalysts and their supported counterparts while scrutinizing the effects of incorporated promoters, preparation methods, and synthesis conditions on the resulting performance. In conventional t-CO2RR pathways, CO2 is firstly converted to CO via the RWGS reaction, with the latter being subsequently transformed into hydrocarbons. The study of suitable catalysts has essentially relied on single metal or alloy-based catalysts dispersed on metal oxides. Notwithstanding the role of the support in the activation of CO2 and the suggested support-dependent mechanistic pathways (cf. Section 2.2.3),62 limited catalytic activity has been found over individually evaluated metal oxide systems, e.g., ZnO, MnOx, Al2O3, and CeO2.63 Notably decisive in all cases is the lack of H2 dissociation sites, with CO formation through the RWGS reaction prevailing over the evaluated materials. In contrast, product selectivity is strikingly determined by the incorporated metal, with the reaction being preferentially directed to CH4 or long-chain hydrocarbons in accordance with the emerging catalytic fingerprints of the selected metal element. Over the past decades, Ru, Rh, Pt, Pd, Ni, and Co-based catalysts have been shown to selectively promote the synthesis of CH4 under the selected operating conditions. An extensive line of work has simultaneously focused on optimized CH4 selectivity, enhanced activity, and the stability of these systems. The singularity of iron oxides, on the other hand, has been directed toward the synthesis of long-chain hydrocarbons in a clear parallelism with conventional CO-based FT platforms. Benchmark catalysts, the corresponding catalytic formulations, and the impact of the support and promoters in each case are detailed in the following sections.| Catalyst | Metal (%) | H2/CO2 | LHSV (L g−1 h−1) | P (MPa) | T (°C) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Ce0.96Ru0.04O2 | Ru | 4 | ≈10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 000 h−1 |
0.1 | 450 (55%) | 99 | 70 |
| Highly dispersed Ru NP/TiO 2 | Ru (0.80) | 4 | 4.18 | 0.1 | 140 | 100 | 73 |
| Ru NP/TiO2 (“wet” impregnation) | Ru (0.75) | 4 | 4.18 | 0.1 | 200 | 100 | 73 |
| Ru/TiO2 (rutile) reduced at 400 °C | Ru (5) | 4 | 75 | 0.1 | 295 | 100 | 74 |
| Ru/TiO2 (anatase) reduced at 400 | Ru (5) | 4 | 75 | 0.1 | 335 | 100 | 74 |
| Single-atom Ru/CeO2 | Ru (0.89) | 4 | 120 | 0.1 | 295 | 100 | 77 |
| Ru nanoclusters/CeO2 | Ru (2.56) | 4 | 120 | 0.1 | 270 | 100 | 77 |
| Pd/SiO2 | Pd (6.2) | 4 | ≈3300 h−1 | 0.1 | 450 (40.8%) | 10.4 | 87 |
| Pd–Mg/SiO2 | Pd (6.2) | 4 | ≈3300 h−1 | 0.1 | 450 | 95 | 87 |
| Pd–Ni/SiO2 | Pd (6.2) | 4 | ≈3300 h−1 | 0.1 | 450 (50.5%) | 89 | 87 |
| Size-tunable Ni NP supported on surface-modified cage-type mesoporous SiO2 | Ni (21.7) | 1 | 1.2 | 0.1 | 475 (11%) | 65 | 96 |
| Ni-MCM-41 | Ni (1) | 4 | 19.7 | 0.1 | 500 (50%) | 83 | 94 |
| Ni/RHA-Al2O3 | Ni (15) | 4 | 30 | 0.1 | 500 | 90 | 90 |
| Ni/TiO2 | Ni (10) | 4 | 10000 h−1 |
0.1 | 400 | 95 | 100 |
| Ni/Al2O3 | Ni (10) | 4 | 10000 h−1 |
0.1 | 350 | 98 | 100 |
| Ordered mesoporous nickel alumina | Ni (15) | 5 | 91 | 0.1 | 350 | 94 | 92 |
| Ni/CaO–Al2O3 | Ni (23) | 4 | 15000 h−1 |
0.1 | 335 | 100 | 106 |
| Co-Impregnated Ni – 1 wt% MgO/SiO2 | Ni (10) | 4 | 15 | 0.1 | 325 | 98 | 105 |
| Ni/ZrO2–Al2O3 | Ni (12) | 3.5 | 8.1 | 0.1 | 325 | 100 | 98 |
| Ni/Al2O3 | Ni (10) | 4 | 20 | 0.1 | 300 | 100 | 43 |
| Ni@MOF-5 | Ni (10) | 4 | 2000 h−1 | 0.1 | 300 | 100 | 97 |
| Ni/CeO2 | Ni (10) | 4.6 | 22 | 0.1 | 300 (73%) | 100 | 99 |
| NiO–MgO–Al2O3 | Ni (10) | 4 | 15 | 0.1 | 290 | 96 | 107 |
| Ni/CeO2 (hard template synthesis method) | Ni (10) | 4 | 45 | 0.1 | 285 | 100 | 102 |
| Ni/mesoporous nanocrystalline γ-Al 2 O 3 | Ni (20) | 3.5 | 9 | 0.1 | 275 | 100 | 91 |
| Ni/CeO 2 | Ni (10) | 4 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 h
−1 000 h
−1
|
0.1 | 270 | 100 | 100 |
| Ni/CeO 2 | Ni (10) | 4 | 20 | 0.1 | 270 | 100 | 43 |
| Ni/ZrO2 | Ni (10) | 4 | 20 | 0.1 | 260 | 100 | 43 |
| Ni/Sm2O3 | Ni (10) | 4 | 20 | 0.1 | 260 | 100 | 43 |
| Ni/Y 2 O 3 | Ni (10) | 4 | 20 | 0.1 | 240 | 100 | 43 |
| Co/SiO2 | Co (3) | 4 | 1640 h−1 | 0.1 | 277 (13.7%) | 42 | 60 |
| Cobalt foil | Co | 3 | — | 0.1 | 302 (1.55 CH4 molecules/Co site s) | 98 | 119 |
| Co/ZrO2 | Co (10) | 4 | 3.6 | 3 | 400 (92.5) | 99.9 | 123 |
| Co/SiO2 | Co (10) | 4 | 3.6 | 3 | 400 (80.1) | 97.8 | 123 |
| Co/Al2O2 | Co (10) | 4 | 3.6 | 3 | 400 (77.8) | 96.5 | 123 |
| Co/SiC | Co (10) | 4 | 3.6 | 3 | 400 (77.5) | 96.4 | 123 |
| Co/Al2O3 | Co (20) | 4 | 5000 h−1 | 1.5 | 300 | — | 125 |
| Co4N/Al2O3 | Co4N (20) | 4 | 5000 h−1 | 1.5 | ca. 230 | 98 | 125 |
Ruthenium and rhodium-based catalysts. When making comparisons between benchmark materials experimentally and theoretically investigated in the past decades, Ru and Rh-based catalysts unveiled remarkable CH4 selectivity, and catalytic activities, even near room temperature.47,66,67 The mode in which transient carbonyl species are bonded with ruthenium is claimed to play a key role in their reactivity, and is believed to depend on the Ru oxidation state at the time of interaction, the particle size and under-coordinated sites, the catalytic temperature, and the composition of the reacting stream.67,68 Although works developed over other supports deserve equal attention,69,70 fast-forward three decades from the seminal contribution of Grätzel, Ru/TiO2 remains widely recognized as one of the most attractive catalytic formulations in CO2RR attaining complete CH4 selectivity under atmospheric pressure and temperatures under 200 °C.64 The increase in activity with increasing metal–support reflects earlier reports in CO methanation over these catalysts. At 473 and 523 K, Ru/TiO2 achieved superior activities as compared with MgO, Al2O3, SiO2, and ZrO2-supported counterparts.71
In the past thirty years, significant work on TiO2-supported materials has shed much light on the performance of Ru. Mechanistically, FTIR experiments provide evidence for the formation of adsorbed CO species (Rux–CO, Run+(CO)x, (TiO2)Ru–CO), and CO species linearly bonded to the reduced Ru crystallites (Rux–CO) were suggested to be reactive surface intermediates.72 The size and morphology of Ru species has been proved to depend on the crystal structure of TiO2. At 180 °C, the size of Ru nanoparticles (NPs) on anatase-type TiO2 determined the hydrogenation activity, with a maximal value with the smallest particles evaluated in this study (2.5 nm diameter).73 Reflected in the formation of a Ru–O–Ti bond assessed by X-ray absorption measurements and H2-TPR experiments, a rutile TiO2-based interaction not only promotes the highly efficient dispersion of Ru NPs to a great extent, but also hinders their subsequent aggregation.74 Sassoye found that Ru-based P25 commercial TiO2, composed of 20% rutile and 80% anatase phases, exhibited superior performance as compared to pure anatase or the rutile-counterparts.75 The authors later assessed the impact of mixing anatase and rutile TiO2 in different ratios and at different stages of the catalyst preparation, i.e., before or after RuO2 deposition, or after annealing.76 Following thermal reduction under H2, the interaction between RuO2 NPs and TiO2 phases during the annealing step was shown to dictate the resulting catalytic performance. Characterization suggested the migration and subsequent epitaxial stabilization of RuO2 NP over the rutile TiO2 phase. The NPs could then be reduced with high dispersion and specific activity. Incorporated anatase was claimed to be beneficial to impede the negative effect of support sintering and the subsequent loss of accessible Ru active sites.
In light of a number of exploratory works on the effect of the support (cf. Section 2.2.3), assessing the strong metal–support interactions (SMSI) that arise has also been of key interest. Following the works of Abe, Zhang and colleagues recently investigated the effect of the size of Ru domains in Ru/CeO2 nanowires from atomically-dispersed Ru centers, to Ru nanoclusters (ca. 1.2 nm), and Ru NPs (ca. 4.0 nm), utilizing ex situ and in situ spectroscopic techniques and density functional theory calculations (Fig. 6).77 Interestingly, the nanoclusters showed superior CO2RR activity, with 98–100% CH4 selectivity, and a turnover frequency (TOF) of 7.41 × 10−3 s−1 at 190 °C. Ce3+–OH sites and Ru sites near the metal–support interfaces were demonstrated to be responsible for CO2 dissociation and for carbonyl hydrogenation, respectively. The strongest SMSI and H-spillover effects with the corresponding suppression of the activation of metal carbonyls and the dehydration of the support surfaces were found in single Ru atoms and the Ru NPs, respectively. Conversely, competitive SMSI and H-spillover reached a balance in CeO2-supported Ru nanoclusters, with the resulting superior CO2RR activity for selective CH4 formation (Fig. 6e).
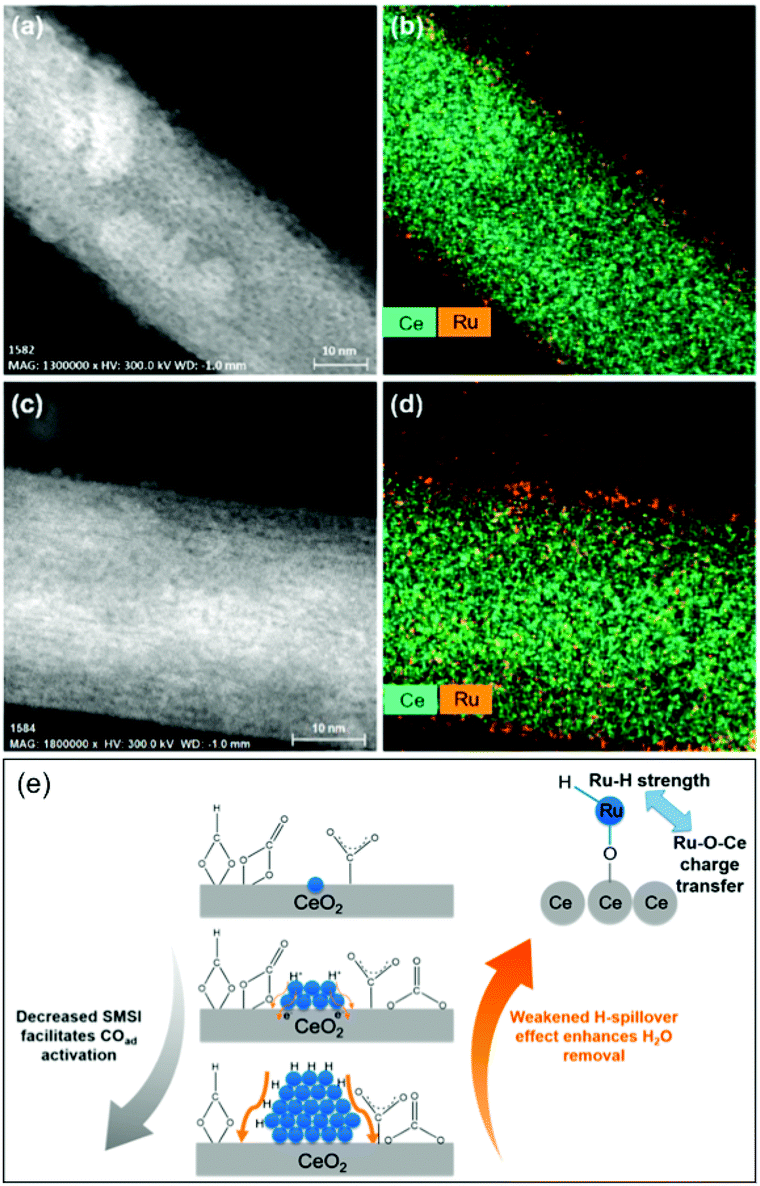 | ||
| Fig. 6 Cs-Corrected (a and c) HAADF-STEM images and (b and d) the corresponding Ce and Ru element maps of (a and b) nanoclusters and (c and d) single-atom Ru on CeO2 nanowires. (e) Proposed competitive strong metal–support interactions and H-spillover effects leading to the competing CO activation and surface dehydration over the Ru single-atoms, nanoclusters, and nanoparticles. Readapted with permission from ref. 77. Copyright 2018 American Chemical Society. | ||
High CH4 selectivity and superior activity have been emphasized in an extensive line of works using Rh/TiO2.78–81 Over Ru/γ-Al2O3 with metal NPs within a wide range of 3.6 to 15.4 nm, larger NPs unveiled a 4-fold superior activity at low temperatures (135–150 °C) whereas, at 200 °C, the turnover frequencies were found to be similar for all samples.82 The same authors later reported that over TiO2, the rate normalized by surface Rh atoms could be increased with an increase in the NP size up to ca. 7 nm, with a continuous size increase being shown to not play a significant role in the catalytic performance.83 Kinetic parameters such as the apparent activation energy and reaction orders with respect to H2 and CO2 were also demonstrated to depend strongly on the Rh cluster size. These variations may arise from the disparity of the selected operating conditions and characterization of the selected supports, but they further pinpoint the NP size effect as one of the most debated issues in the development of supported catalysts in t-CO2RR.
Palladium and platinum-based catalysts. Pd and Pt have emerged as promising candidates for t-CO2RR, with Pt-based catalysts unveiling the most promising conversion levels at analogous operating conditions at the expense of a lower cost-effectiveness.84 Comparatively, Pd shows negligible conversion below 300 °C and a preferential formation of CO over CH4 in the said temperature ranges.84 Favoring the RWGS or CH4 production was, however, early confirmed to be alternately achieved over large or finely dispersed Pd NPs, respectively.85 Shape-controlled Pd-nanocubes and nanopolyhedra particles revealed an activity and selectivity dependence on the exposed Pd facets (100) and (111), which were presumed to control the adsorption strength of intermediate CO*.86 DFT studies on Pd on MgO/SiO2 suggested the activation of CO2 over MgO through the formation of magnesium carbonate species, whereas a supply of atomic hydrogen was essential for further hydrogenation of magnesium carbonate to CH4.87,88
Nickel-based catalysts. Notwithstanding their excellent hydrogenation performance, the large-scale industrial application of noble metals is limited by their high cost. Fast forwarding nearly 30 years since Falconer assessed the premise of Ni under a CO2 atmosphere,52 Ni-based systems are today the most widely studied catalysts for CO2RR due to their attractive cost-effectiveness. The slow adsorption of CO2, believed to result in a higher H2:CO surface ratio, has been ascertained to favor selective CH4 production over long-chain hydrocarbons. Ni-Based catalysts, however, exhibit lower activity when compared with the representative Ru and Pd-based counterparts (Table 1), and a rather rapid deactivation due to carbon deposition, poisoning, and Ni sintering under high temperatures (cf. Section 2.4). For these reasons, the design of novel Ni-based catalysts, screening a wide spectrum of supports of choice with high surface area, e.g., Al2O3, SiO2, TiO2, and CeO2, attempts not only to favor enhanced metal–support interactions but also to hinder the deactivation of the Ni active centers.
Following a series of works on Ni-based amorphous SiO2 catalysts,89 Chang demonstrated that Al incorporation, with an enhancement of the acidity of the support, could crucially increase the overall hydrogenation activity.90 At 500 °C, an optimized yield (ca. 58%) and desired methane selectivity (ca. 90%) were obtained. Over Ni supported on mesoporous γ-Al2O3, experimental results have shown that both t-CO2RR activity and CH4 selectivity could be continuously increased within a 10 to 20 wt% Ni loading range, whereas a 25 wt% Ni content led to larger crystallite size and inferior catalytic activities.91 The evaluated loading range is additionally representative of most literature reports to date in the study of Ni-based catalysts (Table 1). As extensively discussed in Section 2.2.3, incorporating metal NPs into mesoporous and mesostructured materials is considered highly advantageous.92 Ni embedded within a three-dimensional mesoporous SBA-15 unveiled higher activity, lower coke formation, and higher thermal stability in the 600–800 °C range as compared with conventional SiO2 and γ-Al2O3 supports.93 Highly dispersed and thermally stable metallic Ni over siliceous MCM-41 has shown enhanced CH4 selectivity (up to 96%) in a low conversion range at 300 °C, superior to that of Ni/SiO2 and even to a Ru/SiO2 reference.94 Over mesostructured silica nanoparticles (MSN), with extended intra- and inter-particle mesoporosity and a BET area of 1051 m2 g−1, dispersed Ni active centers showed higher activity compared to a conventional tubular-shaped MCM-41 architecture or a microporous 3D HY-zeolite-supported counterpart.95 Taking advantage of the dimensionality, pore-size, and geometrical features of structured architectures is a well-established approach in catalytic platforms. Recently, ultra-small Ni NPs in a 2.7 to 4.7 nm-range, depending on the Ni content, were supported in the cage-type mesopores of –COOH-functionalized-silica SBA-16 via wet impregnation followed by thermal reduction.96 At pH = 9, deprotonation of the carboxylic acid groups on the cage-type mesopore surfaces induced the effective incorporation of Ni2+ precursors via favorable electrostatic interactions, toward high dispersion of the resulting particles.
The manipulation of the interactions between the metal and the support while taking advantage of the micro and mesoporosity, remains a leading strategy for enhancing metallic dispersion.97 Ni-Supported ZrO2–Al2O3 showed higher catalytic activity and better stability than its γ-Al2O3 counterpart, reflecting the acidic/basic properties and enhanced CO2 adsorption of zirconia.98 The highly dispersed ZrO2 was claimed to inhibit the formation of nickel aluminate-like phases, enhancing the dispersion of Ni species, and facilitating the reduction of NiO. In comparison, Ni incorporation in ceria has showcased unprecedented activities and CH4 selectivity close to 100% at 340 °C and atmospheric pressure.99,100 The superior performance can be quickly gauged in Fig. 7 when CeO2-supported Ni catalysts are compared with other state-of-the-art materials (cf.Table 1 for detailed information on the results in Fig. 7). The activity boost over CeO2-supported Ni has been ascribed to its reducibility, the presence of oxygen vacancies and oxygen storage capacity, and the emerging interactions with Ni determine the morphology, size, dispersion, and electronic properties of the incorporated NPs. DFT calculations have suggested the superior activity of CeO2(110) as compared to both (100) and (111) phases since oxygen vacancy formation is more likely over the former.101 The presence of Ce3+/Ce4+ ion pairs and oxygen vacancies effectively promotes CO2 activation;102 these assumptions were corroborated by in situ FT-IR and in situ XPS results evidencing the promoted generation of active CO intermediate species.99 The preparation of NiO-included mixtures of metal oxides, e.g., mesostructured NiO–CeO2 synthesized using a surfactant template method, using SBA-15 as a hard template, or directly mixing NiO with hard template-individually prepared CeO2, has served as an approach in the synthesis of Ni-based metal oxides.99,103,104 The hindered sintering of the resulting narrow metal Ni, formed from the reduction of NiO, was ascribed to their strong interactions with the ceria support.
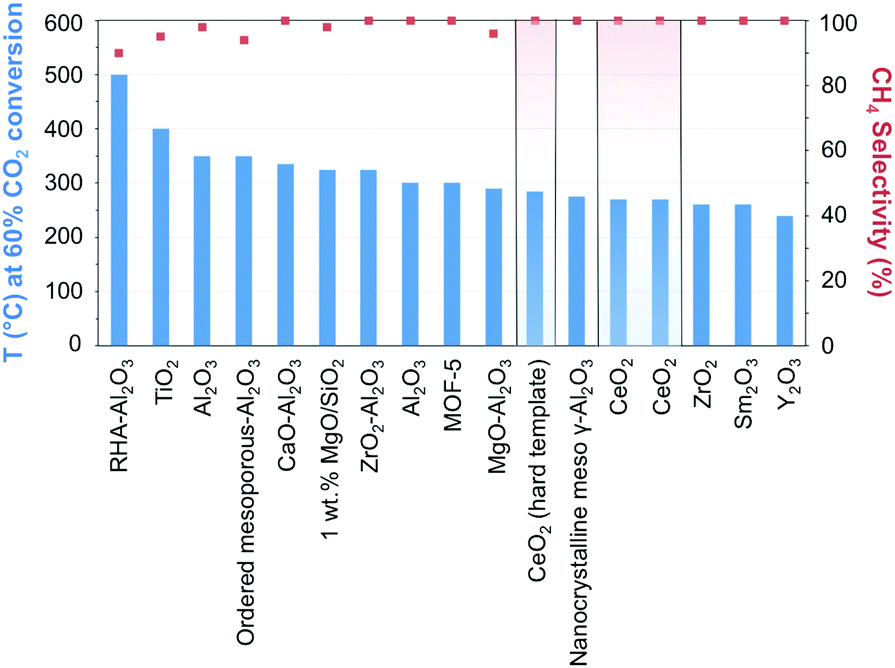 | ||
| Fig. 7 Performance of Ni-based catalysts in t-CO2RR to CH4, tentatively compared on the basis of the temperature necessary to achieve ca. 60% CO2 conversion. The highlighted results indicate the overall superior performance of Ni/CeO2 catalysts in the literature. Further details and respective references have been conveniently summarized in Table 1. | ||
The above findings conspicuously highlight the mechanistic implications of the interactions between the metal and the selected support. While recently assessing the suitability of less explored oxides, Muroyama reported CH4 yields at 250 °C following the order Ni/Y2O3 > Ni/Sm2O3 > Ni/ZrO2 > Ni/CeO2 > Ni/Al2O3 > Ni/La2O3.43 The notability of these catalytic systems, in particular, Ni/Y2O3, is underscored by their activity, which is superior to even Ni/CeO2. The nature of the formed intermediates examined by in situ infrared spectroscopy was claimed to depend on the selected support. In detail, CO species could be detected over Ni/Al2O3, whereas only carbonates were found over Ni/La2O3. Over Ni/Y2O3, considering the higher catalytic activity for CO2 methanation and the rapid decomposition of formate species, the latter were proposed to serve as intermediates in the reaction (Fig. 3).
Basic and redox active promoters have been shown to improve CO2 adsorption in Ni-based catalysts to prevent sintering of the active material and to increase its reducibility.105,106 The incorporation of alkaline Mg species into an ordered mesoporous Al2O3-based framework (Mg/Al molar ratios in a wide 0–10% range) via one-pot evaporation-induced self-assembly (EISA) unveiled increased surface basicity, which could intensify the chemisorption and activation of CO2 and enhance low-temperature catalytic activity.107 Despite finding suitable application toward the production of long-chain hydrocarbons (Section 2.2.2), Fe has been proposed as a second metal to improve the performance of Ni in CO methanation, according to both experimental and density functional theory calculations.108–110 In line with the above, Al2O3-supported Ni, Cu, Fe and corresponding Ni-based alloys prepared with the same total metal loading and varying Ni:Cu and Ni:Fe ratios were recently investigated for CO2 methanation.111 The resulting alloys exhibited lower particle diameter and sharper particle size distribution. At Ni:Cu and Ni:Fe ratios of 3:1, these catalysts showed opposite activity trends when compared with a Ni/Al2O3 reference, with Ni:Fe-based Al2O3 showing a 3-fold enhancement of the reaction rate.
Cobalt-based catalysts. As representative materials in the conventional LTFT using syngas, Co-based catalysts were considered candidates in CO2RR toward long carbon-chains. When switching the reaction feed to a mixture of CO2 and H2, traditional Co-based FT catalysts were, however, shown to not lead to the anticipated ASF product distribution obtained under syngas atmosphere. Instead, products with low C/H ratio, and a favored CH4 formation with a decline in chain growth, have been reported due to the slow adsorption rates of the thermodynamically and chemically stable CO2 on the surface of the catalyst.112 With a relatively low RWGS activity for CO generation,113,114 the equilibrium constraints for the reverse CO shift reaction require severe operating conditions and relatively high temperatures.115 Observed CO vibrations in FT-IR studies were postulated to solely reflect transition states during the conversion of CO2 to –(CH2)n– rather than formed CO species.116 The high temperatures applied in the first step of the reaction (RWGS) lead, however, to the primary formation of CH4 over Co-based catalysts. Conversely, the possibility of working at lower temperatures to favor the extension of the carbon-chain results in lower overall reaction rates, requiring larger reactor volumes and extended gas recycling rates. Literature findings have thus underlined not only the importance of the incorporated components to direct the RWGS for CO production but also the required optimization of the operating conditions for each reaction step in the overall mechanism, when the atmosphere is skewed toward CO2. The latter is particularly difficult with different feed gas ratios of H2 and CO2 (3
:1, 2:1, and 1:1) and operating pressures (ranging from 450 to 150 psig) showing only minor effects in directing the desired product distribution.112 Despite the suitability of the Fe-based catalysts introduced in Section 2.2.2 toward long-chain hydrocarbons, mixtures of Fe and Co and Fe–Co alloys have shown limited enhancement compared to the individual metals.117,118
Following early reports on Co/SiO2 prepared by incipient wetness impregnation60 and studies on high-purity Co foil,119,120 the application of Co-catalysts has since emerged as a valuable and cost-effective route to producing CH4 with high activity and selectivity. In line with the advantageous acid/basic properties of ZrO2 outlined above,98 Song has recently claimed the emergence of a new Co–Zr phase with a thickness of ca. 1–2 nm at the Co–ZrO2 interface.121 These claims were corroborated by representative TEM photographs unveiling lattice fringes with no correlation with ZrO2, Co0, or CoxOy, and XPS results suggesting the presence of oxygen vacancies in ZrO2-supported materials but not in the Al2O3 counterparts (Fig. 8). The bonding between Co species and surface Zr atoms was claimed to become stronger, and therefore more stable upon reduction, which, in return, led to a re-dispersion of Co0 on ZrO2. The resulting high activity at 400 °C (92.5%) superior to SiO2 (80.1%), Al2O3 (77.8%), and SiC-supported counterparts (77.5%), a CH4 selectivity of 99.9% and high stability up to 300 h were reported. Other remarkable results were recently reported, with Co-based porous carbon catalysts fabricated using a ZIF-67 zeolitic imidazolate framework as a template, with controlled morphology from cubic to rhombic dodecahedron, and particle size in the 150 nm to 1 μm range.122 The hierarchical structure suppressed the sintering of the 7–20 nm-sized Co NPs, with high CO2 conversion (52.5%) and CH4 selectivity (99.2%) at 72000 mL g−1 h−1 and 270 °C.
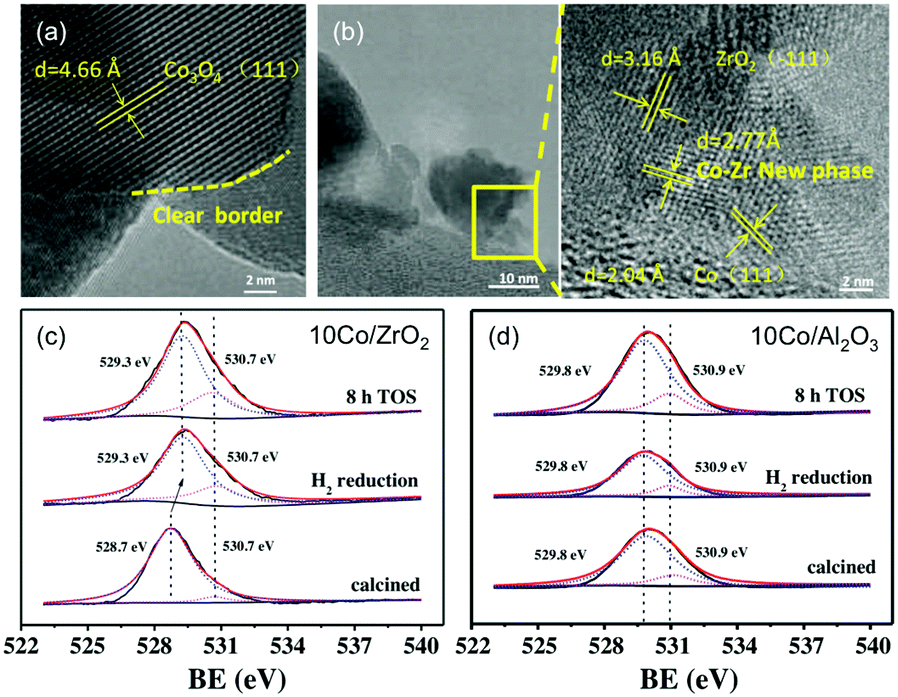 | ||
| Fig. 8 (a) TEM images of calcined catalyst precursors Co3O4/ZrO2 and (b) the corresponding reduced catalyst Co/ZrO2. (c) and (d) are the O 1s XPS spectra for the Co/ZrO2 and Al/Al2O3 catalysts calcined, following H2 reduction, and after 8 h on stream. Readapted with permission from ref. 121. Copyright 2018 Elsevier B.V. | ||
Exploiting synchrotron-based in situ characterization techniques such as XANES and XPS, Somorjai demonstrated that product distribution could be tuned depending on the support and oxidation state of cobalt.123 The authors observed that 10 nm Co NPs on TiO2 or SiO2 could not be reduced at 250 °C, with complete reduction only being achieved at 450 °C. Interestingly, CoO on TiO2 unveiled 10-fold superior CO2RR rates compared to fully reduced cobalt. Inversely, the activity of cobalt supported on SiO2 showed a higher turnover frequency than Co0. In previous works, the authors had reported model Co-based catalysts with particle size in the 3–10 nm range prepared using colloidal chemistry in the absence of trioctylphosphine oxide, a capping ligand claimed to result in phosphorus poisoning of the metal surface.124 Nitride Co4N/γ-Al2O3 samples prepared by NH3-temperature programmed reaction revealed superior activity for CO2 (and CO) conversion compared with Co0-based counterparts.125 The formation of a Co4N phase led to a strong metal–support interaction modulating the metal particle size and dispersion, and enhanced the adsorption capacity of the catalyst, as confirmed by H2, CO, and CO2-TPD.
Establishing synergies in metal-based hybrid catalysts. Structural and chemical effects on the intimacy of Co and other metal NPs as FT catalysts have long been investigated.126,127 An intimate contact between two metals modifying the local band structure, ensemble-type geometric effects, prevention of deactivation by carbonaceous deposits, and improvement in the reducibility of Co have been suggested. A similar strategy has been applied in t-CO2RR using either Pd or Pt co-catalysts. Using in situ NEXAFS spectroscopy, Somorjai suggested that H atoms dissociated on well-defined monometallic Pt NPs deposited near Co centers could be transferred to the latter via long-distance H-spillover and surface diffusion across silica, facilitating Co reduction (Fig. 9).128,129 The close intimacy found between individual Pt and Co NPs could enhance CO2RR rates up to a factor of 6 per Co surface atom. The authors further synthesized CoPt bimetallic NPs with controlled size and composition supported on MCF-17, evidencing Pt segregation under a reducing hydrogen atmosphere for the advantageous application in CO2RR to CH4.130 Recently, a perovskite-structured BaZrO3-supported catalyst corroborated the cooperative H-spillover between the Pt and Co centers, with CO2 preferentially dissociating on Co and H2 dissociating occurring on Pt.131 The interaction could be further enhanced through the atomic decoration of Pt on the surface of Co. Additionally, taking the strong interaction between Co and the BaZrO3 support into account, a 6-fold increase in CH4 formation rate was found in comparison to previously studied γ-Al2O3 supports at 325 °C. At the same temperature, the methane selectivity reached 80%, in comparison to only 43% over γ-Al2O3.
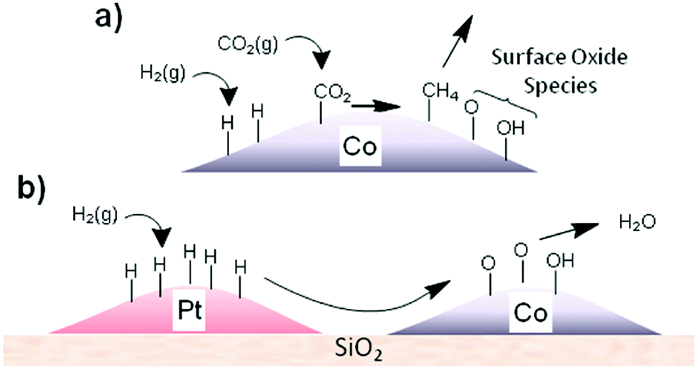 | ||
| Fig. 9 (a) Schematic representation of the production of surface oxide species during CO2 methanation and (b) transfer of adsorbed hydrogen from Pt to Co via a spillover mechanism to re-reduce the cobalt oxide surface, releasing water. Reprinted with permission from ref. 128. Copyright 2014 American Chemical Society. | ||
Notwithstanding the enhanced reducibility of the resulting catalysts with a superior number of metallic sites, the high cost of incorporated noble metals still demands the development of novel synthetic strategies hindering the corresponding metal loadings. The recently reported Pt-promoted Co–Al2O3 (0.03–0.43 wt% Pt) synthesized by double flame spray pyrolysis showed significant improvement in the reducibility of Co3O4 and high t-CO2RR activity as compared to a pristine Co–Al2O3.132 Ruiz et al. demonstrated that a synergistic effect could be found with physical mixtures of Pd (5 wt%) and Rh (2 wt%)-based γ-Al2O3 catalysts at temperatures between 150 and 200 °C.133 Similarly, H2 was activated over Rh/γ-Al2O3, but the activation of CO2, its dissociation to adsorbed CO*, and subsequent hydrogenation to CH4 were believed to take place on both phases. Activated H species on Rh/γ-Al2O3 were claimed to migrate and react with the adsorbed CO* on Pd/γ-Al2O3, leading to selective CH4 formation. A similar strategy was considered by mechanically mixing Rh/γ-Al2O3 and Ni/activated carbon.134 The production of methane was significantly higher than the theoretically predicted values, considering the individual performances, and attributed to an in situ supply of hydrogen by the Ni-based catalyst. The suggested hydrogen spill-over further maintained Rh particles in a metallic state necessary for the reaction.
In this context, Somorjai and Yang have recently highlighted the importance of nanostructured catalysts using well-defined and characterized interfaces. Considering the incompatibility of synthetic conditions of the materials of choice, the integration of all the desired components into one single NP is challenging and requires an elegant synthetic design. The authors, therefore, developed a well-defined CeO2–Pt@mSiO2–Co nanostructured catalyst for converting CO2 into C2–C4 with high selectivity (60%), with no activity decay up to 40 h.135 As suggested above, the Pt/CeO2 interface plays the role of converting CO2 and H2 into CO, which could subsequently be reacted on conventional Co/mSiO2 centers to yield hydrocarbons through an analogous FT process. The successful production of C2–C4via a tandem process on a structurally well-defined catalyst additionally demonstrates the power of sophisticated structure control in multiple-step chemical conversions (Fig. 10). To date, the promising incorporation of optimized functionalities widely extending the library of catalysts in analogous CO2-based FT processes has remained scarce, with the application of Fe-based catalysts being the main focus of the literature, as cautiously reviewed below.
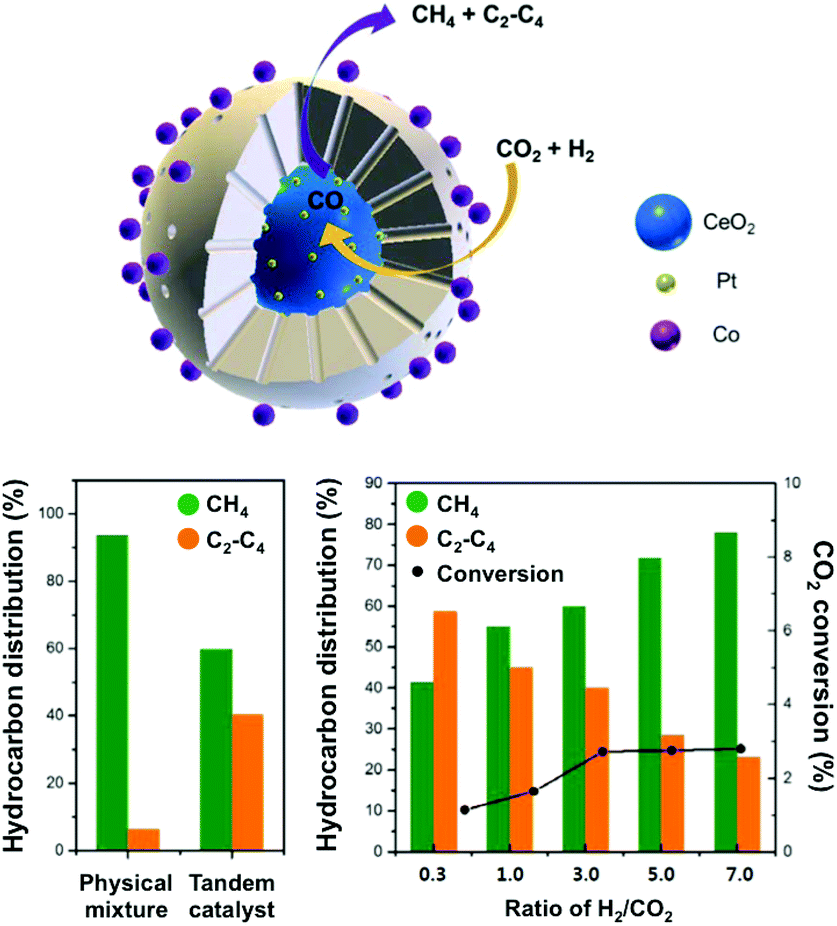 | ||
| Fig. 10 (top) Schematic representation of a well-defined nanostructured catalyst, CeO2–Pt@mSiO2–Co, for t-CO2RR to C2–C4 hydrocarbons using two metal–oxide interfaces. The catalytic performance of the prepared tandem catalyst is compared with a physical mixture of the evaluated components (left). The impact of the H2/CO2 ratio on the hydrocarbon distribution and CO2 conversion (right). Adapted with permission from ref. 135. Copyright 2017 American Chemical Society. | ||
Iron-based catalysts. The application of Fe-based catalysts, possessing the most favorable characteristics for a selective synthesis of long-chain hydrocarbons to date, follows a parallelism with conventional FT platforms under CO2 atmosphere. In the conversion of syngas, Fe-based catalysts are conventionally applied in high-temperature FT synthesis, leading to an olefinic product distribution notably skewed to low carbon number. According to representative syncrude compositions on a mass basis listed by de Klerk, ca. 35% and 33% of the resulting crude corresponds to the C2–C4 fraction (olefins and paraffins) and to the C5–C10 naphthas range.54
The development of these catalysts for CO2RR has followed the guidelines drawn a priori for CO-based platforms. Despite the inherent difficulty in understanding the mechanism and involved species behind CO2RR, the phase reconstruction of Fe catalysts has been well documented to direct the presence of active sites needed to perform the two catalytic reactions in this process, i.e., CO2 and H2 produce CO by the RWGS reaction, which further reacts with H2 by FT synthesis to form hydrocarbons. The phase reconstruction of Fe catalysts, which is the subject of an intense debate, is dependent on the initial Fe2O3 particle size136 and is induced in a series of designated episodes of self-organization, altering the composition and structure of the catalyst and prompting the continuous evolution of the resulting activity and product selectivity (Fig. 11).137 The active centers, a complex mixture of iron phases, are generated when Fe oxide is exposed to the reactants, with the freshly reduced catalyst not being active in FT synthesis. Surface reconstruction unlocks a difficulty in classifying the active centers during the reaction, and most importantly, in quantifying said sites to calculate turnover numbers as a measure of the catalytic activity. Initial iron species, mainly α-Fe and Fe3O4, are believed to undergo phase transformation into an amorphous, and probably oxide, iron phase, active for the RWGS reaction, which strongly interacts with the support. Iron carbides, e.g., Fe5C2, have been suggested as active centers in the hydrogenation of CO via the FT process.138,139 The possibility of Fe5C2 species acting as the active phase in the conventional FT process has been long discussed.140,141 In line with the above findings, attempts have been made to tentatively optimize the ratio between Fe3O4 and Fe5C2 species to adjust the product selectivity.142
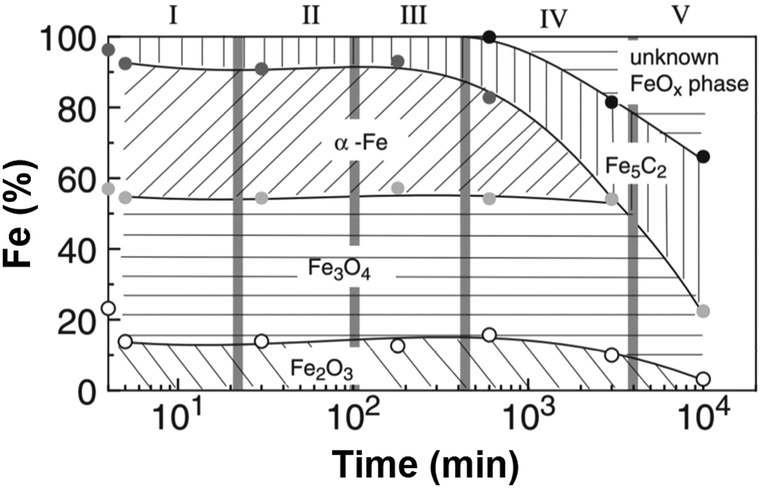 | ||
| Fig. 11 Iron-phase composition as a function of time during the Fischer–Tropsch synthesis under CO2 atmosphere, determined by Mössbauer spectroscopy. Data collected with a Cu–K-doped Fe–Al catalyst at 250 °C, 1 MPa, and H2/CO2 = 3. Reproduced with permission from ref. 138. Copyright 2003 Springer Nature. | ||
The catalytic properties of Fe-based catalysts markedly depend on structural effects and on the strategic incorporation of promoters (e.g. K, Mn, Cu, and Ce) and the selection of supports, toward enhanced activity, product selectivity and stability. The particle size of Fe2O3 NPs was assessed using the confinement effect of an Al2O3 support with varying pore size.143 The catalysts with fixed Fe and K-dopant contents (15 and 10 wt%, respectively) prepared by incipient wetness impregnation exhibited Fe2O3 NP size and distribution determined by the corresponding Al2O3 pore size. Alumina with a pore system in the 7–10 nm-range and corresponding Fe2O3 particle sizes of 5–8 nm were the most active for CO2RR to hydrocarbons. Narrower particle diameters were found to be unsuitable on account of the weak reducibility of smaller iron particles. Recently, bare Fe2O3 prepared via a template-assisted method and precipitation method and with easier reducibility, led to a greater ability for the formation of catalytically active iron carbides.144 Other works of significance have been recently reported using a high surface area iron-based catalyst prepared by the thermal decomposition of ammonium glycolate complexes.142
Despite inducing enhanced light olefin selectivity as compared with SiO2 and Al2O3-supported catalysts, MgO leads to poor dispersion of the incorporated Fe active species. These findings inspired later works on SiO2–MgO and Al2O3–MgO.145,146 Upon the incorporation of MgO up to a 20 wt% loading in Fe–K/Al2O3–MgO catalysts, CO2 conversion, total hydrocarbon selectivity, and the selectivity of C2–C4 olefins and C5+ species displayed a remarkable increase with a continuous increase in the MgO content.146 MgO was confirmed to play a key role in the nature of the Fe species present, determining the resulting product yield. In Fe-based catalysts, the presence of ceria has been conspicuously assessed either as a support or as a promoter. The morphology of supported Fe/CeO2 catalysts markedly determined the CO2 conversion, the methane to hydrocarbon selectivity, and the olefin-to-paraffin ratio,147 with a selective exposure of different crystal planes directing the metal–support interaction and the reactivity of emerging Fe-species. Ceria nanorods exhibited the highest hydrocarbon selectively, whereas higher olefin-to-paraffin ratios were achieved on ceria cubes.
Carbon nanotubes (CNT) have been extensively applied as supports in the conventional CO-based FT synthesis reaction, with a higher selectivity to olefins, improved activity owing to an enhanced catalyst dispersion, and low deactivation rates.148,149 In the conventional FT synthesis, pioneering results were reported over Fe-supported carbon nanofibers with remarkable C2–C4 olefin selectivity.150 Under CO2 conditions, Fe@CNT have also received attention.151 To overcome a number of disadvantages for the industrial applications of CNT in powder form, including a high-pressure drop for gas phase processes and agglomeration, Fe NP–carbon nanotube arrays grown on monoliths were evaluated for CO2RR.152 Detailed studies of the catalyst via HR-TEM, EDX and XRD uncovered the detailed evolution of the Fe NP throughout the process, with the formation of iron carbides, suggesting the presence of magnetite species and the surprising absence of iron oxides.
To increase the yield of hydrocarbons and overcome important drawbacks in the applicability of these materials, a number of dopants (e.g. K, Mn, Cu, and Ce) are often incorporated in the rational design of Fe-based catalytic systems. The extensive research carried out along these lines has additionally benefited from studies carried out under H2 and CO atmosphere in conventional FT synthesis. Therefore, tailored and optimized product distribution and an enhanced RWGS activity – limited a priori when compared with Co-based systems – have been reported. These results have permitted similar performances in the synthesis of hydrocarbons using either CO2 or CO as a feed.115 Potassium has been shown to simultaneously increase the chemisorption of CO2, decrease the adsorption of H2, and enhance the activity for CO formation,153 thus hampering CH4 formation and favoring chain growth.21,114 In addition, K-moieties have been proposed to accelerate the generation and assure the stabilization of the Fe5C2 phase, favoring the modified FT reaction. By simultaneously facilitating the RWGS reaction and carbon-chain propagation, high selectivity (ca. 60%) toward long-chain hydrocarbons has been achieved along with a significant shift in olefin production in the C2–C5+ range up to ca. 80%.58,154–156 Recently, Herskowitz shed additional light on the effect of potassium (2 wt%) on both Fe–Al–O oxide and Fe5C2 carbide materials, formed during CO2 hydrogenation.157 The incorporated potassium enhanced the RWGS rate of reaction on the oxide phase 10-fold as compared to the unpromoted oxide. The results were correlated with an increase in the Fe2+/Fe3+ ratio determined by XPS, hinting at the role of oxygen vacancies as active sites for the first reaction step of t-CO2RR. Conversely, potassium suppressed the methanation rate on the carbide catalyst by 5-fold, while increasing the formation of C2+ hydrocarbons by 1.4-fold. The synergism that emerged in the presence of potassium has been ascribed to the presence of potassium alanate (KAlH4).155 Prior to the reaction, the complete decomposition of KNO3 to other phases e.g., K2O and potassium aluminate (KAlO2) were observed during heat treatments above 500 °C.158 The latter appears to bind hydrogen in its structure to form KAlH4, leading to a lower surface coverage of adsorbed H species suppressing the hydrogenation of olefins to paraffins.
The incorporation of Mn and Cu-dopants improves the reducibility of FeOx species, the distribution of iron species, and the surface basicity.57,159 Mn favors the reduction of iron oxides and the carburization and dispersion of Fe2O3,160 while suppressing CH4 and favoring an increase in the formation of unsaturated high-chain hydrocarbons, with a higher olefin/paraffin ratio.155 However, Mn-loading has been shown to remain effective over a limited concentration range, with a continuous content increase leading to an eventual blockage of iron active centers. Mesoporous Mn–Fe oxide (0.05MnFe) catalysts exhibiting moderately high BET area, showed enhanced performance under fixed conditions (20 bar, 340 °C, CO2:H2:N2 = 23:69:8 (molar)) with reduced CO and CH4 formation and improved selectivity to C2–C5 and C6+ hydrocarbons, accompanied by oxygenates formation.161 Enhanced performance, in terms of hydrocarbon production with copper-promoted samples, has been claimed to be the result of a greater number of carbide species present when the evaluated iron catalysts reach a steady state during operation.162 During the hydrogenation of CO, the incorporation of copper was claimed to provide active sites for hydrogen activation and dissociation, dramatically accelerating the reduction of hematite to magnetite, with a larger number of magnetite nuclei being formed in this case.163 Under CO2 conditions, the presence of a copper surface enriched with hydrogen was however demonstrated to favor CH4 formation, which could be subsequently inhibited with the incorporation of potassium.
In the design of catalytic systems for t-CO2RR, other co-catalysts and dopants have been investigated. In accordance with the high activity of ceria-supported catalysts for CO2 methanation, ceria-promoted Fe-based catalysts have demonstrated enhanced activity and selectivity to C2–C5 olefins.164 As mentioned above, the synergetic effect is solely achieved if the incorporation of ceria, herein reflected in its corresponding particle size, is optimized to minimize the blockage of active Fe sites. Given the mechanistic similarities established between both catalytic processes, catalysts used in CO-based FT (e.g., Ru) have often been considered to be relevant, in an attempt to direct product selectivity to longer chains. Choung claimed that Ru incorporation could increase the probability of α-olefin readsorption in a Fe–K/γ-Al2O3 catalytic system, enhancing the overall activity and skewing product selectivity to higher C5+ hydrocarbons.165
Metal–support interfaces have been established in this review to play a central role in governing both the electronic and chemical properties of the resulting catalytic systems, governing the surface chemistry in t-CO2RR platforms, and directly impacting the charge transfer and mass transport in the activation of reactant molecules.77 Unveiling the nature and effects of these interactions on active site-dependent reaction mechanistic pathways and product selectivity has been covered widely in the literature.175,176In situ studies on Ru-loaded anatase and rutile TiO2 have indicated that the former acts as a better catalyst for CO supply, while rutile TiO2 is more effective in the hydrogenation of adsorbed CO intermediates.47 Zhang recently suggested that adsorbed CO2 could react with the hydroxyl groups of rutile TiO2 to produce CO via the formation of formate species, whereas the dissociation of adsorbed CO could then proceed as assisted by the presence of adsorbed H provided by the incorporated metal NPs.177 In the same year, Wang used operando XANES, IR, and Raman to disclose the generation process and evolution during the reaction of Ce3+, surface hydroxyls, and oxygen vacancies in a Ru/CeO2 catalyst.178 Steady-state isotope transient kinetic analysis-type in situ DRIFT infrared spectroscopy offered proof of CO2 methanation following the formate route (Fig. 3), with the rate-determining step being the dissociation of the latter, catalyzed by oxygen vacancies. However, over an α-Al2O3 supported counterpart and in the absence of oxygen vacancies, CO2 methanation was claimed to undergo the RWGS + CO-hydrogenation pathway (Fig. 3). The reducibility of the metal oxide was thus expected to play a key role in the rate-limiting cleavage of the second C–O bond, known to be dependent on the ratio of co-adsorbed CO* and H*.63 The combined results of H2 temperature-programmed reduction, quantitative H2 and CO chemisorption, in situ DRIFTS, and CO2 methanation kinetics revealed that CO-coverage on a Ru-based catalyst decreased with the support reducibility. For the irreducible Al2O3 support, the Ru particles were quasi-saturated with CO at low temperatures and the reaction was limited by the competitive Langmuir-type co-adsorption of H2. The ZnO support led to the lowest Ru–CO coverage associated with a weak CO adsorption and the reaction was dominated by the reverse water-gas shift reaction. CeO2 showed Ru–CO coverage between Al2O3 and ZnO, ensuring H2 dissociation and sufficiently high Ru–CO adsorption strength. The increasing interest in ceria as an excellent CO2 methanation catalyst has prompted additional studies on other reducible metal–oxides, e.g. In2O3 and Ga2O3.80,171,179,180
Whereas limited catalytic activity was found over individual metal oxide systems as sole t-CO2RR catalysts, a continuous focus on the properties of the support has shed some light on works assessing the properties of new materials as unconventional t-CO2RR catalysts. Transition metal carbides, with desirable behavior for reactions involving CO2 and analogous properties to those of hydrogenating precious metals, are interesting examples. Molybdenum carbide is cost-effective, exhibits the required dual functionality and is expected to behave similarly to reducible oxides.181 Alternatively, Ajayan and collaborators recently reported the use of N-doped graphene quantum dots as metal-free catalysts in the selective synthesis of CH4 above 380 °C.182 The located N-dopants at the edge-sites were claimed to markedly induce the thermocatalytic activity of these carbon-nanostructures, with increasing N-content, leading to lower onset reaction temperatures, higher CO2 conversion, and improved CH4 selectivity. Most importantly, the catalyst performance was crucially dependent on the observed doped N-configurations and corresponding defect density.
2.3. Methanol-mediated routes toward long-chain hydrocarbons
Serving as an alternative to modified Fe-based CO2RR catalysts through an analogous FT process, CO2 has been converted into linear hydrocarbons through intermediate steps involving the synthesis of methanol by methanol-mediated approaches.183,184 In the proposed platforms aligning two individual and well-established reactions, CO2 is first converted to methanol, followed by the conversion of the latter to hydrocarbons.185 Complex catalytic formulations involving Cu-based and/or other metal-based and metal oxide-based components have shown promising performance in methanol synthesis from CO2.186–189 Clear inspiration has been garnered from the typical Cu–ZnO–Al2O3 industrial catalysts for the synthesis of CH3OH from syngas,190 with a highly efficient Cu–ZnO–Al2O3–ZrO2 arising as a promising catalyst.191The conversion of methanol to hydrocarbons has been thoroughly investigated in the literature, with the C–C bond formation being demonstrated over zeolite or zeotype catalysts. The methanol-to-hydrocarbons (MTH) process can be additionally turned into the production of gasoline-rich (methanol-to-gasoline, MTG) or olefin-rich (methanol-to-olefins, MTO) product mixtures by the proper choice of catalyst and reaction conditions. Product selectivity is directly derived from the imposed shape-selectivity created inside the micropores of the selected crystalline structures. How the dimensionality and pore size of selected catalysts controls the product selectivity is a topic of heightened interest that is summarized elsewhere.192 In general, zeolite H-ZSM-5 (MFI topology) with a tri-dimensional interconnected channel system defined by 10-membered ring pore apertures has been utilized in the synthesis of hydrocarbons.193 When the MTO pathway is preferred, the silicoaluminophosphate SAPO-34 with narrow channels (8-membered ring) and wide cages is preferably employed.194
The expectedly effective catalytic conversion of CO2 into gasoline may be attained through a one-pass operation using a serially connected flow-type reactor.183 Such results suggest the promising potential for industrial application but inconveniently rely on consecutive process steps. Coupling both catalytic functionalities in a one-step process has emerged as an alternative that has been receiving increasing attention. In spite of the considerable efforts made in the development of composite catalysts, the results to date have been based on the somewhat disadvantageous design of complicated catalytic systems and remain rather unpromising. Compared with analogous technologies using syngas, the current state-of-the-art methanol-mediated t-CO2RR emphasizes the limiting occurrence of the RWGS reaction leading to undesirably high CO yields (up to 80%) with the prepared bifunctional catalysts. Major drawbacks in both MTG and MTO technologies such as high cost and fast catalyst deactivation are expected during a methanol-mediated t-CO2RR to hydrocarbons. In addition, the formed H2O molecules dramatically affect the stability and activity of the incorporated zeolites during the second step of the consecutive processes. Upcoming research works clearly demand strategies that involve more than the simple incorporation of optimized components for both desired reactions. The desired product selectivity is additionally expected to be considered in the rational design of these hybrid catalytic systems. The effect of the zeolite structure for HY (a tridimensional large pore zeolite with a 12-membered ring pore aperture) and H-ZSM-5 was assessed in Fe–Zn–M-based catalysts toward the maximization of the yield for isoalkane formation.195 The Fe–Zn–Zr/HY catalyst was confirmed to be efficient for the highly selective production of isobutane, whereas its H-ZMS-5 counterpart yielded enhanced C5+ hydrocarbon formation. The latter further unveiled a ratio of branched hydrocarbons over 90%, which is highly suitable for high-octane gasoline.
The production of olefins, which has achieved notable success in analogous technologies using syngas,150,196,197 has unlocked low yields at low overall CO2 conversion levels to date, while alkane formation following alkene hydrogenation has also been reported. Recently, the thermodynamic and kinetic coupling between the components of a ZnZrO/SAPO-34 tandem catalyst, fabricated with a ZnO–ZrO2 solid solution and a Zn-modified SAPO-34 zeolite, achieved a selectivity for lower olefins as high as 80–90% among hydrocarbons at 12.6% CO2 conversion, with only 3% CH4 being formed (Fig. 12).198 Methanol conversion on SAPO-34 or ZnZrO/SAPO-34 produced lower olefins with high selectivity (over 90%), corroborating the assumption that the zeotype phase was the catalyst for MTO conversion. The CO2RR technologies for producing short-chain olefins via methanol-mediated routes remain promising as compared with modified Fe-based FT platforms, being at least partly commercially established (MTO) and directing a more tuned and narrow distribution of the emerging short-chain olefins. In addition, the development of multifunctional catalysts incorporating adjacent active sites combining the fingerprints required over the involving catalytic steps is expected to significantly enhance the potential of these systems in the future.
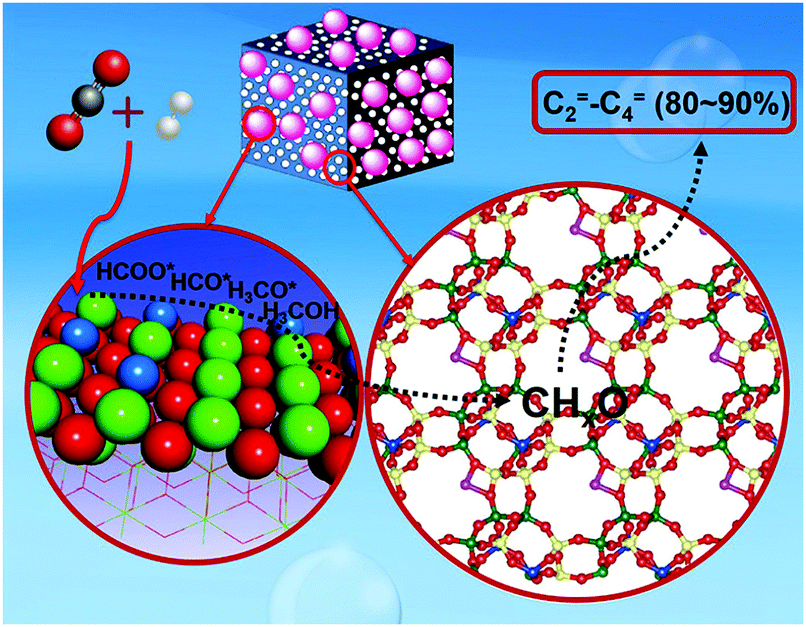 | ||
| Fig. 12 Proposed mechanism for the formation of olefins with high selectivity over a ZnZrO/SAPO-34 catalyst. Adapted with permission from ref. 198. Copyright 2017 American Chemical Society. | ||
In particular, a bifunctional catalytic system incorporating the partially reduced In2O3 and H-ZSM-5 zeolite has recently unveiled pioneering performances in the direct and highly selective production of fuels in a gasoline-relevant range, minimizing the formation of CO and undesirable side-products (Fig. 13).199 C5+ selectivity reached 78.6%, with only 1% for CH4 selectivity at a CO2 conversion of 13.1% under stable conditions over 150 hours. As discussed above, the generated shape selectivity within the pore architecture of the ZSM-5 catalyst facilitated the attained product selectivity. The CO2RR could be manipulated by controlling the surface structure of the In2O3 and the mass ratio between both catalytic phases. The proximity of the two components was additionally shown to play a crucial role in suppressing the undesired RWGS reaction and obtaining a high C5+ selectivity.
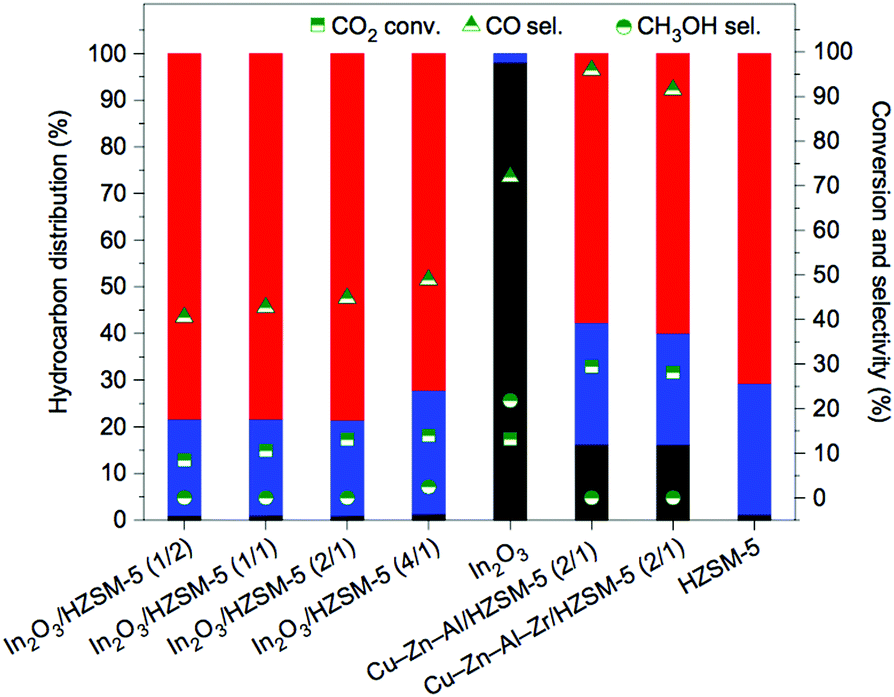 | ||
| Fig. 13 t-CO2RR performance over various bifunctional catalysts containing Cu-based components or In2O3 and HZSM-5 with different mass ratios, as shown in the parentheses, and the stand-alone In2O3 catalyst (reaction conditions, 340 °C, 3.0 MPa, 9000 mL h−1 g−1, H2/CO2/N2 = 73/24/3). Results over HZSM-5 obtained for the direct conversion of methanol (liquid CH3OH 0.855 mL h−1 g−1). Maximum hydrocarbon selectivity to C5+ (red), C2–C4 (blue), and CH4 (black) is maximized over In2O3/HZSM-5. Cu–Zn–Al-based and Cu–Zn–Al–Zr-based catalysts represent materials of choice in the industrial methanol synthesis from syngas and in CO2 hydrogenation, respectively. Reproduced with permission from ref. 199. Copyright 2017 Springer Nature. | ||
2.4. Stability issues
Along with the costs of implementation and production, the lifetime of a catalyst is vital in determining the technological practicality of the thresholds. In the t-CO2RR, metal stability against poisoning, sintering, and coke deposition are the main issues investigated when evaluating the catalytic decay of these platforms. Nonetheless, the stability of these systems further depends on the overall catalyst design (e.g., selected support, choice of the incorporated metal, and eventually added promoters), utilized feedstock (e.g., CO2 purity and partial pressure of the gas) and operating conditions applied during catalytic assessment (e.g., temperature, pressure, and gas hourly space velocity). The poisoning of metal-loaded catalysts derived from the presence of sulfur compounds in industrial feedstocks has prompted decades of research in the use of adsorbents for effective sulfur removal.200,201Large surface areas and the rational design of hierarchical supports have been pinpointed in most attempts to hamper the catalytic decay related to metal sintering and coke deposition. The morphology of the support has been additionally detailed to impact the morphology of the incorporated metal NPs,96 promote NP dispersion, determine the adsorption properties of the resulting catalyst, and to induce product selectivity.174 In methanol-mediated routes for long-chain hydrocarbon synthesis, zeolites and zeotypes with narrow pore size and constraint effects that induce shape-selective properties additionally serve as a bridge in the selective synthesis of the desired products.193 However, the induced confinement of these crystalline microporous aluminosilicates simultaneously prompts severe molecular diffusion limitations, leading to rapid deactivation and compromising the industrial practicality of the zeolite.194 In addition to the dramatic decrease of the full application of the zeolite, pore blockage undesirably affects the selectivity of the catalyst as the reaction is skewed to the surface of the material where shape-selectivity is absent. Reducing the zeolite crystal thickness has been, in this context, reported to be advantageous in reducing diffusion path lengths and remarkably hindering catalytic decay. Inter- or intra-crystalline mesopores have been generated in zeolites by the use of a hard template or a soft template, self-assembly of nanoslabs, packing of nanocrystalline zeolites, or post-synthetic demetallation treatments used to render internal active sites more accessible.202 Along the same line of thought, twenty years of active research on ordered mesoporous materials have found suitable applications in CO2RR toward the facile dispersion of metal NPs with hindered tendency toward deactivation. Larger surface areas and pore volumes, uniform pore distribution and enhanced pore interconnectivity of the resulting architectures have long been highlighted in a wide range of applications toward a better interaction between the metal–oxide and the support, resulting in high metal dispersion. Improved molecular diffusion, suppression of sintering and agglomeration of metal atoms, and hindered coking during the reaction even at high temperatures are to be expected.
Finally, the encapsulation of metallic NPs is an efficient strategy for hindering deactivation due to both metallic particle sintering and coke formation.203–205 Shape-controlled Pd NPs embedded in mesoporous silica shells showed no increase in particle size during the catalytic assessment and had greater stability toward sintering as compared with a Pd/SiO2 counterpart prepared by wetness impregnation.86 The above reports additionally highlight the advantage of encapsulating metal NPs as a valuable pathway to preserving superior catalytic performance. For instance, the activity of CO2 methanation could be significantly enhanced by encapsulating small-sized (111) faceted Ni particles in metal–organic frameworks at 300 °C with high thermal stability demonstrated up to 200 h.206 DFT calculations confirmed that the preserved plane had a lower potential energy barrier (10.0 kcal mol−1) for CO2 dissociation into adsorbed CO* and O* intermediate species when compared with the (200) facet (20.3 kcal mol−1).
The loss of metallic surface has been thoroughly assessed under CO2RR conditions, as it represents a crucial bottleneck in the application of promising catalytic systems. With stability being dependent on the choice of the incorporated metal NPs, Ru-based catalysts emerged as the most resistant to catalytic decay after being assessed over a wide temperature range. Ru NPs on TiO2 prepared by a polygonal barrel-sputtering method were stable at 80 and 140 °C for 24 h, and up to one week at 180 °C.73 TEM imaging revealed no change in the size or size distribution of these incorporated NPs as compared with the as-prepared counterparts. Over a γ-Al2O3 support, Farrauto demonstrated cyclability runs introducing pre-treatment steps under H2 prior to each CO2RR cycle, which could serve as a valuable industrial strategy toward the extended stability of the evaluated catalysts.207
Regarding the significantly less stable Ni-based systems, Mirodatos showed that sintering on a SiO2-supported catalyst can be ascribed to the formation and migration of highly mobile Ni subcarbonyl adspecies due to the interaction of Ni NPs with adsorbed CO*, prompting fast catalytic decay.208 The catalyst, initially presenting a monomodal Ni particle size distribution, evolved toward a bimodal system consisting of small spherical and large faceted crystals, with the selective development of (111) planes under reaction conditions. In contrast, the formation of nickel carbide was suggested to decrease the concentration of the mobile subcarbonyl adspecies. Manipulating the inherent interactions between the (111) faceted incorporated metal NPs and supports serves as a valuable strategy for enhancing the metallic dispersion and hindering catalytic decay due to particle sintering and coke formation. Ce0.72Zr0.28O2 mixed-oxide catalysts with high oxygen storage capacity, for instance, were able to notably enhance Ni dispersion (10 wt%) and further restrain the sintering of the metal, resulting in high stability on stream toward deactivation up to 150 h at 350 °C.209 Similarly, 5 wt% Ni/2 wt% ceria/Al2O3 was reportedly stable after 120 h at 300 °C. A remarkable stability with only a 7% decay after 252 h at 300 °C was reported with a supported Ni nanocatalyst fabricated by the in situ reduction of the NiAl-LDH precursor.210 The superior degree of Ni2+ dispersion in the LDH precursor could induce highly dispersed Ni NPs that are resistant to sintering, with the intrinsic topotactic phase transformation from the LDH matrix to the supported Ni NPs being proposed to ensure a strong anchoring effect.
In the desirable shift to long-chain hydrocarbons, although significant deactivation of Fe-based catalysts is mostly ascribed to the above-discussed issues related to poisoning from gradual coke deposition and obstruction of the active sites, the inherently formed water vapor has been shown to act as a poison in the t-CO2RR, re-oxidizing the catalyst surface and altering the resulting product selectivity.118,211 By removing or reducing these effects, the equilibrium at the Fe-based catalyst surface should favorably skew the production of CO or CH4 to desired intermediates such as olefins.212 An alternative approach is the utilization of in situ permselective silica membranes, as introduced in Section 2.5.213 Fe-Based catalysts undergo inherent deactivation due to the undesirable phase transformation into the stable but non-active carbide (Fe3C) formed by Fe5C2 carburization.214 The optimization of the design of the Fe catalyst and a better understanding of the observed phase transformation is once again highlighted (Fig. 11). It is important to highlight that the stability of the applied catalysts significantly depends on the operating conditions and the reactor design, as discussed in the following section.
2.5. Reactors and process considerations
The t-CO2RR technologies for either selective CH4 production or long-chain hydrocarbons have been designed for the conversion of synthesis gas, according to the obvious parallelism in the proposed reactors when the atmosphere is switched to CO2 as a feedstock. With the acquired knowledge on the catalytic chemistry of the above-discussed possible reaction pathways to convert CO2 and these relatively well-established conventional technologies as a guideline, reprocessing development faces only minor technological barriers requiring engineering solutions for reactor design, implementation, and operation. Playing a role as important as the rational synthesis of optimized catalytic architectures, the control of operating conditions and key parameters in the design of t-CO2RR reactors, i.e., temperature, pressure, and space velocity, have been additionally demonstrated to substantially impact the resulting activity, product selectivity, and stability.142,158 As expected, high reaction pressures, low-temperature operation favoring energy consumption, and high space velocities maximizing the reactor throughput are desired. The thorough energetic integration analysis is also of primary importance in the industrial prospect of thermochemically-driven catalytic platforms.With t-CO2RR being strongly exothermic, efficient heat management and heat removal are crucial requirements for facilitating stable hydrocarbon production to prevent the propensity of the reactor to overheat and lead to rapid catalyst deactivation by sintering and coking (mostly associated to CH4 cracking), and to permit and effective utilization of the released heat.207 Using a transient mathematical model, Simakov mapped a dynamic two-dimensional domain of the two crucial operating parameters, i.e. space velocity and cooling rate, to assess the effect of the selected operating conditions during CO2 methanation in a heat-exchanger type reactor.215 Scanning the reactor performance over extended ranges of space velocities and heat removal rates showed a non-trivial dynamically evolving map; however, the model suggested an enhancement of CH4 yield with controlled thermal management. Interestingly, the high pressures that are favorable for CH4 production and beneficial for practical applications were suggested to lead to a faster catalyst deactivation due to the higher partial pressure of CH4 causing susceptibility to cracking and leading to coke formation.
The emerging carbon deposition has been well documented to deactivate the catalyst by fouling the catalyst surface, blocking catalyst pores, and disintegrating the catalyst support. Heat-exchanger-type reactors are thus of crucial importance, not only toward desirable heat management but also for the energetic reutilization of the generated heat during the reaction. CO2 methanation is preferentially assessed in fixed-bed reactors and reports on microchannel and monolith reactors as design solutions have been valuable. However, directing the reutilization of CO2 to long-chain hydrocarbons has been thoroughly explored in either conventional fixed-bed, fluidized bed, or three-phase slurry configurations (Fig. 14).217 The parallelism found with the conventional FT fluidized bed or three-phase slurry reactors, extensively investigated in the past decades, underlines their numerous advantages (1) toward the effective removal of the generated heat, (2) as a bridge to higher hydrocarbon productivity due to the effective contact between the reacting phases with higher heat and mass transfer rate, and (3) with a facile continuous operation mode compared to the other reactor types.218,219 Over the years, fixed fluidized bed (FFB) reactors have also been preferable as compared to circulating fluidized bed (CFB) reactors. The former require notably smaller dimensions and subsequent lower construction costs up to 40%, offer a wider cooling section allowing the installation of more cooling coils, demand lower rates of removal and replacement with fresh catalyst, and require less maintenance as compared to CFB reactors, which require significant maintenance, particularly in the narrower sections where the very abrasive iron carbide is continuously circulated.216
 | ||
| Fig. 14 Schematic representations of (a) a multi-tubular fixed bed reactor and (b–d) three types of fluidized bed reactors: (b) circulating fluidized bed, (c) ebullated or fixed fluidized bed, and (d) slurry phase bubbling bed. Types (b) and (c) are two-phase systems. In type (d), CO2 and H2 pass through a liquid in which the solid catalyst particles are suspended. Adapted with permission from ref. 216. Copyright 2002 Elsevier B.V. | ||
In the design of t-CO2RR reactors and selected process configuration, the authors highlighted additional points of key relevance: (1) it is crucial to remove water during the catalytic evaluation of iron-based catalysts in modified FT systems, for which membrane reactors, allowing continuous water removal during the reaction, have been proposed to dramatically hinder the observed catalytic decay and improve the performance of the overall process. (2) Emerging process configurations may find higher suitability in the direct RWGS and modified FT synthesis in consecutive reactor volumes under optimized conditions for each reaction. (3) CH4, short-chain alkanes or alkenes, and longer-chain (C5+) hydrocarbons are preferentially recycled in the process in accordance with the desired product selectivity, whereas CO and CO2 are recycled to the RWGS unit (in the case of sequential process configuration).
2.6. Concluding remarks and analytical prospects
The chemical conversion of CO2 into value-added fuels with the assistance of H2 and utilizing inexpensive and stable catalysts is a promising response to the pernicious effects of CO2 accumulation in the atmosphere and to the foreseen shortage of fossil fuels on a global scale. The catalytic t-CO2RR has been known for several decades to directly produce hydrocarbons above methane in high conversion yields. While the production of long-chain hydrocarbons is economically preferable, to date, selectively reducing CO2 to synthetic natural gas has shown superior promise, notwithstanding the lower value of CH4, wider H2 consumption, lower energy per unit volume, and eventual storage difficulties.Ni and Co-based systems, and Fe-based catalytic systems have gained increasing attention due to their promising features regarding the CO2RR to produce CH4 and longer carbon-chains, respectively. The increasingly convergent application of Fe-based CO2RR catalysts in parallel with the conventional high-temperature Fischer–Tropsch platforms utilizing syngas has further highlighted the urgent need for the assessment of novel materials. In addition, the induced reconstruction of Fe-species, which is largely affected by the choice of the selected operating conditions, support and structural promoters, has reflected the lack of well-defined catalysts in which the spatial arrangement of interfaces has been determined; the difficulty in the manipulation of the desired mechanistic pathways is well-known. The broad distribution of the products in the modified FT synthesis directly implies their costly separation and the need for large capacity plants. For these reasons, t-CO2RR through the above-mentioned pathway may require further optimization in the rational design of these catalysts toward enhanced product selectivity.
Showcasing the extensive line of work regarding the t-CO2RR in the past decades, most of the catalysts reviewed herein have exposed the increasingly complex synthesis, with metal catalysts being incorporated on optimized supports and their often further decoration with multiple structural promoters. Most of these catalysts that are synthesized by co-precipitation, impregnation, or physical mixing exhibit large morphological variations in the spatial arrangements of the active sites and subsequent characterization uncertainties. On the one hand, the range of reported preparation methodologies increase the number of catalytic descriptors such as the surface area, particle size, and metal dispersion, leading to clear disparities in the attained catalytic performances. On the other hand, the lack of well-defined catalysts in which the formed interfaces are precisely controlled hinders the development of more efficient catalysts. The in-depth understanding of the reaction mechanism and identification of intermediates further represent a severe limitation in the development of highly efficient catalysts toward the control of the selectivity of each pathway. Notwithstanding the consensus for the presence of CO* as a reaction intermediate, the identification of other descriptors, e.g. oxygen adsorption energy, adsorption configurations of CO2 and key intermediates and activation barriers for key reaction steps, deserves greater focus in the future. It should be additionally noted that CO2 hydrogenation via a modified FT synthesis has been largely demonstrated to give rise to liquid fuels and short-chain olefins, depending on the catalyst and applied promoters, and selected operative conditions.
Along with the above listed practical drawbacks, one of the most defining industrial bottlenecks for the reutilization of CO2 is the need to obtain both CO2 and H2 in an economically favorable manner. In particular, renewable and relatively inexpensive CO2-free H2 production is a requirement for sustainability, and it remains a limitation for the large-scale practical implementation of the t-CO2RR. Although H2 may be obtained from renewable CO2-neutral energy sources (e.g. electrolysis, biomass, WGS, through the use of bacteria, or wind power), to date, 95% of the produced H2 is derived from hydrocarbon-based feedstocks, e.g. steam reforming of CH4, coal gasification and partial oxidation of light oil residues, leading to additional CO2 production.220–222 CO2-free H2 production on a large scale with significantly reduced associated costs plays a key role in the cost-competitive production of gasoline and light olefins. H2 synthesis by alternative (photo)electrocatalytic water splitting is highly regarded in this sense. Similarly, the above listed strategies for a CO2-free H2 production are predicted to follow new developments.
To offset the energetic demand of the reaction, proper energy integration is also required toward the commercialization of the progress outlined herein. Alternative strategies may additionally induce a decrease in the required thermoneutral voltage in the t-CO2RR. Recently, well-dispersed Ru-NPs on exfoliated LDHs have been proposed as highly active heterogeneous catalysts for CO2 methanation under light irradiation in a flow-type photoreactor.223 This is an example of new design ideas for catalysts functionalized by targeting activation to reactants, opening a door to potential industrial solar-to-chemical energy conversion.224
3. Electrochemical CO2 reduction reaction (e-CO2RR)
Over the past decade, a marked decline in renewable electricity prices induced by the steep development of novel energy platforms has extended the possibilities for decreasing the dependence on fossil fuels globally. Herein, we highlight the photovoltaic industry on the basis of the consistent improvement of solar cell efficiencies and the reduction of manufacturing costs.225 Compared with traditional chemical processes driven by fossil energy, electrochemical technologies are environmentally friendly, operate under relatively mild conditions, and can be coupled with renewable electricity sources at remote locations. Transforming CO2 into hydrocarbons is a valuable alternative for leveling the output from these intermittent renewable sources (e.g., wind and solar energy) that are strongly dependent on season and daylight, to serve previously established infrastructures on a global scale.226In principle, CO2 can be electrochemically reduced to give a wide range of products at the surface of solid electrodes. In a typical CO2RR cell, water acts as the source of protons and CO2 is reduced to the target products, e.g., CO, formate, methanol, and methane at the cathode, while oxygen is evolved at the anode. A cost-effective e-CO2RR requires electrocatalysts acting as working electrodes that can operate at low overpotentials and generate desirable products at high current densities over long periods of time (enhanced stability) without the formation of by-products (enhanced selectivity). In practical processes, however, the CO2RR is undesirably skewed to more negative voltage ranges, with the emerging overpotentials being largely attributed to (1) high activation energies needed for the electron transfer during CO2 activation on the surface of the catalyst, (2) ohmic losses related to the electrolyte and electrode conductivity, and (3) observed mass transport limitations. Similarly, high Faradaic efficiencies (FE), defined as the ratio of charges transferred to a certain product relative to the number of total charges passed through the circuit, are a key prerequisites in the synthesis of attractive electrocatalysts.
Electrocatalytically, methane was first reported with the co-formation of CO and CH3OH at significantly low partial currents in the presence of Ru.227 Only in 1985 did the pioneer works of Hori and Suzuki disclose the formation of CH4 and C2H4 as the main products of e-CO2RR on a Cu electrode.228,229 Fast-forward to three decades from the seminal works of Hori, and Cu has remained the sole metal electrocatalyst capable of binding and converting the now well-established adsorbed CO (CO*) intermediate (cf. Section 3.1) to more reduced hydrocarbon products at fairly high efficiencies. To date, the synthesis of hydrocarbons requires the highest overpotentials and lowest FE (Fig. 15).230 Hydrocarbon or alcohol formation should be thermodynamically more favorable than the production of CO, formic acid, formaldehyde or H2.231 Experimentally, slow kinetics are observed as multiple electron transfer (beyond n = 2) is required in both cases (e.g. for CH4, n = 8).232 In addition, with increasingly negative applied potentials the parasitic H2 evolution reaction (HER) becomes more significant, by consuming protons and electrons required in the e-CO2RR.
 | ||
| Fig. 15 Performance map for the e-CO2RR toward commonly reported products in terms of the maximum attained FE and corresponding overpotential for each reported catalyst. A cluster analysis of the points (by kernel density estimation, KDE), demonstrated by the corresponding background color intensity, unequivocally revealed the performance regions associated with each product. Bibliographic references and relevant data for each report were provided by the authors. Adapted with permission from ref. 230. Copyright 2017 American Chemical Society. | ||
3.1. Mechanistic difficulties
The mechanism for the e-CO2RR is an area of intense discord.233 In the presence of heterogeneous electrocatalysts, the reaction takes place at the interface established with a usually CO2-saturated aqueous electrolyte. The rate-limiting chemical adsorption of CO2 is followed by electron transfer and/or proton migration to cleave C–O and form C–H bonds, configuration rearrangement of involved intermediates, and product desorption and diffusion into the electrolyte. In the multi-step e-CO2RR to CH4 involving an eight-electron transfer and seven reaction intermediates, in-depth mechanistic studies demand an understanding of the problem in a seven-dimensional space of adsorbate–surface interaction energies.234 The seminal works from Hori first claimed the intermediate formation of CO,235–237 with adsorbed CO (CO*) being later confirmed by chronopotentiometric evaluation.238,239 To date, reducing CO and CO2 has revealed similar product distribution and potential dependence, thereby reinforcing that the rate-determining step occurs after CO formation.240,241 In light of the complexity of the proposed mechanisms, studies based on other identified representative intermediate species e.g., HCOOH, HCHO, MeOH, and CH3CHO, have become a noteworthy strategy.242Throughout the years, comparative studies have ascribed the activity and selectivity of efficient CO2RR electrocatalysts to the resulting binding energy of the involved intermediates with the selected catalyst.234,243–245 Within the extensive line of examined materials, e.g. ionic liquids, proteins, organometallic complexes, metals, transition-metal oxides, transition metal chalcogenides, and carbon-based materials (N-doped nanostructures in the form of few-layered graphene, carbon nanotubes and carbon fibers), the large majority has been found to be capable of solely yielding CO (n = 2).246–252 To date, the presence of metals is almost universally required for the e-CO2RR to occur to a significant extent,230 with, Al, Si, V, Cr, Mn, Co, Zr, Nb, Mo, Ru, Rh, Hf, Ta, W, Re, and Ir being mostly inactive at −2.2 VSCE in 0.05 M KHCO3.253 Seminal works over metal electrodes (Hg, Au, Pb, Zn, Cd, Sn, and In) with no investigation of emerging gaseous products reported the nearly exclusive production of formate in solution.254,255
The singularity of Cu has been established to arise from an ideal binding energy to the formed intermediates allowing the subsequent reduction of CO2 to CO and to further desired hydrocarbons (Fig. 16). With metals like Sn, Hg, Pb, In, Cd, and Tl, the formed HCOO−, following the activation of CO2 with the transfer of one electron and one proton, desorbs easily. With Au, Ag, and Zn, the presence of a strongly bound HCOO− permits the breaking of the CO bond, but the resulting weakly adsorbed CO is promptly evolved as a reaction product.244 In particular, Au features the highest current density and the lowest onset potential for e-CO2RR. Metals of this group were shown to produce methanol (Au) or both methane and methanol (Ag and Zn) at more negative potentials,245 which further pinpoints the importance of the oxophilicity of the surface in the selectivity between hydrocarbons and oxygenates, with Au being, in this case, the least oxophilic metal of the group. Metals that bind CO* strongly are poisoned by CO or other intermediates, with H2 being evolved as the main product from the competing water reduction. With a high CO* coverage, the reduction of CO* is rate-determining and rather insensitive to the binding strength. For these reasons, metals such as Pt, Ti, Ni and Fe show similar onset potentials as compared to Cu for the limited CH4 production.245
 | ||
| Fig. 16 (a) Volcano plot of partial current density for the e-CO2RR assessed at −0.8 V vs. CO binding strength. (b) Distinct onset potentials plotted vs. CO binding energy for the overall e-CO2RR, and for methane/methanol production. Dashed lines serve as a guide. Reprinted with permission from ref. 245. Copyright 2014 American Chemical Society. | ||
The complexity behind the proposed reaction mechanisms is largely ascribed to the dependence of the catalytic phase and operating conditions.256 Mechanistic difficulties are further aggravated by oxygenate species sharing commonalities with the pathways toward hydrocarbon production, for which the oxophilicity of the surface plays a central role.245 Over the Cu electrode, extending the chain-length could not be achieved merely based on the selected potential range (Fig. 17). The observed product distributions evidenced an increase in the FE to C2H4 and ethanol up to a certain potential range, with the latter seemingly decreasing at more negative potentials. Interestingly, methane and ethylene unveiled different onset potentials, largely suggesting that the formation of C2H4 could surprisingly be attained in less negative ranges as compared to CH4. For CH4 and C2H4, product selectivity has been ascribed to distinct rate determining steps and coverage effects.232,241 In Fig. 18, the overall mechanistic complexity of these products is depicted as intentionally featuring proposed pathways in the synthesis of oxygenates, CO, and formate. Over the Cu cathode, following the rate-determining CO2 activation step and the formation of the generally accepted CO* intermediate, distinct facet-dependent C1 and C2-based mechanisms have been reported (cf. Section 3.2.1);257 on Cu(111) the reduction of CO* to CH4 and C2H4 takes place simultaneously, whereas on Cu(100) a second pathway directs C2H4 at lower potentials without CH4 generation.258 These findings were corroborated by DFT calculations demonstrating that dimerization should have a lower activation barrier on Cu(100) than Cu(111), and that the divergent mechanistic pathways are in fact the reason for the distinct onset potentials found for CH4 and C2H4 in Fig. 17.259 On Cu(111), adsorbed CO species have been proposed to have subsequent proton-electron incorporation to give CHO*, H2CO* and OCH3*.260 With each electrochemical step involving the transfer of a proton from the solution to a species adsorbed on the surface, the CH4 partial current has been shown to be proportional to proton activity.261 At very low and high pH, CH4 formation is mirrored by the HER.262 Herein, Fig. 18 additionally pinpoints the controversy found in the literature reflecting the proposed mechanistic pathways involving either the generation of CHO* or COH* species (Fig. 19).263–265
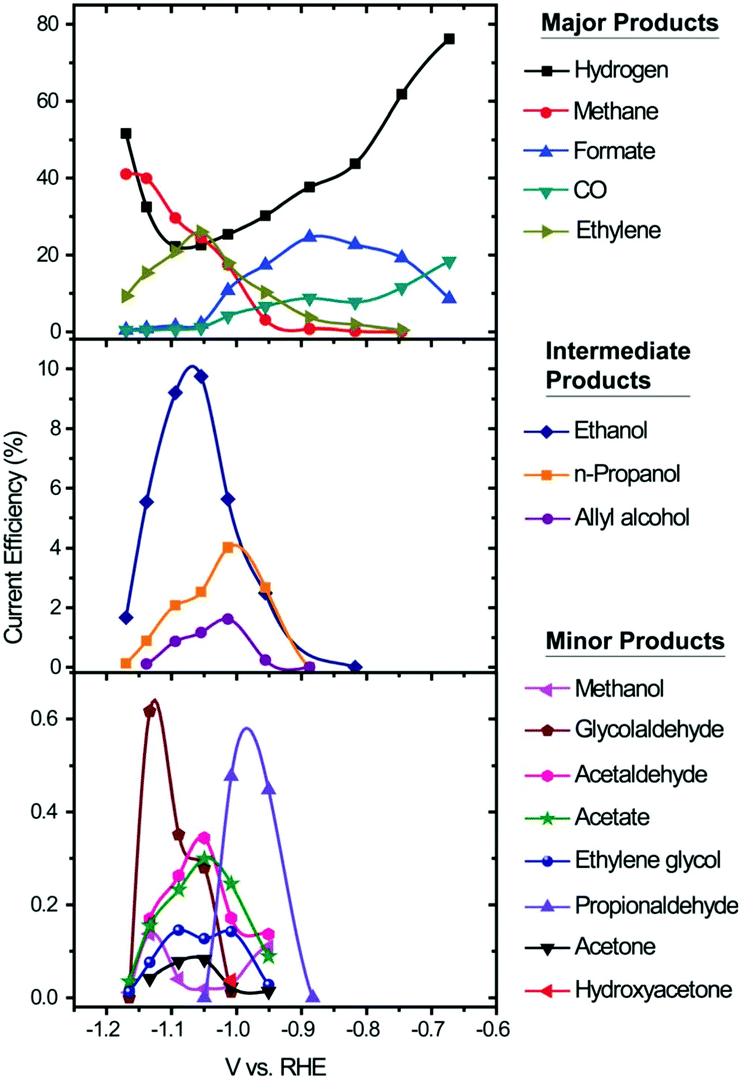 | ||
| Fig. 17 Plots of the current efficiency of emerging e-CO2RR products over the Cu electrode as a function of potential. Products were divided into three panels in which major, intermediate, and minor products have been grouped. The data reported by Jaramillo reflected previous reports on the Cu electrode being extended to a wider overpotential range and therefore shedding light on the current efficiency of several new products detected in this contribution for the first time. Reprinted with permission from ref. 232. Copyright The Royal Society of Chemistry 2012. | ||
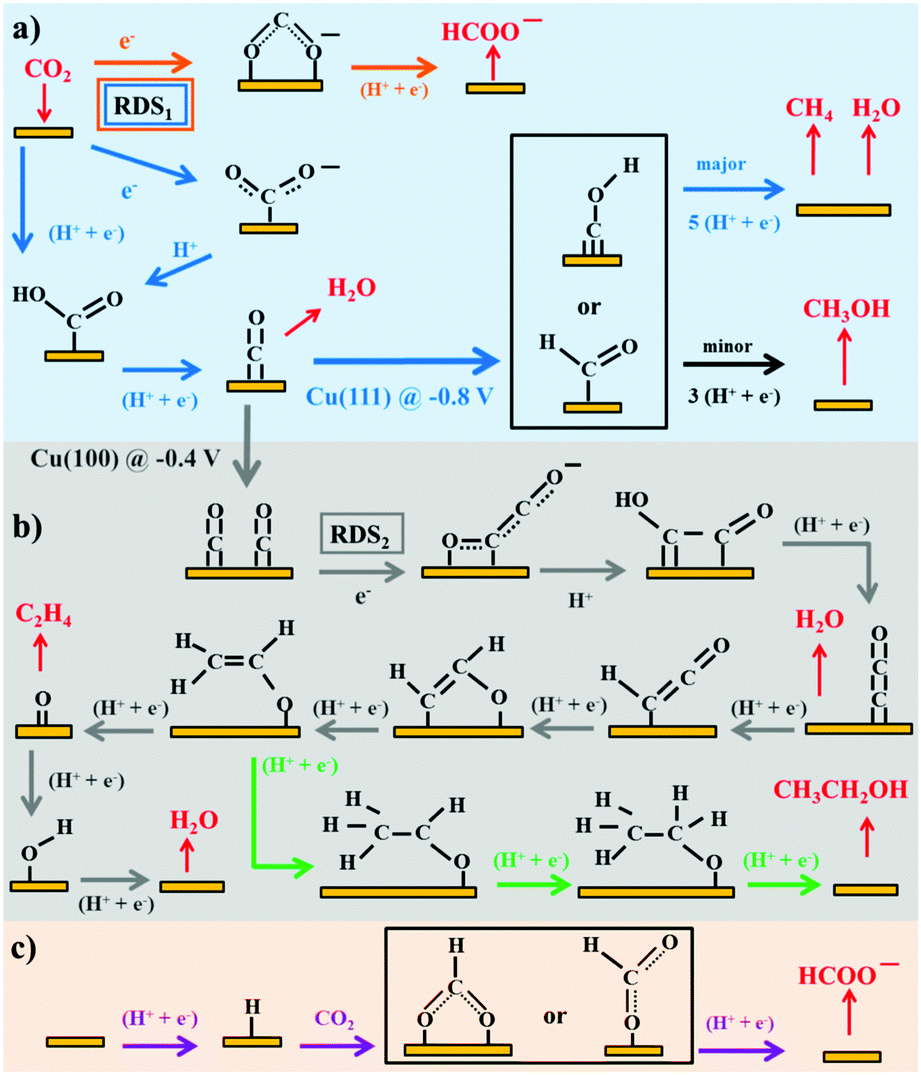 | ||
| Fig. 18 Possible reaction pathways for the e-CO2RR leading to the synthesis of hydrocarbons (CH4 and C2H4) along with CO, formate (orange and purple arrows), and oxygenates (black and green arrows). (a) and (b) are mechanistic pathways toward C1 and C2 species, respectively, while (c) represents the pathway for CO2 insertion into a metal–H bond yielding formate. Species in black are adsorbates and species in red are reactants or products in solution. RDS are rate-determining steps, accounting for the activation of CO2 and the dimerization reaction between CO* adsorbed species. Potentials (VRHE) for Cu(111) and Cu(100) are in accordance with experimental observations. Reprinted with permission from ref. 256. Copyright 2015 American Chemical Society. | ||
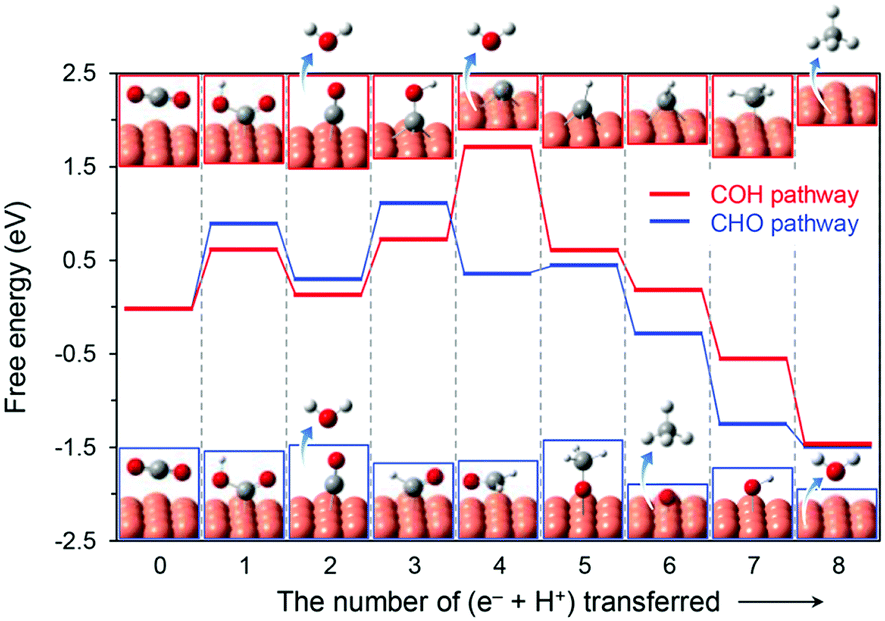 | ||
| Fig. 19 Multi-step conversion of CO2 to CH4, indicating the calculated free energies (at zero external bias) for alternative pathways considering the formation of CHO* (blue) and COH* (red) intermediates.264,265 Molecular species at each step are identified. Reprinted with permission from ref. 263. Copyright 2017 American Chemical Society. | ||
On Cu(100), the proposed mechanisms suggest OCCO* formation by CO* dimerization under relatively low overpotential, which represents a more favorable pathway compared to the rate-determining proton-transfer-dependent CO* hydrogenation. C2H4 is preferentially formed in basic electrolytes, while in acidic or neutral solutions CH4 formation is preferred.262 The rate of CO* dimerization toward the selective formation of C2H4 has been further suggested to benefit from high CO* surface coverage and tensile strain.266,267 For the formation of C2H4 on Cu(100), the formed dimer can be consecutively hydrogenated to OCCHO*, OHCCHO*, and OH2CCH2OH*. The OH2CCH2OH* intermediate can form HOH2CCH2OH* by direct hydrogenation and H2CCH2OH* by breaking the C–O bond, which might be a parallel pathway in the reduction mechanism of the CO dimer on Cu(100). C2H4 is finally formed by dehydroxylation of the HOH2CCH2OH* and H2CCH2OH* intermediates.
3.2. The singularity of Cu: modification of metal electrodes
Since the initial claims by Hori, the extensive literature on the aqueous e-CO2RR to hydrocarbons on Cu has conspicuously unveiled Faradaic yields above 50% at reasonable current densities (ca. −5 mA cm−2). These hydrocarbons are produced at prohibitively high overpotentials (nearly 1 V), reflecting high efficiency losses on the Cu electrode. Fast catalytic deactivation and low product selectivity accompanied by the formation of oxygenates have reflected additional bottlenecks for practical use. The extensive research to enhance the catalyst performance of Cu and its derivatives, e.g. studies on the morphology and surface oxidation state, is reviewed below and conveniently summarized in Table 2. Works on Cu overlayers over a substrate metal of choice are suggested for revision268–270 and not detailed herein.| Catalyst | Electrolyte | pH | Potential (VRHE) | Faradaic efficiency (%) | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| C2H4 | CH4 | Others | ||||||
| a Cu-Based electrodes exhibited remarkable stability levels up to 40 h. b For the plasma-activated Cu, values refer to collected Faradaic selectivities instead of Faradaic efficiencies. | ||||||||
| a | Cu foil | 0.1 M KHCO3 | 6.8 | −1.1 | 20 | 57 | CO (5), H2 (20) | 279 |
| Polycrystalline Cu | 0.1 M KHCO3 | 6.8 | −1.01 | 30.1 | 29.4 | C2+ (3.0) | 238 | |
| CO (2) | ||||||||
| Cu foil | 0.1 M KCl | 5.9 | −1.05 | 47.8 | 11.5 | C3H7OH (3.6), C2H5OH (21.9) | 237 | |
| CO (2.5), H2 (5.9) | ||||||||
| b | Cu(111) | 0.1 M KHCO3 | 6.8 | −1.15 | 8.3 | 46.3 | C2H5OH (2.6) | 271 |
| HCOOH (11.5), CO (6.4), H2 (16.3) | ||||||||
| Cu(100) | 0.1 M KHCO3 | 6.8 | −1.00 | 40.4 | 30.4 | C3H7OH (1.5), C2H5OH (9.7) | 271 | |
| HCOOH (3.0), CO (0.9), H2 (6.8) | ||||||||
| c | 5-fold twinned Cu NW | 0.1 M KHCO3 | 6.8 | −1.25 | 2 | 55 | C2H5OH (3) | 289 |
| HCOO− (3), CO (1) | ||||||||
| Cu NP (2 to 15 nm) | 0.1 M KHCO3 | 6.8 | −1.1 | 3 | 12 | CO (22), H2 (65) | 279 | |
| Cu nanoflower | 0.1 M KHCO3 | 6.8 | −1.6 | 10 | 5 | HCOOH (50) | 278 | |
| 8.1 μm-length Cu NW arrays | 0.1 M KHCO3 | 6.8 | −1.1 | 17.4 | N.D. | C3H7OH (7.8) | 288 | |
| C2H5OH (3.8), C2H6 (2.4) | ||||||||
| HCOOH (17.5), CO (7.6), H2 (44.2) | ||||||||
| Cu/p-NG | 0.5 M KHCO3 | 7.3 | −0.9 | 19 | 0.9 | C2H6 (0.6) | 284 | |
| HCOO− (3.8) | ||||||||
| Cu NP ensembles | 0.1 M KHCO3 | 6.8 | −0.81 | 27 | 0.4 | C3H7OH (5.9), C2H5OH (13.3), C2H6 (0.3) | 287 | |
| HCOO− (8.5), CO (6.6), H2 (35.7) | ||||||||
| Prism-shaped Cu nanocatalyst | 0.1 M KHCO3 | 6.8 | −1.1 | 27.8 | 14 | C3H7OH (4), C2H5OH (3) | 291 | |
| HCOOH (14), CO (1), H2 (33) | ||||||||
| Cu nanocubes (44 nm) | 0.1 M KHCO3 | 6.8 | −1.1 | 41 | 20.2 | C2H6 (0.3) | 275 | |
| HCOO− (4), H2 (60) | ||||||||
| 40 wt% Cu/VC | 0.1 M KHCO3 | 6.8 | −1.39 | 42 | 17 | H2 (17) | 283 | |
| d | Cu foam | 0.5 M NaHCO3 | 7.2 | −0.7 | 14 | N.D. | C2H6 (37) | 292 |
| CO (23), H2 (12) | ||||||||
| Cu mesocrystals | 0.1 M KHCO3 | 6.8 | −0.99 | 27.2 | 1.47 | C2H6 (0.3) | 276 | |
| HCOO− (4), H2 (60) | ||||||||
| Cu NP-covered surface | 0.1 M KClO4 | 6 ± 0.5 | −1.1 | 36 | 1 | C2H6 (1) | 281 | |
| CO (34), H2 (28) | ||||||||
| e | PdCl2/Cu2O-derived Cu | 0.1 M KHCO3 | 6.8 | −1.0 | 3.4 | N.D. | C3H7OH (5), C2H5OH (11), C2H6 (30.1) | 342 |
| H2 (16) | ||||||||
| 8.8 μm Cu2O film | 0.1 M KHCO3 | 6.8 | −0.99 | 21.6 | 0.2 | C2H5OH (5.1), C2H6 (0.2) | 308 | |
| CO (0.5), H2 (67.7) | ||||||||
| Cu2O-Derived Cu | 0.5 M KHCO3 | 7.5 | −1.25 | 22 | 2.5 | CO (4) | 307 | |
| Cu2O-Derived Cu | 0.1 M KHCO3 | 6.8 | −0.98 | 32 | N.D. | C3H7OH (5), C2H5OH (8) | 316 | |
| H2 (34) | ||||||||
| Cu2O-Derived Cu | 0.1 M KHCO3 | 6.8 | −1.0 | 32.1 | N.D. | C3H7OH (4), C2H5OH (16.4) | 342 | |
| H2 (34) | ||||||||
| Cu2O-Derived Cu | 0.1 M KHCO3 | 6.8 | −0.98 | 32.4 | <2 | C3H7OH (5), C2H5OH (8) | 266 | |
| HCOO− (3), H2 (35) | ||||||||
| Anodized Cua | 0.1 M KHCO3 | 6.8 | −1.08 | 38.1 | 1.3 | H2 (36.6) | 343 | |
| Electro-redeposited Cu | 0.1 M KHCO 3 | 6.8 | −1.2 | 39.04 | 0.03 | C 2 H 5 OH (7.8) | 320 | |
| HCOO − (16.30), CO (1.16), H 2 (31.56) | ||||||||
| Plasma-activated Cu nanocube | 0.1 M KHCO3 | 6.8 | −1.0 | 45 | N.D. | C3H7OH (9), C2H5OH (22) | 323 | |
| CO (2) | ||||||||
| Boron-doped Cu | 0.1 M KHCO 3 | 6.8 | −1.1 | 52 | 0.08 | C 2 H 5 OH (27), H 2 (20) | 321 | |
| Nano dendritic Cu catalysts | 0.1 M KBr/Br 2 | 2.5 | −1.30 | 57 | 9 | CO (8), H 2 (9) | 322 | |
| Plasma-activated Cu | 0.1 M KHCO 3 | 6.8 | −0.9 | 60 | 2 | HCOO − (2), CO (2) | 309 | |
| f | Chloride-induced Cu2O–Cu | 0.1 M KCl | N.A. | −1.6 | 22 | 1.4 | C4H10 (0.9) | 325 |
| C3H6 (0.9), C3H8 (1), C3H7OH (8.7) | ||||||||
| C2H5OH (24), C2H6 (1), CH3OH (3) | ||||||||
| HCOO− (3) | ||||||||
| Cu foil | 0.1 M KHCO3 + 0.3 M KI | 6.8 | −1.0 | 58 | 22 | HCOO− (6), CO (<1), H2 (12) | 326 | |
| g | Au3Cu alloy nanocrystals | 0.1 M KH2PO4/K2HPO4 | 7.0 | −0.98 | 7 | 36 | HCOO− (9), CO (27), H2 (21) | 335 |
Finding a surface that favors C–C coupling pathways toward C2H4 formation not only underlines the economic interest of ethylene over methane, but it is also of importance to unlock the pathways to desired multicarbon products. Works focusing on the nano-structuralization of Cu have disclosed enhanced performance on single crystal Cu(100) surfaces and even more on cleaved Cu(100) substrates with high-indexed planes.271 Cu nanocubes easily synthesized in situ and predominantly exposing (100) facets were claimed to be among the most selective for C2H4 over CH4.274 Onset potentials for C2H4 production in 0.1 M KHCO3 over these Cu nanocubes, Cu(100), Cu(Poly), Cu(211) and Cu(111) decreased in the order −0.60 > −0.73 > −0.74 > −0.79 > −0.96 VRHE, respectively. Analogous results suggested a key role played by the edge sites of these nanocubes.275,276 When coupled with density functional theory (DFT) calculations, the approach has further shed light on the determination of the energetics of the initial C–C coupling steps on varying Cu facets.277
These findings have stimulated the rational design of creative Cu-based nanostructures as a strategic path to enhance the catalytic activity and attain desired product selectivity.278 CO2RR on Cu NPs was shown to depend on the particle size, the population of low-coordinated surface sites, and the chemisorption strength of intermediate and hydrocarbon species.279,280 In agreement, several studies have been reported on the abundance of weakly coordinated surface sites on Cu, e.g. sputtering,281 electropolishing,281 electrodeposition,282 and anodization.282 Baturina claimed that over Cu NPs supported on carbon (carbon black, single-wall carbon nanotubes, and Ketjen Black) the ratio of C2H4 to CH4 could be enhanced by decreasing particle sizes, which was also ascribed to an increase in the fraction of under-coordinated sites, e.g. corners, edges, and defects.283 Monodisperse Cu NPs assembled on a pyridinic-N rich graphene (p-NG) support showed mass- and particle size-dependent catalysis for the selective e-CO2RR to C2H4 through C–C coupling over CH4, with corresponding FE of 19 and 0.9% at −0.9 VRHE, respectively.284 Despite a still important formate formation (FE of 3.6%), the localized electron pair of the pyridinic-N with stronger Lewis basicity was claimed to facilitate the rate-determining formation of the CO2˙− anion radical, and its protonation to COOH*.
Corroborated by reaction-diffusion modeling, additional reports on Cu NPs revealed that mesoscale phenomena such as interparticle reactant diffusion and readsorption of intermediates could be expected to play a defining role in product selectivity.285 Agglomerates of ca. 15 nm sized Cu nanocrystals showed the promising formation of propanol at current densities ca. 25-fold larger than those found on Cu0 NPs at the same applied potential.286 Alternatively, ensembles of densely packed Cu NPs were shown to undergo structural transformations during electrolysis into electrocatalytically active cube-like particles inter-mixed with smaller nanoparticles (Fig. 20).287 Enhanced C2H4 formation along with ethanol and n-propanol was found at an onset potential at −0.53 VRHE, with a FE to C2–C3 species of ca. 50% at −0.75 VRHE. Although the observed in situ structural evolution of Cu-based electrocatalysts is thoroughly discussed in the following subsections, a greater understanding of how NPs and the atoms within evolve under electrically biased and chemically relevant conditions is necessary. Nonetheless, using NPs as precursors to an active architecture is presumed to serve as a valuable pathway to extend the catalyst library in future exploratory works.
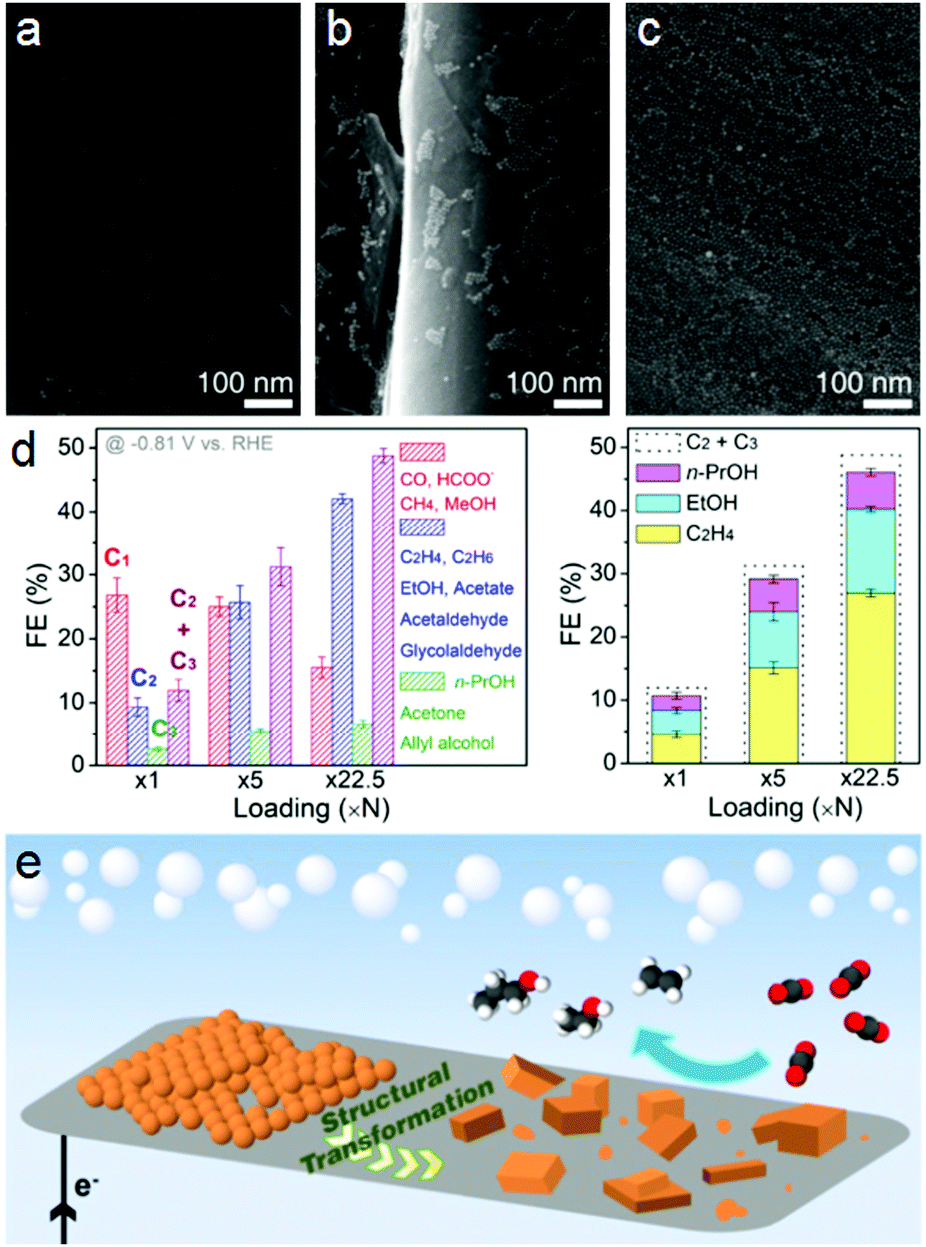 | ||
| Fig. 20 Representative SEM images of Cu NPs loaded on carbon-paper support at corresponding (a) ×1 loading, (b) ×5 loading, and (c) ×22.5 loadings. (d) FE for C1, C2, and C3 products, and detailed values for ethylene, ethanol, and n-propanol depending on the loading of Cu NPs in each sample. These measurements were carried out at −0.81 VRHE, using CO2-saturated 0.1 M KHCO3. (e) Schematic representation of the proposed transformation process of Cu NP-ensembles into an active catalyst for C2–C3 formation.287 | ||
Over Cu nanowire array electrodes, the hydrocarbon selectivity at a fixed potential could be tuned by systematically altering the Cu nanowire length and density.288 Recently, 5-fold twinned ∼20 nm-thin Cu nanowires were found to exhibit high CH4 selectivity over other carbon products, reaching 55% FE at −1.25 VRHE.289 Additional wrapping with graphene oxide (GO) preserved both the morphology and reaction selectivity of these nanostructures. These findings further agree with the enhanced stability and facile electron transfer reported elsewhere between rGO and Cu NPs, believed to increase localized electron concentrations.290
Recently, highly defective prism-shaped Cu catalysts prepared using a one-step electrodeposition method showed FE to C2H4 up to 27.8% for 12 h.291 The improved activity for C2H4 production was reflected in a 4-fold higher partial current density (−11.8 mA cm−2), as compared to that of the planar Cu sample (−2.8 mA cm−2). The presence of a significant fraction of defect sites on the prism Cu surface plays a role in improving C2H4 production. The report is an example of assessing the impact of surface roughness as a strategic pathway to enhance the number of active sites reflected in recent years. For instance, Broekmann designed mesoporous Cu foams with FE to C2 reaching 55%, by a template-assisted electrodeposition process occurring during selected HER-conditions.292 Highest efficiencies were reached for C2H6 at −0.7 VRHE (37%) and C2H4 at −0.8 VRHE (20%). The synthesis of Cu foams emphasized the importance of high surface roughness, hierarchical porosity, and the confinement of reactive species toward an increase of the hydrocarbon chain length, but it remains a challenge to assess the exact role of morphology taking into account the high variability in size, tortuosity, and crystal structure of the nanofabricated electrodes explored to date.292,293 In addition and as detailed in the following sections, the morphology can further affect both local pH and flow field, accelerating the C–C coupling reaction and prolonging retention times.294
It is of current general consensus that surface reconstruction during e-CO2RR is a key factor in the activity and selectivity of Cu electrodes, despite attempts to optimize the rational design of these catalysts. With a weak cohesive energy, simple polycrystalline Cu held at a fixed negative potential (−0.9 VSHE) in 0.1 M KOH can undergo stepwise surface reconstruction, first to Cu(111), and then to Cu(100), highlighting the dynamic nature of the electrocatalytic surfaces.295 In addition to physical conditions including temperature, pressure, and electrical potential, reversible surface reconstruction induced by adsorbates and reactants have been proposed. Reflecting the difficulty in witnessing the said phenomenon by ex situ methods, Waegele has presented, for the first time, clear evidence of the reversible reconstruction of Cu surfaces induced by the CO* intermediate.296 Using attenuated total internal reflection infrared and surface-enhanced Raman spectroscopies, the reversible formation of nanoscale metal clusters on the electrode was revealed by the appearance of a new C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O absorption band characteristic of CO bound to undercoordinated copper atoms and by the strong enhancement of the surface-enhanced Raman effect.
O absorption band characteristic of CO bound to undercoordinated copper atoms and by the strong enhancement of the surface-enhanced Raman effect.
Excellent results have been reported in the application of metal oxides with tunable layer thickness in the CO2RR.298 Copper oxides, in particular, have been the object of numerous studies since the seminal work of Frese.282,299,300 In principle, however, CH4 is thermodynamically more favorable on a pure Cu surface, whereas CH3OH is preferred on Cu2O or partially oxidized Cu electrodes due to the reverse free-energy distribution.299 In fact, by introducing a moderate amount of oxygen (either oxidized surface or hydroxyl species), the reduction product has been shown to switch from CH4 to CH3OH over Cu-based electrodes.301 It was in this sense unexpected when in recent years highly nanostructured surface electrocatalysts derived from the reduction of copper oxides showed unprecedented energy efficiency toward ≥C2 species. Whether the oxide layer plays a role in the mechanism behind the reactivity trends, or whether it only acts as a promoter of the resulting metal-based architectures has remained under intense discussion.302,303 Importantly, these findings are notably derived from the stability of the formed Cu oxide surfaces.304 Kanan suggested that Cu2O layers formed by annealing and subsequent reduction to Cu particles during e-CO2RR were able to efficiently catalyze the CO2 reduction process at comparatively lower overpotentials.305 The team reported the notable suppression of CH4 in favor of C2H4, largely corroborated by distinct reports on C2H4 to CH4 ratios up to ca. 100 over electrodeposited Cu2O.306–308
In line with the discussion in the previous section, the impact of the resulting density of steps and edges of these oxide-derived nanostructures is anticipated.308 Highly roughened surfaces with low-coordinated sites have been proposed to bind oxygen more strongly, helping to stabilize oxides during the reaction, with the highly porous architecture serving as a favorable oxygen reservoir.309 In addition, it is expected that during the catalytic reaction on these rough surfaces, there is a significant rise in the local pH, suppressing the pH-dependent CO* protonation that leads to methane formation. The pH-independent pathway via CO dimerization is, however, not affected, resulting in high ethylene selectivities (cf. Section 3.1).309 Similarly, reported disparate proton coupling requirements for CO and H2 production have established a mechanistic basis for the CO2RR selectivity toward CO.310 The effect of the electrolyte concentration and the importance of the local pH are, in fact, believed to be a strategy of interest in upcoming research works (cf. Section 3.5).311
With e-CO2RR being sensitive to electrolyte polarization, which causes gradients in pH and the CO2 concentration near the cathode surface and therefore impacts the intrinsic kinetics of the reduction reaction, it is therefore desirable to measure the concentration of reaction-relevant species in the immediate vicinity of the cathode. Whereas conventional analytical methods only sample products from the bulk electrolyte, Bell and colleagues proposed the use of differential electrochemical mass spectrometry (DEMS) to measure the composition, i.e. CO2 and reaction products of the local reaction environment.312 This capability could be achieved by strategically coating the electrocatalyst directly onto the pervaporation membrane used to transfer volatile species into the mass spectrometer, enabling in this sense species to be directly sampled from the electrode–electrolyte interface during both e-CO2RR assessments over Ag and Cu.
However, as pointed out by Cuenya and colleagues, observed lower onset potentials for C2H4 production could not be solely attributed to a local pH effect.309 Grain boundaries in oxide-derived Cu have been suggested to support unique surface sites, which could show enhanced activity for e-CO2RR. In line with their results in the electroreduction of CO, Kanan believed that engineering the grain boundaries by altering the oxide reduction method or by using an alternative preparation of nanocrystalline materials could yield catalysts with higher activity and selectivity for long-chain products.313,314 Aggregates of smaller particles were believed to give rise to a higher surface population of catalytically active sites situated on grain boundaries, defects and particle–particle junctions. These findings agreed with the presence of strongly chemisorbed CO* on sites supported by grain boundaries, uncovered by Verdaguer-Cassadevall.314 Linear correlations found between the surface area-normalized activity for CO2RR to CO and the grain boundaries density on Au electrodes significantly add to the hypothesis.315 With analogous concluding remarks, Yeo claimed that with oxide-derived Cu NPs, the crystallite size decreased from 41 to 18 nm, and the FE of C2H4 formation could be enhanced from 10 to 43%.316
The nature of the remaining Cuδ+ active catalytic sites has been thought to play a key role in the resulting catalytic performance, as recently demonstrated over oxidized Cu electrocatalysts prepared through facile and tunable plasma treatments.309 The presence of these Cuδ+ species, rather stable under reaction conditions, was demonstrated through operando X-ray absorption spectroscopy and cross-sectional scanning transmission electron microscopy. The exemplary claims by Cuenya and colleagues, not only emphasized the importance of characterization to unlock unanswered mechanistic details but also attained an outstanding Faradaic selectivity (defined as the ratio between the target product amount and the total detected products amount) to C2H4, up to 60%. The result linked in analogous reports to the presence of subsurface oxygen species has received increasing attention in recent literature.317,318 With the help of DFT calculations, Pettersson demonstrated that the presence of subsurface oxygen species could enhance the adsorption energy of CO on Cu(100), with effects extending to at least the third layer.319 With an improved coverage of adsorbed CO on the Cu catalyst, the greater probability of CO dimerization, which is a rate-determining step toward the production of ethylene, is to be expected.
Notwithstanding the clear difficulty in disentangling the contributions of specific crystallographic facets, surface roughness, nano-structuralization, local pH, and the presence of Cuδ+ and subsurface oxygen species, efforts have permitted the establishment of important guidelines in upcoming research works. In situ time-resolved tracking of the Cu oxidation state under e-CO2RR conditions through soft X-ray absorption (sXAS) spectroscopy analysis shared by Sargent's team earlier this year showed the presence of remaining Cuδ+ species even at highly negative potentials below −1.0 VRHE (Fig. 21a).320 These findings were ascribed to the fact that the selected sol–gel copper oxychloride (Cu2(OH)3Cl) precursor was able to slow the electrochemical reduction of the resulting electro-redeposited Cu-based catalyst, further enabling control over its nanoscale morphology. The authors reported a rapid transition from Cu2+ to Cu+ (within 5 min), with the transition from Cu+ to Cu0 being much slower. Surprisingly, for over an hour under e-CO2RR conditions, 23% of the catalyst was shown to exist as Cu+ under an applied bias as low as −1.2 VRHE. Under these conditions, a FE of 54% for C2+ products (ethylene, acetate, ethanol) was attained. The work shed light on the morphology evolution of these catalysts, suggesting a simultaneous dissolution and redeposition of copper ions from the sol–gel copper oxychloride precursor, and enabling a wide spectrum of emerging architectures of varying sharpness to be grown from within the pristine bulk material (Fig. 21b). In light of the above findings, highly selective C2 products from e-CO2RR could be obtained on B-doped Cu with stable electron localization.321 The attained selectivity of e-CO2RR to C2 hydrocarbons could be similarly ascribed, theoretically and experimentally, to the oxidation state of copper. In detail, at an average copper valence state of +0.35, the FE for C2 hydrocarbons and C2H4 attained ca. 80% and 52%, respectively. Under these conditions, C1 products (CH4, CO, and HCOOH) could be additionally completely suppressed in both gas and liquid products.
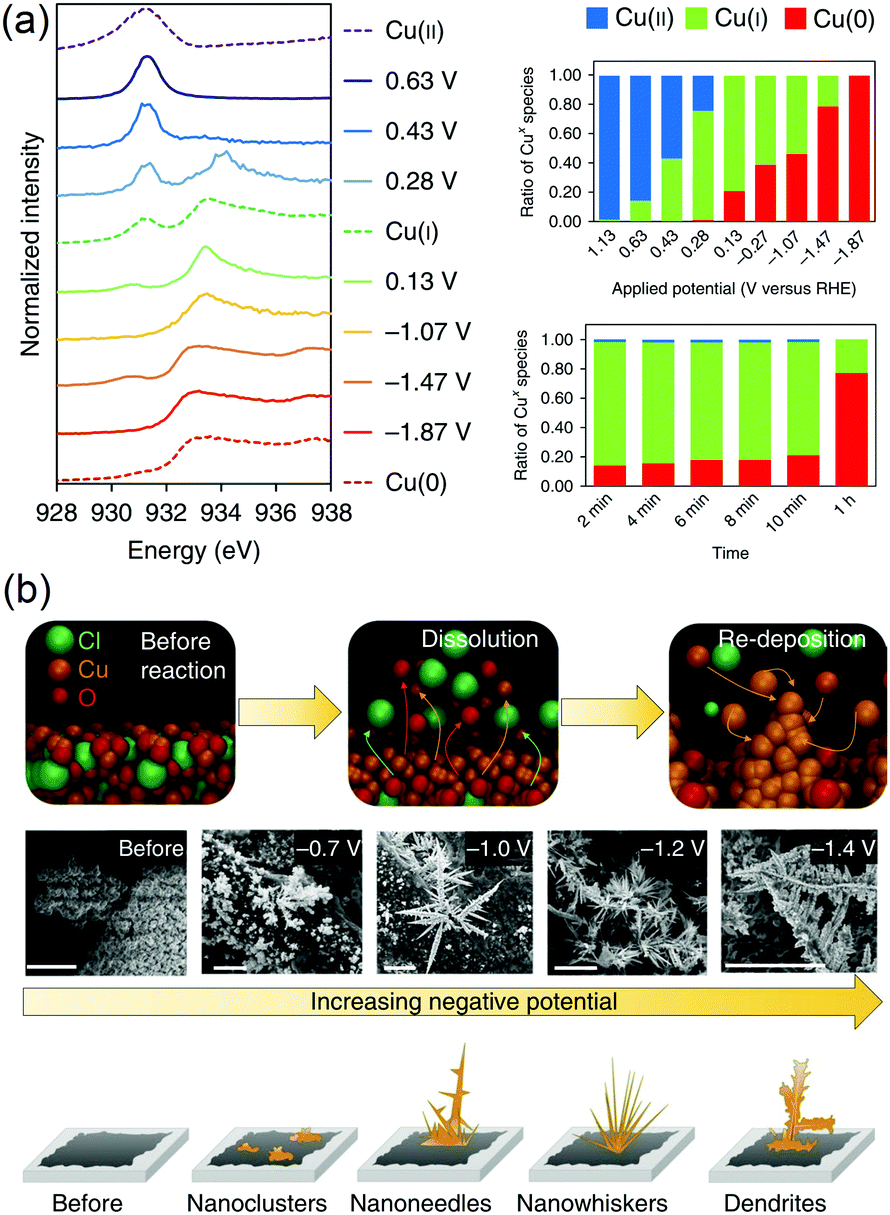 | ||
| Fig. 21 (a) In situ Cu L3-edge X-ray spectra of electro-redeposited Cu at different applied potentials and references for Cu metal, Cu2O, and CuO. On the right side, calculated ratios of Cu oxidation states from linear combination fitting as a function of the applied potential or time. (b) Schematic representation of the growth of these nanostructures following simultaneous dissolution and redeposition of Cu. Representative SEM images (scale bars = 5 μm) collected at specific applied potentials after at least one hour of reaction. At −1.0 VRHE, sharp needles of 5 to 10 μm in length could be witnessed, whereas at −1.2 VRHE, sharp nanowhiskers of 5 to 10 μm with high length-to-diameter ratios predominated. At the most negative applied potential (−1.4 VRHE), dendrites with rounded tips were reported. Adapted with permission from ref. 320. Copyright 2018 Springer Nature. | ||
Induced surface modification: additional studies. In a rather similar fashion to the works of Broekmann, Cu nanodendrites, obtained during the in situ electrodeposition in acidic medium (pH = 2.5) under high overpotential (<−2 VAg/AgCl) following local sintering and oxidation processes, showed an impressive FE to C2H4 of 57% at a current density of 170 mA cm−2.322 Contrary to the above-cited works, here the corrosive conditions under which catalysis was performed (using KBr, or mixtures of KBr and Br2) were found to be responsible for the emergence of CuBr and Cu2O-based needle-like dendrites. Of equal significance is the synthesis of highly active and selective Cu nanocubes with tunable Cu(100) facets as discussed above, but incorporating oxygen/chlorine ion content by low-pressure plasma pretreatments.323 In this case, these electrodes displayed lower overpotentials and higher ethylene, ethanol, and n-propanol selectivity, resulting in a maximum FE of ca. 73% for C2 and C3 products.
Initial works by Hori evidencing changes in the product selectivity by changing the cationic species (Li+, Na+, K+, and Cs+) in bicarbonate solutions have recently inspired further works toward the surface modification of Cu-electrodes.324 It was argued that the larger cations (e.g. Cs+) had a greater specific adsorption, which caused a higher local pH, inhibiting CH4 formation. Recently C2 and ≥C3 species were promoted over in situ generated Cl-induced bi-phasic Cu2O–Cu using potassium chloride (KCl) electrolyte.325 FE of ≥C3 species attained values up to 11.5% at −1.6 VRHE. Because of the chemoaffinity between chloride (Cl−) and Cu, a uniquely shaped phase was observed during the application of a cathodic potential. An abundance of Cu+ that could strongly bind and preserve the reaction intermediates for a longer time on the surface was demonstrated to be a key factor for the efficient synthesis of longer carbon chains. Similarly, Strasser showed that aside from an induced morphological evolution, the interaction of Cl−, Br− and, in particular, I− with the Cu surface could lead to surface changes on the Cu interface as supported by DFT studies.326
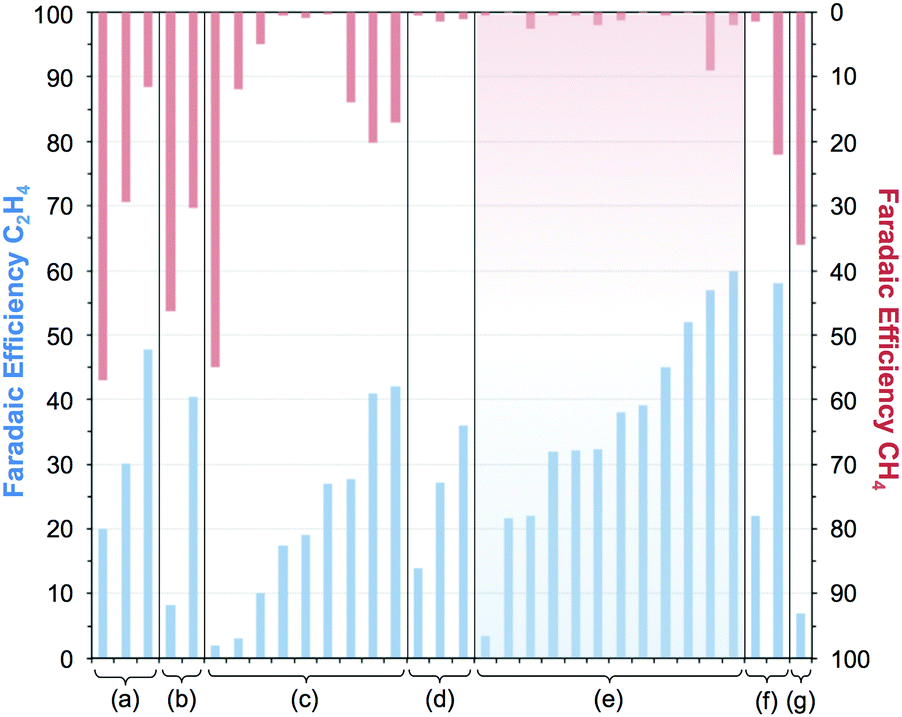 | ||
| Fig. 22 Reported Faradaic efficiencies for CH4 and C2H4 over Cu-based electrocatalysts. Representative reports were organized by the underlying strategy in the synthesis of the evaluated materials, and compared with (a) commonly reported Cu foil/polycrystalline Cu references. (b) Single crystal electrodes, (c) nanostructures, (d) enhanced surface roughness, (e) Cu2O-derived electrodes, (f) surface modification studies (e.g. chlorine-induced), and (g) Cu-based alloys. Data are compared on the basis of a maximization of the C2H4 to CH4 ratios in each study. Further details and respective references have been conveniently summarized in Table 2. | ||
3.3. Alternatives to Cu electrocatalysts
In the design of an efficiency-enhanced electrocatalytic system, Peterson suggested that Cu-alloying is preferential with metals with higher oxygen affinity, allowing CHO* to bind to the surface through both the carbon and oxygen atoms, thus increasing its stability without affecting the stability of CO.234 A comprehensive investigation of Cu-alloy electrodes was carried out considering Ni, Pb, Sn, Zn, Cd, and Ag.332 Limtrakul assessed the formation of CH4 on Cu-based alloy (Cu3X) stepped surfaces, with X = Ag, Au, Co, Ni, Pd, Pt, Rh, and Ir, using density functional theory calculations.333 With the exception of Cu3Pd and Cu3Pt, the alloys were shown to be more energetically favorable toward CH4 as compared to methanol. Alloying Cu with high OH affinity metals (i.e. Co, Ni, Ir, and Rh) showed an increasing OH* surface poisoning as compared to the pure Cu surface, while possibly suppressing HER. Conversely, alloys with Au, Ag, Pd, and Pt, improved the OH* removal step while promoting the HER.333 Peterson rationalized a high selectivity toward CH4 on Cu–Pt alloys based on the presence of Pt centers, which was believed to facilitate the protonation of CO* adsorbed on adjacent Cu active sites.334
Research focused on Au–Cu alloys showed enhanced formate, methane and ethylene formation over Au3Cu nanocrystals as compared with monometallic Au counterparts,335 the results being in line with an enhanced selectivity toward methanol and ethanol, claimed elsewhere.336 Comparatively, Cu seems to have a more suitable binding strength for both of the adsorbed intermediates to facilitate the conversion toward hydrocarbons with superior FE.327 Cu–Ag alloys showed high selectivity for the formation of C2 compounds, but maximum FE to CH3CHO (24%), C2H5OH (17%), and C2H4 (13%) recorded with a Cu/Ag ratio of 28/72 in CO2-saturated 0.1 KHCO3 reflected a relatively poor product selectivity.337 Similarly, the analysis of Cu–Co thin films covering the complete compositional range revealed a shift in selectivity toward C2 products (ethylene and ethanol).338 Between 5 and 15 at% Co, the change in adsorption energies induced a change from CHO* to COH* intermediates, or to a favored CO–CO dimerization, both believed to be advantageous toward C2. The above-listed results reflect the limitations in improving Cu-based catalysts through alloying strategies.
More interesting results were recently obtained with non-Cu containing alloy-based studies on nickel–gallium films (NiGa, Ni3Ga, and Ni5Ga3) in e-CO2RR to C2H4 and C2H6.339 The onset potential at low current densities was >250 mV more positive than that of polycrystalline copper, and approximately equal to that of single crystals of copper. Similar results were obtained upon CO-based RR. Alternatively, Kortlever focused on a rationally designed bimetallic catalyst combining a metal that binds CO* strongly, Pd, with a metal that binds CO weakly, Au, in an attempt to tune the binding energy of CO.340 The non-Cu containing catalyst was claimed to produce a mixture of C1–C5 hydrocarbons and soluble products at an onset potential of −0.8 VRHE, tentatively formed through the polymerization of –CH2 groups adsorbed on the catalyst surface.
It is, however, important to note that in single-atom systems there is a lack of adjacent active centers to stabilize intermediate species in C–C coupling reaction pathways believed essential in the synthesis of C2 and C3 hydrocarbons. In this sense, and contrary to homogeneous element dimers, trimers, or clusters, isolated active centers may be expected to inhibit CO–CO dimerization mechanisms and selectively produce C1 species. For instance, 4% Cu–CeO2 nanorods recently exhibited a FE of ca. 58% for CH4 at −1.8 VRHE, claimed to be the highest reported efficiency of CH4 production in e-CO2RR using H-type cells.349 Fitted EXAFS results of these cathodes yielded an unconventionally low coordination number of Cu as 4.8 ± 0.5. The high electrocatalytic activity and selectivity were attributed to the synergetic effect of the resulting multiple (≈3) oxygen vacancy-bound single-atomic Cu active centers, where a single CO2 molecule is strongly adsorbed, activated, and gradually reduced, as further corroborated by DFT calculations.
Conversely, while directing the synthesis of C2H4 may be of imposed difficulty, these authors believe focusing on this domain may further unveil analogous strategic paths of heightened importance. For instance, assessing the premise of structurally well-defined heterogeneous Cu–X–Cu bridged architectures (X being a guest atom), or alternatively, heterogeneous dual-atom catalysts incorporating adjacently placed single-atoms are suggested herein. In the latter case, the resulting selectivity is presumed to reflect the difference in the bonding strength of intermediates, according to the selected elements incorporated in the hybrid structure. Rigorous characterization and spectroscopic measurements (XANES & EXAFS) would be decisive in fully elucidating the undisclosed chemistry of the designed systems while shedding light on the corresponding mechanistic pathways.
A recently assessed strategy focuses on the homogeneous molecular structures and the practical handling of heterogeneous catalysts to tackle innovative possibilities for e-CO2RR. Works of significance in homogeneous catalysis (summarized elsewhere and recommended for revision) may provide valuable inspiration for the rational design of heterogeneous catalysts.354 A molecular copper–porphyrin complex deposited on a carbon electrode unlocked high activity, with a partial current density of 21 mA cm−2 for hydrocarbon production at −0.976 VRHE, corresponding to turnover frequencies of 4.3 CH4 and 1.8 C2H4 molecules site−1 s−1.355 This result represents the highest catalytic activity for hydrocarbon production over a molecular e-CO2RR, which will foment additional interest in immobilized molecule catalysts in the future. In line with the above discussion focusing on the restructuration of electrodes as a crucial platform to assess structure–reactivity correlations, Wang and co-workers have recently assessed the restructuration of Cu complexes in CO2 methanation through in situ and operando X-ray absorption spectroscopy (XAS) characterization of representative Cu complex structures under electrochemical reaction conditions.356
The singularity of Cu and copper oxide-derived catalysts has only very recently faced examples of metal-free catalysts prone to catalyze e-CO2RR through reaction pathways involving more than two electrons. The said results have reflected analogous approaches toward enhanced CO formation in carbon nanotubes and graphene oxide, in which the acidic CO2 molecule was confirmed to find preferential adsorption onto incorporated Lewis basic pyridinic N groups.251,252 By combining a high inherent defect density, achieved by tuning the dimensions and morphology of carbon nanostructure into nm-scale, with foreign N-doping, Wu et al. achieved multi-carbon hydrocarbons and oxygenates formation at high FE, high current densities and low overpotentials (Fig. 23).357 The synthesized N-doped graphene quantum dots (NGQDs) showed high FE to CO2RR up to 90%, with a C2H4 and ethanol selectivity reaching 45%, and a C2 and C3 product distribution and production rate comparable to those over copper (Fig. 23). With lower formation energy toward N-doping at the edge sites compared to basal planes, and a roughly estimated 44% of exposed edge sites on these quantum dots, catalytic site proximity was herein claimed to favor C–C coupling pathways toward C2 product formation. Additional theoretical insights into the reduction pathways of CO2 on these NGQDs were later published.358 Studies focusing on microporous carbons of high nitrogen-content and incorporating chemically heterogeneous nitrogen species of varying reactivity are therefore expected to emerge in the future.359
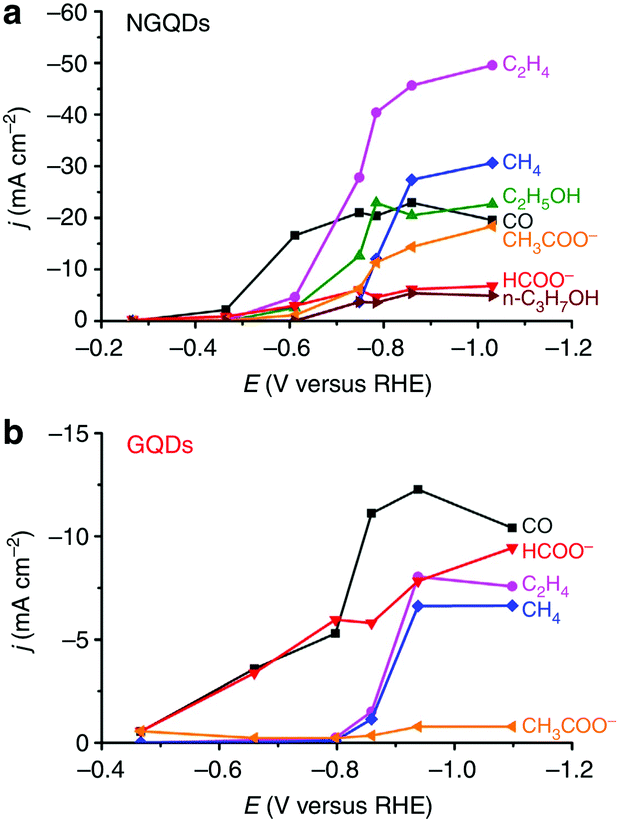 | ||
| Fig. 23 Partial current densities of emerging CO2RR products as a function of the applied potential over (a) N-doped graphene quantum dots (NGQDs) and (b) GQDs cathodes. Adapted with permission from ref. 357. Copyright 2016 Springer Nature. | ||
3.4. Stability issues
In the development of CO2RR electrocatalysts, the stability of the reported systems remains a significantly underdeveloped research area, even when extended to CO2RR value-added products, e.g. formic acid attaining FE above 90% and current densities of 200 mA cm−2, currently the closest to a potential commercialization break. While long-term tests are yet to be performed, assessing the stability of the reported electrocatalysts and mapping emerging drawbacks in function of the selected operating conditions are scarcely practiced.Contextually, the durability of electrosynthesis reactors is usually measured in years, with stationary fuel cells having proven lifetimes as high as 40000 h.360 Following the parallelism with H2 technologies, current values below 100 h are insufficient and require a dramatic enhancement for practical application in the future. In particular, over the Cu electrode, a notable deactivation of e-CO2RR to hydrocarbons and subsequent HER dominance within a two-hour range was earlier evidenced in representative reports.240,361,362 The nature of the species responsible for blocking and poisoning the catalytic active surface, leading to rapid activity degradation, has remained debatable in the literature. In 2005, Hori proposed that the activity decay could be derived from heavy metal impurities in the electrolyte being cathodically reduced and deposited at the electrode, as metal electrodes other than Cu are inactive toward hydrocarbon production.363 On the basis of the observed potential of the anodic peaks, identified Fe2+ and Zn2+ in chemical reagents of the best quality were proposed as the cause of the observed catalytic decay. Suppression of the effects of metal deposition could be attained through prior purification of the electrolyte solution via pre-electrolysis or ion-complexation of trace metal impurities using ethylenediaminetetraacetic acid or solid-supported iminodiacetate resins over Au, Ag, and Cu electrodes.364
Several research groups proposed the deactivation of the Cu cathode by graphitic carbon species formed via the decomposition of reaction intermediates.240,278 The accumulation of carbonaceous species on the catalytic surface has parallelism with metals that strongly bind CO*, e.g. Pt, Ti, Ni and Fe, evolving only H2 from the competing water reduction. Over the Cu surface, Mul claimed that emerging COH* species in CH4-directed pathways prompted a rapid deterioration of the hydrocarbon production rates, whereas favoring ethylene production through CO–CO dimerization could substantially hinder the activity decay up to several hours.365 The results are of particular interest as they extend the previously discussed effects of the selected operating conditions, oxidation states, catalyst morphology, and local pH to the stability of the working electrode.311
As outlined in previous sections, an increasing number of reports over Cu–O combinatorial catalytic systems have demonstrated an enhancement of the FE of C2H4 on account of the presence of mixed Cu oxidation states and possibly subsurface oxygen. The performances of catalysts previously referenced in this review remain limited when probed from a stability point of view, e.g. Cu2O film (1 h), plasma-activated Cu (5 h), and electro-redeposited Cu (1 h).308,309,320 Alternatively, while assessing the effect of the selected pretreating reduction operating conditions, Hwang reported FE for C2H4 of ca. 40% and a remarkably superior stability up to 40 h over anodized copper Cu(OH)2 catalysts prepared by a simple electrochemical synthesis method.343 Equally surpassing stability levels reported to date, the boron-doped copper electrode developed by Sargent's team unveiled superior stability up to 40 h under continuous operation at −1.1 VRHE, confirming that the Cuδ+ sites induced by boron doping were highly stable at the high applied potential during the CO2RR process.321 Comparatively, on morphology-based investigations, the ensembles of Cu NPs reported by Yang were stable for 10 h.287
Mechanistic uncertainties and correspondingly unaccounted poisonous carbonaceous species reflect additional drawbacks to this problem. Augustynski proposed an activation treatment, repeated every 5 min during the course of the electrolysis, involving the periodic anodic stripping of poisoning species from the active surface of the electrode.366 Despite intervention and potentially mandatory modification on the reactor level, these results showed a facile method to extend the activity to 48 h with no activity decay being virtually witnessed. Alternatively, Wang developed surface-restructuring-based Cu electrodes with remarkable stability in the e-CO2RR, even in electrolytes without pre-purification.367 Pre-deposited Pd atoms on the Cu working electrode were shown to induce continuous morphological and compositional restructuration of the Cu metallic surface, inducing a stable catalytic activity up to 16 h. The strategy is expected to be extended to other foreign atoms, as demonstrated in this work upon the replacement of Pd with Rh.
3.5. Reaction cells and electrolyte-related effects
Increasing reports have focused on the implications of process variables and operating conditions, e.g. the effect of temperature, pressure, and CO2 solubility on the rates of e-CO2RR and product distribution.243,368 Exploratory works have revealed the effect of the adsorption of anions and cations on the electrode surface, the presence of impurities, changes in the surface wettability and pH, or the impact of ion-pairing interactions with reactants and products.365,369,370 The preferential CO2 solubility in organic aprotic solvents has further sparked a number of increasing research works using various metallic electrodes.371During their initial works, Hori alluded to the impact of the nature and composition of the electrolyte on the e-CO2RR activity and product selectivity,237,238 with the stabilization of the CO2 radical being known to be impacted by the choice of the electrolyte.372 For instance, in Hori's seminal works, the HER could be favored at FE above 70% in KH2PO4/K2HPO4, or hindered below 10% in KClO4 utilizing the same catalyst. Formation of C2H4 and alcohols was favored in KCI, K2SO4, KClO4 and diluted HCO3− solutions, while CH4 was preferentially produced in relatively concentrated HCO3− and phosphate solutions. Since CO2 forms bicarbonate and carbonate species in alkaline solutions, selected electrolytes have also been preferentially limited to neutral and acidic pH ranges. However, with increasing the local pH following increasing proton consumption at the cathode surface, an emerging reactivity between CO2 and evolving OH− species is presumed in the proximity of the cathode. According to the findings of Gupta, the local pH can be up to 6 units higher, depending on the electrolyte buffer capacity, the thickness of the diffusion layer, and operating current density.373 As the product selectivity toward CH4 and C2H4 is pH-dependent, i.e., it depends on the availability of hydrogen or protons on the electrode interface,238 in electrolytes such as KClO4 with low buffer capacities, high local pH is expected, enhancing the C2 selectivity. Conversely, in buffered electrolytes like phosphate, CH4 is favored over C2H4 due to a local pH close to neutral.311 This understanding encourages the rational manipulation of the CO2 concentration near the CO2 reduction sites to give interesting results.374
H2 production from water splitting and e-CO2RR share several conceptual and technological features, both being cathodic in nature and the latter being ideally complemented by the oxygen evolution reaction (OER) on the anode. This has led to comparable designs for CO2RR and water electrolysers, as reflected in the use of similar membranes, electrode types, and bipolar plates.360 While CO2 electrolysis in traditional electrochemical H-cells is widely appropriate for the fundamental research and screening of novel electrocatalysts, practical applications require the superior complexity of the working systems. Continuous exploratory works on the design of reaction cells have outlined the importance of separating the anodic and cathodic half-cells, e.g. using a cation exchange membrane (Nafion 117) to further prevent the reoxidation of CO2 reduction products in the reaction cell. A schematic representation is given in Fig. 35 for photoelectrochemical systems. In general, experiments carried out in H-type cells do not effectively report how CO2 electrocatalysts behave in flow reactors that are more pertinent to scalable CO2 electrolyzer systems.
Alternatively, flow reactors have been shown to provide enhanced control over reagent delivery, enabling the use of a gaseous CO2 feed to the cathode of the cell, and generating much higher current densities by hindering mass transport issues inherent in H-type cells (Fig. 24). In representative configurations, stainless steel plates serve as current collectors and provide electrical contact between the GDE and an external potentiostat.375 In addition, a skewed focus from H-type to flow cells is predicted to better account for the dynamic nature of flow parameters, ion and electron transfer/transport, and the resulting electrocatalytic performance.41 The resulting boost in the performance of e-CO2RR platforms has been recently reflected in works on Cu dendrites, Cu nanoparticles, and electro-redeposited Cu, attaining absolute partial C2H4 current densities of 96.9, 150, and 161 mA cm−2, respectively.320,322,375 By the end of 2017, the collected number of papers published since 2007 on metal-based e-CO2RR systems in continuous flow reactors remained negligible when compared with reports on research carried out in H-cells (Fig. 24a).41
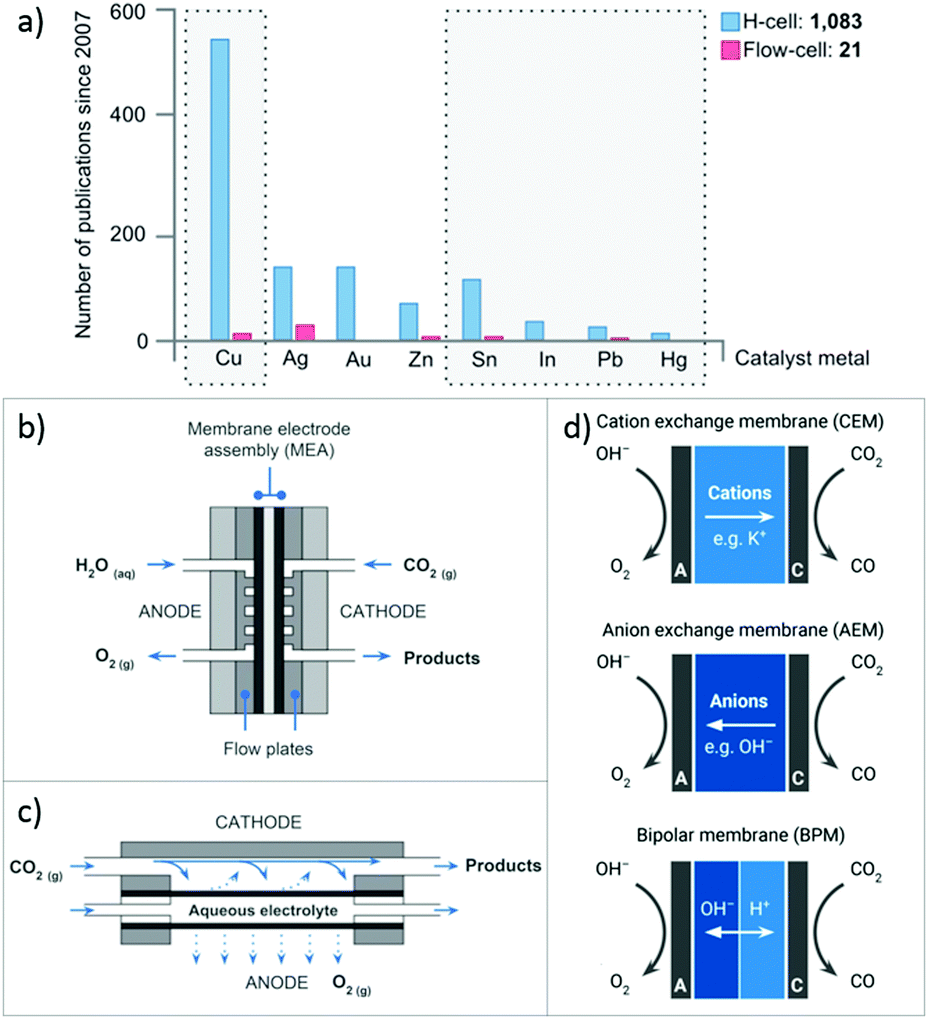 | ||
| Fig. 24 (a) The number of papers published since 2007 on selected metal-based electrocatalysts for the CO2RR in H-cells and in continuous flow reactors (data collected 29/12/2017). Cross-sectional diagrams of (b) a membrane-based reactor and (c) a microfluidic reactor. The former contains a membrane electrode assembly (MEA) consisting of the anode and cathode gas diffusion electrodes (GDEs) on either side of a polymer electrolyte membrane (PEM). In the schematized microfluidic reactor, the liquid electrolyte flows in the channel between the anode and cathode GDE materials, with CO2 being supplied on the cathode side of the cell where it diffuses to the electrocatalyst supported on the cathode GDL. (d) Overview of different ion transport pathways between the anode and the cathode labeled “A” and “B”, respectively, depending on the type of selected polymer electrolyte membrane. Readapted with permission from ref. 41. Copyright 2018 American Chemical Society. | ||
Reported flow reactors in the e-CO2RR are classified into two primary systems, i.e., membrane-based flow cells and microfluidic reactors, with both architectures permitting an adaptive design to enhance current densities through the delivery of CO2 to the cathode in the gas phase rather than dissolved in a liquid electrolyte. Fig. 24b and c provide schematic representations of these configurations with detailed information. Over membrane-based reactors, the choice of cation-exchange, anion-exchange, or bipolar membranes plays a key role in the kinetics of ion transport pathways between the anode and the cathode sides and the range of applicable electrolyte conditions (Fig. 24d).376 In microfluidic cells, extensive studies have been performed concerning the properties of porous carbon gas diffusion layers, materials that are relevant to membrane reactors.375,377 The recent contribution of Berlinguette shed light on the influence of noncatalytic components and feedstock characteristics (e.g., gas vs. liquid, flow rate, electrolyte supply) on the overall CO2RR performance in flow cells (e.g., current density, prolonged stability, operating voltage, and selectivity).41
3.6. Concluding remarks and analytical prospects
We have shed light on the development of electrocatalysts toward the enhancement of the carbon chain, and the impact of factors such as the working potential, the nature of the electrolyte, and selected operating conditions (e.g. temperature, pressure, and resulting CO2 solubility) on the resulting product selectivity. The underlying attention to ethylene provides insight into the C–C coupling reaction step required for the formation of multicarbon products, and the rational design of said catalysts. Being the most studied materials in the synthesis of hydrocarbons, the ability of Cu and derivatives to produce highly reduced products is a consequence of the balance of CO binding energy and further protonation of said adsorbed species as compared to other transition metals. Notwithstanding this unique capability and cost-effectiveness, prohibitively high (nearly 1 V) overpotentials, low product selectivity competing with the hydrogen evolution side reaction, and fast catalytic deactivation remain barriers to application. In addition to the limited selectivity toward long-chains, discerning the pathways that lead to the formation of alcohols, e.g. methanol, ethanol, or propanol, over Cu-based catalysts has remained ambiguous.The FE toward CH4 and C2H4 are particularly low as compared with CO and formate, both of which are above 90%, with the latter attaining high current densities of 200 mA cm−2, highlighting the closest e-CO2RR could lead to a potential breaking of the commercialization barrier (Fig. 15).374,378–382 The production of ethylene and ethane show similar degrees of development, which is a step ahead of that of CH3OH or C2H5OH.360 For C2H4 production, multiple reports have claimed FEs greater than 40%, with nanostructuralization approaches reaching 41%, and a remarkable Faradaic selectivity of 60% belonging to a plasma-activated Cu oxide-derived surface configuration. However, on the basis of the currently observed accelerated growth toward target levels (95% FE for CO2RR with 90% C2 selectivity, according to technical and economic models), it is expected that these records will be broken in the future.
Specific crystallographic facets, surface roughness, the effects of nano-structuralization, induced local pH, and the presence of Cu+ species and/or subsurface oxygen have been shown to play key roles in lowering the overpotential and enhancing the formation of C2H4 through dimerization reactions. Disentangling the contributions and determining the relative importance of each of them has remained an area of intense discord, with the dynamic character of Cu-based electrocatalysts prone to surface reconstruction significantly adding to the complexity of the systems discussed herein.323 Evidence of simultaneous morphology and oxidation state evolution during reaction further underline the prominence of available in situ techniques expected to continue to lead the way to results of striking importance in this field, e.g. in situ synchrotron-based XRD and X-ray absorption techniques and eventually environmental TEM studies. The effect of the selected initial reduction conditions on the structure and chemical states of these Cu-based electrodes has been highlighted in recent reports. Using anodized copper Cu(OH)2 catalysts, Hwang and colleagues proposed that harsh pretreatment reduction conditions at very negative potentials (−4.0 VAg/AgCl) could significantly enhance the selectivity and stability of C2H4 over the resulting O–Cu combination catalysts.343 The results were attributed to the presence of mixed valences, with the harsh treatment being proposed to tune the surface properties of the oxygen-containing copper surface.
Highly efficient, robust and relatively cheap catalysts would be an effective way to push forward the e-CO2RR on its way to industrial application. Exemplary research outcomes in the works of Cuenya and Sargent additionally highlight the remarkable progress in moving toward higher C2H4 to CH4 ratios, which represent a desirable extension of the hydrocarbon chains of anticipated renewable alternative fuels.309,320 Results are given in Table 2, emphasizing the number of recent studies close to and eventually surpassing C2H4 to CH4 ratios of 100. Cu2O-Derived electrocatalysts have proven to be able to enhance the selectivity to C2H4 and also dramatically reduce CH4 yields (highlighted surface in Fig. 22). Works that may further foment the selectivity within the ≥C2 range are of additional interest in the future. High paraffin selectivity has been attained upon the incorporation of a hydrogenating co-catalyst phase (e.g. PdClx), but works focusing on this angle have remained limited to date.342 Nonetheless, the uniqueness of previously reported catalysts indicate the difficulty in establishing valuable guidelines in the design of analogous systems. Most importantly, the singularity of Cu and copper oxide-derived catalysts has only very recently faced heightened interest, indicating the need for new strategies. In-depth studies using microbial electrolysis cells (MEC) with electricity as an energy source and microorganisms as the catalyst are needed.383–385 An additional alternative based on the well-established assumption of CO as a reaction intermediate could be to focus on two-step cascade processes, in which CO2 is initially reduced to CO, with the latter being sequentially reduced toward long-chain hydrocarbons under optimized near-surface CO concentration (and consequently *CO surface coverage) conditions.
Fast forwarding three decades from the seminal works of Hori, reported studies on the e-CO2RR still largely rely on time-consuming GC, HPLC and NMR analytical approaches that require the separate analysis of the formed products. Emerging real-time detection techniques are expected to accelerate the chances of discovery of noteworthy findings and bring the e-CO2RR a step forward on the path toward feasibility.312,386,387 In the design of new catalytic systems, the above-cited works often reflect nanostructure-engineering approaches leading to the formation of novel active sites capable of manipulating the reaction mechanistic paths to significantly improve the catalytic performance with high FE and low overpotentials. Alternatively, the use of redox mediators that can transfer both protons and electrons is appealing, given the multi-proton multi-electron nature of the e-CO2RR. Striking technological advances are expected for the process to achieve economic viability. Attained current densities determine the size of the reactor and the corresponding capital cost of the process. Mass and charge transport properties remain a prerequisite in the design of the reaction cells to accompany the optimization of the active sites developed in the catalyst toward high performance.
4. Photochemical CO2 reduction reaction (p-CO2RR)
The emergence of solar radiation as a clean and sustainable energy source has unlocked promising pathways to address the problems of global warming and replace traditional fossil energy in a number of chemical processes including water splitting, production of fuels, and environmental remediation.388–390 Solar-to-fuel synthesis through the reaction of CO2 and H2O is considered a promising technology for the storage of solar energy in the form of chemical bonds. The light-induced CO2RR to formic acid, formaldehyde, methanol, and methane was first demonstrated by Honda in 1979 over a library of semiconductors, e.g., TiO2, ZnO, CdS, GaP, SiC, and WO3, in the presence of water.391 In 1987, Grätzel demonstrated that the reaction rate for CO2RR to CH4 at room temperature and atmospheric pressure using highly dispersed Ru/RuOx on TiO2 could be sharply enhanced through photo-excitation of the support material.64Fast-forwarding nearly four decades since the works of Honda, an extensive line of works has ascertained the promising character of light-responsive catalysts by the direct use of solar energy. However, the p-CO2RR is a complex multi-step process in which light harvesting and charge separation and transfer (i.e., electron and hole transport to surface reaction sites) play, in addition to surface reactions, a crucial role in the overall process efficiency. In addition to limited light harvesting and charge separation hindering improved process efficiencies, most solar-active catalysts have been shown to suffer from inefficient CO2RR selectivity, the inability to hinder the competing HER, and catalytic instability. In-depth studies on the p-CO2RR are still scarce, with emerging strategies to enhance CO2 activation and the subsequent reduction lacking clarity and effectiveness. The design of highly efficient photocatalytic systems with meaningful selectivity toward C2H4 or longer carbon-chains remains a particular challenge. Below, we discuss the recent progress and opportunities in the development of relevant heterogeneous electrocatalysts reflecting the energetic efficiency and attained product selectivity.
4.1. Mechanistic difficulties
The CO2RR has parallelism to water splitting technologies, which have largely accelerated the search for suitable alternatives to the common drawbacks of these solar-driven systems. Enhanced efficiency in p-CO2RR, semiconductors with a wide optical absorption region and appropriate band edges are desirable.392 Since the seminal works of Grätzel, an extensive semiconductor-library has been assessed in recent years to overcome the limitations of light harvesting and electron–hole recombination. Strategies to extend the light absorption under visible and near-infrared (NIR) illumination are of importance. Additionally, electron–hole recombination significantly limits the number of charge carriers available for surface reactions. Over the benchmark TiO2, the rate of recombination of photogenerated charge-carrier pairs has been reported to reach 90% within 10 ns after their generation.393While most reported CO2RR mechanistic studies have been conducted on TiO2, drawn conclusions are often extended to other materials. Nonetheless, the limited number of in-depth exploratory works on the photocatalytic mechanism, potential pathways, and CO2 activation underscore the need for greater focus on these research fields.394 Valuable strategies for enhancing the adsorption and activation of CO2 and the factors determining the reaction pathways and selectivity for reduction products have been recently reviewed by Gong and colleagues.395 An open discussion on e-CO2RR, the activation of CO2, is the subject of discord in light of the number of intermediate species in solution; e.g., hydrated CO2 molecules, carbonic acid (H2CO3), bicarbonate (HCO3−) and carbonate (CO32−) ions. As recently assessed through isotopic experiments using 13CO2 and in situ infrared spectroscopy studies in a NaHCO3 aqueous solution (0.10 M), dissolved CO2 was confirmed to be the intermediate species over a ZnGa2O4/Ga2O3 catalyst, whereas at corresponding pH values, H2CO3 and CO32− were present only at very low concentrations.396 HCO3− derived from NaHCO3 was shown to not contribute to the reduction of CO2 but to function as a buffer to enhance CO2 dissolution. Most importantly, the single-electron reduction potential of CO2 to the highly unstable radical anion CO2˙− at −1.9 VNHE is much more negative than the conduction band (CB) of candidate semiconductors, as shown in Fig. 25. Virtually no semiconductor provides sufficient potential to directly transfer a single photogenerated electron to a free CO2 molecule, thus making this step highly improbable.392 Instead, proton reduction at 0.0 VNHE is more favorable, competing with CO2 for electrons. This problem additionally aligns with (1) a preferential H2O adsorption onto the catalyst surface as compared to CO2, and (2) a facile transfer of only two electrons for the HER, while from a kinetics point of view, the production of the simplest hydrocarbon demands the transfer of eight electrons. These issues, therefore, highlight one of the major concerns in the p-CO2RR, with the strongly competing HER leading to a low efficiency and selectivity.
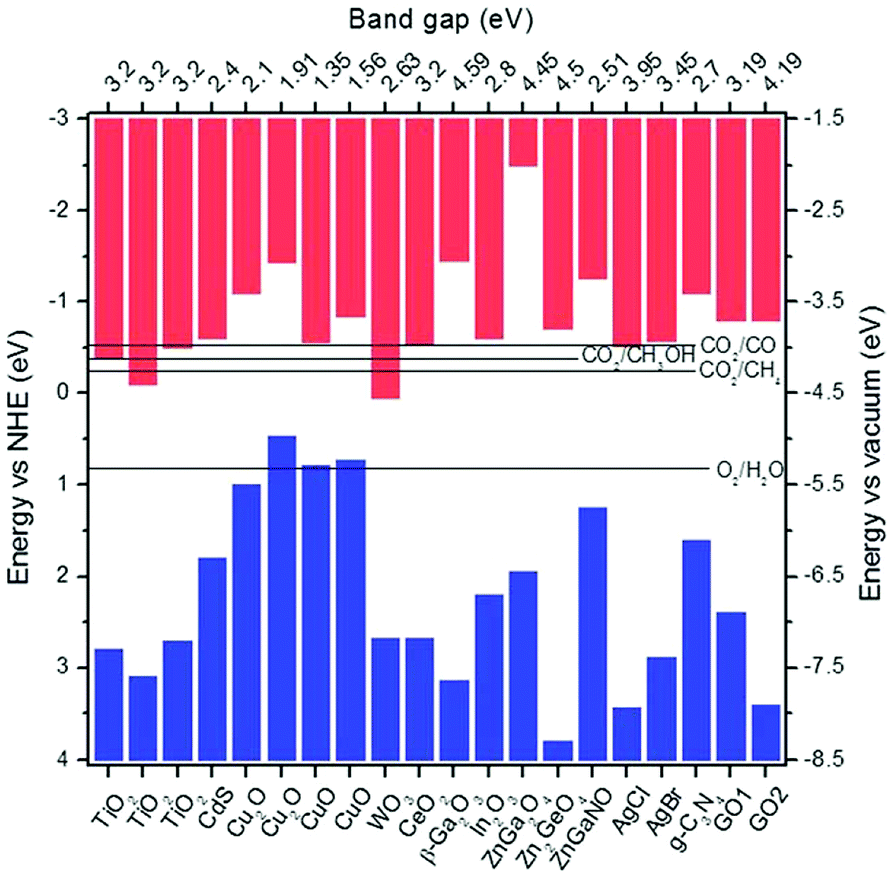 | ||
| Fig. 25 Band edge positions for selected photocatalysts in the p-CO2RR. Results summarized by Jaroniec and colleagues were based on references listed in the review. Particularly, for the listed TiO2, Cu2O, CuO, and GO, the reader is referred to the published works.397–402 Reproduced with permission from ref. 392. Copyright The Royal Society of Chemistry 2015. | ||
 | ||
| Fig. 26 Possible binding modes of CO2 on the surface of Zn2GeO4. Reprinted with permission from ref. 403. Copyright 2012 American Chemical Society. | ||
Following CO2 activation, the subsequent electron transfer initiates a series of chemical reactions, determining product distribution and the overall efficiency of the process. Different proposed pathways for this multistep reduction process involving a minimum of eight electrons and protons, with cleavage of C–O and formation of C–H bonds, have been recently summarized by Stolarczyk and colleagues (Fig. 27).404 If bidentate binding occurs through both oxygen atoms to two Ti atoms, preferential attachment of a hydrogen atom to the carbon atom occurs with consecutive formate anion and formic acid formation in the absence of C–O bond cleavage. Conversely, monodentate binding through one of the oxygen atoms to a titanium atom or binding through the carbon atom to a surface-bridging O atom generally favors the formation of an adsorbed carboxyl radical, COOH. The designated formaldehyde-pathway proposes the recombination of a hydrogen radical with a carboxyl radical to form formic acid, which subsequently undergoes repeated reduction steps toward the synthesis of methane.405 Methanol emerges here as an intermediate instead of a side product, in clear disagreement with the parallel formation of methanol and methane reported by Kočí.406 In an alternative carbine pathway, the attachment of the hydrogen radical to the oxygen atom of the adsorbed CO2− intermediate prompts the immediate cleavage of the C–O bond, as opposed to what is proposed in the formaldehyde pathway where this cleavage occurs very late in the process.406,407 The emergence of CO as a reaction intermediate is not surprising, with further hydrogenation toward hydrocarbon formation being observed. The proposed mechanism additionally accounts for the formation of a carbon residue on the surface and, subsequently, CH radicals, carbine, and a methyl radical prior to the formation of either methane or methanol. A glyoxal-based pathway involving the formation of a unique glyoxal intermediate through the dimerization of adsorbed formyl species is shown in Fig. 27. CO evolution from the aldehyde is a by-product that is commonly observed in the p-CO2RR. Methyl radicals can also be formed from acetaldehyde along a different pathway, if the aldehyde is oxidized to acetic acid.
 | ||
| Fig. 27 Proposed formaldehyde, carbine, and glyoxal pathways for the CO2RR to produce CH4. Readapted with permission from ref. 404. Copyright 2013 John Wiley and Sons. | ||
4.2. Semiconductors: library and engineering
Variations in the crystal structures delineate the band gaps of anatase (3.2 eV) and rutile (3.0 eV), requiring minimal photon energies of 389 and 413 nm, respectively, and hindering recombination rates on anatase up to ten times lower than rutile.412,413 With visible and NIR light absorption exclusion accounting for ca. 45% and 50% of the total solar spectrum, respectively, the efficiency of TiO2-based processes remains low. With limited photo-responsivity for large-scale application, efforts to extend the optical absorption edge of TiO2-based photocatalysts have been pursued.414 Besides its role in generating and separating electron–hole pairs during photo-excitation, the rational structural and morphological control of TiO2 may further induce CO2 activation through facile electron transfer from the surface of the semiconductor to CO2.415–417 Analogously, surface defects such as oxygen vacancies and step edges have been claimed to be among the most reactive sites on the surface of metal oxides.413,418 Through scanning tunneling microscopy studies on a TiO2(110) surface, the surface oxygen vacancies were shown to lower the activation barrier and enhance the trapping and activation of CO2.419 To enhance CO2 activation on surface electron centers or Lewis basic sites, surface modification through the introduction of functional basic sites has been explored.
In order to enhance light harvesting and favor CO2 activation, strategic steps have included the incorporation of foreign metal/non-metal species420–422 and upconversion NPs,423,424 bulk single-electron-trapped oxygen vacancies425 and self-doping by Ti3+ and oxygen vacancies,426,427 coupling with other semiconductors,390 addition of hole scavengers during catalytic evaluation,428 NaOH loading,429 and the simple incorporation of carbon-based moieties.430–432 N-Doping has been shown to facilitate CO2 adsorption and promote photogenerated electron transfer, leading to superior p-CO2RR rates. Numerous oxygen vacancies on synthesized conductive black TiO2 can remarkably enhance the formation rate of CH4 up to 14.3 μmol g−1 h−1.433 The incorporation of Mg in TiO2 photocatalysts synthesized by the sol–gel method revealed an enhancement of the presence of Ti3+ species and oxygen vacancies, ascribed to a 4.5 fold greater CH4 formation.434 The results further corroborate reports by Xie where an 11 μmol CH4 gTi−1 h−1 production rate in Pt–MgO/TiO2 systems was attained, in which MgO was proposed to strikingly promote the activation of CO2.435 The same authors claimed that a modification of the Pt–TiO2 using a range of basic oxides, e.g. SrO, CaO, BaO, La2O3 and Lu2O3, could increase the amount of CH4 and inhibit CO formation.
Nano-structuralization. Numerous TiO2 morphologies e.g. NPs,436 nanosheets,437 nanotubes,438 brookite quasi-nanocubes,439 and hollow structures440 with controlled crystal phases, particle sizes and surface structures have been assessed in the CO2RR. TiO2 NPs in the 4.5 to 29 nm-range showed an optimal average size of 14 nm for enhanced CH4 formation activity, attributed to an optimized surface area, charge-carrier dynamics and light-harvesting efficiency.436 Nano-structuralization may also serve as the ideal groundwork to elucidate the effect of exposed TiO2 facets.437 Although surfactants commonly used to control the crystal morphology often induce surface contamination, creative synthesis strategies have gained some attention. TiO2 nanoplates, nano-octahedrons and nanorods reaching 90% of exposed low-index (001), (101) and (001) facets, respectively, unveiled decreasing CH4 yields in the order of (010) > (101) > (001).441 As the ability of electron–hole separation was confirmed to decrease in the opposite order (010) < (101) < (001), the results agreed with previous studies unveiling a higher CB edge for the (010)-dominant TiO2 nanorods, and stronger CO2 and H2O interactions with (010) facets.440,442 Appropriate manipulation of the ratio of co-existing facets can further boost the activity of CO2RR to CH4.443–445 Recently, TiO2 nanocrystals with co-exposed (101) and (001) facets showed enhanced CO2RR activity as a result of the promoted spatial separation of photogenerated charges toward the different facets of TiO2 nanocrystals.
Nanoporous supports. Works by Anpo established the significance of highly dispersed TiO2 on zeolite frameworks toward the development of systems with high catalytic efficiency and selectivity.446,447 Later, mesoporous and mesostructured materials, exhibiting well-dispersed active centers and large pores suitable for the facile diffusion of sterically hindered molecules, were suggested as potential host candidates in photocatalytic systems. These have included hierarchical silica materials and corresponding aluminosilicates; e.g., MCM-41, MCM-48, SBA-15, KIT-6, and FSM-16 have been commonly reported.448–451 CH4 product yields commonly reported per wt of Ti attained values notably superior to the P25 reference. A 100 μmol CH4 gTi−1 h−1 production rate with Ti-SBA-15 remains the highest reported value, although a calculation inaccuracy may arise from the reported Si/Ti ratio (270) in this case.450 The development of TiO2 mesoporous and macroporous architectures and more recently other metal oxide mesostructures of relevance has emerged in recent years as an expected follow-up to these studies.
Results discussed herein have been summarized in Table 3, providing a direct comparison of corresponding CH4 production rates. Improvements in the efficiency of TiO2 as evidenced in this section appear, however, to remain mostly ineffectual toward desirable CH4 formation rates and denote the need to study other semiconductors and develop innovative catalytic systems. In addition, several of the reported strategies fail to uniquely promote the CO2RR, favoring the undesirable HER and reducing the overall efficiency of the process.
| Catalyst | Co-catalyst | Light source | Rate (μmol g−1 h−1) | Ref. | |
|---|---|---|---|---|---|
| a | TiO2 (pellets) | — | 4.8 W UVC (253.7 nm) | 0.001 | 409 |
| Anatase TiO2 | — | 300 W Xe lamp | 0.04 | 413 | |
| P25 | — | 100 W Hg lamp (>250 nm) | 0.37 | 447 | |
| Anatase TiO2 (presence of 2-propanol as a hole scavenger) | — | 350 nm light source | 0.88 | 428 | |
| TS-1 | — | 100 W Hg lamp (>250 nm) | 1.27 | 447 | |
| P25 | — | 300 W Xe lamp (<700 nm) | 2.52 | 417 | |
| b | Anatase TiO2(001) | — | 300 W Hg lamp | 0.19 | 441 |
| Anatase TiO2(101) | — | 300 W Hg lamp | 0.74 | 441 | |
| Anatase TiO2(010) | — | 300 W Hg lamp | 1.25 | 441 | |
| c | TiO2 nanocubes | — | 100 W Hg vapor (365 nm) | 3.6 μmol m−2 h−1 | 438 |
| TiO2 cuboids | — | 300 W Xe lamp (>300 nm) | 8 ppm g−1 h−1 | 437 | |
| Anatase TiO2 nanosheets, 95% (100) facets | — | 300 W Xe lamp (>300 nm) | 35 ppm g−1 h−1 | 437 | |
| Fluorinated anatase TiO2 NS, (001):(101) of 72:28 |
— | 18 W Hg lamp (245 nm) | 0.21 | 443 | |
| Anatase TiO2 nanoparticles (14 nm) | — | Hg lamp (254 nm) | 0.40 | 436 | |
| Hollow anatase TiO2 single crystals | 1 wt% RuO2 | 300 W Xe lamp | 1.2 | 440 | |
| Anatase TiO2, co-exposed (001) and (101) facets | — | 300 W Xe lamp | 1.35 | 444 | |
| Titania (Ti0.91O2) nanosheets | — | 300 W Xe lamp | 1.43 | 534 | |
| Hollow anatase TiO2 mesocrystals, dominant (101) facets | 1 wt% RuO2 | 300 W Xe lamp | 1.75 | 440 | |
| Nano-sized anatase rods, dominant (010) facets | 1 wt% Pt | 300 W Xe lamp | 2.5 | 442 | |
| Cubic anatase TiO2 nanocrystals (100 ± 13 nm) | — | 300 W Xe lamp | 4.6 | 445 | |
| d | Ti-BEA zeolite (synthesis under OH− conditions) | — | 100 W Hg lamp (>250 nm) | 5.76 | 447 |
| Ti-MCM-48 zeolite (Si/Ti = 80) | — | 100 W Hg lamp (>250 nm) | 7.5 | 450 | |
| Ti-MCM-48 zeolite | 1 wt% Pt | Hg lamp (>280 nm) | 12.26 | 449 | |
| e | Anatase-rutile TiO2 (sintered in air at 773 K) | — | 450 W Hg lamp | 35.4 | 487 |
| Rutile TiO2 NP decorated on anatase TiO2 nanorods | — | 300 W Hg lamp (365 nm) | 2.36 | 489 | |
| f | Brookite TiO2 (treated under He at 493 K) | — | 300 W Xe lamp | 0.31 | 413 |
| Anatase TiO2 (treated under He at 493 K) | — | 300 W Xe lamp | 0.42 | 413 | |
| 0.2% MgO–TiO2 | — | 550 W Xe lamp, AM 1.5G filter | 1.0 | 434 | |
| 3% NaOH-surface modification TiO2 (ST01) | — | 300 W Xe lamp | 8.7 | 429 | |
| 1.0% MgO–TiO2 | 2 wt% Pt | 100 W Xe lamp (320–780 nm) | 11.0 | 435 | |
| Crystalline core–amorphous shell black TiO2 (TiO2@TiO2−x) | — | Solar irradiation | 14.3 | 433 | |
| g | 4 nm PbS quantum dots sensitized-TiO2 | 2 wt% Cu | 300 W Xe lamp | 0.58; 0.31 (C2H6) | 492 |
| 1 at% CdSe quantum dots sensitized-TiO2 | 0.5 at% Pt | 300 W Xe lamp (>420 nm) | 0.6 | 491 | |
| h | TiO2 | 0.5:0.5 wt% Cu:Fe |
UVA (320–500 nm) | 0.91; 0.58 (C2H4) | 511 |
| TiO2 | Pt–Cu2O | 300 W Xe lamp (300–400 nm) | 1.42 | 455 | |
| TiO2 | 0.5% Ag | 100 W Xe lamp (320–780 nm) | 2.1 | 507 | |
| TiO2 | 0.5% Au | 100 W Xe lamp (320–780 nm) | 3.1 | 507 | |
| TiO2 | 0.5% Rh | 100 W Xe lamp (320–780 nm) | 3.5 | 507 | |
| TiO2 | 0.5% Pd | 100 W Xe lamp (320–780 nm) | 4.3 | 507 | |
| TiO2 | 0.5% Pt | 100 W Xe lamp (320–780 nm) | 5.2 | 507 | |
| TiO2 | 1.5 wt% Cu | 200 W Xe lamp (320–780 nm) | 8.7 | 514 | |
| TiO2–SiO2 | 0.5 wt% Cu | Xe lamp | 10 | 509 | |
| TiO2 | 0.89 wt% Pt | 200 W Xe lamp (320–780 nm) | 11 | 514 | |
| TiO2 | 12.5 wt% Pd7Cu1 | 300 W Xe lamp | 19.6 | 513 | |
| TiO2 | 0.92:1.7 wt% Pt:Cu |
200 W Xe lamp (320–780 nm) | 33 | 514 | |
| TiO2 | 1.0 wt% Pd | 150 W Hg lamp | 118.2 | 508 | |
| TiO 2 (thin film) | 1.5 wt% Au | 1000 W Xe lamp (AM 1.5) | 210 | 407 | |
| TiO 2 (thin film) | 1.5 wt% Cu | 1000 W Xe lamp (AM 1.5) | 280 | 407 | |
| SrTiO 3 /TiO 2 | Au 3 Cu | 300 W Xe lamp | 3770 (CO) | 538 | |
| 421.2 | |||||
| 190.1 (C 2 H 6 ) | |||||
| 73.3 (C 2 H 4 ) | |||||
| 40.8 (C 3 H 6 ) | |||||
| 1D TiO 2 single crystals | 0.94 at% Pt (1.04 nm) | 400 W Xe lamp (250–388 nm) | 1361 | 397 | |
| TiO 2 (thin film) | 1.5 wt% Au 1 Cu 2 | 1000 W Xe lamp (AM 1.5) | 2200 | 407 | |
| i | TiO2-Activated carbon | — | 15 W UVA (365 nm) | 4.31 | 535 |
| Carbon@TiO2 hollow structure | — | Simulated solar light (>200 nm) | 4.79 | 536 | |
| TiO2–graphene 2D sandwich-like hybrid nanosheets | — | 500 W Xe lamp | 8; 16.8 (C2H6) | 537 | |
| TiO2–multi-walled carbon nanotube | — | 15 W UVA (365 nm) | 11.74 | 535 | |
Strategies followed for the enhancement of the performance of TiO2 have been applied in other semiconductors. Carbon and nitrogen co-doped sodium titanate nanotubes showed enhanced CH4 formation (9.75 μmol g−1 h−1) that was 2.7 times higher as compared to the un-doped counterpart (3.7 μmol g−1 h−1).460 Oxygen-vacancy-rich ultrathin W18O49 nanowires up to several micrometers long with unique optical properties, i.e. NIR absorption and blue-light emission, revealed an unexpected ability to photochemically reduce carbon dioxide to methane as a result of the defect structure.461 Nano-structuralization approaches have been shown to facilitate efficient charge separation and transportation, and superior p-CO2RR performance,462–465 as recently reviewed by Kang and colleagues.466 A resulting advantage is to tune different crystal facets to optimize the reactivity of a photocatalyst for targeted reactions. Although quasi-cubic-like WO3 crystals with nearly equal percentages of (002), (200) and (020) facets remained relatively inactive for the p-CO2RR, rectangular sheet-like WO3 crystals with predominant (002) facets and CB elevated to ca. 0.3 eV were able to generate CH4 in the presence of H2O vapor.467
Among the non-titania evaluated photocatalysts, Zn2GeO4 and ZnGa2O4 have shown great promise to facilitate the p-CO2RR to CH4 due to their high CB positions (Fig. 25). When compared with TiO2, however, both materials exhibited significantly larger band gaps, 4.5 and 4.45 eV, respectively, limiting their light absorption. To tentatively narrow the band gap of these materials and improve light harvesting, several approaches have been pursued.468,469 Ultra-long 7 nm-thick 20–50 nm-wide Zn2GeO4 nanobelts were shown to greatly enhance the activity to CH4 in the presence of water vapor,470 with the results being ascribed to an enhancement of its intrinsically low specific surface area as later corroborated by analogous studies.471,472 Similarly porous Ga2O3 with a 2-fold enhanced specific surface area unveiled a 3-fold CO2 uptake and a 4-fold CH4 yield, as compared to commercial counterparts.473 CO2 adsorption on Zn2GeO4(001) was shown to be higher than on (010) facets and, in particular, near surface oxygen vacancy centers, where CO2 dissociates to CO filling the vacancy with its oxygen atom.403 Single-crystalline ultrathin ZnGa2O4 nanosheets with exposed (110) facets attained a 35% enhanced CH4 yield with an apparent quantum efficiency (AQE) of 0.035% at a wavelength of 280 ± 15 nm as compared to meso-ZnGa2O4, despite the latter possessing a 2-fold larger surface area.474 Similarly, Yan demonstrated the impact of surface energetics as compared with sole surface area enhancement.475 ZnGa2O4 nanocubes with preferentially exposed (100) facets exhibited improved performance in the p-CO2RR to produce CH4 (6.7-fold yield enhancement), or water splitting into H2, when compared with a mesoporous counterpart with a 10-fold larger specific surface area. In accordance with results previously discussed for N-doped TiO2, the incorporation of nitrogen over Ga and Ge-based semiconductors has been shown to dramatically narrow the band gap to ca. 2.6 eV, enhancing light absorption in the visible region.476–478
A well-known limitation of using sulfides is reflected in their poor photostability and short lifetimes. However, attractive native candidates for visible light-driven photocatalysis, e.g. cadmium sulfide (CdS) with a narrow bandgap, have shown adequate thermodynamic requirements for water splitting, CO2RR, and dye degradation.479 The CB edge position is relatively more negative than that of TiO2, indicating that the photogenerated electrons possess stronger reduction power in the reaction. In addition to the observed tendency toward photocorrosion, deficient adsorption capacity toward reactants, and interparticle aggregation, the photocatalytic activity of the pure CdS semiconductor material remains far from the requirements for practical application.480 Following an obvious parallelism to the incorporation of oxygen vacancies on oxide materials, sulfide-based photocatalysts have shown enhanced p-CO2RR activity in the presence of induced sulfur vacancies on the surface acting as adsorptive sites for the CO2 molecule.481 Vertically oriented Cu2S and CuS nanowalls, nanoleafs and rods bearing three-dimensional (3D) dendritic nanobranches were additionally assessed under AM 1.5 simulated light irradiation in the p-CO2RR toward CH4 formation with a production rate as high as 38 μmol m−2 h−1.482
The slightly more reductive CB niobates, with a perovskite structure, share several properties with titanates (nontoxicity, stability, indirect wide band gap).483,484 NaNbO3 nanowires with a diameter of about 100 nm and a length up to several tens micrometers showed enhanced activity as compared with microstructured-counterparts for CH4 production, possibly due to crystallinity, surface-to-volume ratio and anisotropic aspect.485 Conversely, lamellar perovskite niobates showed great promise for the separation and transfer of photoexcited charges, deposition of co-catalysts, and selective intercalation of reactants. Because of the hydrophilic nature and Brønsted acidity, 50 nm-thick, 300–500 nm-wide acidic HNb3O8 nanobelts prepared by a hydrothermal method also showed a two-fold and ten-fold enhanced photocatalytic activity toward CH4 formation as compared with a KNb3O8-counterpart and P25 particles, respectively.486
 | ||
| Fig. 28 Schematic representation of formed heterojunctions in the p-CO2RR. (a) Sensitization, (b) staggered type heterojunctions, and Z-scheme systems (c) in the absence of a mediator or (d) in the presence of conductive solid mediators (e.g. Au nanoparticles). | ||
In quantum confined spherical NPs, or quantum dots (QDs), the quantum confinement effect gives rise to new properties that are unachievable in traditional bulk materials.490 Acting as an antenna in the adsorption of visible light, the incorporation of QDs with tunable size (and therefore tunable band gaps) has been assessed in the p-CO2RR. Toward the enhancement of Pt–TiO2 photocatalytic systems under visible light irradiation (λ > 420 nm), the incorporation of CdSe QDs primarily yielded CH4.491 The sensitization mechanism was achieved through the transfer of generated electrons from the CB of CdSe to that of TiO2, as the quantum confinement shifts the CB of CdSe QDs to higher energies, facilitating charge injection (Fig. 28a). The authors later incorporated PbS QDs within a 3 to 5 nm range over Cu–TiO2, with similar CH4 formation rates (below 1 μmol g−1 h−1) being obtained in both studies.492 In addition, on the basis of its well-known propensity to photocorrosion, the PbS QD-incorporated photocatalyst became inactive after ca. 8 h of continuous visible light illumination. With increasing application in the development of solar cells, photodetectors and biological labels, PbS QD are nonetheless expected to benefit from recent works toward efficient synthesis methods within a wide size range employing lower cost, less toxicity and safer precursors.493 Recently, halide perovskite CsPbBr3 QDs under AM 1.5G simulated illumination showed steady generation and injection of electrons into CO2 with a selectivity of over 93.3% and a CO2 reduction up to 23.7 μmol g−1 h−1.494 The rate of electron consumption could be increased by 25.5% upon incorporation into a graphene oxide support, due to enhanced electron extraction and transport.
In staggered-type heterojunctions, the photogenerated electrons in the higher CB of one semiconductor are transferred to the lower CB of the other semiconductor, while holes are transferred from the lower valence band (VB) to the higher VB (Fig. 28b). In composite semiconductors with a p–n junction mechanism, charge carrier separation results in the additional separation of occurring redox reactions, which prevents back and side reactions and benefits the product yield. In addition to appropriate band-gaps, high surface areas and enhanced surface basicity to improve CO2 activation are desirable features in the selection of these semiconductors. Cases of interest have been reported in recent years.495,496 Z-scheme structures, composed of two semiconductors with the staggered alignment of band structures, have received increasing attention in water splitting research. The CB and VB of the two semiconductors do not satisfy the redox potential requirements for an overall reaction alone, but they can separately perform reduction or oxidation half reactions. The semiconductor-based system performs in the absence of a mediator, or alternatively in the presence of a reversible redox mediator (Fig. 28c) or conductive solid mediators e.g. Au NPs or graphene (Fig. 28d).497
4.3. Co-catalyst effect
Previous sections have highlighted how the reported activity of semiconductor materials for p-CO2RR, although promising, remains relatively low (significantly below 10 μmol CH4 g−1 h−1 for individually evaluated TiO2), with poor selectivity toward methane and, in particular, longer carbon-chains being reported. Water splitting-inspired incorporation of co-catalysts has emerged as a promising strategy to promote the quantum yield and enhance the activity of the resulting photocatalysts. Upon illumination, electrons are expected to flow from the semiconductor to the metallic surface having a larger work function at the interface and therefore act as an electron-sink, facilitating CO2 activation for the formation of bent CO2˙− species (Fig. 29a).498 With enriched electron density, metal co-catalysts are prone to promote CH4 formation, which, although being thermodynamically more feasible than the formation of CO, requires a higher number of transferred electrons.499 While possibly participating in the reaction, it is also important to highlight that the co-catalyst may further compete for light absorption, direct product selectivity, or impact the overall process efficiency in an undesirable way, e.g. by promoting the HER.500 Because the co-catalyst can act as a light absorber and the catalyst, questions arise regarding the possibility of the photoexcitation of carriers in the metal being central to the activity, with the semiconductor acting only as a support. While this issue has been assessed in the literature, it deserves further attention in upcoming studies.501 | ||
| Fig. 29 (a) Schematic representation of light-induced CO2RR in the presence of a co-catalyst acting as an electron-sink, with electron flow from the semiconductor to the metallic surface having a larger work function at the interface. (b) Plasmon-enhanced p-CO2RR as typically explained by the direct charge transfer mechanism and local electric field enhancement. | ||
While assessing the size effect of Pt NPs coated on films with a unique one-dimensional structure of TiO2 single crystals, the activity of these catalysts under UV light irradiation was shown to be optimal at a particle size of 1.04 ± 0.08 nm, with a calculated quantum yield of CH4 of 2.41%, a methane production rate of ca. 1361 μmol g−1 h−1, and a dramatic enhancement as compared with a pristine TiO2-film and a P25 reference.397 With smaller particle sizes, the energy level of Pt clusters was hinted to increase to a level higher than the CB edge of TiO2, thus hindering the transfer of photogenerated electrons. The proposed mechanism and strategy are depicted in Fig. 30.
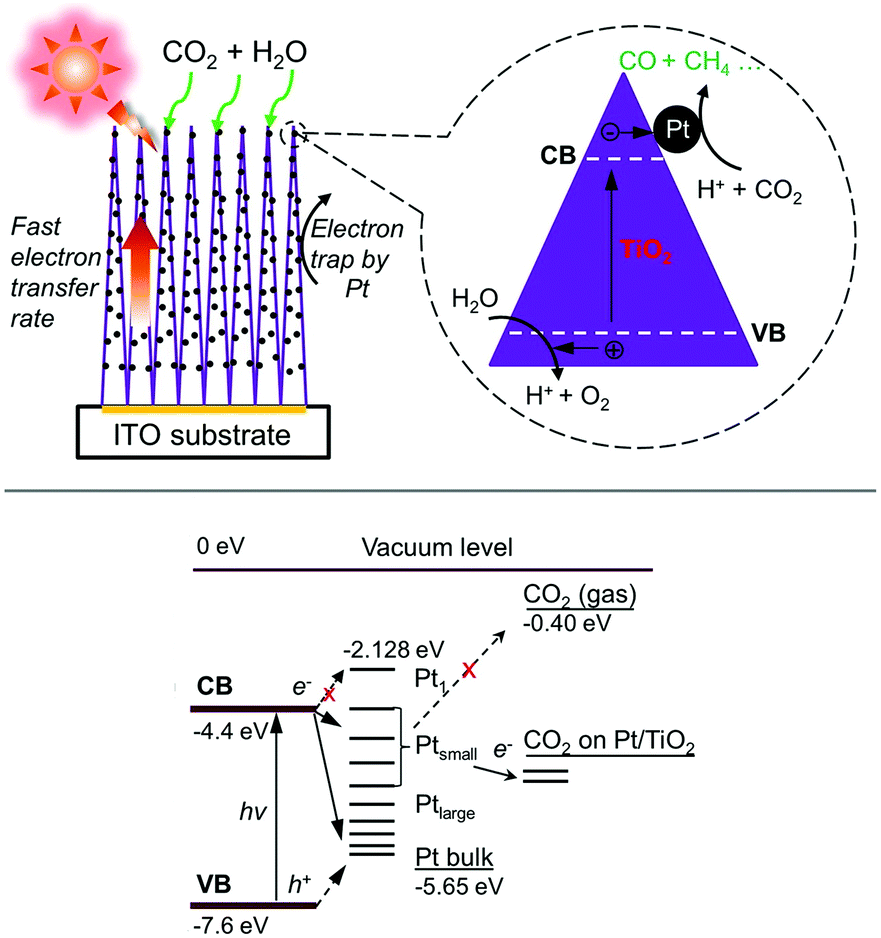 | ||
| Fig. 30 Schematic diagram of the p-CO2RR in the presence of Pt–TiO2 nanostructured films with a remarkable CH4 production rate (1361 μmol g−1 h−1). Photogenerated electrons are proposed to move fast inside the highly oriented TiO2 single crystals, flowing to the Pt deposits where the redox reaction occurs to convert CO2 into CH4. Readapted with permission from ref. 397. Copyright 2017 American Chemical Society. | ||
Cu (identified to be Cu2O by XPS) incorporated on TiO2–SiO2 systems509 and Cu-moieties on the surface of nitrogen-doped TiO2 nanotubes revealed enhanced CH4 formation.510 The presence of Fe as a co-dopant in Cu/TiO2 systems was found to synergistically reduce CO2 to C2H4 (0.58 μmol g−1 h−1), while suppressing the production of CH4 (0.91 μmol g−1 h−1).511 CH4, C2H4 and C2H6 were obtained as major hydrocarbon products (total production rate of 574 mmol cm−2 h−1) over bimetallic Cu–Pt co-catalysts (Cu0.33–Pt0.67) loaded on periodically modulated double-walled TiO2 nanotube arrays.512 Similar highly isolated Cu sites in a Pd lattice were shown to generate selective sites for CH4 synthesis (19.6 μmol g−1 h−1).513 According to synchrotron–radiation characterizations and theoretical simulations, the isolation of Cu atoms in the Pd7Cu1–TiO2 photocatalyst was claimed to (1) provide active sites for enhanced CO2 adsorption and suppressed HER and (2) elevate the d-band center of Cu sites for improved CO2 activation. The Cu/Pt/TiO2 catalyst prepared by stepwise photodeposition gave a significantly enhanced CH4 formation rate and was characterized using XPS, AES and high-sensitivity low-energy ion scattering spectroscopy.514 These findings suggest the formation of a Pt@Cu2O core–shell structure with optimal Cu content when the latter reached 1.7 wt%. The Cu2O shell was believed to provide the active sites for the activation of CO2, whereas the Pt core mainly worked for the extraction of photogenerated electrons from TiO2.
In the photochemical society, the integration of plasmonic metal NPs on the surface of semiconductors has been largely assessed.520 Most importantly, it has been shown that in contrast to semiconductor-based photocatalysts, photocatalytic quantum efficiencies on these plasmonic metal nanostructures can be enhanced by selected light intensities and operating temperatures.521 The resulting plasmon-enhanced performance is typically explained either by the direct charge transfer mechanism or local electric field enhancement (Fig. 29b).501 SPR excitation can be alternatively used to transfer photon energy in an indirect photocatalytic approach, via a plasmon-mediated electron-transfer pathway to molecular photocatalysts or other adjacent metals,522 or directly utilized by the reactants and reaction intermediates adsorbed on the metal surface.523 Over the surface of metal NPs, proposed mechanistic paths for CO2RR have been recently presented by Jain and colleagues, as depicted schematically in Fig. 31.263 Establishing relationships between the electronic structures of adsorbate species and plasmonic photocatalytic signatures have been pinpointed as a requirement for optimizing the rational design of these systems toward the unique control of the reaction selectivity.524 Of equal importance, when CO2RR occurs directly over the surface of these metal NPs, the geometry of the plasmonic nanostructure, which is well established to determine their SPR wavelength and primary plasmon decay mechanism, has not yet been evaluated in detail regarding the resulting efficiency, selectivity, and stability of the overall photocatalytic system. In each case nonetheless, the presumed existence of abundant charge-carriers has been expected to enhance the likelihood for the occurrence of multiple e−/H+ reactions. The assumption remains, however, under examination in the literature. Very recently, Linic highlighted a plausible skepticism for the presumed correlation between multiple electronic excitations required in multi-electron reactions (with at least 8 for the simplest hydrocarbon) utilizing plasmonic nanostructures, based on the limited occurrence of single-electron excitations taking place in these nanostructures, the corresponding short lifetimes of the photoinduced charge carriers, and the relatively long period between excitation events, particularly at low light intensities.525 Despite the need for further investigation of the mechanistic utilization of the properties of plasmonic nanostructures, emerging examples have continuously fomented interest in this domain. Recently, Ag-loaded TiO2 nanotube arrays showed enhanced performance in the CO2RR to produce CH4 under visible light, with only minor amounts of CH3OH being detected.526 This unique SPR effect and underlying hot electron generation induced by Ag NPs was further ascribed to coupling with the light scattering enhancement within the TiO2 nanotube arrays effectively increasing light absorption and utilization by the Ag NP deposited in the inner spaces of these nanotubes. Similarly, Au has found suitable use in a wide range of photo and electrocatalytic applications, assuring cost-effectiveness on the basis of low required loadings and the durability of the materials toward photocorrosion. Coupling Au NPs with TiO2 in the design of visible-light-responsive nanohybrid architectures with synergistically strong SPR effects has been shown to enhance charge separation and photocatalytic efficiency.527 Experimental and calculated results based on Au@TiO2 yolk–shell hollow spheres have even demonstrated the formation of C2H6.528 Excluding resonance photon scattering and the direct transfer of hot electrons from Au NPs to TiO2, in agreement with a lack of intimate contact between both components to allow the SPR-excited hot electrons to overcome the Schottky barrier between the metals and semiconductors, Zou and coworkers have attributed the observed results to local electromagnetic field enhancement. Equally remarkable, a commercial P25 modified by Au–Cu alloy NPs as a thin film unveiled a CH4 formation rate greater than 2200 μmol g−1 h−1, resulting in a 44-fold and a 10-fold enhancement as compared to bare TiO2 films and corresponding Au/TiO2 and Cu/TiO2, respectively.407 The selectivity toward CH4 was claimed to arise from the presence of Cu bonding to the reaction intermediates (CO*), while the surface plasmon band of Au could introduce a visible light photoresponse. In agreement, hot electrons on TiO2 favored proton reduction through the HER, and the hot-electron-induced reduction of Cu2O to metallic Cu promoted selective activation of CO2 to CH4. A schematic representation of the overall mechanism is depicted in Fig. 32.
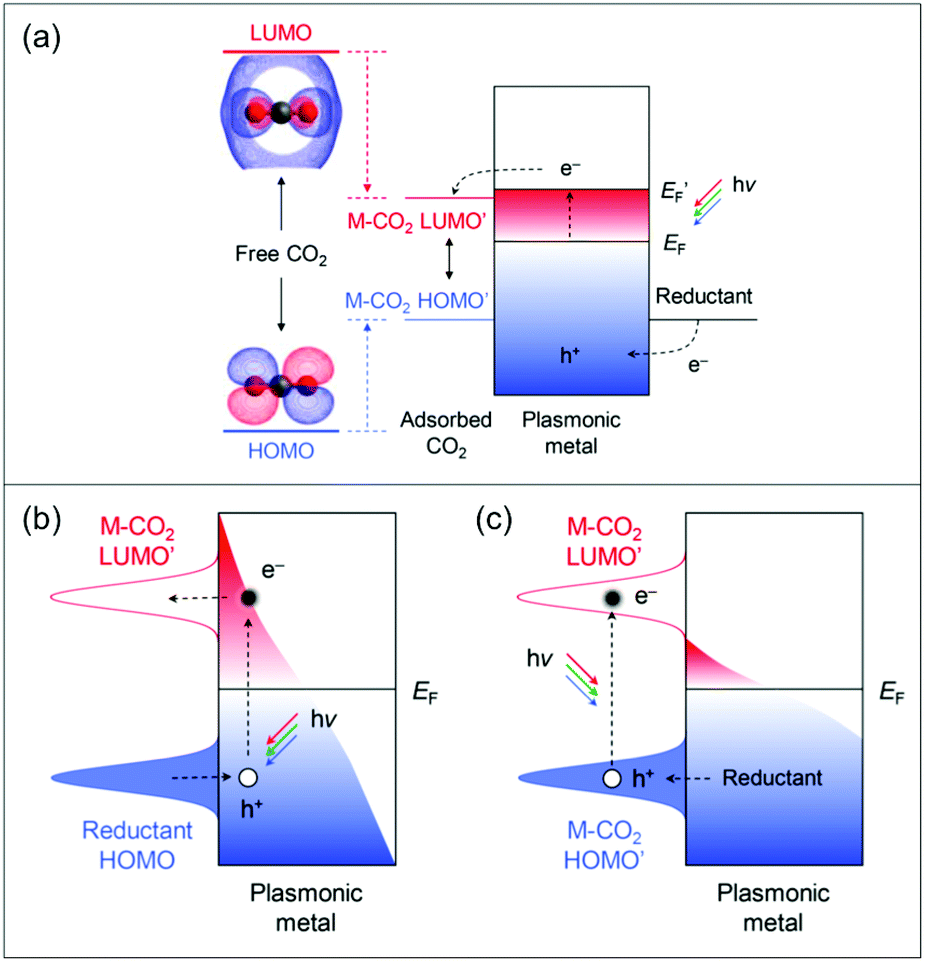 | ||
| Fig. 31 Potential mechanistic scenarios for CO2RR by plasmonic photocatalysis, as summarized by Jain and colleagues. Adsorption of CO2 onto the surface of a metal NP prompting orbital hybridization, with a reduction of its HOMO–LUMO energy gap from the gas-phase value to a new HOMO′–LUMO′ gap, which can be excited under visible light irradiation. (a) Under steady-state light irradiation, the resulting electron injection/hole removal cathodically charges the NP, elevating the Fermi level to a quasi-Fermi level (EF′). Energetic electrons occupying states above the EF′ are thus injected into the LUMO′ of the adsorbed CO2. (b) LSPR excitation followed by dephasing or energy relaxation within the metal NP transiently produces hot electrons above the EF. A fraction of these hot electrons has been proposed to be scattered into the unoccupied LUMO′ states of the CO2 adsorbate, thus prompting a vibrational CO2 activation on the surface of the NPs. (c) Direct HOMO′–LUMO′ photoexcitation of the M–CO2 complex is facilitated in accordance with the reduction of the HOMO′–LUMO′ gap of the M–CO2 complex. Readapted with permission from ref. 263. Copyright 2017 American Chemical Society. | ||
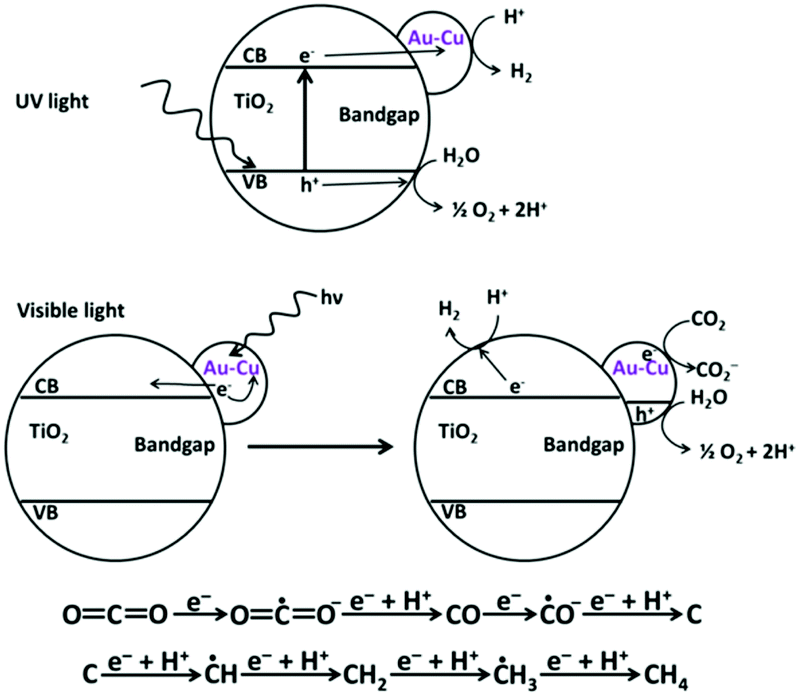 | ||
| Fig. 32 Schematic representation of the proposed mechanism under UV and visible light for Au–Cu alloy NPs decorated on TiO2 as photocatalysts, to rationalize the influence of the irradiation wavelength range on the product distribution. A proposed route for CO2RR to CH4 in water is represented and in accordance with the carbine mechanism given in Fig. 27. Reprinted with permission from ref. 407. Copyright 2014 American Chemical Society. | ||
Electrons in the CB of TiO2 are believed to easily migrate to graphene and to still possess enough reduction potential to promote p-CO2RR.531 Solvent-exfoliated graphene on GO–TiO2 was reported to exhibit improved p-CO2RR activity, especially under visible light.532 Alternatively, the material dimensionality has been claimed to play a key role in determining the spectral response, charge transfer and reaction specificity of carbon–TiO2 nanohybrid composites.533–535 Carbon@TiO2 hollow spherical structures with enhanced visible-light absorption and CO2 adsorption have unveiled an improved photogenerated charge transfer efficiency and a CH4 formation rate of 4.79 μmol g−1 h−1.536 Ascribed to facilitated charge carrier transfer, sandwich-like TiO2–graphene hybrid nanosheets showed a remarkable C2H6 production of 16.8 μmol g−1 h−1 (Fig. 33).537
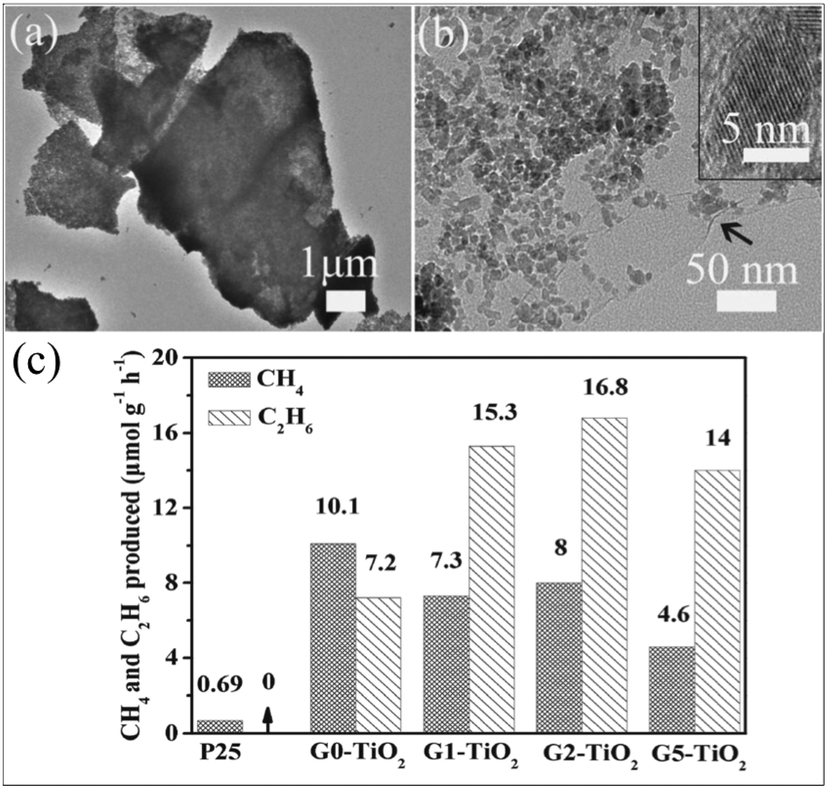 | ||
| Fig. 33 (a and b) TEM photographs of sandwich-like TiO2–graphene hybrid nanosheets (G2–TiO2). The inset of (b) shows an HR-TEM image evidencing the lattice fringes of the TiO2 nanoparticles, corresponding to the (101) plane of anatase TiO2. (c) Catalytic assessment results showing a remarkable C2H6 production. The weight contents of graphene were designated as x (wt%) in these Gx–TiO2 nanocomposites, with the evaluated samples having 0, 1%, 2%, and 5% graphene. Readapted with permission from ref. 537. Copyright 2013 John Wiley and Sons. | ||
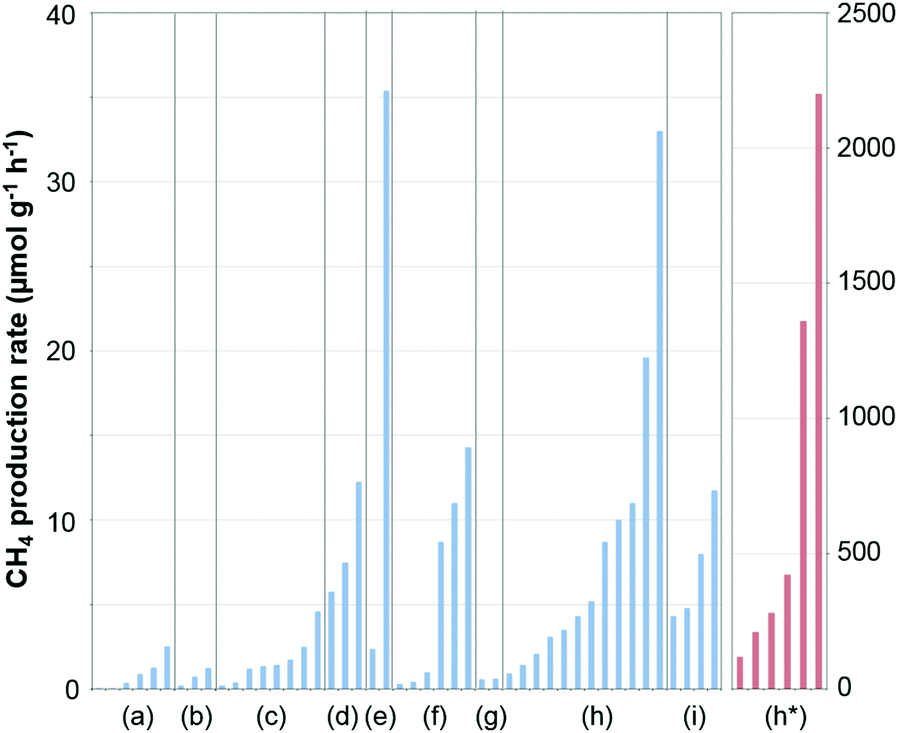 | ||
| Fig. 34 Reported CH4 production rates (μmol g−1 h−1) on TiO2-based catalytic systems. Reports were organized by attempted strategy and compared with (a) commonly reported pristine TiO2 references. (b) Single crystal electrodes, (c) nanostructures, (d) highly dispersed TiO2 on aluminosilicate frameworks, (e) phase junctions between TiO2 polymorphs, (f) following surface modification, (g) quantum-dot sensitization, (h) co-catalyst incorporation, and (i) carbon-incorporation. Results for (h*) represent exceptionally high reports in the literature following co-catalyst incorporation, with results being discussed in corresponding sections. Details have been summarized in Table 3. | ||
4.4. The photoelectrocatalytic (PEC) CO2RR
Despite the evident advantages of electrocatalysis as a renewable source of energy in the reduction of CO2, the above-discussed section on the e-CO2RR has largely underlined the large amounts of electricity required to overcome the high-energy barriers for the reutilization of CO2. PEC materials rationally integrate light-responsive materials prompting a significant reduction in electricity consumption and higher overall process efficiency. The use of PEC systems comprising a working photoelectrode, a counter electrode and a reference electrode has gained traction as an attractive alternative in analogous H2 technologies. The PEC system is expected to attain higher efficiency levels with the application of an external bias to promote the separation of photogenerated charge carriers. Semiconductors have been reported as photoanodes, photocathodes, or simultaneously applied in designed PEC systems. As photocathodes, p-type semiconductors have been featured in a number of exploratory works with Pt or carbon rods as dark anodes of choice. Here, the photoelectrode harvests light to generate charge electrons and holes and performs a half-cell reaction to reduce CO2. Assessed materials listed in recent comprehensive reviews include p-GaP, p-Si, p-GaAs, p-InP, p-Si, p-ZnTe, p-NiO, p-Co3O4, N-Ta2O5, and CuO.539 Nonetheless, along with stability concerns, the efficiency toward CO2RR has remained low as the reduction of water is prone to preferentially occurring over the surface of p-type semiconductors. In addition, on photocathode-driven PEC systems, the selectivity toward HCOOH and CO is notably superior to that of the CH4, which emerges as a by-product with low FE values.Alternatively, the integration of n-type semiconductor photoanodes with superior cost-effectiveness as compared to p-type cathodes has emerging potential.540 In particular, photoanode-driven systems, e.g. WO3, TiO2, BiVO4, Ge-doped GaN, may in a seemingly facile manner couple with the above-listed electrocatalysts of choice for the e-CO2RR (cf. Section 3). In 1996, Ichikawa reported a TiO2 photoanode-driven single unit PEC system operated under pulse bias that was claimed to have superior activity and selectivity toward a stable hydrocarbon formation over the Cu cathode with FE of 44% and 24% for CH4 and C2H4, respectively.541 A PEC system incorporating TiO2 photoanodes illuminated with 100 mW cm2 AM 1.5G light and a Cu2O cathode was reported by Chang for CH4 synthesis, attaining FE of 54.6%.542 In a similar fashion, Grimes and colleagues reported a PEC photocathode comprised of p-type Si nanowire arrays, with and without copper sensitization, paired with n-type TiO2 nanotube array films as the photoanode. Along with CO and substantial rates of hydrogen generation due to water photoelectrolysis, CH4, C2H4 and even traces of C3–C4 hydrocarbons could be observed at an applied potential of 1.5 VAg/AgCl.543 A Cu cathode coupled with visible-light responsive WO3 as the photoanode unveiled superior energy efficiency and FE toward CH4 and C2H4 of 42.3% and 4.0% at 0.55 VRHE, or alternatively 67.0% and 0.6% at 0.75 VRHE.544 Prior e-CO2RR reports on a Cu cathode include the works of Lee and colleagues using a carbonate-coordinated cobalt/BiVO4/WO3 photoanode exhibiting unique effectiveness as a water oxidation activity catalyst, comparable to state-of-the-art values.545 Most importantly, the PEC system showed stable photocurrent and a FE of 51.9% for CO and hydrocarbons in the C1–C2 range, balanced by an exclusive H2 formation.
Although the above reports and the underlying FE attained for CH4 and C2H4 may emerge as pale comparisons to the exemplary e-CO2RR reports scrutinized in Section 3, exploring the energetic advantages of PEC cells as compared to electrocatalytic platforms deserves more attention in the future. In particular, the knowledge acquired to date on Cu-based electrocatalysts could serve as a valuable guideline toward notably superior efficiencies of the systems discussed herein.
4.5. Stability issues
To date, exploratory works for the development of CO2RR photocatalysts have only sparingly assessed the durability of the reported materials. Measured stability ranges and ascribed bottlenecks either related to the material itself or the selected operating conditions have received little attention as compared to their electrocatalytic CO2RR counterparts. The number of available reports is further narrowed when solely reports regarding the synthesis of hydrocarbons are considered. In photocatalytic applications, the stability is measured in continuous cycles of 6 to 8 hours. Among the current listed state-of-the-art catalysts having competitive results, Au–Cu NPs on SrTiO3/TiO2 coaxial nanotube arrays were stable for five cycles during a 34 h test.538 Garcia corroborated the stability of the reported 1.5 wt% Au1Cu2/TiO2 (thin film) for a period of 46 h of irradiation based on the linearity found for the temporal evolution of H2 and CH4 production.407 For possible practical applications, the development of technologies for prolonging the catalyst lifetime is imperative.The photocorrosion of selected photocatalysts has emerged as one of the main issues in the development and application of durable materials in p-CO2RR systems. As the benchmark photocatalyst with high catalytic efficiency in p-CO2RR, anatase TiO2 exhibits high stability and durability with a low tendency to undergo deactivation and photocorrosion.408 In attempts to enhance light harvesting, doping with transition metals in minute quantities can replace titanium atoms in the lattice and introduce empty orbitals at energies below the CB of pure TiO2. This has been shown to lead to the reorganization of the crystal lattice, resulting in the leaching of the dopant metal out of the crystal framework, undesirable photodissolution, and enhanced catalytic decay.546 Alternatively, doping with non-metallic elements, e.g., N, S, and C, results in the introduction of occupied orbitals above the energy of the VB, narrower energy gaps and a proposed enhancement in the durability of TiO2-based photocatalysts.547
Along with studies utilizing TiO2-based materials, most reported architectures exhibit pronounced catalytic decay within the first hours of catalytic assessment. In parallel water splitting applications, preventing photocatalysts from coming into contact with the electrolyte through the introduction of dense coatings has been shown to lead solely to a limited enhancement in the resulting stability. In p-CO2RR, an example worth highlighting involves metal sulfides with photogenerated holes in their valence band not being energetic enough to oxidize water and resulting instead in their irreversible photocorrosion. Here, hole scavengers may serve as a simplified approach to extend their stability. Serving as electron sink and thus enhancing the activity of photocatalytic platforms, the incorporation of metal co-catalysts should be cautiously pondered in slowing down the photocorrosion of semiconductors by enhancing the timely consumption of photogenerated charges. The stability of the incorporated co-catalyst deserves additional attention. With plasmon-enhanced nanostructures, the stability of the incorporated noble metals depends on their redox potential, with Au emerging as the most promising candidate in these studies due to the absence of photocorrosion. Although at an underdeveloped stage, the utilization of plasmonic Au photocatalysts, with the reaction occurring directly over the surface of these metal NPs, could promising in the near future.524
4.6. Photocatalytic reactors
Although solar energy conversion continues to emerge as a valuable pathway to replace traditional fossil energy, the design and underlying viability of photocatalytic reactors are topics of concern. In a photocatalytic system, CO2 can be reduced over the photocatalyst suspended on a CO2-containing electrolyte in a rather simplified reactor, with both p-CO2RR and water oxidation occurring on different active sites present in the surface of the said photocatalyst. In the absence of hole scavengers, e.g. H2O2 and Na2SO3, which require additional costs and significantly contribute to implementation limitations, the formed p-CO2RR products are susceptible to re-oxidation by photogenerated holes or the evolved oxygen. PEC cells and, as discussed for e-CO2RR, the possibility of designing photoreactors with two separate compartments, e.g. using a cation exchange membrane (e.g. Nafion 117) are highly desirable. Schematic representations of photoelectrocatalytic reactors in two-compartment cells separated by proton-exchange membranes have been presented in Fig. 35. They reflect the configurations corresponding to the commonly utilized H-type cells in the e-CO2RR, and in PEC systems discussed in Section 4.4, for the utilization of semiconductors as photoelectrodes, photocathodes, or both. Reducing the chance of back reactions not only enhances the overall efficiency of the process but also facilitates product separation, reducing energy costs in subsequent purification processes.548 In addition to separation cost motivations, on a larger scale, physically separating the production of O2 and H2 in the reactor is necessary for safety reasons. Toward a facile process implementation however, photocatalytic-based platforms have a greater potential to be applied on a large scale as compared to photoelectrocatalytic technologies.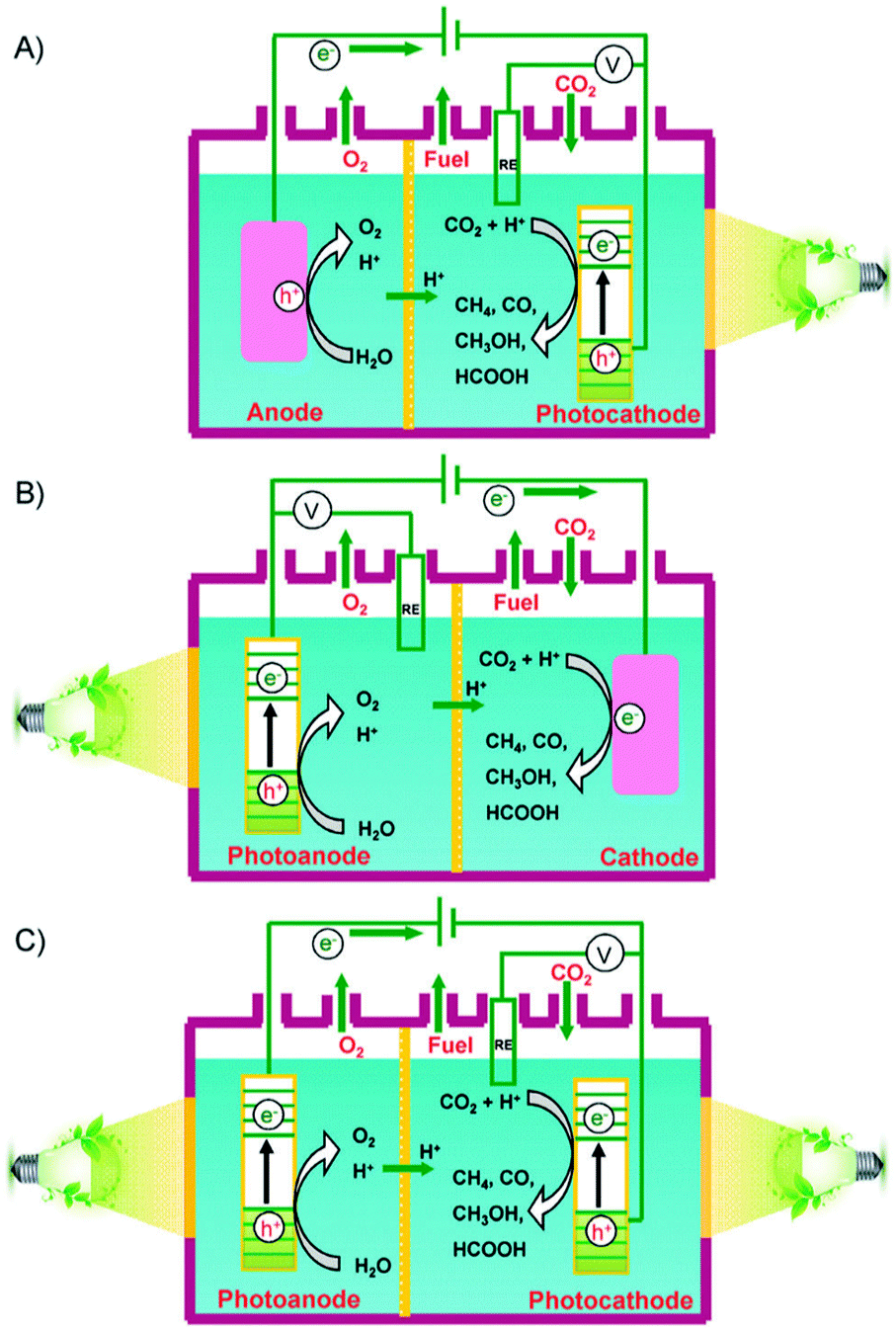 | ||
| Fig. 35 Schematic representation of photoelectrocatalytic reactors in two-compartment cells separated by proton-exchange membranes. CO2RR and water oxidation occur over the cathode and the anode sides, respectively. The alternative or simultaneous utilization of photocatalysts under light irradiation on the cathode and anode side is represented in each case. Reprinted with permission from ref. 539. Copyright The Royal Society of Chemistry 2016. | ||
The investigation of alternative photo-reactors, in addition to reactor sizing and selected configurations, catalyst loading, mass transfer considerations and selected operating conditions, has remained insufficient. Recent studies, carried out in an innovative photoreactor operating under high pressure, have shed light on the complex mechanistic pathways toward both liquid and gas phase p-CO2RR products.549 The design of photocatalytic reactors often falls into two main categories, i.e. the most commonly used slurry reactor and the fixed bed reactor. In slurry-type reactors, the selected photocatalyst is vigorously stirred to attain high dispersion and prevent sedimentation as scattering properties largely depend on time-dependent agglomeration phenomena and porosity. In the development and validation of photocatalytic tests, the reactor should be additionally designed to allow the optimized exposure of catalytically active sites to light for enhanced light harvesting efficiency. The effect of selected illumination sources and lamp position are to be taken into account in the reported rates, along with a detailed investigation of the resulting radiation field and the impact of irradiation on the reaction temperature. The importance of such requirements is directly reflected in the variations found with reference catalysts as discussed above. In addition to the reactivity of the catalyst in terms of molecules of CO2 converted, the quantum yield or efficiency, which requires measurement of the quantity of photons absorbed by the catalysts, is a measure of the effectiveness of the evaluated photocatalysts. A cautious estimation of the total transmitted radiation, transmitted non-scattered radiation, and photon back-scattered radiation exiting the system has been assessed in a limited number of works.550
4.7. Concluding remarks and analytical prospects
The production of hydrocarbons from CO2 using solar energy is a promising alternative for tackling the current climate and energy problem. To date, the design and development of p-CO2RR platforms have substantially followed the growth of water splitting technologies and the corresponding strategic pathways, which have prominently accelerated the first two mutual process steps, namely photon absorption and charge separation and transfer (i.e. electron and hole transport to surface reaction sites). Notwithstanding the exciting developments with the incorporation of visible light and NIR-responsive components, the low efficiency in the utilization of solar light in the process is still of marked concern. Similarly, H2 technologies have guided the design of CO2RR photocatalysts within a noteworthy range of materials, which is largely in contrast to the converging focus on Cu or copper oxide-derived catalysts in the e-CO2RR. Over TiO2-based photocatalysts, the most commonly applied photo-responsive material, CH4 (and alternatively CH3OH) can be the prevalent product. However, as reflected in this review, no concrete guidelines have been produced toward the synthesis of long-chain hydrocarbons to date. Reports on the formation of C2H4 or C2H6 offer limited groundwork in the establishment of analogous strategies and often fail to provide concrete mechanistic assumptions. Metal oxides and compound semiconductors with non-porous or porous nanostructures with meaningful photocatalytic activity and long-term stability remain scarce. 1D or 2D architectures can accelerate the transfer of generated carriers, with corresponding research focus being expected in the future.539 Great progress is needed in the clarification of the reaction mechanism toward the rationalization of product distribution and enhancement of the catalytic activity in the design of innovative systems.With limitations similar to the e-CO2RR systems, final gaseous and liquid photoreduction products are commonly identified and quantified separately through GC and HPLC analyses, respectively. As suggested by Gong and co-workers, a number of approaches, e.g. EPR spectroscopy, in situ DRIFTS, transient absorption spectroscopy (TAS), and scanning tunneling microscopy (STM), are expected to accelerate the chances for the discovery of results of significance by unlocking unknown mechanistic details.395 In p-CO2RR systems, the overall process effectiveness may be drawn in terms of photon efficiency or based on the amount of loaded photocatalyst. To date, however, there is no single measure to establish a direct and unambiguous comparison between increasingly complex systems. In mapping the current literature, it is hard to find a suitable baseline among the variables of the type of light, pH of the solution, CO2 pressure, and selected experimental conditions. As with other photocatalytic applications, p-CO2RR suffers from a broad selection of light sources (sunlight, solar simulators or visible light), position and distance, and a lack of comparison with results under dark conditions. In each study, detailed mass balance, product distribution and catalytic stability assessments are requested.
The results assessed herein confirm that the photo-driven production of hydrocarbons from CO2 is still far from practical consideration. The current average CH4 productivities remain below the 102 μmol g−1 h−1 mark under light irradiation, requiring an increase by orders of magnitude. To date, only a few reports have attained (limiting) production rates up to 103 μmol g−1 h−1, although the said findings and underlying conclusions are insufficient to dictate a clear direction for upcoming research works. Establishing synergetic effects in rationally designed architectures bridging a number of research strategies has appeared meaningful to date. The incorporation of co-catalysts and the effective enhancement of plasmonic nanostructures in these multi-electron processes have been underscored in this review. Nonetheless, the utilization of charge scavengers considered in the majority of the exploratory works revised here and beneficial in stability issues, impose important limitations. From the engineering and technological point of view, hole scavengers demand additional costs and significantly contribute to the projected implementation constraints. In addition, it is important to establish that the reported reaction products do not arise from the incorporated hole scavenger species, as recently outlined by Linic and colleagues.525 Finally, (often undisclosed) solar-to-fuel energy conversion efficiencies unveil the technological underdevelopment of these systems as compared with water-splitting HER systems exhibiting remarkably higher solar-to-hydrogen values.551–553 Significant improvements in the attained low conversion efficiency, product selectivity and quantum yields are required to bring p-CO2RR in aqueous solutions closer to practical application.
5. Conclusions and outlook
In this review, the recent developments and future perspectives in CO2 reutilization for its sustainable conversion to hydrocarbons have been outlined. Tentatively closing the carbon cycle through the catalytically assisted thermochemical, electrochemical, and photochemical CO2RR has unveiled promising results that may provide an answer to the growing environmental concerns and a source of alternative fuels for a sustainable future. The synthesis of longer chain hydrocarbons, toward typical gasoline and diesel carbon-chain ranges, provides the promise of a smooth transition in the utilization of alternative liquid fuels in already established infrastructures currently served by fossil fuels. Ground-breaking outcomes in this research field have, however, remained subtle to date. Mechanistic uncertainties and unidentified activation barriers for key reaction steps represent fundamental limitations to all the technologies discussed herein. Significant light is required in the identification of the nature and adsorption configuration of involved intermediate species through, for example, DFT calculations in the establishment of structure–property descriptors required in the rational design of novel catalytic systems. In addition, while the optimization of the activity, selectivity, and stability of designed catalytic systems represent decisive steps toward the eventual commercialization of such catalyst-based platforms, both CO2 capture and distillation, along with the availability of large-volume sources of pure CO2 remain acute drawbacks to the economic viability of these technologies.Although only briefly assessed, it is important to further establish potential synergies between the technologies presented herein. Within the possibilities for potential commercial breaks and large-scale application of the introduced CO2RR systems, electro- and photo-driven platforms may be valuable in the development of conventional thermochemical processes. Technical and economical analyses may be required to establish the premise of utilizing photovoltaic energy or renewable electricity to produce H2 from water to be used in t-CO2RR systems, or to directly reduce CO2 in water in either electro- or photocatalytic reactors. Although the former pathway may require up to three consecutive steps in the transformation of CO2 to hydrocarbons, the final long-term production goal should be to develop one step with the eventual facile integration of CO2 capture from the atmosphere. Evidently, this will markedly depend on the development of the technologies reviewed herein and parallel water splitting platforms as the most promising sources of renewable H2.
Fig. 36 establishes a comparison of the current the state-of-the-art thermochemical, electrochemical, and photochemical CO2RR technologies, their levels of progress, and the promise of their development. Please note that the selected scales are a mere qualitative comparison of said parameters for the three technologies and represent the authors’ analysis of the discussed problems. Currently, thermochemically-driven CO2RR processes have the greatest potential to be applied on a large scale, offering the highest reaction rates and product yields, and being receptively compatible with the existing industrial plants as compared to the photo- or electrocatalytic platforms. These platforms have unveiled the most favorable shifts to long-chain hydrocarbons with high activity and selectivity in CO2 methanation. In addition, benefiting from decades of thoroughly documented research and light shed from conventional FT works, we have illustrated the promising features of Ni and Co-based, and Fe-based catalytic systems on CO2RR to methane and long-chain hydrocarbons, respectively. Most of the current catalytic libraries reflect the increasing complexity of synthesis and therefore the need to urgently inspect new approaches to the design of novel catalytic systems. Energetic demands and associated costs to the production of renewable H2 from additional sources represent the most notable limitations to the economically sustainable implementation of t-CO2RR. The production of CO2-neutral renewable fuels simultaneously demands the production of H2 from a sustainable source, with H2O, the most naturally abundant source of hydrogen that is inexpensive and readily available, being the ideal candidate. While thermochemical approaches have largely established the use of H2 from alternative sources, photochemical and electrochemical approaches aspire to the in situ supply of hydrogen from water. However, the simultaneous activation of two very stable molecules in the synthesis of hydrocarbons reflects extensive energy consumption. Insufficient socio-economic driving forces and a clear lack of industrial commitment and investment incentives are additional drawbacks to the technological development of the CO2RR.
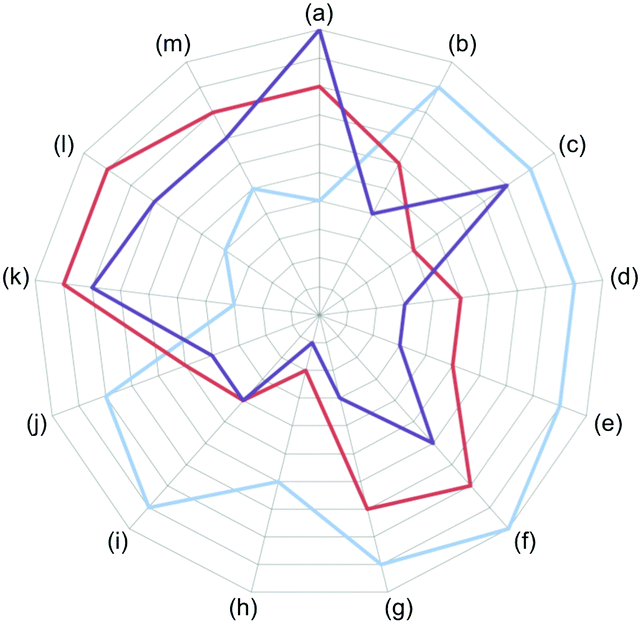 | ||
| Fig. 36 A comparison of catalytically assisted thermochemical (blue), electrochemical (red), and photochemical (purple) CO2RR platforms according to (a) the corresponding ideally predicted energy efficiency of these technologies, (b) the potential breakdown of the commercial barriers based on current reports, (c) relative scalability, (d) compared activity of state-of-the-art catalysts, (e) attained shifts to longer chain hydrocarbons, reported selectivity levels toward (f) methane, (g) olefins, and (h) long-chain hydrocarbons, (i) currently reported catalyst stability, and (j) current mechanistic clarity driving the design of novel catalytic systems. (k and l) Compare recent catalyst development and the emergence of published breakthroughs of high impact. (m) Reflects the current number of reports involving the facile synthesis of the employed catalyst. Please note that the selected scales are a mere qualitative comparison of the above-listed technological metrics for each platform and represent the authors’ analysis of the discussed problems based on the extensive list of exploratory works included in this review article. | ||
Considering the use of renewable photovoltaic or wind-derived electricity, in the electrochemical reduction of CO2 noteworthy breakthroughs have been achieved in recent years toward the enhancement of the formation of C2H4 at the expense of reduced CH4 yields, combining lower overpotentials with increasingly high Faradaic efficiencies. This effort toward highly selective C2H4 to CH4 ratios further evades costly paraffin–olefin separation processes.554 We have highlighted the significance of works carried out with Cu2O-derived electrodes and the derived number of currently intertwined possible contributions to these findings, e.g. grain boundaries, subsurface oxygen species and surface roughness. The conversion of CO2 to C2–C4 olefins is a major pursuit in clean energy research, further believed herein to unlock the desirable mechanistic pathways toward C–C bonding required for the production of long-chain hydrocarbons. In addition, the electrochemical production of valuable ethylene and n-propanol products is preferable toward CH4 production, since these are in strong price competition with the abundant and cheap natural gas. To date, however, steps toward the envisioned commercialization still require practical reaction rates above 300 mA cm−2, superior FE, and catalytic stability, which currently remains below the 50 h mark.
The solar-driven reutilization of CO2 is ideal from an economic point of view and offers significant potential in scalability as compared to electro- and photoelectrocatalytic platforms. Although the state-of-the-art production of fuels or chemicals from CO2 using solar energy is still far from a practical consideration, rich knowledge has been accumulated for understanding the key factors that determine the catalytic performances. Following analogous studies in H2 technologies, the research on the photo-driven CO2RR is wide and urgently requires unifying guidelines such as mechanistic details and enhanced material characterization. The need for better quantification, i.e. CH4 formation rates and solar-to-fuel efficiencies, toward the possible comparison of current advances is desirable. To date, such alternative approaches in the CO2RR to high-energy-content fuels remain rather primary and face major drawbacks, with reported catalysts failing to yield adequate conversion efficiencies or produce long-chain products. Research on both the photo- and electro-driven reduction of CO2, currently based on traditional trial and error approaches, would largely benefit from investments in experimental in situ and operando techniques and model systems, complemented with theoretical efforts to unveil performance descriptors in the rational design of new catalytic systems. Equally, in both fields, efforts are expected in the development of suitable reactors and operating conditions, reflecting a parallelism with water splitting technologies to serve as a major guideline in these research fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Research Fellowship Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2017H1D3A1A02054206), and the mid-career researcher program (2017R1A2A1A05022387).Notes and references
- M. Sudiro and A. Bertucco, Energy, 2009, 34, 2206 CrossRef CAS.
- M. Sudiro, A. Bertucco, F. Rugger and M. Fontana, Energy Fuels, 2008, 22, 3894 CrossRef CAS.
- C. Knocke and J. Vogt, Eng. Life Sci., 2009, 9, 96 CrossRef CAS.
- P. J. le, B. Williams and L. M. L. Laurens, Energy Environ. Sci., 2009, 3, 554 Search PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16 CrossRef PubMed.
- BP, BP Statistical Review of World Energy June 2015, 2015.
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719 CrossRef CAS PubMed.
- IPCC 2014, Climate Change 2014: Mitigation of Climate Change, Cambridge University Press, UK and New York, NY, USA, 2014 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- D. Roemmich, J. Church, J. Gilson, D. Monselesan, P. Sutton and S. Wijffels, Nat. Clim. Change, 2015, 5, 240 CrossRef.
- A. Bernstein, E. H. Sargent, A. Aspuru-Guzik, R. Cogdell, G. R. Fleming, R. Van Grondelle and M. Molina, Nature, 2016, 538, 30 CrossRef PubMed.
- IEA, World Energy Investment Outlook 2014 Factsheet, 2014.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426 CrossRef CAS.
- W. M. Budzianowski, Energy efficient solvents for CO2 capture by gas-liquid absorption, Springer, 2017, pp. 1–11 Search PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243 CrossRef CAS.
- IEA, Energy Technology Perspectives 2012—How to Secure a Clean Energy Future, 2012.
- B. I. McNeil, R. J. Matear, R. M. Key, J. L. Bullister and J. L. Sarmiento, Science, 2003, 299, 235 CrossRef CAS PubMed.
- T. Coffey, D. R. Hardy, G. E. Besenbruch, K. R. Schultz, L. C. Brown and J. P. Dahlburg, Def. Horiz., 2003, 36, 1 Search PubMed.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884 RSC.
- H. Kolbe and E. Lautemann, Justus Liebigs Ann. Chem., 1869, 113, 125 CrossRef.
- E. Solvay, US Pat., 263981 A, 1882 Search PubMed.
- B. Carl and M. Wilhelm, US Pat., 1429483 A, 1922 Search PubMed.
- Z. Xie, X. Zhang, Z. Zhang and Z. Zhou, Adv. Mater., 2017, 29, 1605891 CrossRef PubMed.
- S. Yang, Y. Qiao, P. He, Y. Liu, Z. Cheng, J. Zhu and H. Zhou, Energy Environ. Sci., 2017, 10, 972 RSC.
- Y. Qiao, J. Yi, S. Wu, Y. Liu, S. Yang, P. He and H. Zhou, Joule, 2017, 1, 359 CrossRef CAS.
- Z. Yang, X. Gao and Z. Liu, Curr. Opin. Green Sustain. Chem., 2016, 1, 13 CrossRef.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995 RSC.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804 CrossRef PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed.
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326 RSC.
- H. M. T. Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130 CrossRef.
- S. Lewandowski, 4th Annual Asia Chemical Conference, 2016.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536 RSC.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910 CrossRef CAS PubMed.
- U.S. Energy Information Administration: http://www.eia.gov.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739 CrossRef CAS PubMed.
- G. A. Mills and F. W. Steffgen, Catal. Rev.: Sci. Eng., 1974, 8, 159 CrossRef.
- S. Eckle, H.-G. Anfang and R. J. Behm, J. Phys. Chem. C, 2011, 115, 1361 CrossRef CAS.
- M. R. Prairie, A. Renken, J. G. Highfield, K. R. Thampi and M. Grätzel, J. Catal., 1991, 129, 130 CrossRef CAS.
- S.-J. Choe, H.-J. Kang, S.-J. Kim, S.-B. Park, D.-H. Park and D.-S. Huh, Bull. Korean Chem. Soc., 2005, 26, 1682 CrossRef CAS.
- A. Trovarelli, C. Mustazza, G. Dolcetti, J. Kaspar and M. Graziani, Appl. Catal., 1990, 65, 129 CrossRef CAS.
- C.-K. Kuei and M.-D. Lee, Can. J. Chem. Eng., 1991, 69, 347 CrossRef CAS.
- M. J. L. Ginés, A. J. Marchi and C. R. Apesteguía, Appl. Catal., A, 1997, 154, 155 CrossRef.
- J. L. Falconer and A. E. Zaǧli, J. Catal., 1980, 62, 280 CrossRef CAS.
- A. de Klerk, Green Chem., 2008, 10, 1249 RSC.
- A. de Klerk, Energy Environ. Sci., 2011, 4, 1177 RSC.
- A. Borgschulte, N. Gallandat, B. Probst, R. Suter, E. Callini, D. Ferri, Y. Arroyo, R. Erni, H. Geerlings and A. Züttel, Phys. Chem. Chem. Phys., 2013, 15, 9620 RSC.
- R. A. Fiato, E. Iglesia, G. W. Rice and S. L. Soled, Stud. Surf. Sci. Catal., 1998, 114, 339 CrossRef.
- S.-R. Yan, K.-W. Jun, J.-S. Hong, S.-B. Lee, M.-J. Choi and K.-W. Lee, Korean J. Chem. Eng., 1999, 16, 357 CrossRef CAS.
- T. Riedel, G. Schaub, K.-W. Jun and K.-W. Lee, Ind. Eng. Chem. Res., 2001, 40, 1355 CrossRef CAS.
- U. Rodemerck, M. Holena, E. Wagner, Q. Smejkal, A. Barkschat and M. Baerns, ChemCatChem, 2013, 5, 1948 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1984, 87, 352 CrossRef CAS.
- E. Iglesia, S. C. Reyes and R. J. Madon, J. Catal., 1991, 129, 238 CrossRef CAS.
- B. Zhao, Y.-X. Pan and C. Liu, Catal. Today, 2012, 194, 60 CrossRef CAS.
- J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H.-L. Sit and W. Y. Teoh, Appl. Catal., B, 2017, 219, 715 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Grätzel, Nature, 1987, 327, 506 CrossRef CAS.
- X. Su, J. Xu, B. Liang, H. Duan, B. Hou and Y. Huang, J. Energy Chem., 2016, 25, 553 CrossRef.
- P. J. Lunde and F. L. Kester, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 27 CrossRef CAS.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282 CrossRef CAS.
- N. M. Gupta, V. S. Kamble, V. B. Kartha, R. M. Iyer, K. Ravindranathan Thampi and M. Grätzel, J. Catal., 1994, 146, 173 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1981, 68, 67 CrossRef CAS.
- S. Sharma, Z. Hu, P. Zhang, E. W. McFarland and H. Metiu, J. Catal., 2011, 278, 297 CrossRef CAS.
- S. Takenaka, T. Shimizu and K. Otsuka, Int. J. Hydrogen Energy, 2014, 29, 1065 CrossRef.
- P. Panagiotopoulou, Appl. Catal., A, 2017, 542, 63 CrossRef CAS.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315 RSC.
- Q. Lin, X. Y. Liu, Y. Jiang, Y. Wang, Y. Huang and T. Zhang, Catal. Sci. Technol., 2014, 4, 2058 RSC.
- A. Kim, C. Sanchez, G. Patriarche, O. Ersen, S. Moldovan, A. Wisnet, C. Sassoye and D. P. Debecker, Catal. Sci. Technol., 2016, 6, 8117 RSC.
- A. Kim, D. P. Debecker, F. Devred, V. Dubois, C. Sanchez and C. Sassoye, Appl. Catal., B, 2018, 220, 615 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203 CrossRef CAS.
- F. Solymosi, I. Tombácz and J. Koszta, J. Catal., 1985, 95, 578 CrossRef CAS.
- Z. Zhang, A. Kladi and X. E. Verykios, J. Catal., 1994, 148, 737 CrossRef CAS.
- C. Deleitenburg and A. Trovarelli, J. Catal., 1995, 156, 171 CrossRef CAS.
- S. Ma, W. Song, B. Liu, H. Zheng, J. Deng, W. Zhong, J. Liu, X.-Q. Gong and Z. Zhao, Catal. Sci. Technol., 2016, 6, 6128 RSC.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237 CrossRef CAS.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141 CrossRef CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, Appl. Catal., A, 2008, 344, 45 CrossRef CAS.
- M. A. Vannice and R. L. Garten, Ind. Eng. Chem. Process Des. Dev., 1979, 18, 186 CAS.
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot and D. Uzio, Catal. Commun., 2015, 58, 11 CrossRef CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92 CrossRef CAS.
- H. Y. Kim, H. M. Lee and J.-N. Park, J. Phys. Chem. C, 2010, 114, 7128 CrossRef CAS.
- F.-W. Chang, M.-T. Tsay and S.-P. Liang, Appl. Catal., A, 2001, 209, 217 CrossRef CAS.
- F.-W. Chang, M.-S. Kuo, M.-T. Tsay and M.-C. Hsieh, Appl. Catal., A, 2003, 247, 309 CrossRef CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 1346 CrossRef CAS.
- A. Aljishi, G. Veilleux, J. A. H. Lalinde and J. Kopyscinski, Appl. Catal., A, 2018, 549, 263 CrossRef CAS.
- J.-K. Lee, B.-I. An, D. Kim, S.-H. Min, J.-S. Jung and S.-H. Lee, Bull. Korean Chem. Soc., 2007, 28, 121 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359 CrossRef CAS.
- C.-S. Chen, C. S. Budi, H.-C. Wu, D. Saikia and H.-M. Kao, ACS Catal., 2017, 7, 8367 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, Chem. Commun., 2015, 51, 1728 RSC.
- M. Cai, J. Wen, W. Chu, X. Cheng and Z. Li, J. Nat. Gas Chem., 2011, 20, 318 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Appl. Surf. Sci., 2016, 383, 248 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527 CrossRef CAS.
- Z. Cheng, B. J. Sherman and C. S. Lo, J. Chem. Phys., 2013, 138, 14702 CrossRef PubMed.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108 CrossRef CAS.
- L. Atzori, M. G. Cutrufello, D. Meloni, R. Monaci, C. Cannas, D. Gazzoli, M. F. Sini, P. Deiana and E. Rombi, Int. J. Hydrogen Energy, 2017, 42, 32 CrossRef.
- L. Atzori, M. G. Cutrufello, D. Meloni, C. Cannas, D. Gazzoli, R. Monaci, M. F. Sini and E. Rombi, Catal. Today, 2018, 299, 183 CrossRef CAS.
- M. Guo and G. Lu, Catal. Commun., 2014, 54, 55 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J.-D. Grunwaldt, J. Catal., 2015, 327, 48 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, H. Yang, D. Nie, L. Qi and X. Lian, RSC Adv., 2017, 7, 18199 RSC.
- A. L. Kustov, A. M. Frey, K. E. Larsen, T. Johannessen, J. K. Nørskov and C. H. Christensen, Appl. Catal., A, 2007, 320, 98 CrossRef CAS.
- D. Tian, Z. Liu, D. Li, H. Shi, W. Pan and Y. Cheng, Fuel, 2013, 104, 224 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. C. Jung, D. J. Koh, H. Lim, C. Byun and I. K. Song, J. Ind. Eng. Chem., 2012, 18, 243 CrossRef CAS.
- K. Ray and G. Deo, Appl. Catal., B, 2017, 218, 525 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams, B. H. Davis and H. D. Willauer, Energy Fuels, 2009, 23, 4190 CrossRef CAS.
- H. Schulz, E. van Steen and M. Claeys, Top. Catal., 1995, 2, 223 CrossRef CAS.
- J. Patzlaff, Y. Liu, C. Graffmann and J. Gaube, Appl. Catal., A, 1999, 186, 109 CrossRef CAS.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S.-S. Nam, K.-W. Jun, M.-J. Choi, G. Kishan and K.-W. Lee, Appl. Catal., A, 1999, 186, 201 CrossRef CAS.
- C. G. Visconti, L. Lietti, E. Tronconi, P. Forzatti, R. Zennaro and E. Finocchio, Appl. Catal., A, 2009, 355, 61 CrossRef CAS.
- F. Tihay, A. C. Roger, G. Pourroy and A. Kiennemann, Energy Fuels, 2002, 16, 1271 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, J. CO2 Util., 2013, 3–4, 102 CrossRef CAS.
- J. Lahtinen, T. Anraku and G. A. Somorjai, Catal. Lett., 1994, 25, 241 CrossRef CAS.
- M. Voß, G. Fröhlich, D. Borgmann and G. Wedler, J. Catal., 1999, 187, 348 CrossRef.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397 CrossRef CAS.
- W. Li, A. Zhang, X. Jiang, C. Chen, Z. Liu, C. Song and X. Guo, ACS Sustainable Chem. Eng., 2017, 5, 7824 CrossRef CAS.
- G. Melaet, W. T. Ralston, C.-S. Li, S. Alayoglu, K. An, N. Musselwhite, B. Kalkan and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 2260 CrossRef CAS PubMed.
- V. Iablokov, S. K. Beaumont, S. Alayoglu, V. V. Pushkarev, C. Specht, J. Gao, A. P. Alivisatos, N. Kruse and G. A. Somorjai, Nano Lett., 2012, 12, 3091 CrossRef CAS PubMed.
- R. Razzaq, C. Li, M. Usman, K. Suzuki and S. Zhang, Chem. Eng. J., 2015, 262, 1090 CrossRef CAS.
- F. Diehl and A. Y. Khodakov, Oil Gas Sci. Technol., 2009, 64, 11 CrossRef CAS.
- T. N. Phaahlamohlaka, D. O. Kumi, M. W. Dlamini, R. Forbes, L. L. Jewell, D. G. Billing and N. J. Coville, ACS Catal., 2017, 7, 1568 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 9898 CrossRef CAS PubMed.
- S. K. Beaumont, S. Alayoglu, C. Specht, N. Kruse and G. A. Somorjai, Nano Lett., 2014, 14, 4792 CrossRef CAS PubMed.
- S. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. Zheng, V. Iablokov, Z. Liu, J. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778 CrossRef CAS.
- H. H. Shin, L. Lu, Z. Yang, C. J. Kiely and S. McIntosh, ACS Catal., 2016, 6, 2811 CrossRef CAS.
- M. Schubert, S. Pokhrel, A. Thomé, V. Zielasek, T. M. Gesing, F. Roessner, M. Lutz and M. Bäumer, Catal. Sci. Technol., 2016, 6, 7449 RSC.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799 CrossRef CAS.
- C. Swalus, M. Jacquemin, C. Poleunis, P. Bertrand and P. Ruiz, Appl. Catal., B, 2012, 125, 41 CrossRef CAS.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798 CrossRef CAS PubMed.
- K. Cheng, M. Virginie, V. V. Ordomsky, C. Cordier, P. A. Chernavskii, M. I. Ivantsov, S. Paul, Y. Wang and A. Y. Khodakov, J. Catal., 2015, 328, 139 CrossRef CAS.
- H. Schulz, T. Riedel and G. Schaub, Top. Catal., 2005, 32, 117 CrossRef CAS.
- T. Riedel, H. Schulz, G. Schaub, K.-W. Jun, J.-S. Hwang and K.-W. Lee, Top. Catal., 2003, 26, 41 CrossRef CAS.
- N. Utsis, R. Vidruk-Nehemya, M. V. Landau and M. Herskowitz, Faraday Discuss., 2016, 188, 545 RSC.
- C. Yang, H. Zhao, Y. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134, 15814 CrossRef CAS PubMed.
- R. A. Fiato, S. L. Soled, G. W. Rice and S. Miseo, US Pat., 5140049, 1992 Search PubMed.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti, P. Forzatti, G. Iaquaniello, E. Palo, B. Picutti and F. Brignoli, Appl. Catal., B, 2017, 200, 530 CrossRef CAS.
- T. Xie, J. Wang, F. Ding, A. Zhang, W. Li, X. Guo and C. Song, J. CO2 Util., 2017, 19, 202 CrossRef CAS.
- M. Albrecht, U. Rodemerck, M. Schneider, M. Bröring, D. Baabe and E. V. Kondratenko, Appl. Catal., B, 2017, 204, 119 CrossRef CAS.
- N. G. Gallegos, A. M. Alvarez, M. V. Cagnoli, J. F. Bengoa, S. G. Marchetti, R. C. Mercader and A. A. Yeramian, J. Catal., 1996, 161, 132 CrossRef CAS.
- G. Kishan, M.-W. Lee, S.-S. Nam, M.-J. Choi and K.-W. Lee, Catal. Lett., 1998, 56, 215 CrossRef CAS.
- L. Torrente-Murciano, R. S. L. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496 RSC.
- M. C. Bahome, L. L. Jewell, D. Hildebrandt, D. Glasser and N. J. Coville, Appl. Catal., A, 2005, 287, 60 CrossRef CAS.
- P. Serp and E. Castillejos, ChemCatChem, 2010, 2, 41 CrossRef CAS.
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835 CrossRef PubMed.
- D. Mattia, M. D. Jones, J. P. O’Byrne, O. G. Griffiths, R. E. Owen, E. Sackville, M. McManus and P. Plucinski, ChemSusChem, 2015, 8, 4064 CrossRef CAS PubMed.
- D. R. Minett, J. P. O’Byrne, S. I. Pascu, P. K. Plucinski, R. E. Owen, M. D. Jones and D. Mattia, Catal. Sci. Technol., 2014, 4, 3351 RSC.
- P. H. Choi, K.-W. Jun, S.-J. Lee, M.-J. Choi and K.-W. Lee, Catal. Lett., 1996, 40, 115 CrossRef CAS.
- M.-J. Choi, J.-S. Kim, H.-K. Kim, S.-B. Lee, Y. Kang and K.-W. Lee, Korean J. Chem. Eng., 2001, 18, 646 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Appl. Catal., A, 2010, 373, 112 CrossRef CAS.
- L. Xu, Q. Wang, D. Liang, X. Wang, L. Lin, W. Cui and Y. Xu, Appl. Catal., A, 1998, 173, 19 CrossRef CAS.
- M. Amoyal, R. Vidruk-Nehemya, M. V. Landau and M. Herskowitz, J. Catal., 2017, 348, 29 CrossRef CAS.
- T. Numpilai, T. Witoon, N. Chanlek, W. Limphirat, G. Bonura, M. Chareonpanich and J. Limtrakul, Appl. Catal., A, 2017, 547, 219 CrossRef CAS.
- S.-R. Yan, K.-W. Jun, J.-S. Hong, M.-J. Choi and K.-W. Lee, Appl. Catal., A, 2000, 194–195, 63 CrossRef CAS.
- T. Li, Y. Yang, C. Zhang, X. An, H. Wan, Z. Tao, H. Xiang, Y. Li, F. Yi and B. Xu, Fuel, 2007, 86, 921 CrossRef CAS.
- M. Al-Dossary, A. A. Ismail, J. L. G. Fierro, H. Bouzid and S. A. Al-Sayari, Appl. Catal., B, 2015, 165, 651 CrossRef CAS.
- S. Li, A. Li, S. Krishnamoorthy and E. Iglesia, Catal. Lett., 2001, 77, 197 CrossRef CAS.
- T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 311, 66 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Catal. Commun., 2011, 15, 88 CrossRef CAS.
- S.-C. Lee, J.-H. Jang, B.-Y. Lee, J.-S. Kim, M. Kang, S.-B. Lee, M.-J. Choi and S.-J. Choung, J. Mol. Catal. A: Chem., 2004, 210, 131 CrossRef CAS.
- A. Erdöhelyi, M. Pásztor and F. Solymosi, J. Catal., 1986, 98, 166 CrossRef.
- F. Solymosi, A. Erdöhelyi and T. Bánsági, J. Catal., 1981, 68, 371 CrossRef CAS.
- M. A. Henderson and S. D. Worley, J. Phys. Chem., 1985, 89, 1417 CrossRef CAS.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89 CrossRef CAS.
- M. Yamasaki, H. Habazaki, K. Asami, K. Izumiya and K. Hashimoto, Catal. Commun., 2006, 7, 24 CrossRef CAS.
- C. de Leitenburg, A. Trovarelli and J. Kaspar, J. Catal., 1997, 166, 98 CrossRef CAS.
- H. Song, J. Yang, J. Zhao and L. Chou, Chin. J. Catal., 2010, 31, 21 CrossRef CAS.
- N. Takezawa, H. Terunuma, M. Shimokawabe and H. Kobayashib, Appl. Catal., 1986, 23, 291 CrossRef CAS.
- A. M. Abdel-Mageed, D. Widmann, S. E. Olesen, I. Chorkendorff, J. Biskupek and R. J. Behm, ACS Catal., 2015, 5, 6753 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170 CrossRef CAS.
- C. K. Vance and C. H. Bartholomew, Appl. Catal., 1983, 7, 169 CrossRef CAS.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298 CrossRef CAS PubMed.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Int. J. Hydrogen Energy, 2014, 39, 10090 CrossRef CAS.
- Q. Sun, J. Ye, C. Liu and Q. Ge, Greenhouse Gases: Sci. Technol., 2014, 4, 140 CrossRef CAS.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705 CrossRef CAS PubMed.
- J. Wu, C. Wen, X. Zou, J. Jimenez, J. Sun, Y. Xia, M.-T. Fonseca Rodrigues, S. Vinod, J. Zhong, N. Chopra, I. N. Odeh, G. Ding, J. Lauterbach and P. M. Ajayan, ACS Catal., 2017, 7, 4497 CrossRef CAS.
- T. Inui, H. Hara, T. Takeguchi, K. Ichino, J. B. Kim, S. Iwamoto and S. B. Pu, Energy Convers. Manage., 1997, 38, S385 CrossRef CAS.
- O. Martin and J. Pérez-Ramírez, Catal. Sci. Technol., 2013, 3, 3343 RSC.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155 RSC.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969 CrossRef CAS PubMed.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296 CrossRef CAS PubMed.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834 CrossRef CAS PubMed.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061 CrossRef CAS PubMed.
- P. Gao, F. Li, H. Zhan, N. Zhao, F. Xiao, W. Wei, L. Zhong, H. Wang and Y. Sun, J. Catal., 2013, 298, 51 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311 CrossRef CAS.
- Y. Wang, S.-L. Chen, Y.-L. Gao, Y.-Q. Cao, Q. Zhang, W.-K. Chang and J. B. Benziger, ACS Catal., 2017, 7, 5572 CrossRef CAS.
- Y. Tan, M. Fujiwara, H. Ando, Q. Xu and Y. Souma, Ind. Eng. Chem. Res., 1999, 38, 3225 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065 CrossRef CAS PubMed.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84 CrossRef CAS PubMed.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019 CrossRef CAS PubMed.
- R. A. Dalla Betta, A. G. Piken and M. Shelef, J. Catal., 1975, 40, 173 CrossRef CAS.
- T. Szailer, É. Novák, A. Oszkó and A. Erdőhelyi, Top. Catal., 2007, 46, 79 CrossRef CAS.
- R. Ryoo, K. Cho and F. Marques Mota, in Zeolites in Sustainable Chemistry: Synthesis, Characterization and Catalytic Applications, ed. F.-S. Xiao and X. Meng, 2015, pp. 101–148 Search PubMed.
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Chaumonnot, D. Uzio, T. S. Nguyen and L. Piccolo, Appl. Catal., A, 2015, 504, 504 CrossRef CAS.
- F. Goodarzi, L. Kang, F. R. Wang, F. Joensen, S. Kegnæs and J. Mielby, ChemCatChem, 2018, 10, 1566 CrossRef CAS.
- Y. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 17420 RSC.
- W. Zhen, F. Gao, B. Tian, P. Ding, Y. Deng, Z. Li, H. Gao and G. Lu, J. Catal., 2017, 348, 200 CrossRef CAS.
- C. Janke, M. S. Duyar, M. Hoskins and R. Farrauto, Appl. Catal., B, 2014, 152–153, 184 CrossRef CAS.
- M. Agnelli, M. Kolb and C. Mirodatos, J. Catal., 1994, 148, 9 CrossRef CAS.
- F. Ocampo, B. Louis and A.-C. Roger, Appl. Catal., A, 2009, 369, 90 CrossRef CAS.
- S. He, C. Li, H. Chen, D. Su, B. Zhang, X. Cao, B. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 1040 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Catal. Today, 2015, 251, 34 CrossRef CAS.
- H. D. Willauer, R. Ananth, M. T. Olsen, D. M. Drab, D. R. Hardy and F. W. Williams, J. CO2 Util., 2013, 3–4, 56 CrossRef CAS.
- M. P. Rohde, D. Unruh and G. Schaub, Ind. Eng. Chem. Res., 2005, 44, 9653 CrossRef CAS.
- S.-C. Lee, J.-S. Kim, W. C. Shin, M.-J. Choi and S.-J. Choung, J. Mol. Catal. A: Chem., 2009, 301, 98 CrossRef CAS.
- D. Sun, F. M. Khan and D. S. A. Simakov, Chem. Eng. J., 2017, 329, 165 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276 CrossRef.
- S. T. Sie and R. Krishna, Appl. Catal., A, 1999, 186, 55 CrossRef CAS.
- J.-S. Kim, S. Lee, S.-B. Lee, M.-J. Choi and K.-W. Lee, Catal. Today, 2006, 115, 228 CrossRef CAS.
- D. B. Levin and R. Chahine, Int. J. Hydrogen Energy, 2010, 35, 4962 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62 RSC.
- S. Saeidi, F. Fazlollahi, S. Najari, D. Iranshahi, J. J. Klemeš and L. L. Baxter, J. Ind. Eng. Chem., 2017, 49, 1 CrossRef CAS.
- J. Ren, S. Ouyang, H. Xu, X. Meng, T. Wang, D. Wang and J. Ye, Adv. Energy Mater., 2017, 7, 1601657 CrossRef.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, J. Am. Chem. Soc., 2014, 136, 6798 CrossRef CAS PubMed.
- N. M. Haegel, R. Margolis, T. Buonassisi, D. Feldman, A. Froitzheim, R. Garabedian, M. Green, S. Glunz, H.-M. Henning, B. Holder, I. Kaizuka, B. Kroposki, K. Matsubara, S. Niki, K. Sakurai, R. A. Schindler, W. Tumas, E. R. Weber, G. Wilson, M. Woodhouse and S. Kurtz, Science, 2017, 356, 141 CrossRef CAS PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451 CrossRef CAS.
- K. W. Frese Jr. and S. Leach, J. Electrochem. Soc., 1985, 132, 259 CrossRef.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897 CrossRef CAS.
- G. O. Larrazábal, A. J. Martín and J. Pérez-Ramírez, J. Phys. Chem. Lett., 2017, 8, 3933 CrossRef PubMed.
- S. Back, H. Kim and Y. Jung, ACS Catal., 2015, 5, 965 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 16105 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Chem. Lett., 1987, 1665 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309 RSC.
- Y. Hori, A. Murata and Y. Yoshinami, J. Chem. Soc., Faraday Trans., 1991, 87, 125 RSC.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1 CrossRef CAS.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1989, 136, 1982 CrossRef CAS.
- Y. Hori, in Handbook of Fuel Cells – Fundamentals, Technology and Applications, ed. W. Vielstich, H. A. Gasteiger and A. Lamm, John Wiley & Sons, Ltd, 2010 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112 RSC.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 CrossRef.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466 CrossRef CAS PubMed.
- M. Watanabe, M. Shibata, A. Katoh, M. Azuma and T. Sakata, Denki Kagaku, 1991, 59, 508 CAS.
- T. E. Teeter and P. Van Rysselberghe, J. Chem. Phys., 1954, 22, 759 CrossRef CAS.
- K. Ito, T. Murata and S. Ikeda, Bull. Nagoya Inst. Technol., 1975, 27, 209 CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073 CrossRef CAS PubMed.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902 RSC.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311 RSC.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075 CrossRef CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53 CrossRef CAS.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058 CrossRef CAS.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354 CrossRef CAS.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56 CrossRef CAS.
- R. Reske, M. Duca, M. Oezaslan, K. J. P. Schouten, M. T. M. Koper and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 2410 CrossRef CAS.
- A. S. Varela, C. Schlaup, Z. P. Jovanov, P. Malacrida, S. Horch, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. C, 2013, 117, 20500 CrossRef CAS.
- D. Friebel, F. Mbuga, S. Rajasekaran, D. J. Miller, H. Ogasawara, R. Alonso-Mori, D. Sokaras, D. Nordlund, T.-C. Weng and A. Nilsson, J. Phys. Chem. C, 2014, 118, 7954 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., 2013, 125, 7423 CrossRef.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789 CrossRef CAS PubMed.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161 RSC.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111 CrossRef.
- J.-F. Xie, Y.-X. Huang, W.-W. Li, X.-N. Song, L. Xiong and H.-Q. Yu, Electrochim. Acta, 2014, 139, 137 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978 CrossRef CAS PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76 RSC.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682 CrossRef CAS.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1 CrossRef CAS.
- H. Mistry, F. Behafarid, R. Reske, A. S. Varela, P. Strasser and B. Roldan Cuenya, ACS Catal., 2016, 6, 1075 CrossRef CAS.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20 CrossRef CAS PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312 CrossRef CAS PubMed.
- M. N. Hossain, J. Wen and A. Chen, Sci. Rep., 2017, 7, 3184 CrossRef PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796 CrossRef CAS PubMed.
- Y.-G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, Langmuir, 2014, 30, 15053 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337 CrossRef CAS.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853 CrossRef CAS PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68 CrossRef CAS PubMed.
- K. W. Frese Jr., J. Electrochem. Soc., 1991, 138, 3338 CrossRef.
- J. Xiao, A. Kuc, T. Frauenheim and T. Heine, J. Mater. Chem. A, 2014, 2, 4885 RSC.
- Y.-J. Zhang and A. A. Peterson, Phys. Chem. Chem. Phys., 2015, 17, 4505 RSC.
- H. Shibata, J. A. Moulijn and G. Mul, Catal. Lett., 2008, 123, 186 CrossRef CAS.
- S. Lee and J. Lee, ChemSusChem, 2015, 9, 333 CrossRef PubMed.
- X. Nie, G. L. Griffin, M. J. Janik and A. Asthagiri, Catal. Commun., 2014, 52, 88 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231 CrossRef CAS PubMed.
- R. Kas, R. Kortlever, A. Milbrat, M. T. M. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194 RSC.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824 RSC.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. Roldan Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585 CrossRef CAS PubMed.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606 CrossRef CAS PubMed.
- A. D. Handoko, C. W. Ong, Y. Huang, Z. G. Lee, L. Lin, G. B. Panetti and B. S. Yeo, J. Phys. Chem. C, 2016, 120, 20058 CrossRef CAS.
- A. Engelbrecht, M. Hämmerle, R. Moos, M. Fleischer and G. Schmid, Electrochim. Acta, 2017, 224, 642 CrossRef CAS.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285 CrossRef CAS PubMed.
- C. Liu, M. P. Lourenço, S. Hedström, F. Cavalca, O. Diaz-Morales, H. A. Duarte, A. Nilsson and L. G. M. Pettersson, J. Phys. Chem. C, 2017, 121, 25010 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorovic, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103 CrossRef.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974 CrossRef CAS PubMed.
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Adv. Energy Mater., 2017, 7, 1602114 CrossRef.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. R. Cuenya, ACS Nano, 2017, 11, 4825 CrossRef CAS PubMed.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., 2015, 127, 14914 CrossRef.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- M. Watanabe, M. Shibata, A. Katoh, T. Sakata and M. Azuma, J. Electroanal. Chem. Interfacial Electrochem., 1991, 305, 319 CrossRef CAS.
- J. He, K. E. Dettelbach, D. A. Salvatore, T. Li and C. P. Berlinguette, Angew. Chem., 2017, 129, 6164 CrossRef.
- Z. P. Jovanov, H. A. Hansen, A. S. Varela, P. Malacrida, A. A. Peterson, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2016, 343, 215 CrossRef CAS.
- Z. Yin, D. Gao, S. Yao, B. Zhao, F. Cai, L. Lin, P. Tang, P. Zhai, G. Wang, D. Ma and X. Bao, Nano Energy, 2016, 27, 35 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Katoh, M. Azuma and T. Sakata, J. Electrochem. Soc., 1991, 138, 3382 CrossRef CAS.
- P. Hirunsit, W. Soodsawang and J. Limtrakul, J. Phys. Chem. C, 2015, 119, 8238 CrossRef CAS.
- X. Guo, Y. Zhang, C. Deng, X. Li, Y. Xue, Y.-M. Yan and K. Sun, Chem. Commun., 2015, 51, 1345 RSC.
- W. Zhao, L. Yang, Y. Yin and M. Jin, J. Mater. Chem. A, 2014, 2, 902 RSC.
- F. Jia, X. Yu and L. Zhang, J. Power Sources, 2014, 252, 85 CrossRef CAS.
- S. Ishimaru, R. Shiratsuchi and G. Nogami, J. Electrochem. Soc., 2000, 147, 1864 CrossRef CAS.
- J.-P. Grote, A. R. Zeradjanin, S. Cherevko, A. Savan, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, J. Catal., 2016, 343, 248 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100 CrossRef CAS.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. M. Koper, Chem. Commun., 2016, 52, 10229 RSC.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119, 26875 CrossRef CAS.
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681 CrossRef CAS PubMed.
- X.-F. Li, Q.-K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X.-H. Zhang, Z.-Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218 CrossRef CAS PubMed.
- H. Bin Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140 CrossRef.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63 CrossRef.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893 RSC.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113 CrossRef CAS.
- X. Wang, A. S. Varela, A. Bergmann, S. Kühl and P. Strasser, ChemSusChem, 2017, 10, 4642 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935 RSC.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef CAS PubMed.
- F. Marques Mota, D. L. T. Nguyen, J.-E. Lee, H. Piao, J.-H. Choy, Y. J. Hwang and D. H. Kim, ACS Catal., 2018, 8, 4364 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. S. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- X. Zou, M. Liu, J. Wu, P. M. Ajayan, J. Li, B. Liu and B. I. Yakobson, ACS Catal., 2017, 7, 6245 CrossRef CAS.
- W. Li, N. Fechler and T. J. Bandosz, Appl. Catal., B, 2018, 234, 1 CrossRef CAS.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114 RSC.
- S. Wasmus, E. Cattaneo and W. Vielstich, Electrochim. Acta, 1990, 35, 771 CrossRef CAS.
- G. Kyriacou and A. Anagnostopoulos, J. Electroanal. Chem., 1992, 328, 233 CrossRef CAS.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354 CrossRef CAS.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479 CrossRef CAS.
- R. Kas, R. Kortlever, H. Yilmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2014, 2, 354 CrossRef.
- B. Jermann and J. Augustynski, Electrochim. Acta, 1994, 39, 1891 CrossRef CAS.
- Z. Weng, X. Zhang, Y. Wu, S. Huo, J. Jiang, W. Liu, G. He, Y. Liang and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 13135 CrossRef CAS PubMed.
- H. Li and C. Oloman, J. Appl. Electrochem., 2006, 36, 1105 CrossRef CAS.
- B. Kim, S. Ma, H.-R. M. Jhong and P. J. A. Kenis, Electrochim. Acta, 2015, 166, 271 CrossRef CAS.
- R. Kortlever, K. H. Tan, Y. Kwon and M. T. M. Koper, J. Solid State Electrochem., 2013, 17, 1843 CrossRef CAS.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Energy Fuels, 2006, 20, 409 CrossRef CAS.
- S. Verma, X. Lu, S. Ma, R. I. Maseld and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075 RSC.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C.-T. Dinh, F. Fan, C. Cao, F. P. Garcia de Arquer, T. Saberi Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 527, 382 CrossRef PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219 CrossRef CAS.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149 CrossRef CAS.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 10, 728 RSC.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- T. Saberi Safaei, A. Mepham, X. Zheng, Y. Pang, C.-T. Dinh, M. Liu, D. Sinton, S. O. Kelley and E. H. Sargent, Nano Lett., 2016, 16, 7224 CrossRef CAS PubMed.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107 CrossRef CAS.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953 CrossRef CAS PubMed.
- M. C. A. A. Van Eerten-Jansen, A. Ter Heijne, C. J. N. Buisman and H. V. M. Hamelers, Int. J. Energy Res., 2012, 36, 809 CrossRef CAS.
- T. Mogi, T. Ishii, K. Hashimoto and R. Nakamura, Chem. Commun., 2013, 49, 3967 RSC.
- A. H. Wonders, T. H. M. Housmans, V. Rosca and M. T. M. Koper, J. Appl. Electrochem., 2006, 36, 1215 CrossRef CAS.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261 RSC.
- J. Zhang and Z. Wang, in Nanomaterials for Photocatalytic Chemistry, ed. Y. Sun, World Scientific, 2016, pp. 229–288 Search PubMed.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Chem. Commun., 1997, 841 RSC.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177 RSC.
- K. Teramura, K. Hori, Y. Terao, Z. Huang, S. Iguchi, Z. Wang, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2017, 121, 8711 CrossRef CAS.
- W.-N. Wang, W.-J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276 CrossRef CAS PubMed.
- H. Xu, S. Ouyang, L. Liu, D. Wang, T. Kako and J. Ye, Nanotechnology, 2014, 25, 165402 CrossRef PubMed.
- X. An, K. Li and J. Tang, ChemSusChem, 2014, 7, 1086 CrossRef CAS PubMed.
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen, Y.-C. Lin and K.-H. Chen, Nanoscale, 2013, 5, 262 RSC.
- J. Yu, J. Jin, B. Cheng and M. Jaroniec, J. Mater. Chem. A, 2014, 2, 3407 RSC.
- S.-I. In, D. D. Vaughn II and R. E. Schaak, Angew. Chem., Int. Ed., 2012, 51, 3915 CrossRef CAS PubMed.
- L. Liu, W. Fan, X. Zhao, H. Sun, P. Li and L. Sun, Langmuir, 2012, 28, 10415 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- N. Sasirekha, S. J. S. Basha and K. Shanthi, Appl. Catal., B, 2006, 62, 169 CrossRef CAS.
- K. Kočí, L. Obalová and O. Šolcová, Chem. Process. Eng., 2010, 31, 395 Search PubMed.
- S. Neatu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217 RSC.
- S. S. Tan, L. Zou and E. Hu, Sci. Technol. Adv. Mater., 2007, 8, 89 CrossRef.
- T. Mizuno, K. Adachi, K. Ohta and A. Saji, J. Photochem. Photobiol., A, 1996, 98, 87 CrossRef CAS.
- S. Kaneco, Y. Shimizu, K. Ohta and T. Mizuno, J. Photochem. Photobiol., A, 1998, 115, 223 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara, J. Electroanal. Chem., 1995, 396, 21 CrossRef.
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817 CrossRef CAS.
- W. Wang, W. Huang, Y. Ni, C. Lu and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 340 CrossRef CAS PubMed.
- H. He, P. Zapol and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6196 RSC.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745 RSC.
- F. Saladin, L. Forss and I. Kamber, J. Chem. Soc., Chem. Commun., 1995, 533 RSC.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211 RSC.
- J. Lee, D. C. Sorescu and X. Deng, J. Am. Chem. Soc., 2011, 133, 10066 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24 CrossRef CAS PubMed.
- R. Boppella, S. Thomas Kochuveedu, H. Kim, M. J. Jeong, F. Marques Mota, J. H. Park and D. H. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 7075 CrossRef CAS PubMed.
- Z. Xu, M. Quintanilla, F. Vetrone, A. O. Govorov, M. Chaker and D. Ma, Adv. Funct. Mater., 2015, 25, 2950 CrossRef CAS.
- H. Kwon, F. Marques Mota, K. Chung, Y. J. Jang, J. K. Hyun, J. Lee and D. H. Kim, ACS Sustainable Chem. Eng., 2018, 6, 1310 CrossRef CAS.
- J. Li, M. Zhang, Z. Guan, Q. Li, C. He and J. Yang, Appl. Catal., B, 2017, 206, 300 CrossRef CAS.
- F. Zuo, K. Bozhilov, R. J. Dillon, L. Wang, P. Smith, X. Zhao, C. Bardeen and P. Feng, Angew. Chem., 2012, 124, 6327 CrossRef.
- R. Boppella, J.-E. Lee, F. Marques Mota, J. Y. Kim, Z. Feng and D. H. Kim, J. Mater. Chem. A, 2017, 5, 7072 RSC.
- G. R. Dey, A. D. Belapurkar and K. Kishore, J. Photochem. Photobiol., A, 2004, 163, 503 CrossRef CAS.
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670 RSC.
- W. J. Lee, J. M. Lee, S. T. Kochuveedu, T. H. Han, H. Y. Jeong, M. Park, J. M. Yun, J. Kwon, K. No, D. H. Kim and S. O. Kim, ACS Nano, 2012, 6, 935 CrossRef CAS PubMed.
- L. N. Quan, Y. H. Jang, K. A. Stoerzinger, K. J. May, Y. J. Jang, S. T. Kochuveedu, Y. Shao-Horn and D. H. Kim, Phys. Chem. Chem. Phys., 2014, 16, 9023 RSC.
- G. Yin, Q. Bi, W. Zhao, J. Xu, T. Lin and F. Huang, ChemCatChem, 2017, 9, 4389 CrossRef CAS.
- M. Manzanares, C. Fàbrega, J. O. Ossó, L. F. Vega, T. Andreu and J. R. Morante, Appl. Catal., B, 2014, 150–151, 57 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Fan, W. Deng and Y. Wang, Chem. Commun., 2013, 49, 2451 RSC.
- K. Kočí, L. Obalová, L. Matějová, D. Plachá, Z. Lacný, J. Jirkovský and O. Šolcová, Appl. Catal., B, 2009, 89, 494 CrossRef.
- H. Xu, S. Ouyang, P. Li, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 1348 CrossRef CAS PubMed.
- K. L. Schulte, P. A. DeSario and K. A. Gray, Appl. Catal., B, 2010, 97, 354 CrossRef CAS.
- K. Li, T. Peng, Z. Ying, S. Song and J. Zhang, Appl. Catal., B, 2016, 180, 130 CrossRef CAS.
- W. Jiao, L. Wang, G. Liu, G. Q. (Max) Lu and H.-M. Cheng, ACS Catal., 2012, 2, 1854 CrossRef CAS.
- L. Ye, J. Mao, T. Peng, L. Zan and Y. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 15675 RSC.
- J. Pan, X. Wu, L. Wang, G. Liu, G. Q. (Max) Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 8361 RSC.
- Z. He, L. Wen, D. Wang, Y. Xue, Q. Lu, C. Wu, J. Chen and S. Song, Energy Fuels, 2014, 28, 3982 CrossRef CAS.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839 CrossRef CAS PubMed.
- Q. Xu, J. Yu, J. Zhang, J. Zhang and G. Liu, Chem. Commun., 2015, 51, 7950 RSC.
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, Y. Ichihashi, S. G. Zhang, D. R. Park, S. Ehara, S.-E. Park, J.-S. Chang and J. W. Yoo, Stud. Surf. Sci. Catal., 1998, 114, 177 CrossRef CAS.
- K. Ikeue, H. Yamashita and M. Anpo, J. Phys. Chem. B, 2001, 105, 8350 CrossRef CAS.
- S. G. Zhang, Y. Fujii, H. Yamashita, K. Koyano, T. Tatsumi and M. Anpo, Chem. Lett., 1997, 659 CrossRef.
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang, Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327 CrossRef CAS.
- J.-S. Hwang, J.-S. Chang, S.-E. Park, K. Ikeue and M. Anpo, Top. Catal., 2005, 35, 311 CrossRef CAS.
- M. Hussain, P. Akhter, G. Saracco and N. Russo, Appl. Catal., B, 2015, 170–171, 53 CrossRef CAS.
- M. S. Khan, M. N. Ashiq, M. F. Ehsan, T. He and S. Ijaz, Appl. Catal., A, 2014, 487, 202 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- P. Dias, M. Schreier, S. D. Tilley, J. Luo, J. Azevedo, L. Andrade, D. Bi, A. Hagfeldt, A. Mendes, M. Grätzel and M. T. Mayer, Adv. Energy Mater., 2015, 5, 1501537 CrossRef.
- Z. Xiong, Z. Lei, C.-C. Kuang, X. Chen, B. Gong, Y. Zhao, J. Zhang, C. Zheng and J. C. S. Wu, Appl. Catal., B, 2017, 202, 695 CrossRef CAS.
- S. Zhu, S. Liang, Y. Wang, X. Zhang, F. Li, H. Lin, Z. Zhang and X. Wang, Appl. Catal., B, 2016, 187, 11 CrossRef CAS.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008 CrossRef CAS PubMed.
- K. Katsumata, K. Sakai, K. Ikeda, G. Carja, N. Matsushita and K. Okada, Mater. Lett., 2013, 107, 138 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O’Hare and T. Zhang, Adv. Mater., 2015, 27, 7824 CrossRef CAS PubMed.
- S. K. Parayil, A. Razzaq, S.-M. Park, H. R. Kim, C. A. Grimes and S.-I. In, Appl. Catal., A, 2015, 498, 205 CrossRef CAS.
- G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, Angew. Chem., 2012, 124, 2445 CrossRef.
- Y. Y. Lee, H. S. Jung, J. M. Kim and Y. T. Kang, Appl. Catal., B, 2018, 224, 594 CrossRef CAS.
- Y.-X. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y.-L. Men, F.-F. Cao, S.-H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123 CrossRef CAS PubMed.
- Y. Zhou, Z. Tian, Z. Zhao, Q. Liu, J. Kou, X. Chen, J. Gao, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2011, 3, 3594 CrossRef CAS PubMed.
- H. Cheng, B. Huang, Y. Liu, Z. Wang, X. Qin, X. Zhanga and Y. Daib, Chem. Commun., 2012, 48, 9729 RSC.
- Y. Y. Lee, H. S. Jung and Y. T. Kang, J. CO2 Util., 2017, 20, 163 CrossRef CAS.
- Y. P. Xie, G. Liu, L. Yin and H.-M. Cheng, J. Mater. Chem., 2012, 22, 6746 RSC.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400 CrossRef CAS PubMed.
- Q. Liu, Y. Zhou, W. Tu, S. Yan and Z. Zou, Inorg. Chem., 2014, 53, 359 CrossRef CAS PubMed.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385 CrossRef CAS PubMed.
- N. Zhang, S. Ouyang, P. Li, Y. Zhang, G. Xi, T. Kako and J. Ye, Chem. Commun., 2011, 47, 2041 RSC.
- V. B. R. Boppana, N. D. Hould and R. F. Lobo, J. Solid State Chem., 2011, 184, 1054 CrossRef CAS.
- H. Park, J. H. Choi, K. M. Choi, D. K. Lee and J. K. Kang, J. Mater. Chem., 2012, 22, 5304 RSC.
- Q. Liu, D. Wu, Y. Zhou, H. Su, R. Wang, C. Zhang, S. Yan, M. Xiao and Z. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 2356 CrossRef CAS PubMed.
- S. Yan, J. Wang, H. Gao, N. Wang, H. Yu, Z. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2012, 23, 758 CrossRef.
- Q. Liu, Y. Zhou, Z. Tian, X. Chen, J. Gao and Z. Zou, J. Mater. Chem., 2012, 22, 2033 RSC.
- S. Yan, H. Yu, N. Wang, Z. Liab and Z. Zou, Chem. Commun., 2012, 48, 1048 RSC.
- N. Zhang, S. Ouyang, T. Kako and J. Ye, Chem. Commun., 2012, 48, 1269 RSC.
- Q. Li, X. Li, S. Wageh, A. A. Al-Ghamdi and J. Yu, Adv. Energy Mater., 2015, 5, 1500010 CrossRef.
- P. Praus, O. Kozák, K. Kočí, A. Panáček and R. Dvorský, J. Colloid Interface Sci., 2011, 360, 574 CrossRef CAS PubMed.
- H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270 CrossRef CAS.
- P. Kar, S. Farsinezhad, X. Zhang and K. Shankar, Nanoscale, 2014, 6, 14305 RSC.
- H. Shi and Z. Zou, J. Phys. Chem. Solids, 2012, 73, 788 CrossRef CAS.
- P. Li, S. Ouyang, G. Xi, T. Kako and J. Ye, J. Phys. Chem. C, 2012, 116, 7621 CrossRef CAS.
- H. Shi, T. Wang, J. Chen, C. Zhu, J. Ye and Z. Zou, Catal. Lett., 2011, 141, 525 CrossRef CAS.
- X. Li, H. Pan, W. Li and Z. Zhuang, Appl. Catal., A, 2012, 413–414, 103 CrossRef CAS.
- G. Li, S. Ciston, Z. V. Saponjic, L. Chen, N. M. Dimitrijevic, T. Rajh and K. A. Gray, J. Catal., 2008, 253, 105 CrossRef CAS.
- H. Zhao, L. Liu, J. M. Andino and Y. Li, J. Mater. Chem. A, 2013, 1, 8209 RSC.
- P.-Q. Wang, Y. Bai, J.-Y. Liu, Z. Fan and Y.-Q. Hu, Catal. Commun., 2012, 29, 185 CrossRef CAS.
- K. Wu and T. Lian, Chem. Soc. Rev., 2016, 45, 3781 RSC.
- C. Wang, R. L. Thompson, J. Baltrus and C. Matranga, J. Phys. Chem. Lett., 2010, 1, 48 CrossRef CAS.
- C. Wang, R. L. Thompson, P. Ohodnicki, J. Baltrus and C. Matranga, J. Mater. Chem., 2011, 21, 13452 RSC.
- S. Chan, M. Liu, K. Latham, M. Haruta, H. Kurata, T. Teranishi and Y. Tachibana, J. Mater. Chem. C, 2017, 5, 2182 RSC.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660 CrossRef CAS PubMed.
- G. Xi, S. Ouyang and J. Ye, Chem. – Eur. J., 2011, 17, 9057 CrossRef CAS PubMed.
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637 CrossRef CAS.
- J. Jin, J. Yu, D. Guo, C. Cui and W. Ho, Small, 2015, 11, 5262 CrossRef CAS PubMed.
- J. Rascó, Catal. Lett., 1998, 56, 11 CrossRef.
- K. Kočí, K. Matějů, L. Obalová, S. Krejčíková, Z. Lacný, D. Plachá, L. Čapek, A. Hospodková and O. Šolcová, Appl. Catal., B, 2010, 96, 239 CrossRef.
- O. Ishitani, C. Inoue, Y. Suzuki and T. Ibusuki, J. Photochem. Photobiol., A, 1993, 72, 269 CrossRef CAS.
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929 CrossRef CAS.
- J. C. Hemminger, R. Carr and G. A. Somorjai, Chem. Phys. Lett., 1978, 57, 100 CrossRef CAS.
- G. Guan, T. Kida and A. Yoshida, Appl. Catal., B, 2003, 41, 387 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863 CrossRef CAS PubMed.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594 CrossRef CAS PubMed.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492 RSC.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644 CrossRef CAS.
- N. Li, M. Liu, B. Yang, W. Shu, Q. Shen, M. Liu and J. Zhou, J. Phys. Chem. C, 2017, 121, 2923 CrossRef CAS.
- Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386 CrossRef CAS.
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731 CrossRef CAS PubMed.
- T.-V. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112 CrossRef CAS.
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732 CrossRef CAS PubMed.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486 CrossRef CAS PubMed.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776 CrossRef CAS PubMed.
- S. Zeng, D. Baillargeat, H.-P. Ho and K.-T. Yong, Chem. Soc. Rev., 2014, 43, 3426 RSC.
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702 CrossRef CAS PubMed.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133 RSC.
- Y. H. Jang, Y. J. Jang, S. Kim, L. N. Quan, K. Chung and D. H. Kim, Chem. Rev., 2016, 116, 14982 CrossRef CAS PubMed.
- C. H. Choi, K. Chung, T. T. H. Nguyen and D. H. Kim, ACS Energy Lett., 2018, 3, 1415 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247 CrossRef CAS PubMed.
- S. Linic, P. Christopher, H. Xin and A. Marimuthu, Acc. Chem. Res., 2013, 46, 1890 CrossRef CAS PubMed.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116 CrossRef CAS.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656 CrossRef.
- J. Low, S. Qiu, D. Xu, C. Jiang and B. Cheng, Appl. Surf. Sci., 2018, 434, 423 CrossRef CAS.
- Z. Zhang, Z. Wang, S.-W. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939 CrossRef CAS.
- W. Tu, Y. Zhou, H. Li, P. Li and Z. Zou, Nanoscale, 2015, 7, 14232 RSC.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 RSC.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353 CrossRef CAS.
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426 RSC.
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865 CrossRef CAS PubMed.
- Y. T. Liang, B. K. Vijayan, O. Lyandres, K. A. Gray and M. C. Hersam, J. Phys. Chem. Lett., 2012, 3, 1760 CrossRef CAS PubMed.
- W. Tu, Y. Zhou, Q. Liu, Z. Tian, J. Gao, X. Chen, H. Zhang, J. Liu and Z. Zou, Adv. Funct. Mater., 2012, 22, 1215 CrossRef CAS.
- X.-H. Xia, Z.-J. Jia, Y. Yu, Y. Liang, Z. Wang and L.-L. Ma, Carbon, 2007, 45, 717 CrossRef CAS.
- W. Wang, D. Xu, B. Cheng, J. Yu and C. Jiang, J. Mater. Chem. A, 2017, 5, 5020 RSC.
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743 CrossRef CAS.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841 CrossRef CAS PubMed.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35 RSC.
- N. C. D. Nath, S. Y. Choi, H. W. Jeong, J.-J. Lee and H. Park, Nano Energy, 2016, 25, 51 CrossRef.
- S. Ichikawa and R. Doi, Catal. Today, 1996, 27, 271 CrossRef CAS.
- X. Chang, T. Wang, P. Zhang, Y. Wei, J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2016, 128, 8986 CrossRef.
- T. J. LaTempa, S. Rani, N. Bao and C. A. Grimes, Nanoscale, 2012, 4, 2245 RSC.
- G. Magesh, E. S. Kim, H. J. Kang, M. Banu, J. Y. Kim, J. H. Kim and J. S. Lee, J. Mater. Chem. A, 2014, 2, 2044 RSC.
- J. H. Kim, G. Magesh, H. J. Kang, M. Banu, J. H. Kim, J. Lee and J. S. Lee, Nano Energy, 2015, 15, 153 CrossRef CAS.
- M. I. Litter and J. A. Navío, J. Photochem. Photobiol., A, 1996, 98, 171 CrossRef CAS.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049 CrossRef CAS.
- W.-H. Lee, C.-H. Liao, M.-F. Tsai, C.-W. Huang and J. C. S. Wu, Appl. Catal., B, 2013, 132–133, 445 CrossRef CAS.
- F. Galli, M. Compagnoni, D. Vitali, C. Pirola, C. L. Bianchi, A. Villa, L. Prati and I. Rossetti, Appl. Catal., B, 2017, 200, 386 CrossRef CAS.
- M. Salaices, B. Serrano and H. I. de Lasa, Chem. Eng. J., 2002, 90, 219 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593 CrossRef CAS PubMed.
- L. Gao, Y. Cui, R. H. J. Vervuurt, D. van Dam, R. P. J. van Veldhoven, J. P. Hofmann, A. A. Bol, J. E. M. Haverkort, P. H. L. Notten, E. P. A. M. Bakkers and E. J. M. Hensen, Adv. Funct. Mater., 2016, 26, 679 CrossRef CAS.
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
- A. Van Miltenburg, W. Zhu, F. Kapteijn and J. A. Moulijn, Chem. Eng. Res. Des., 2006, 84, 350 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |