
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation paths of manganese-based MOF materials in a model oxidative environment: a computational study†
Elena V.
Khramenkova
a,
Mikhail V.
Polynski
*a,
Alexander V.
Vinogradov
a and
Evgeny A.
Pidko
 *ab
*ab
aTheoMAT group, International Laboratory “Solution Chemistry of Advanced Materials and Technologies”, ITMO University, Lomonosova str. 9, St. Petersburg 191002, Russia. E-mail: polynskimikhail@gmail.com
bInorganic Systems Engineering group, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: E.A.Pidko@tudelft.nl
First published on 18th June 2018
Abstract
Stability is the key property of functional materials. In this work we investigate computationally the degradative potential of a model Mn-BTC (BTC = benzene-1,3,5-tricarboxylate) metal–organic framework (MOF) building block in aqueous solutions under oxidative conditions. Model density functional theory calculations have shown that the direct hydrolysis of the Mn-containing moieties is more difficult than their decomposition via oxidation-induced paths. While the interaction with H2O2 species is of non-covalent nature and requires O–O-bond breaking to initiate Mn-center oxidation, open-shell O2 species readily oxidize radical Mn-centers and form bonds of σ-, π-, or δ-symmetry with the metal. The oxidative transformations of di-Mn paddle-wheel carboxylate structure-forming units are accompanied with substantial distortions of the coordination polyhedra that, together with the increased Lewis acidity of the oxidized metal centers, facilitates the hydrolysis leading to the degradation of the structure at a larger scale. Whereas such a mechanism is expected to hamper the catalytic applications of such Mn-MOFs, the associated structural response to oxidizing and radical species can create a basis for the construction of Mn-MOF-based drug delivery systems with increased bio-compatibility.
1. Introduction
Metal–organic frameworks (MOFs) are crystalline nanoporous structures, which consist of metallic coordination centers and organic ligands.1,2 The key features of these materials are high crystallinity and well-defined highly porous structure along with unprecedented structural and functional tunability.3,4 MOF-based materials and devices show great promise for various applications, ranging from gas sorption5 and separation of enantiomers6 to photonics7 and catalysis.8,9 Long-term stability and the ability to preserve the well-defined structure of the framework under operating conditions are the necessary features in the large-scale implementation of MOF-based systems and the lack of these may hamper their wide application.10–12Fundamentally different requirements to material properties arise for biomedical applications. There is a growing interest in the utilization of inorganic chemistry polymers, including MOFs, in the development of multifunctional biomedical materials.13,14 Here, the intrinsic instability of the hybrid organic–inorganic framework can be regarded as an advantageous feature enabling programmed degradation of the material.15,16 The possibility of the programmed decomposition of an inorganic matrix into molecular building blocks17,18 facilitates its removal from the organism as well as driving the controlled and targeted release of bio-active materials.19,20 Therefore, MOFs are considered promising for the construction of responsive matrices and carriers for smart drug delivery systems.21,22 Their predictable and tunable chemistry on the molecular level creates a possibility to design chemical response mechanisms to varying environmental conditions, i.e.,23 one can design these materials to respond to specific changes in the microenvironment of pathological tissues. Acidosis, elevated tissue temperature, and formation of reactive oxygen species (ROS) are the specific signals that can direct the targeted delivery of an active pharmaceutical ingredient (API) to a pathologic area.24–26 MOFs readily satisfy the essential requirements for smart drug delivery platforms, which are controllable degradation, biocompatibility and simplicity of chemical functionalization. They may be synthesized from biocompatible components as non-toxic metals and organic ligands. The medical use of widely explored Fe- and Cr-based MOFs is limited due to the toxicity issues, although such MOFs are able to upload high amounts of API.27 In this context, Mn-based inorganic systems are particularly appealing owing to their low toxicity combined with Mn-rich coordination chemistry. While being toxic only at a high dosage,28 Mn regulates biological processes and is a component of many enzymes.29–31
Mn-Based MOFs are also considered as catalysts for a number of important chemical processes. In particular, such Mn-MOFs as [Mn3(atpa)3(dmf)2] and [Mn2(tpa)2(dmf)2] were investigated as effective Lewis acid catalysts for the cyanosilylation of acetaldehyde (atpa = 2-aminoterephthalate, tpa = terephthalate, dmf = dimethylformamide).32 The oxidative degradation of phenol catalyzed by bimetallic Fe/Mn-MOF-71 was studied by Sun et al.33 According to Pereira et al.,34 Mn-based PIZA, RPM and ZJU MOF families are catalytically active for alkane oxidation.
The behavior of Mn-based MOFs in oxidative environments has been a subject of several experimental studies. Depending on the nature of the ligands and the MOF structure, the intrinsic structural stability under harsh reaction conditions may vary substantially.35–37 Zuluaga et al. have reported Mn-MOF-74's (Mn2(DOBDC), DOBDC = 2,5-dioxido-1,4-benzenedicarboxylic acid) susceptibility to hydrolysis that led to the degradation of the structure and limited the sorption capacity of the material.38 On the other hand, the report by Wu et al. demonstrated the perfect structural stability of a defected Mn-MOF-74 when exposed to water for up to 7 days.39 The same framework material has been further employed for the catalytic oxidation of alkyl aromatics in an O2 flow.40 Hansen et al. reported an Mn-based catalyst featuring 2,2′:6′,2′′-terpyridine structure-forming ligands as an active oxidation catalyst capable of retaining structural integrity in the catalytic oxidative environment.41 MOF structures featuring Mn(3+)-porphyrin catalytic motifs have been reported as active and highly durable olefin oxidation catalysts.42 An alternative IRMOF-3(Mn) catalyst made of Mn-carboxylate units was also found to be stable under the conditions of the gas-phase selective oxidation of alkenes.43 Importantly, a wide range of catalytic studies have explored the reactivity of MOFs in non-aqueous media.42–46 In biological systems, the aqueous environment may potentially make either direct or oxidation-induced hydrolysis possible as the routes for controlled decomposition of Mn-MOF. The stability of materials in oxidative environments has been the subject of computational studies as well.47–49
The interaction of MOF-based materials with the oxidizing species is thus of key importance for pharmaceutical chemistry and catalysis. The knowledge of the corresponding mechanisms on the molecular level may help to determine whether, on the one hand, a MOF material under consideration will irreversibly interact and (controllably) degrade under oxidative conditions. The understanding of controllable degradation mechanisms will allow for the construction of new targeted drug delivery systems. On the other hand, the material may be stable towards oxidizing species, reacting reversibly and activating them. Therefore, the understanding of the MOF-oxidizing species’ interaction mechanism may direct new oxidation catalyst development as well.
The focus of the present computational study is the reactivity of the Mn-BTC framework (BTC = benzene-1,3,5-tricarboxylate, Fig. 1a) towards oxidizing species (O2 and H2O2) in aqueous media. We show that the oxidative transformations of the Mn centers in MOFs can potentially be employed as the driver for controlled degradation of Mn-MOF-based nanocontainers in response to increased levels of oxidants produced in pathologic tissues. We computationally evaluated the energetics of the reaction mechanisms corresponding to the MOF–O2- and MOF–H2O2-interactions in aqueous media. The results provide guidance towards the further design of responsive structures for a wider range of applications, including smart drug delivery systems. Besides, the insights into the processes determining the structural (in)stability of Mn-BTC frameworks under oxidative conditions are crucial for their utilization in oxidation catalysis.
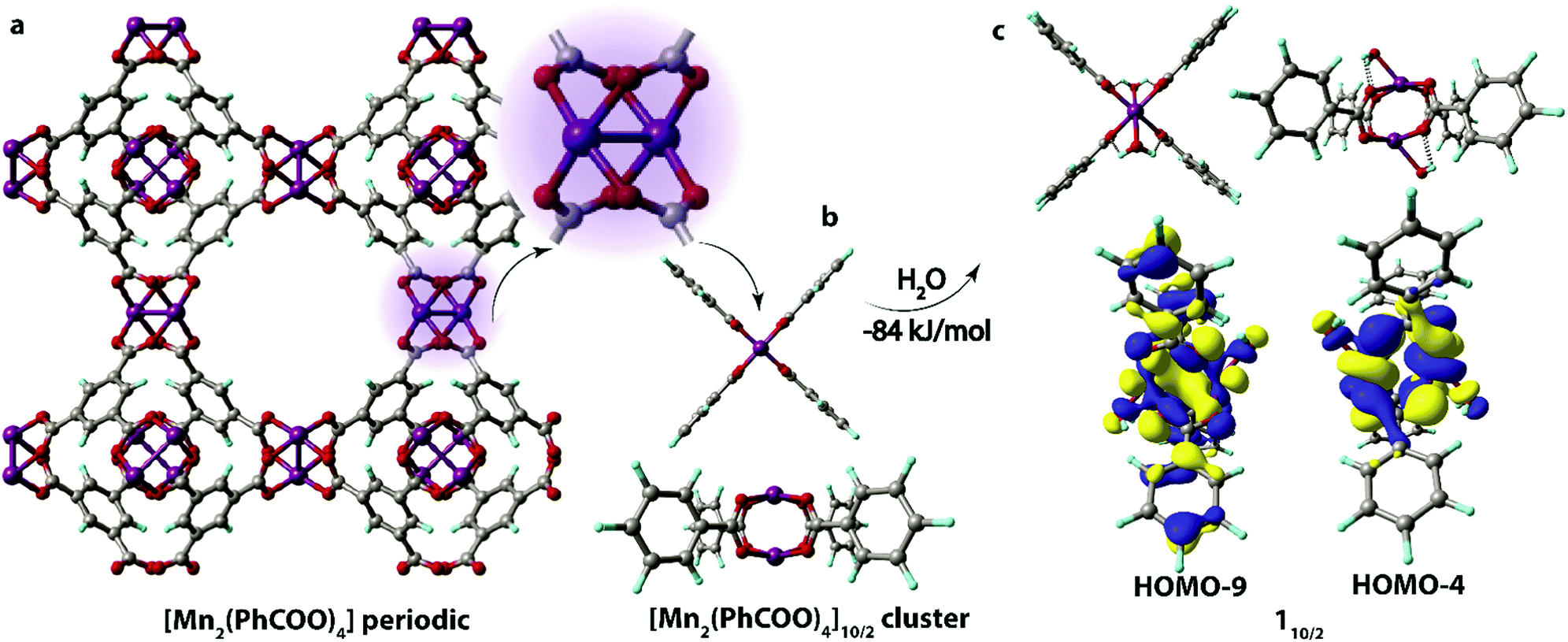 | ||
| Fig. 1 The periodic structure of (a) Mn-BTC MOF and (b) the corresponding [Mn2(PhCOO)4] cluster model in S = 10/2 spin state; (c) the 110/2 aqua complex along with the MOs representing the relevant molecular orbitals responsible for the Mn–H2O binding (isosurface thresholds are 0.012 and 0.018 for HOMO−4 and HOMO−9 respectively). (b) and (c) optimized at the PBE-D3/6-31G(d,p) level of theory. Oxygen atoms are red, manganese atoms are purple, carbon atoms are grey, and hydrogen atoms are blue. | ||
2. Computational methods
Density functional theory (DFT) calculations were performed using the Gaussian 09 D.0150 program suite to study the structural properties of Mn-BTC MOF and elucidate its stability towards the oxidizing species (O2 and H2O2) in water. Similar to our previous work on an MOF-based drug delivery system,51 all calculations were carried out at the PBE-D3/6-311++G(d,p)//PBE-D3/6-31G(d,p) level of theory.52,53 Dispersion attraction was accounted for using the empirical correction scheme proposed by Grimme.54 Bulk solvent effects (water) were accounted for with the PCM solvation model55 applied to single point calculations at the PBE-D3/6-311++G(d,p) level while the short-range interaction of Mn centers in Mn-BTC with H2O molecules were accounted for by the explicit inclusion of H2O species in the model systems. Vibrational frequencies were calculated analytically to evaluate the nature of the optimization-obtained stationary points.The interaction with O2 and H2O2 molecules and local structural deformations that could initiate the material degradation processes were investigated in the cluster-continuum approach. The cluster model representing the Mn-carboxylate paddle-wheel configuration was cut from a periodic structure of Mn-BTC MOF (Fig. 1a). The resulting cluster fragment of [Mn2(PhCOO)4] was then coordinatively saturated by the inclusion of two water molecules. The geometries of all cluster models were optimized without any geometry constraints to satisfactorily represent the substantial geometrical flexibility and relaxation potential of the coordination polymers.56,57 The effect of possible steric inflexibility was tested by the set of constrained optimizations, in which the positions of the para-H atoms of the PhCOO− units were fixed. These constrained test optimizations were carried out on the configurations corresponding to the most stable (in terms of exothermicity of formation) and distorted complexes obtained through the unconstrained optimizations. The computed energetics and optimized structures are summarized in the Supporting Information for the article (Fig. S1, S2 and Table S1, ESI†). These tests evidence the key role of the structural flexibility for the preservation of the optimal coordination environment of the Mn centers. The excessive geometrical strain imposed by the fixed positions of the para-H-atoms in PhCOO− ligands gave rise to the decoordinaton of Mn-centers in some cases.
All L[Mn2(PhCOO)4]L model complexes (where L is H2O, H2O2, or O2) were considered in various spin states (namely, S = 6/2, 8/2, 10/2) while in the case of H2O[Mn2(PhCOO)4]O2 and O2[Mn2(PhCOO)4]O2 complexes, S = 12/2 and 14/2 spin states were also considered. Initial test calculations indicated that the other electronic configurations (including the lower-spin and broken symmetry configurations, see below) are characterized by excessively high energies and are therefore not relevant to the processes considered here.
The concurrent adsorption energies (Eads) of the oxidizing species were calculated according to the formula:
| Eads = En−OX-Mn-BTC + nEH2O − EAQUA − nEOX |
The molecular orbital isosurfaces used for the analysis of bonding in the adsorption complexes were constructed with the ChemCraft program.58
3. Results and discussion
3.1. Starting configurations
The structure unit of the Mn-BTC metal–organic framework (Fig. 1a) was modeled as the [Mn2(PhCOO)4] cluster (Fig. 1b). The structure of Mn-BTC is represented by a periodic array of dimeric Mn2+ paddle-wheel centers linked by four benzene-1,3,5-tricarboxylic ligands (BTC). A number of spin states are accessible to the dimeric [Mn2(PhCOO)4] species since the carboxylate ligand are formally considered to be of weak-field class. Particularly, 6/2, 8/2, 10/2 spin configurations as well as the broken symmetry singlet state were considered. In the case of the [Mn2(PhCOO)4] cluster, high-spin S = 10/2 state with the square-planar coordination of the Mn2+ cations having the paddle-wheel structure was the most energetically favorable. The lower spin-states of S = 6/2 and 8/2 were less favorable (by 10 and 19 kJ mol−1), respectively. To ensure the unlikeliness of the singlet state with the antiferromagnetic coupling of the unpaired electrons in the two metal centers, broken symmetry calculations were performed. The configuration resulting from the computation at the PBE-D3/6-311++G(d,p)//PBE-D3/6-31G(d,p) level (PCM solvation included) had 238 kJ mol−1 higher electronic energy compared to the lowest-lying 10/2 state and the antiferromagnetic coupling was confirmed by the analysis of the Mulliken spin densities, which had values of 2.6 and −2.9 a.u. on the Mn centers.In aqueous solution, the open axial coordination sites are likely to be occupied by H2O molecules. Among the H2O[Mn2(PhCOO)4]H2O complexes, the one with S = 10/2 spin state, the 110/2 complex (Fig. 1c), was energetically preferred, and the low-spin S = 8/2 (18/2) and 6/2 (16/2) states had higher energies (43 and 54 kJ mol−1, respectively). Thereby, 110/2 formation from the [Mn2(PhCOO)4] complex in the 10/2 spin state is exothermic by −84 kJ mol−1. The formation of the antiferromagnetic state was unlikely in the case of the H2O[Mn2(PhCOO)4]H2O complex as well since the broken-symmetry computation at the PBE-D3/6-311++G(d,p)//PBE-D3/6-31G(d,p) level (PCM solvation included) showed that the singlet state had 332 kJ mol−1 higher energy compared to the 10/2 state. The Mulliken spin densities in the Mn-centers in the singlet state were 4.5 and −4.5 a.u.
Water-bound cluster 110/2 (Scheme 1) was selected as the reference point and starting configuration for further modeling the interaction of Mn-BTC with the oxidizing species. It is worth noting that the overlap between H2O and [Mn2(PhCOO)4] fragments in 110/2 is negligible (Fig. 1c), although the binding is highly exothermic (ΔEads = −84 kJ mol−1). The coordination of H2O molecules results in the formation of the four hydrogen bonds between OH groups and the oxygen atoms of the carboxylate ligands. Thus, the highly exothermic effect of H2O binding may be caused by the formation of strong hydrogen bonds and the reduction of electrostatic repulsion between Mn2+ cations owing to the countering electrostatic field of H2O dipoles.
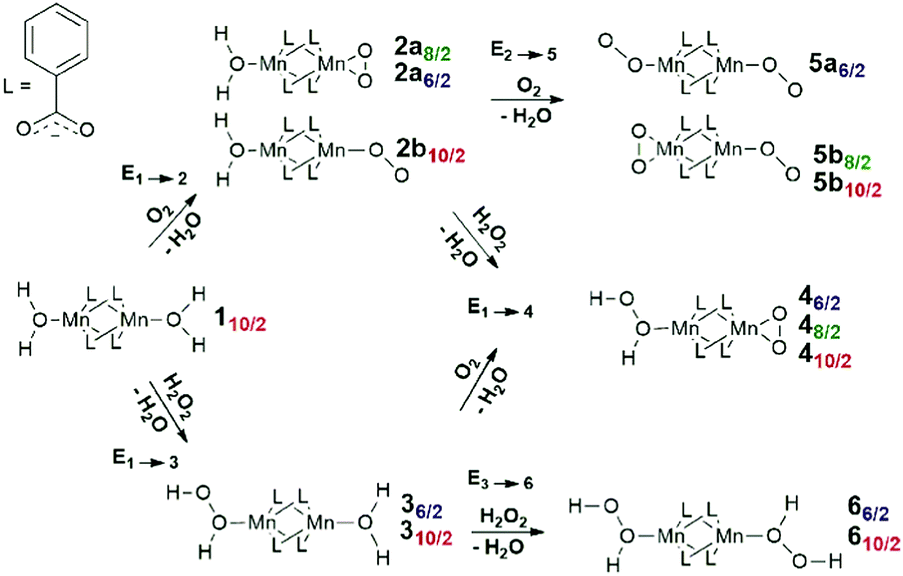 | ||
| Scheme 1 A stepwise path for coordination of oxidizing species to 110/2 complex. | ||
Scheme 1 summarizes the chemical transformations that are considered here as potential pathways for the oxidative degradation of Mn-BTC in the presence of O2 or H2O2 species. The model oxidants are relevant to both biological and catalytic potential applications of Mn- and carboxylate-based metal–organic frameworks. Note that, depending on the spin-state, coordination complexes of different structures are formed; Scheme 1 explicitly indicates them with the appropriate labels. Fig. 2 graphically summarizes the relative energies of the formation of the oxidated complexes. These oxidative transformations are discussed in more detail in the subsequent sections.
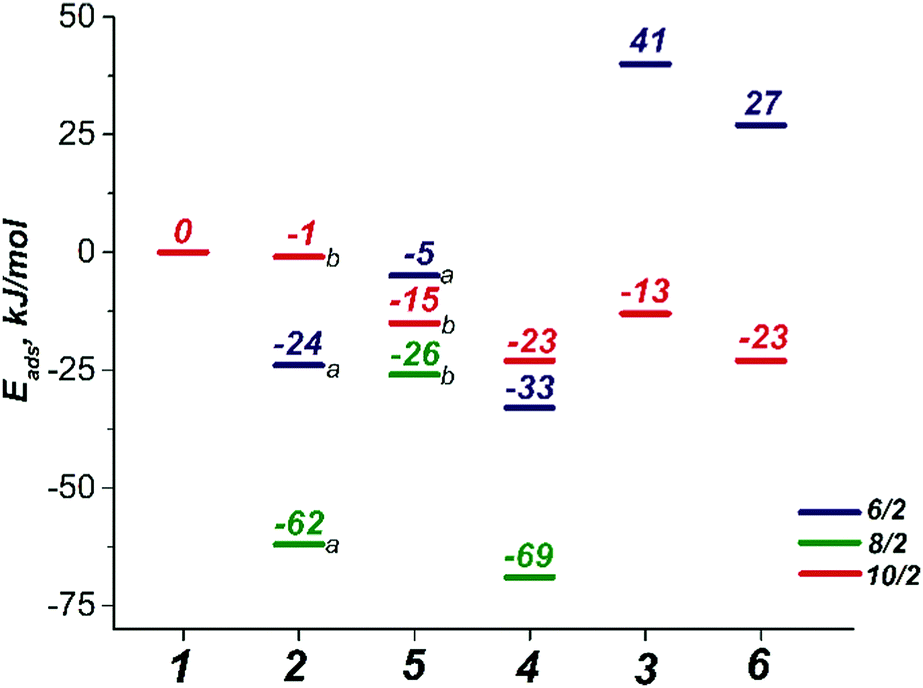 | ||
| Fig. 2 The concurrent adsorption energies (Eads, see Section 2) of the O2 and H2O2 oxidizing species to H2O[Mn2(PhCOO)4]H2O complex. | ||
3.2. O2 binding
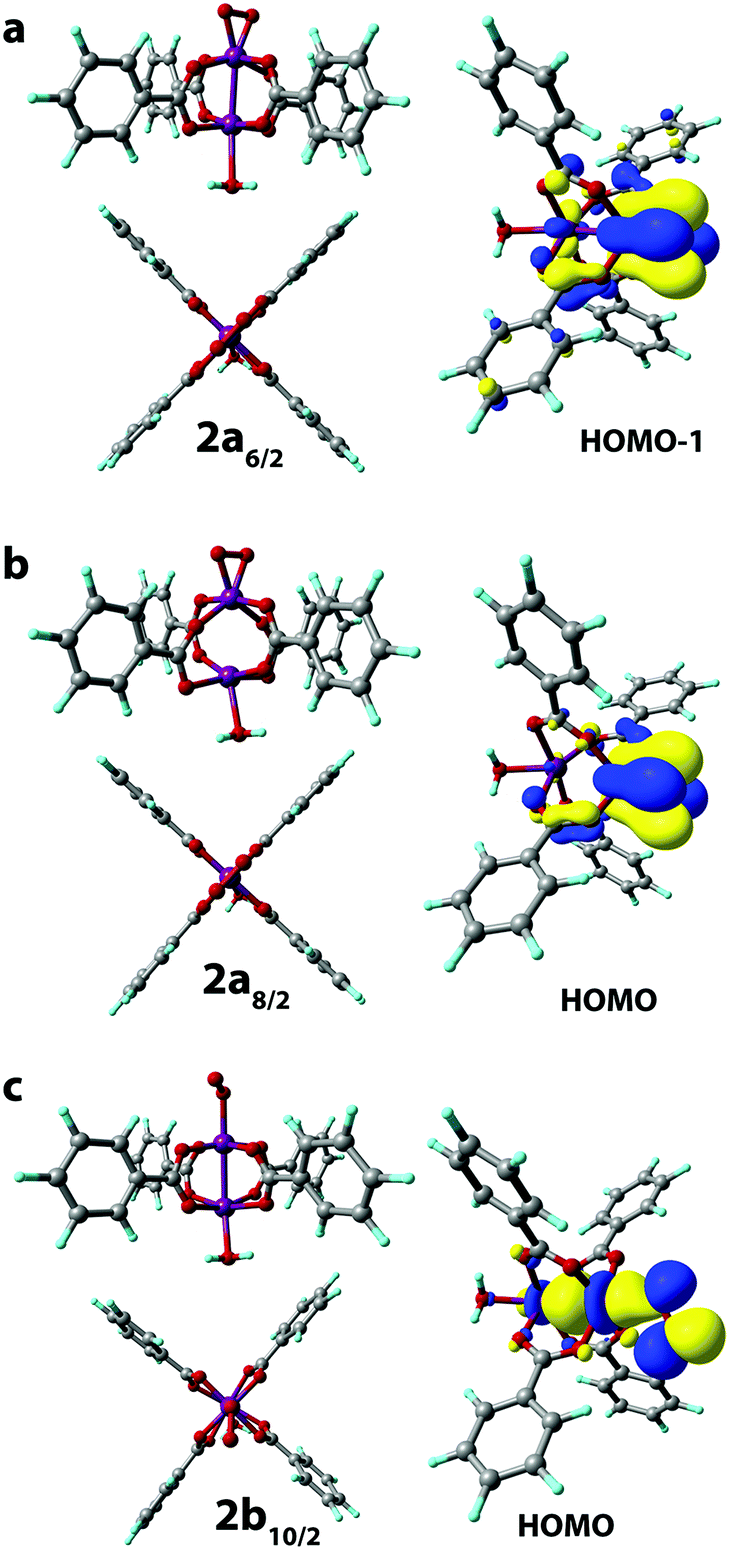
The binding of an O2 molecule to the coordinately unsaturated Mn centers in Mn-BTC (1 → 2, Scheme 1) may result in two isomeric structures that are in the end-on (η1) and side-on (η2) configurations. The end-on configuration is the result of the σ-type bonding of the O2 molecule with the Mn2+ cation while the side-on configuration is the complex having δ-type bond that corresponds to the binding of both oxygen atoms to the Mn2+ center. It should be noted that metal–ligand δ-symmetry bonding is somewhat unusual since it is usually observed in transition metal complexes with quadruple metal–metal bonds.59The binding in both coordination modes results from the overlap between singly-occupied O2 π* orbitals and 3d orbitals of Mn that are singly occupied as well, and the formation of the particular configuration leads to different spin states. Whereas the formation of the η1 complex preserves the spin state (with the corresponding structure being 2b10/2, see Scheme 1), the formation of δ complexes with η2-bound O2 ligand leads to the electron pairing and the formation of 2a6/2 and 2a8/2 complexes in 6/2 and 8/2 spin states, respectively. The formation of the latter is strongly exothermic. The reaction energies computed for water exchange 1 → 2 are −24 and −62 kJ mol−1 for the formation of 2a6/2 and 2a8/2, respectively (Fig. 2). The formation of the σ-type bound 2b10/2 complex in the 1 → 2 process has a weak exothermic effect of −1 kJ mol−1. The chemical irrelevance of the high-spin 12/2 state was ensured with the computed binding energy of 42 kJ mol−1 (see Fig. S3a, ESI†).
The complexation with O2 distorts the initial square pyramidal geometry of the Mn centers in 110/2 to form a distorted octahedron in 2a6/2 and trigonal bipyramid in 2a8/2 of O2-coordinated Mn sites. The water-coordinated site in 2a6/2 has octahedral geometry while the H2O-bonded Mn2+ center in 2a8/2 has severely distorted trigonal-bipyramidal geometry with the cleaved Mn–Mn bond. Mn centers in 2b10/2 form highly distorted octahedrons (Fig. 3c).
 | ||
| Fig. 3 Geometries were optimized at the PBE-D3/6-31G(d,p) level of theory (front and side views are given). The key bonding MOs of the O2[Mn2(PhCOO)4]H2O complexes in S = 6/2 (a), 8/2 (b), and 10/2 (c) spin states are depicted with isosurface thresholds eq. 0.023. | ||
The end-on-bound O2 molecule in 2b10/2 may formally be considered as a superoxide (O2−) ligand, which implies the formal oxidation of Mn(2+) to Mn(3+). The formal oxidation state of oxygen-bound Mn cations in 2a6/2 and 2a8/2 complexes should then be considered as Mn(4+) with the side-on bound peroxide (O22−) ligands.
The changes in the Mn oxidation states are in line with the Mulliken spin density changes in reaction 1 → 2 (Scheme 1). The O2 binding in the case of 2b10/2 leads to the decrease of the spin density on the interacting Mn (from 4.80 to 3.56) and the asymmetric decrease from (1, 1) to (0.70, 0.82) of the spin densities on O atoms in the O2 molecule (see Table S2, ESI†). Based on the analysis of the spatial distribution of Kohn–Sham orbitals, σ-symmetry bonding is evident when a single O atom is bound to Mn. This implies the formation of an Mn center in the formal (3+) oxidation state.
The δ binding of the O2 species proceeded via the electron density donation by Mn d electrons to the π* orbitals of O2 molecules resulting in the weakening of the O–O bond. This is reflected in the significant increase of the O–O bond length in 2a8/2 and 2a6/2 structures (r(O–O) = 1.411 Å and 1.373 Å, respectively) when compared to that in the dioxygen molecule (r(O–O) = 1.228 Å). The analysis of the electron density distribution in 2a6/2 shows the decrease of spin density on oxygen atoms in the O2 molecule (from (1, 1) to (0.15, 0.15) a.u.) and the reduction of spin density values on the corresponding Mn site (from 3.16 to 2.33). A similar trend was found in 2a8/2 where spin densities on the Mn site and the coordinated O2 decreased from 3.28 to 2.94 and from (1, 1) to (0.11, 0.10), respectively (see Table S2, ESI†). These changes indicate the formation of the Mn(4+) formal oxidation state in both cases.
The reaction of 2a and 2b complexes with a second O2 molecule leads to the formation of two isomers (end-on-end-on and side-on-end-on) of the three possible ones (2 → 5 step on the Scheme 1). The formation of the side-on-side-on O2[Mn2(PhCOO)4]O2 isomer was not observed owing to the significant steric strain of the [Mn2(PhCOO)4] core imposed by the side-on binding of O2 species (see the discussion above and Fig. 3a–c).
The σ-type bonding of two oxygen molecules leads to an end-on-end-on configuration with the octahedral coordination of both Mn cations. The formation of the corresponding 5a6/2 complex proceeded with a low exothermic effect of −5 kJ mol−1 (Fig. 4a). The side-on-end-on configuration corresponds to the simultaneous binding of O2 species in δ- and σ-modes and the formation of 5b8/2 and 5b10/2 complexes is exothermic by −26 and −15 kJ mol−1, respectively (Fig. 4b and c). Here, both Mn centers adopt square pyramidal geometry.
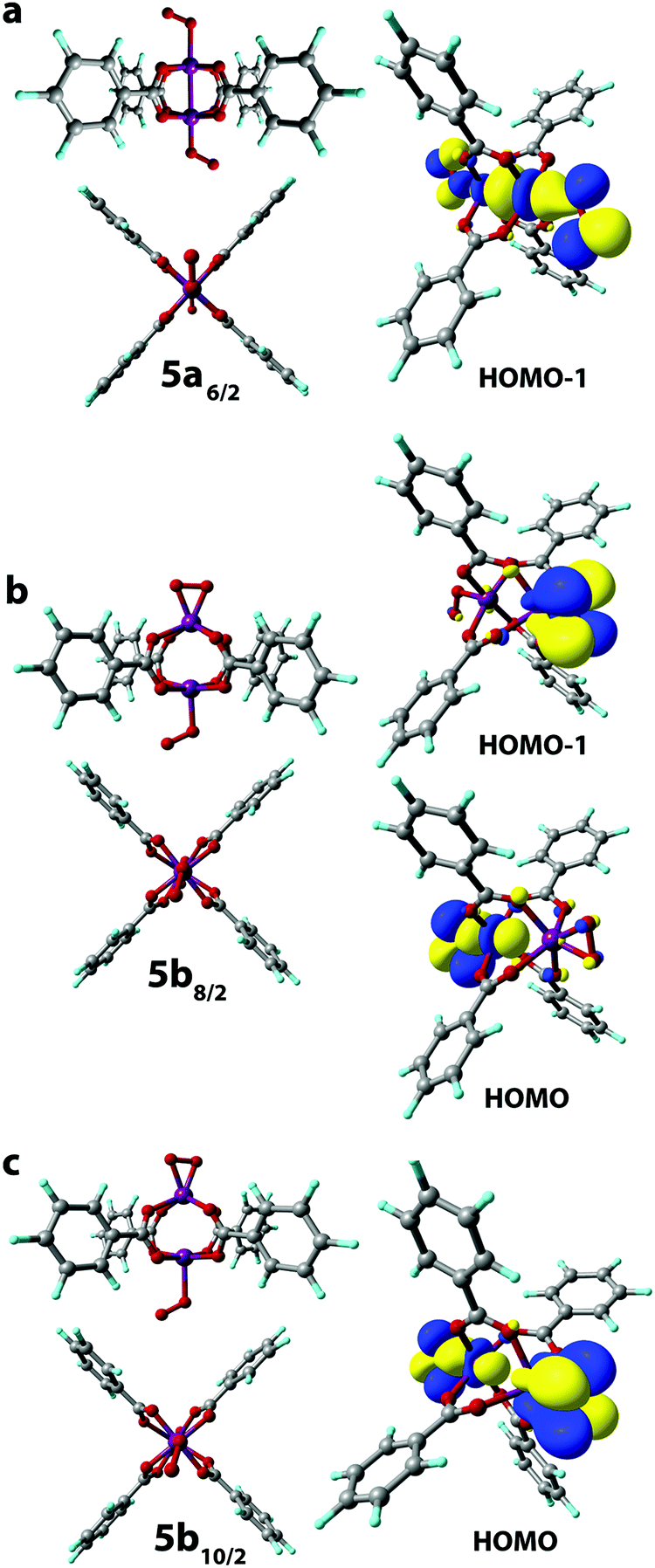 | ||
| Fig. 4 Geometries were optimized at the PBE-D3/6-31G(d,p) level of theory (front and side views are given). The key bonding MOs of the O2[Mn2(PhCOO)4]O2 complexes in S = 6/2 (a), 8/2 (b), and 10/2 (c) spin states are depicted with isosurface thresholds eq. 0.023. | ||
The Mn centers having an η2-bound O2 ligand in 5b8/2 and 5b10/2 isomers were in Mn(4+) oxidation state as follows from the significant alternation of the spin densities upon their formation that is the spin density on the O atoms of the ligand changes from (1, 1) to (−0.27, −0.27) and (0.15, 0.15), respectively. At the same time, the spin density at the η1-bound O2 ligand in 5b8/2 and 5b10/2 changed from (1, 1) to (0.38, 0.46) and (0.78, 0.93), respectively, suggesting the oxidation of the metal center to the Mn(3+) state (see Table S2, ESI†).
The 514/2 spin isomer corresponds to weak non-covalent bonding. Particularly, the lack of close Mn–O contacts is evident in the optimized structures (see Fig. S3b, ESI†) and the corresponding reaction energy is equal to 84 kJ mol−1.
The Mn(3+) state is known to easily disproportionate leading to Mn(2+) and Mn(4+) formation in aqueous solutions.60 Accordingly, our calculations show that the formation of η1-bound O2 complexes, which are the superoxide-bound (O2−) Mn(3+) cations in L[Mn2(PhCOO)4]L, is the least exothermic reaction (5a6/2 and 2b10/2 formation) among the alternative pathways. Therefore, we conclude that the oxidation of Mn-BTC MOF with O2 forming Mn(3+) centers is less likely than the two-electron oxidation paths resulting in the oxidized structures with more stable Mn(4+) centers.
Compared to the exothermic step 1 → 2, in which the first O2 molecule binds, the binding of the second O2 molecule is strongly endothermic. The formation of 5a6/2 and 5b8/2 increases the energies of the systems by 19 and 36 kJ mol−1 compared to 2a6/2 and 2a8/2, respectively. Only formation of the σ-type bound complex 2b10/2 is exothermic by −14 kJ mol−1. The unfavorable binding of the second O2 molecule in the cases of δ complexes is supposedly a result of the distorted and, consequently, more sterically strained structure of the oxidized species. The results above on the facile oxidation and substantial structural distortions of the coordination sphere of Mn centers upon O2 coordination imply that the degradation of Mn-BTC MOFs can already be induced at low O2 concentrations, thus creating an opportunity to develop O2-selective response matrices as a basis for new smart drug delivery platforms.
3.3. H2O2 binding
Next, we computationally investigated the interaction of the Mn-BTC structure-forming unit 110/2 with H2O2 species. The binding results in the formation of two spin isomers (namely, 36/2 and 310/2) via the 1 → 3 process. The isomers have qualitatively close structures with the H2O2 molecule bound in end-on configuration. The formation of the low-spin complex with octahedral structure of Mn centers (36/2, Fig. 5a) was highly endothermic with the reaction energy of 41 kJ mol−1. The formation of the high-spin complex 310/2 was exothermic by −13 kJ mol−1 and the Mn center acquired a severely distorted trigonal-bipyramidal structure (Fig. 5b). The negligible change of the Mulliken spin density on the Mn centers (from 3.16 to 3.14) and (from 4.80 to 4.77) for 36/2 and 310/2, respectively, illustrates a purely non-covalent nature of H2O2 binding that does not directly induce the oxidation of the Mn centers (see Table S2, ESI†).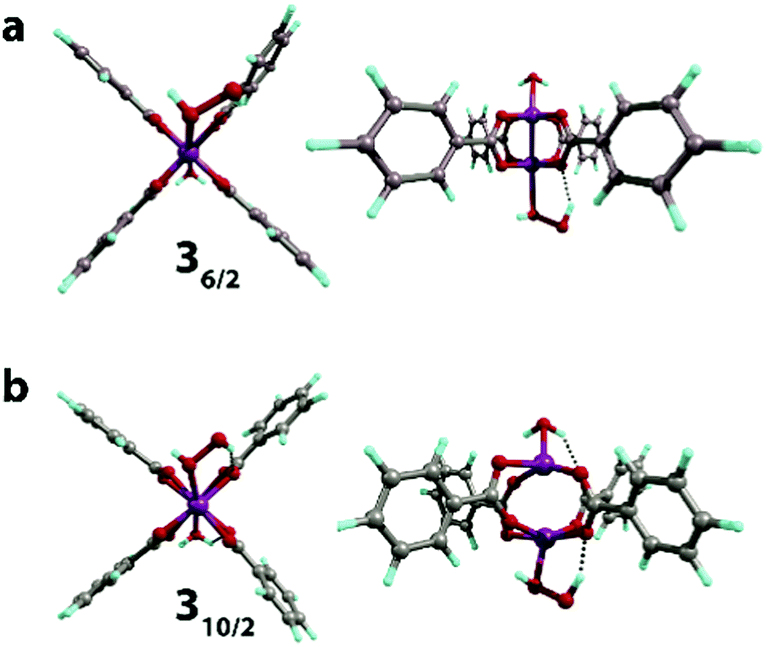 | ||
| Fig. 5 The geometries (front and side views) of the H2O[Mn2(PhCOO)4]H2O2 complex in S = 6/2 (a) and 10/2 (b) spin states optimized at the PBE-D3/6-31G(d,p) level of theory. | ||
The binding of the second H2O2 molecule via the 3 → 6 process proceeded with the retention of the coordination polyhedra and led to the formation of 66/2 and 610/2 (Fig. 6). These reactions had thermal effects of 27 and −23 kJ mol−1, respectively.
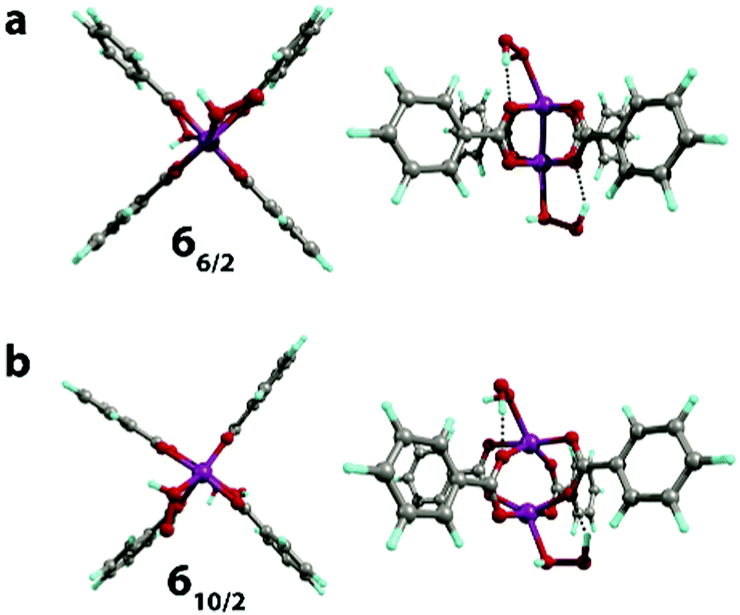 | ||
| Fig. 6 The geometries (front and side views) of the H2O2[Mn2(PhCOO)4]H2O2 complex in S = 6/2 (a) and 10/2 (b) spin states optimized at the PBE-D3/6-31G(d,p) level of theory. | ||
The exchange of two water ligands with H2O2 leads to the coordination bond, which is accompanied by a negligible decrease of the Mulliken spin density on the Mn centers in both 16/2 → 66/2 and 110/2 → 610/2 transformations (from 3.16 to 3.14 and from 4.80 to 4.77, respectively; see Table S2, ESI†). These changes clearly show that the binding of H2O2 is not accompanied by the oxidation of the Mn centers. The final complexes in the 1 → 3 and 3 → 6 steps are stabilized via the formation of the five-membered cycles with the hydrogen bonds between oxygen atoms of the carboxylate ligands and the OH groups of the bound H2O2 species (r(OH⋯O) = 1.7–2.0 Å). In the case of 66/2 and 610/2, two H2O2 molecules bound to both Mn sites form two five-member cyclic structures contributing to their increased stability, resulting in the thermodynamic favorability of the ligand exchange.
3.4. Combined O2 and H2O2 binding
Finally, we considered the possibility of the simultaneous binding of O2 and H2O2 species via the 1 → 4 process. The geometry optimization procedures resulted in a structure of only one structural type to be formed having η2-bound O2 and weakly (covalently) bound H2O2 molecules. The O2-coordinated Mn sites in all 4 complexes adopted a trigonal-bipyramidal configuration (Fig. 7). The high-spin 410/2 complex showed the highest degree of structural distortion upon formation. Notably, the formation of all three possible complexes 46/2, 48/2, and 410/2 was exothermic with reaction energies of −33 kJ mol−1, −69 kJ mol−1 and −23 kJ mol−1, respectively. The oxidation states of the Mn centers in the formed H2O2[Mn2(PhCOO)4]O2 complexes are (2+, 4+), (2+, 4+), and (2+, 4+), respectively. The Kohn–Sham orbital and the Mulliken spin density analyses clearly illustrate the (2+, 4+) formal oxidation states in both 46/2 and 48/2 spin isomers. Spin density values on O atoms in the O2 ligand are notably reduced from (1, 1) to (0.15, 0.15) and (0.11, 0.11) in 46/2 and 48/2, respectively (Table S2, ESI†). The spin density values on the O2-bound Mn site decreased upon formation of 46/2 (from 3.16 to 2.35) and 48/2 (from 3.28 to 2.91). Less pronounced spin density changes were observed in the 110/2 → 410/2 process. Particularly, the change from 4.80 to 3.88 a.u. was observed on the Mn cation. In the case of the O2 ligand, the observed change was from (1, 1) to (0.42 to 0.61) a.u. (Table S2, ESI†). The key bonding orbital analysis (Fig. 7) corroborates the π-symmetry bonding and the formation of the Mn center in the (+4) formal oxidation state. The lower exothermicity of the formation of 410/2 compared to the 46/2 and 48/2 isomers is in line with the lower orbital overlap (no δ-symmetry bond formed).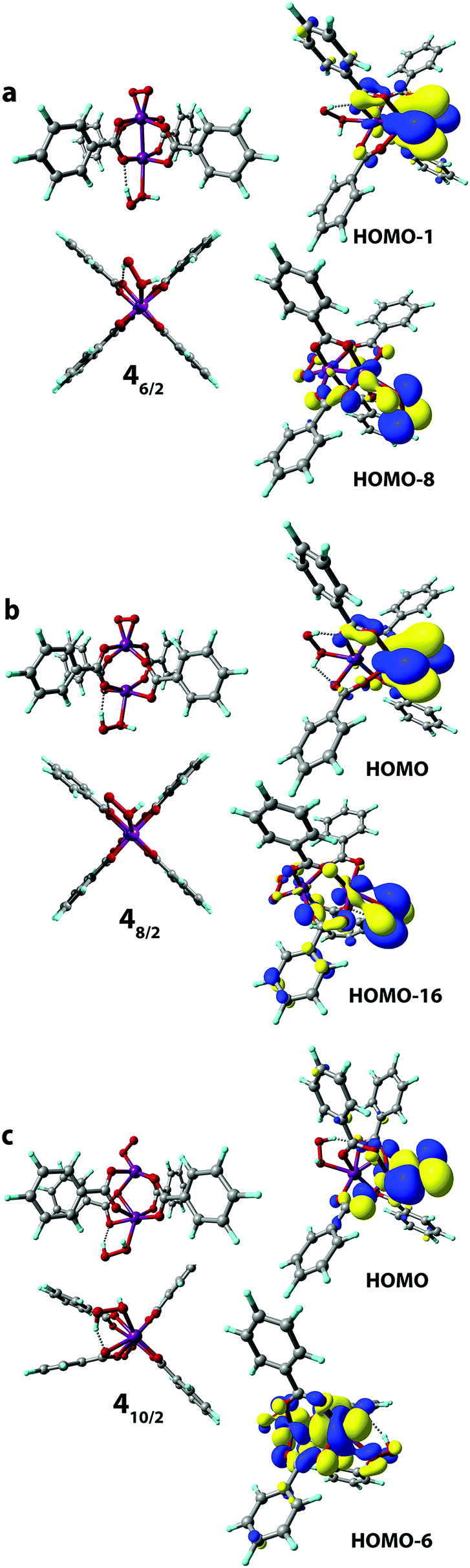 | ||
| Fig. 7 Geometries were optimized at the PBE-D3/6-31G(d,p) level of theory (front and side views are given). The key bonding MOs of the H2O2[Mn2(PhCOO)4]O2 complex in S = 6/2 (a), 8/2 (b), and 10/2 (c) spin states are depicted (isosurface thresholds are 0.023 (a), 0.023 for HOMO and 0.018 for HOMO−16 (b), and 0.023 for HOMO and 0.018 for HOMO−6 (c)). | ||
3.5. Reactivity
Degradation of the MOF material in aqueous solutions may potentially occur either via the direct hydrolysis of the metal-carboxylate moieties or via the oxidation of the metal-containing nodes. Whereas our calculations suggest that the latter mechanism should dominate the structural response of Mn-BTC to the oxidative conditions in the presence of molecular O2, the mechanism of MOF degradation in the presence of H2O2 as the oxidant requires the combination of the metal oxidation with a hydrolytic process. Indeed, no significant deformation of the [Mn2(PhCOO)4] structural unit was observed upon H2O2 binding. The coordinatively unsaturated metal centers in the [Mn2(PhCOO)4] cluster tend to bind strongly with the open-shell O2 species owing to the radical nature of the former. The degradation of Mn-BTC via the oxidation by H2O2 closed-shell species may have a more complex mechanism. The adsorption complexes 6 discussed above thus become the starting point for the computational assessment of the feasibility of such a complex oxidation-assisted hydrolysis mechanism.The oxidation-assisted hydrolysis thus starts with the substitution of the water molecules in the solvated 110/2 structure discussed in detail previously and the formed 66/2 and 610/2 intermediates undergo further oxidation (Fig. 6). The transition state on the potential energy surface with 10/2 spin state is denoted as TS10/2 and corresponds to a barrier of 21 kJ mol−1. The computed imaginary frequency of the TS10/2 structure has the value of i365 cm−1, corresponding to the O–O-bond stretching in the H2O2 moiety and the neighbouring Mn site has the trigonal-bipyramidal coordination polyhedron. After passing TS10/2, the relative order of the potential energy surfaces with S = 10/2 and 6/2 changes and the system may undergo a spin crossing.
For the high-spin complex 710/2, the O–O bond cleavage in the peroxide moiety leads to Mn(4+) center formation, which has octahedral geometry and bears two terminal OH ligands. The oxidation is accompanied with the reduction of the Mulliken spin density value in the Mn center (4.78 to 3.90 a.u.; Table S3, ESI†). The reaction energy for the latter process is −87 kJ mol−1. The subsequent hydrolysis leads to H2O binding to the Mn(4+) site through the formation of the hydrogen bonds between H atoms of H2O molecule and O sites of the hydroxyl and benzoate ligands. The corresponding intermediate is denoted as 810/2 (Fig. 8). The non-valent H2O molecule coordination does not alter the octahedral geometry of the Mn(4+) centers.
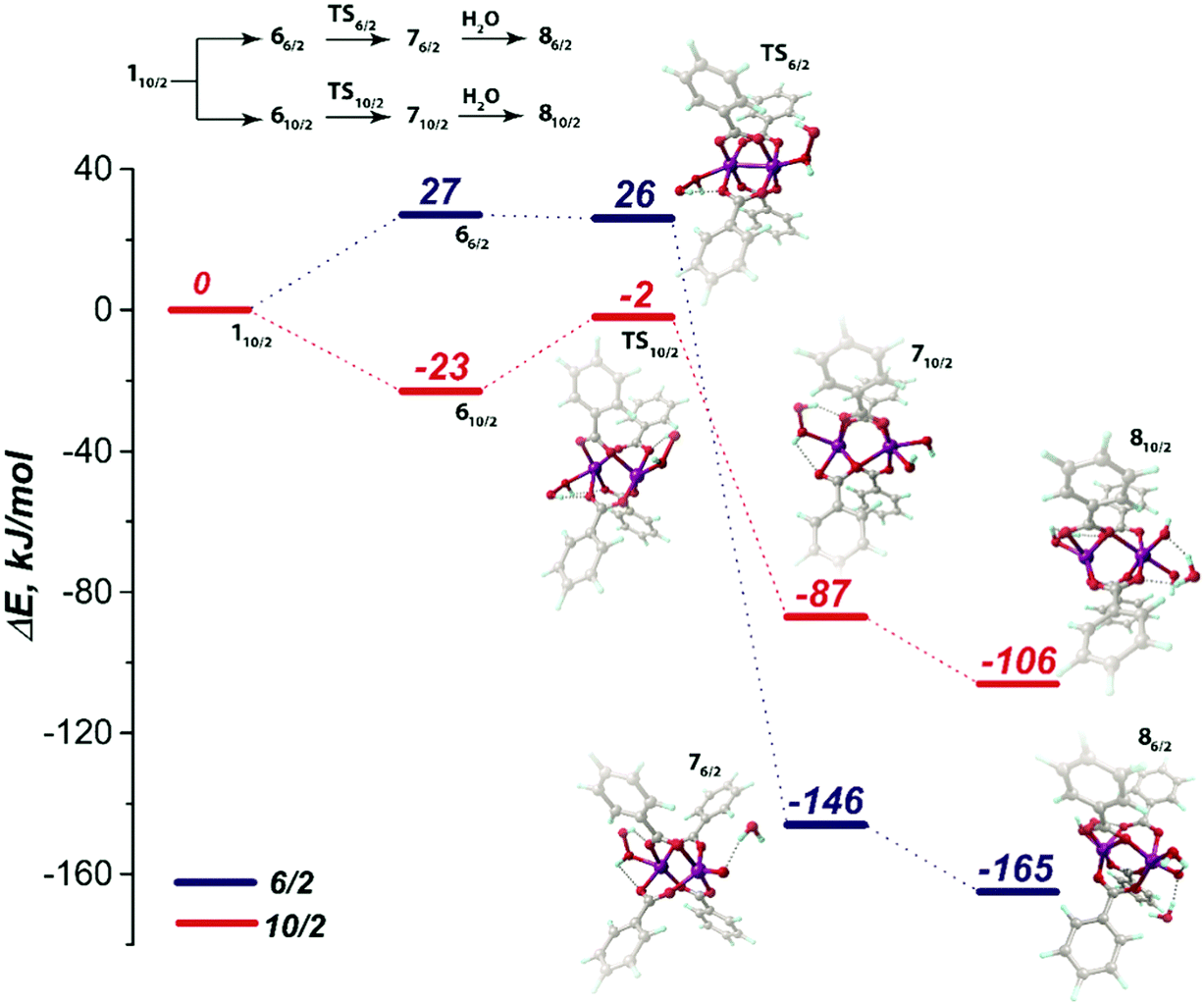 | ||
| Fig. 8 Reaction energy profiles for the oxidation-induced hydrolysis of 66/2 and 610/2. The optimized structures of the initial (66/2, 610/2), transition (TS6/2, TS10/2), final oxidized (76/2, 710/2) and hydrolyzed states (86/2, 810/2) are presented next to the corresponding energy levels. All geometries were optimized at the PBE-D3/6-31G(d,p) level of theory. | ||
The Mn-BTC degradation via the 6/2 pathway was thermodynamically preferred according to the modeling results. The 76/2 intermediate had −146 kJ mol−1 lower energy relative to the 110/2 level and −59 kJ mol−1 lower relative to the 710/2 counterpart. We expect the 66/2 to 76/2 transition to be barrierless. Particularly, the “barrier” of −1 kJ mol−1, which was computed at the PBE-D3/6-311++G(d,p)//PBE-D3/6-31G(d,p) level with the PCM solvation model, could be an inaccuracy caused by the solvation model that was applied in post-optimization single-point energy refinement. The imaginary mode in the TS6/2 structure corresponded to O–O bond stretching and had the frequency of i261 cm−1 as computed at the PBE-D3/6-31G(d,p) level of theory. The oxidized Mn(4+) center in the 76/2 intermediate had a trigonal-bipyramidal structure bearing the oxo-ligand. The spin density value decreased from 3.14 to 2.33 a.u. in 66/2 → 76/2 (Table S3, ESI†). Then oxidized 76/2 species underwent a strongly exothermic (ΔE = −165 kJ mol−1) hydrolysis step. The Mn center adopted the octahedral coordination in the 86/2 intermediate formed upon the hydrolysis.
The structural flexibility of the unconstrained cluster model provides sufficient space for the relaxation upon the interaction with the oxidative species needed to ensure the overall stability of the extended periodic structure composed of such units. Here we infer that the potential degradation paths are initiated when the ROS binding induces severe structural deformations that substantially alter the coordination polyhedra of the Mn sites. Although the introduction of the hard geometric constraints decreased the exothermicity of the formation of the adsorption complexes (Fig. S1 and Table S1, ESI†), the effects were minor and did not affect the main conclusions made on the basis of the fully relaxed models that have been discussed above. In some cases, we, however, observed the enhancement of the reactivity of the Mn sites owing to the excessive strain provided by the constrained benzoate ligands, which resulted in some cases in decoordination events and structural alteration of the otherwise stable geometries (Fig. S2, ESI†). These effects appear to be highly model-dependent and are not expected to be representative to the sufficiently flexible MOF structures that are captured by the relaxed cluster models.
4. Conclusions
Density functional theory calculations have been performed to investigate the interactions of common oxidants (O2 and H2O2) with a cluster model representing the key structural fragment of Mn-BTC MOF. The modeling was performed to assess potential material degradation paths under the oxidizing conditions commonly encountered in pathogenic tissues of living organisms. Such an environment-induced degradation of the nanoporous material was discussed in the context of the development of new approaches for targeted drug delivery.The calculations indicated the sufficient stability of the Mn-carboxylate structure-forming units towards direct hydrolysis while the interaction with the O2 and H2O2 species led to a facile oxidation of the Mn(2+) to the more thermodynamically stable Mn(4+) state. The oxidation was proceeded with a substantial deformation of the coordination sphere of the transition metal centers and, accordingly, facilitation of the subsequent hydrolysis of the coordination bonds.
DFT calculations showed a facile exchange of non-covalently bound H2O ligands at the Mn(2+) centers in the Mn-BTC with molecular O2 resulting in covalent σ, π, or δ bonding. The latter path was more favorable and it resulted in the oxidation of the Mn center to the formal (4+) state and the formation of a peroxide adduct. The formation of the Mn(3+) state was the result of the O2 ligand complexation in the σ bonding mode and was likely the intermediate state in the formation of the more stable Mn(4+) counterpart.
On the contrary, hydrogen peroxide molecules bind weakly to the Mn sites. The respective ligand exchange reaction with the starting aqua complex was an exothermic process for the high-lying spin state. The H2O2-induced degradation of Mn-BTC in aqueous media involved the cleavage of the O–O bond in the coordinated H2O2 molecules, resulting in the oxidation of the Mn(2+) center to the (4+) state, which strongly facilitated the subsequent hydrolysis. Importantly, we note the importance of spin transitions for all reaction paths considered in this study. This has to be properly accounted for in further studies on the stability of Mn-based MOF materials. Our current results clearly show the limited potential of Mn-carboxylate building blocks for oxidation catalysis or gas-sorption applications where the interactions with potential ROS species will inevitably result in the structural distortion of the framework and the long-term degradation of the functional materials. Nevertheless, the favorable reaction paths identified here may be utilized for engineering a new mechanism of the structural response of Mn-containing nanocontainers to pathology-induced alterations of biological environments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education and Science of the Russian Federation (Project 11.1706.2017/4.6). E. A. P. acknowledges partial support from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 725686). A. V. V. acknowledges the support from the Government of the Russian Federation (Grant 08-08). The Netherlands Organization for Scientific Research (NWO) is acknowledged for providing access to the supercomputer facilities.References
- P. W. Siu, J. P. Siegfried, M. H. Weston, P. E. Fuller, W. Morris, C. R. Murdock, W. J. Hoover, R. K. Richardson, S. Rodriguez and O. K. Farha, Boron Trifluoride Gas Adsorption in Metal-Organic Frameworks, Inorg. Chem., 2016, 55, 12110–12113 CrossRef PubMed.
- T. Simon-Yarza, S. Rojas, P. Horcajada and C. Serre, The Situation of Metal-Organic Frameworks in Biomedicine, Elsevier, 2017 Search PubMed.
- J. S. Choi, W. J. Son, J. Kim and W. S. Ahn, Microporous Mesoporous Mater., 2008, 116, 727–731 CrossRef.
- N. Wei, Y. Zhang, L. Liu, Z. B. Han and D. Q. Yuan, Pentanuclear Yb(III) cluster-based metal-organic frameworks as heterogeneous catalysts for CO2 conversion, Appl. Catal., B, 2017, 219, 603–610 CrossRef.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés, I. Xamena and J. Gascon, Metal-organic framework nanosheets in polymer composite materials for gas separation, Nat. Mater., 2015, 14, 48–55 CrossRef PubMed.
- Z.-G. Gu, S. Grosjean, S. Bräse, C. Wöll and L. Heinke, Enantioselective adsorption in homochiral metal–organic frameworks: the pore size influence, Chem. Commun., 2015, 51, 8998–9001 RSC.
- L. R. Mingabudinova, V. V. Vinogradov, V. A. Milichko, E. Hey-Hawkins and A. V. Vinogradov, Metal–organic frameworks as competitive materials for non-linear optics, Chem. Soc. Rev., 2016, 45, 5408–5431 RSC.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Multifunctional metal–organic framework catalysts: synergistic catalysis and tandem reactions, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- L. Zhu, X. Q. Liu, H. L. Jiang and L. B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef PubMed.
- D. Damasceno Borges, G. Maurin and D. S. Galvão, Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries, MRS Adv., 2017, 2, 519–524 CrossRef.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, Virtual high throughput screening confirmed experimentally: Porous coordination polymer hydration, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef PubMed.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chemical and thermal stability of isotypic metal-organic frameworks: Effect of metal ions, Chem. – Eur. J., 2011, 17, 6437–6442 CrossRef PubMed.
- C. Y. Sun, C. Qin, C. G. Wang, Z. M. Su, S. Wang, X. L. Wang, G. S. Yang, K. Z. Shao, Y. Q. Lan and E. B. Wang, Chiral nanoporous metal-organic frameworks with high porosity as materials for drug delivery, Adv. Mater., 2011, 23, 5629–5632 CrossRef PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef PubMed.
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef.
- E. Bellido, M. Guillevic, T. Hidalgo, M. J. Santander-Ortega, C. Serre and P. Horcajada, Understanding the colloidal stability of the mesoporous MIL-100(Fe) nanoparticles in physiological media, Langmuir, 2014, 30, 5911–5920 CrossRef PubMed.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, Postsynthetic modifications of iron-carboxylate nanoscale metal-organic frameworks for imaging and drug delivery, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef PubMed.
- J. Della Rocca, D. Liu and W. Lin, Nanoscale metal-organic frameworks for biomedical imaging and drug delivery, Acc. Chem. Res., 2011, 44, 957–968 CrossRef PubMed.
- R. Ananthoji, J. F. Eubank, F. Nouar, H. Mouttaki, M. Eddaoudi and J. P. Harmon, Symbiosis of zeolite-like metal–organic frameworks (rho-ZMOF) and hydrogels: Composites for controlled drug release, J. Mater. Chem., 2011, 21, 9587 RSC.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging, Nat. Mater., 2010, 9, 172–178 CrossRef PubMed.
- C.-Y. Sun, C. Qin, X.-L. Wang and Z.-M. Su, Metal-organic frameworks as potential drug delivery systems, Expert Opin. Drug Delivery, 2013, 10, 89–101 CrossRef PubMed.
- S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Adv. Energy Mater., 2017, 7 Search PubMed.
- P. Canepa, K. Tan, Y. Du, H. Lu, Y. J. Chabal and T. Thonhauser, Structural, elastic, thermal, and electronic responses of small-molecule-loaded metal–organic framework materials, J. Mater. Chem. A, 2015, 3, 986–995 RSC.
- J. a Kellum, M. Song and J. Li, Science review: extracellular acidosis and the immune response: clinical and physiologic implications, Crit. Care, 2004, 8, 331–336 CrossRef PubMed.
- P. J. Barnes, Free Radical Biol. Med., 1990, 9, 235–243 CrossRef PubMed.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Reactive Oxygen Species in Inflammation and Tissue Injury, Antioxid. Redox Signaling, 2014, 20, 1126–1167 CrossRef PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Flexible porous metal-organic frameworks for a controlled drug delivery, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef PubMed.
- K. S. Egorova and V. P. Ananikov, Toxicity of Metal Compounds: Knowledge and Myths, Organometallics, 2017, 36, 4071–4090 CrossRef.
- G. C. Dismukes, Manganese Enzymes with Binuclear Active Sites, Chem. Rev., 1996, 96, 2909–2926 CrossRef PubMed.
- N. A. Law, M. T. Caudle and V. L. Pecoraro, Manganese Redox Enzymes and Model Systems: Properties, Structures, and Reactivity, Adv. Inorg. Chem., 1998, 46, 305–440 CrossRef.
- G. D. Lawrence and D. T. Sawyer, The chemistry of biological manganese, Coord. Chem. Rev., 1978, 27, 173–193 CrossRef.
- T. Ladrak, S. Smulders, O. Roubeau, S. J. Teat, P. Gamez and J. Reedijk, Manganese-based metal-organic frameworks as heterogeneous catalysts for the cyanosilylation of acetaldehyde, Eur. J. Inorg. Chem., 2010, 3804–3812 CrossRef.
- Q. Sun, M. Liu, K. Li, Y. Han, Y. Zuo, F. Chai, C. Song, G. Zhang and X. Guo, Synthesis of Fe/M (M = Mn, Co, Ni) bimetallic metal organic frameworks and their catalytic activity for phenol degradation under mild conditions, Inorg. Chem. Front., 2017, 4, 144–153 RSC.
- C. Pereira, M. Simoes, J. Tome and F. Almeida Paz, Porphyrin-Based Metal-Organic Frameworks as Heterogeneous Catalysts in Oxidation Reactions, Molecules, 2016, 21, 1348 CrossRef PubMed.
- Y. Ming, J. Purewal, J. Yang, C. Xu, R. Soltis, J. Warner, M. Veenstra, M. Gaab, U. Müller and D. J. Siegel, Kinetic stability of MOF-5 in humid environments: Impact of powder densification, humidity level, and exposure time, Langmuir, 2015, 31, 4988–4995 CrossRef PubMed.
- P. Guo, D. Dutta, A. G. Wong-Foy, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 2651–2657 CrossRef PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Metal–organic frameworks as heterogeneous catalysts for oxidation reactions, Catal. Sci. Technol., 2011, 1, 856 RSC.
- S. Zuluaga, E. M. A. Fuentes-Fernandez, K. Tan, F. Xu, J. Li, Y. J. Chabal and T. Thonhauser, Understanding and controlling water stability of MOF-74, J. Mater. Chem. A, 2016, 4, 5176–5183 RSC.
- D. Wu, W. Yan, H. Xu, E. Zhang and Q. Li, Defect engineering of Mn-based MOFs with rod-shaped building units by organic linker fragmentation, Inorg. Chim. Acta, 2017, 460, 93–98 CrossRef.
- Y. Kuwahara, Y. Yoshimura and H. Yamashita, Liquid-phase oxidation of alkylaromatics to aromatic ketones with molecular oxygen over a Mn-based metal–organic framework, Dalton Trans., 2017, 46, 8415–8421 RSC.
- R. E. Hansen and S. Das, Biomimetic di-manganese catalyst cage-isolated in a MOF: robust catalyst for water oxidation with Ceiv, a non-O-donating oxidant, Energy Environ. Sci., 2014, 7, 317–322 RSC.
- D. H. Lee, S. Kim, M. Y. Hyun, J.-Y. Hong, S. Huh, C. Kim and S. J. Lee, Controlled growth of narrowly dispersed nanosize hexagonal MOF rods from Mn(III)–porphyrin and In(NO3)3 and their application in olefin oxidation, Chem. Commun., 2012, 48, 5512 RSC.
- S. Bhattacharjee, D.-A. Yang and W.-S. Ahn, A new heterogeneous catalyst for epoxidation of alkenes via one-step post-functionalization of IRMOF-3 with a manganese(ii) acetylacetonate complex, Chem. Commun., 2011, 47, 3637 RSC.
- J. Ye and C. Liu, Cu3(BTC)2: CO oxidation over MOF based catalysts, Chem. Commun., 2011, 47, 2167 RSC.
- K. Brown, S. Zolezzi, P. Aguirre, D. Venegas-Yazigi, V. Paredes-García, R. Baggio, M. A. Novak and E. Spodine, [Cu(H2btec)(bipy)]∞: a novel metal organic framework (MOF) as heterogeneous catalyst for the oxidation of olefins, Dalton Trans., 2009, 1422 RSC.
- K. K. Tanabe and S. M. Cohen, Engineering a metal-organic framework catalyst by using postsynthetic modification, Angew. Chem., Int. Ed., 2009, 48, 7424–7427 CrossRef PubMed.
- M. Zhang, X. Huang and Y. Chen, DFT insights into the adsorption of NH3-SCR related small gases in Mn-MOF-74, Phys. Chem. Chem. Phys., 2016, 18, 28854–28863 RSC.
- P. Maitarad, S. Namuangruk, D. Zhang, L. Shi, H. Li, L. Huang, B. Boekfa and M. Ehara, Metal-porphyrin: A potential catalyst for direct decomposition of N2O by theoretical reaction mechanism investigation, Environ. Sci. Technol., 2014, 48, 7101–7110 CrossRef PubMed.
- P. Maitarad, J. Meeprasert, L. Shi, J. Limtrakul, D. Zhang and S. Namuangruk, Mechanistic insight into the selective catalytic reduction of NO by NH3 over low-valent titanium-porphyrin: A DFT study, Catal. Sci. Technol., 2016, 6, 3878–3885 RSC.
- M. J. Frisch et al. , Gaussian 09, Revis. D.01, 2009 Search PubMed.
- V. V. Vinogradov, A. S. Drozdov, L. Mingabudinova, E. M. Shabanova, N. Kolchina, E. I. Anastasova, A. A. Markova, A. Shtil, V. Milichko, G. L. Starova, R. Precker, A. Vinogradov, E. Hey-Hawkins and E. Pidko, Composites Based on Heparin and MIL-101(Fe): The Drug Releasing Depot for Anticoagulant Therapy and Advanced Medical Nanofabrication, J. Mater. Chem. B, 2018, 6, 2450–2459 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef.
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Complexity behind CO2 capture on NH2-MIL-53(Al), Langmuir, 2011, 27, 3970–3976 CrossRef PubMed.
- J. Van Den Bergh, C. Gücüyener, E. A. Pidko, E. J. M. Hensen, J. Gascon and F. Kapteijn, Understanding the anomalous alkane selectivity of ZIF-7 in the separation of light alkane/alkene mixtures, Chem. – Eur. J., 2011, 17, 8832–8840 CrossRef PubMed.
- Chemcraft, http://www.chemcraftprog.com, accessed 5 January 2018.
- L. R. Falvello, B. M. Foxman and C. A. Murillo, Fitting the Pieces of the Puzzle: The δ Bond, Inorg. Chem., 2014, 53, 9441–9456 CrossRef PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education Limited, Harlow, 4th edn, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp00397a |
| This journal is © the Owner Societies 2018 |