
Cluster-model DFT simulations of the infrared spectra of triazine-based molecular crystals†
 a
Yuanchun
Zhao
*a
and
a
Yuanchun
Zhao
*a
and
Abstract
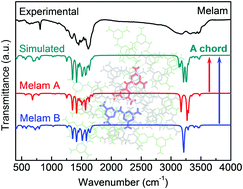
Understanding the intermolecular interactions in the context of crystal packing is of fundamental significance in molecular materials science. Infrared (IR) spectroscopy can provide complementary structural information; however, it still remains a great challenge to accurately predict the molecular IR vibrations in the crystalline phase. Here we report a cluster-model approach to simulate the IR spectra of triazine-based molecular crystals via density functional theory (DFT) calculations. In the properly designed cluster models, the molecular IR vibrations are expressed by a representative unit, while the nearest-neighbouring molecules are treated as a “frozen shell” to mimic the surrounding crystallographic environments. Much smaller clusters can be built by considering the crystallographic equivalence in the unit cell, which are able to perform DFT calculations on more complicated crystal structures with endurable computational costs. The simulated spectra show excellent consistencies with the experimental ones, particularly providing an in-depth understanding of the vibrational modes closely related to hydrogen bonding. Most importantly, the selectively built clusters based on the crystallographically independent molecules in the unit cell allow us to perform specific IR-spectral simulations, by which their distinct hydrogen-bonding environments have been clearly revealed for the first time.
 Please wait while we load your content...
Please wait while we load your content...

