
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical ruthenium-catalyzed alkyne annulations by C–H/Het–H activation of aryl carbamates or phenols in protic media†
Ruhuai
Mei
,
Julian
Koeller
and
Lutz
Ackermann
 *
*
Institute for Organic and Biomolecular Chemistry, Georg-August-Universität Göttingen, Tammannstr. 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 19th October 2018
Abstract
Electrooxidative peri-C–H activation was accomplished by versatile ruthenium(II) catalysis in terms of C–H/N–H and C–H/O–H functionalization. Thus, alkyne annulations proved viable with ample scope by organometallic C–H activation. The sustainable electrocatalysis exploited electricity, thereby avoiding the use of toxic transition metals as sacrificial oxidants. The robust ruthenium(II)-electrocatalysis was operative in a protic alcohol/H2O reaction medium with excellent levels of position-, regio- and chemo-selectivity.
Transition metal-catalyzed oxidative C–H activation1 has been recognized as a powerful tool for the assembly of π-conjugated heterocycles, with major potential for molecular syntheses and material sciences.2 Despite of considerable progress, these oxidative C–H functionalizations heavily rely on stoichiometric amounts of expensive and toxic metal salts as the sacrificial oxidants.3 Electrosynthesis4 has emerged as an increasingly viable platform for sustainable transformations, which enables the use of inexpensive electricity as the terminal oxidant.5 In this context, major advances were achieved, exploiting transition-metals based on inter alia palladium,6 cobalt,7 ruthenium,8 rhodium,9 copper,10 or iridium.11
Recently, we have introduced a versatile ruthenium12 catalyst for oxidative C–H/O–H annulation of alkynes13 by 1-naphthols to access fused pyran derivatives.14 Despite considerable experimentation, these transformations were restricted to the use of anti-bacterial copper(II) oxidants. Within our program on sustainable C–H activation,15 we have now developed the first electrochemical, organometallic peri C–H activation with synthetically meaningful arylcarbamates,16 on which we report herein (Fig. 1). Notable features of our finding include (a) the first electrooxidative ruthenium-catalysed peri C–H activation, (b) C–H/N–H annulation by electrooxidative ruthenium catalysis, (c) weakly coordinating17 naphthol for C–H/O–H alkyne annulations with electricity as the terminal oxidant, and (d) mechanistic insights into electrochemical peri C–H activation by experiments. It is furthermore noteworthy that our electrooxidative strategy also set the stage for the first ruthenium-catalyzed C–H/N–H activation/alkyne annulation of naphthylcarbamates.
 | ||
| Fig. 1 Electrochemical C–H/Het–H activation with alkynes. | ||
At the outset of our studies, we tested various reaction conditions for the envisioned electrooxidative annulation of alkyne 2a by easily-accessible ethyl naphthalen-1-ylcarbamate (1a) (Table 1 and Table S-1 in the ESI†).18 Among a set of representative solvents, DMF and t-AmOH furnished promising results (entries 1 and 2), while a solvent mixture of t-AmOH and H2O,19 along with KOAc as the additive, proved to be ideal (entries 3–6). The catalytic efficacy could be further improved when the electrolysis was conducted at a constant current regime of 1.5 mA (entries 7–9). Control experiments demonstrated the outstanding performance of the ruthenium(II) carboxylates (entries 9–11), while typically used rhodium(III) catalysts fell short in delivering the desired product 3aa (entry 12).
|
|
|||
|---|---|---|---|
| Entry | [TM] | Solvent | Yield [%] |
| a General reaction conditions: 1a (0.60 mmol), 2a (0.30 mmol), [TM] (10 mol %), KOAc (0.60 mmol), n-Bu4NPF6 (0.18 mmol), solvent (4.0 mL), 100 °C, 16 h, under N2, constant current electrolysis (CCE) at 4.0 mA, undivided cell, RVC anode, Pt-plate cathode. Isolated yields. b NaOPiv (0.60 mmol) instead of KOAc. c n-Bu4NClO4 (0.18 mmol) instead of n-Bu4NPF6. d 10 mA, 2.5 h. e 2.0 mA, 16 h. f 1.5 mA, 24 h. g Without current, 24 h. | |||
| 1 | [RuCl2(p-cymene)]2 | DMF | 35 |
| 2 | [RuCl2(p-cymene)]2 | t-AmOH | 51 |
| 3 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 61 |
| 4 | [RuCl2(p-cymene)]2 | DMF/H2O (3/1) | 28 |
| 5 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 57b |
| 6 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 65c |
| 7 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 36c,d |
| 8 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 77c,e |
| 9 | [RuCl 2 (p-cymene)] 2 | t-AmOH/H 2 O (3/1) | 82 , |
| 10 | [RuCl2(p-cymene)]2 | t-AmOH/H2O (3/1) | 31c,g |
| 11 | — | t-AmOH/H2O (3/1) | <5c,f |
| 12 | [Cp*RhCl2]2 | t-AmOH | <5 |

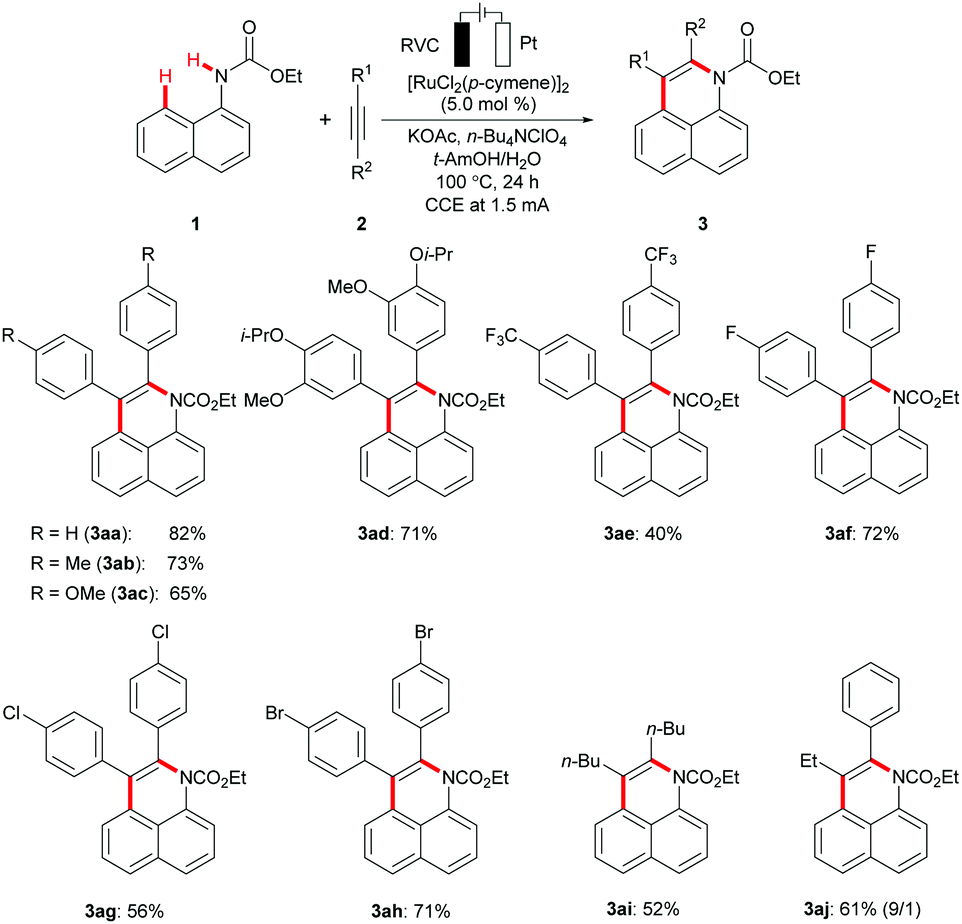
With the optimized ruthenium electrocatalysis being established, we explored its versatility with a set of representative internal alkynes (Scheme 1). Thus, the broadly applicable ruthenium(II) catalyst enabled the efficient conversion of diaryl- and dialkyl-substituted alkynes 2a–2i. The robust rutheniumelectrocatalysis displayed a remarkable tolerance of valuable functional groups, such as ester, fluoro, chloro, or bromo substituents, setting the stage for further late-stage diversifications. Unsymmetrical alkyne 2j delivered the desired product 3aj with high levels of regio-control, placing the aromatic moiety in proximity to the nitrogen heteroatom.
 | ||
| Scheme 1 Electrochemical C–H/N–H annulation of alkynes 2. | ||
Subsequently, a variety of arylcarbamates 1 were probed, highlighting an outstanding selectivity for the peri C–H activation to deliver benzoquinoline derivatives 3 (Scheme 2). Ethyl naphthalen-1-ylcarbamates 1 bearing either electron-withdrawing or electron-donating groups on the naphthyl ring provided the corresponding benzoquinolines 3 with high efficacy. It is worth noting that the ruthenium(II) electrocatalysis was likewise applicable to heterocyclic substrate ethyl quinolin-5-ylcarbamate 1d, selectively furnishing the annulated product 3da. Likewise, the extended aromatic substrate fluoranthen-3-ylcarbamate 1f was smoothly converted to give the π-conjugated polyheterocycles 3fa, featuring unique fluorescence emission.
 | ||
| Scheme 2 Electrochemical C–H/N–H activation with amides 2. | ||
The ruthenium(II) electrocatalysis was not limited to C–H/N–H activation with arylcarbamates 1. Indeed, highly effective annulations of alkynes 2 proved to be viable with the weakly coordinating17 naphthol derivatives 4 (Scheme 3, and Table S-2 in the ESI†).18 Thus, a series of fluorescent pyrans were accessed in a step-economical fashion via the chemo-selective C–H/O–H functionalization, featuring electricity as the sacrificial oxidant.
 | ||
| Scheme 3 Electrochemical C–H/O–H activation with naphtholes 4. | ||
Given the unique site- and regio-selectivity of the electro-chemical ruthenium(II) catalysis, we became interested in its modus operandi. To this end, a catalytic C–H transformation in the presence of D2O employing an excess of substrate 1a resulted in a significant H/D exchange in the peri-position of the recovered starting material [D1]-1a (Scheme 4a). This observation provided strong evidence for a reversible, organometallic C–H ruthenation step. In good agreement with this finding, a kinetic isotope effect (KIE) was not observed, when comparing the initial rates of transformations with substrates 1a and [D7]-1a (Scheme 4b). Furthermore, we performed intermolecular competition experiments between differently substituted arylcarbamates 1 and alkynes 2, which revealed electron-deficient alkynes and arylcarbamates to be preferentially converted (Schemes 4c and d).
 | ||
| Scheme 4 Summary of key mechanistic findings. | ||
Based on our mechanistic studies, we propose a plausible catalytic cycle to commence by a facile organometallic C–H activation (Scheme 5). Thereby, ruthena(II)cycle 7 is generated, along with two equivalents of carboxylic acid. Thereafter, migratory alkyne insertion furnishes the seven-membered ruthena(II)cycle 9, which rapidly undergoes reductive elimination to deliver the ruthenium(0) sandwich complex 10. The key reoxidation of the thus-formed ruthenium(0) complex 10 is finally accomplished by anodic oxidation, while cathodic reduction generates molecular hydrogen as the sole stoichiometric byproduct.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
In summary, we have reported on the first electrocatalytic organometallic C–H activation with aromatic carbamates and phenols. Hence, a versatile ruthenium(II) carboxylate catalyst enabled electrooxidative C–H/N–H and C–H/O–H activation/alkyne annulations with ample scope. The C–H activation employed electricity as the sacrificial oxidant, which avoids the use of toxic metals as terminal oxidants, generating molecular hydrogen as the only byproduct. Mechanistic studies provided strong support for an organometallic C–H ruthenation, along with an efficient electro-reoxidation of the key ruthenium(0) intermediate by environmentally-benign electricity that is amenable for renewable forms of energy. The electrochemical C–H/Het–H activation was operative in protic aqueous reaction media.
Generous support by the DFG (Gottfried-Wilhelm-Leibniz Award) and CSC (fellowship to R. M.) is most gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.References
- (a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (b) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (c) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (d) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (e) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC; (f) J. He, M. Wasa, K. S. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2016, 117, 8754–8786 CrossRef PubMed; (g) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (h) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (j) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- (a) J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed; (b) L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS; (c) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (d) L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed; (e) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS.
- Selected reviews: (a) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC; (b) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 624–655 CrossRef PubMed.
- (a) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (c) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (e) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2017, 118, 4485–4540 CrossRef PubMed; (f) Z.-W. Hou, Z.-Y. Mao and H.-C. Xu, Synlett, 2017, 1867–1872 CAS; (g) M. Khrizanforov, S. Strekalova, V. Grinenko, V. Khrizanforova, T. Gryaznova and Y. H. Budnikova, Russ. Chem. Bull., 2017, 66, 1446–1449 CrossRef CAS; (h) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed; (i) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- For reviews, see: (a) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (b) M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (c) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (d) N. Sauermann, T. H. Meyer and L. Ackermann, Chem. – Eur. J., 2018, 24 DOI:10.1002/chem.201802706.
- (a) A. Shrestha, M. Lee, A. L. Dunn and M. S. Sanford, Org. Lett., 2018, 20, 204–207 CrossRef CAS PubMed; (b) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed; (c) C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC; (d) Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905–2908 CrossRef CAS PubMed; (e) M. Konishi, K. Tsuchida, K. Sano, T. Kochi and F. Kakiuchi, J. Org. Chem., 2017, 82, 8716–8724 CrossRef CAS PubMed; (f) F. Saito, H. Aiso, T. Kochi and F. Kakiuchi, Organometallics, 2014, 33, 6704–6707 CrossRef CAS; (g) H. Aiso, T. Kochi, H. Mutsutani, T. Tanabe, S. Nishiyama and F. Kakiuchi, J. Org. Chem., 2012, 77, 7718–7724 CrossRef CAS PubMed; (h) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed; (i) C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- (a) T. H. Meyer, J. C. A. Oliveira, S. C. Sau, N. W. J. Ang and L. Ackermann, ACS Catal., 2018, 8, 9140–9147 CrossRef CAS; (b) L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C.-W. Pao, J.-L. Chen, J.-F. Lee and A. Lei, ACS Catal., 2018, 8, 5448–5453 CrossRef CAS; (c) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., 2018, 130, 2407–2411 CrossRef; (d) S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed; (e) N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed; (f) R. Mei, N. Sauermann, J. C. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 57, 7719–7723 Search PubMed; (g) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed; (h) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed.
- (a) F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS; (b) Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., 2018, 130, 5920–5924 CrossRef.
- Y. Qiu, W. J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed.
- Y. Qiu, M. Stangier, T. H. Meyer, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14179–14183 CrossRef CAS PubMed.
- (a) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC; (b) M. Jeganmohan and R. Manikandan, Chem. Commun., 2017, 53, 8931–8947 RSC; (c) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744–5767 RSC; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (e) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS PubMed; (f) L. Ackermann, Top. Organomet. Chem., 2007, 24, 35–60 CrossRef CAS.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (b) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379–6382 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed.
- L. Ackermann, Synlett, 2007, 507–526 CrossRef CAS.
- (a) X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4830–4833 CrossRef CAS PubMed; (b) J. D. Dooley, S. Reddy Chidipudi and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829–10836 CrossRef CAS PubMed; (c) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem. – Asian J., 2010, 5, 847–851 CrossRef CAS PubMed.
- (a) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- For detailed information, see the ESI†.
- For reviews, see: (a) J. Sherwood, Angew. Chem., Int. Ed., 2018, 57, 14286–14290 CrossRef CAS PubMed; (b) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC; (c) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07732k |
| This journal is © The Royal Society of Chemistry 2018 |