
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature methoxylation in zeolite H-ZSM-5: an operando DRIFTS/mass spectrometric study†
Santhosh K.
Matam
 *ab,
Russell F.
Howe
c,
Adam
Thetford
ab and
C. Richard A.
Catlow
abd
*ab,
Russell F.
Howe
c,
Adam
Thetford
ab and
C. Richard A.
Catlow
abd
aDepartment of Chemistry, University College London, London, WC1E 6BT, UK
bThe UK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, OX11 0FA, UK. E-mail: santhosh.matam@rc-harwell.ac.uk; Web: http://www.ukcatalysishub.co.uk/
cChemistry Department, University of Aberdeen, AB24, UK
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, CF10 3AT, UK
First published on 22nd October 2018
Abstract
At high loading, methanol reacts under ambient conditions with acidic hydroxyls of H-ZSM-5 to methoxylate framework oxygen; while a significant proportion remains hydrogen bonded to the framework with a protonated geometry. The findings not only explain the data which have been published so far, but also pave a way forward for potential unravelling of the initial reaction steps in the relevant chemical processes.
Methoxylation is a key intermediate step for hydrocarbon synthesis through alkylation in different petro- and fine chemical processes, especially methanol to gasoline, catalysed by the zeolite ZSM-5.1–7 However, there remain many uncertainties concerning the methoxylation reaction (eqn (1)) in this catalyst.4–11
| CH3–OH + Z–OH → Z–OCH3 + H2O | (1) |
Spectroscopic studies, mainly by solid state nuclear magnetic resonance (NMR) and infrared (IR), suggest that methoxylation occurs only at elevated temperatures (>150 °C).9–11 Moreover, IR studies to date appear to indicate that, under ambient conditions, methanol forms only hydrogen bonded species,12 in line with which, several computational studies have suggested that methoxylation could have a significant activation energy and hence require higher temperatures.11,13,14 However, simulations also suggest decreased energy barrier for methoxylation with more than one methanol molecule per unit cell6,11,13–18 and also hint at the influence of other factors, such as the local reaction environment in the zeolite pores or the reaction conditions, on methoxylation.11,13 Interestingly, inelastic neutron scattering (INS) studies showed the occurrence of methoxylation at room temperature (RT), with, however, no indication of hydrogen bonded methanol in ZSM-5 pores.8 The INS study on the one hand substantiates the reduced energy barrier for methoxylation as indicated by simulations, and on the other hand leaves the issue still for debate due to the failure to detect hydrogen bonded species, which are observed by IR. The latter could be related to the sensitivity of the INS technique. The present study addresses this key problem by applying operando diffuse reflectance infrared Fourier transformed spectroscopy (DRIFTS) and mass spectrometry (MS) which simultaneously capture the methoxylation reaction by probing surface adsorbed species and reaction products, respectively.
We find that the dehydration of H-ZSM-5 resulted in well resolved characteristic O–H stretching bands between 3750 and 3550 cm−1 while, zeolite framework modes (overtone and combination modes) dominate the spectrum below 2000 cm−1 (Fig. S1 of ESI†). The band at 3736 cm−1 is attributed to silanol groups on the external surface of the zeolite, while the band at 3600 cm−1 is assigned to Brønsted acidic hydroxyls.19,20 The band at 3650 cm−1 is often ascribed to Al–OH groups.20
DRIFTS difference spectra of zeolite ZSM-5 with a methanol pulse of 7 molecules per acidic site injected at RT, which is similar to our earlier INS study,8 are shown in Fig. 1. In agreement with earlier IR studies, we see that the spectra are dominated by hydrogen bonded methanol species that are unambiguously characterised by the triplet (Fig. 1A), which falls between 1500 and 3500 cm−1,12,19 plus the corresponding CH stretching modes of this species. The region below 1500 cm−1 does provide additional insight. However, a potential contribution from the framework modes to difference spectra needs to be considered for bands below 1500 cm−1. A negative band at 871 cm−1 followed by positive bands at around 937, 990–1000, 1185 and 1285 cm−1 are present, besides gas phase methanol P–Q–R bands between 1012 and 1050 cm−1 (Fig. 1B and Table S1, ESI†). The negative band at 871 cm−1 suggests the reaction of methanol with the acid sites to form methoxy groups.19 The bands at 937 and 1185 cm−1 evolved with time not only regarding intensity but also with the position of the bands under He flow for 30 min. The band at 937 cm−1 is assigned to C–O stretch of the methoxy species with the corresponding methyl rock band at 1185 cm−1,20,21 indicating the occurrence of methoxylation at RT.8 In the first few minutes of the reaction, the characteristic P–Q–R bands of gas phase methanol are observed at 1052, 1032 and 1012 cm−1 (Fig. 1B), resulting in a complex combination of bands between 980 and 1050 cm−1.
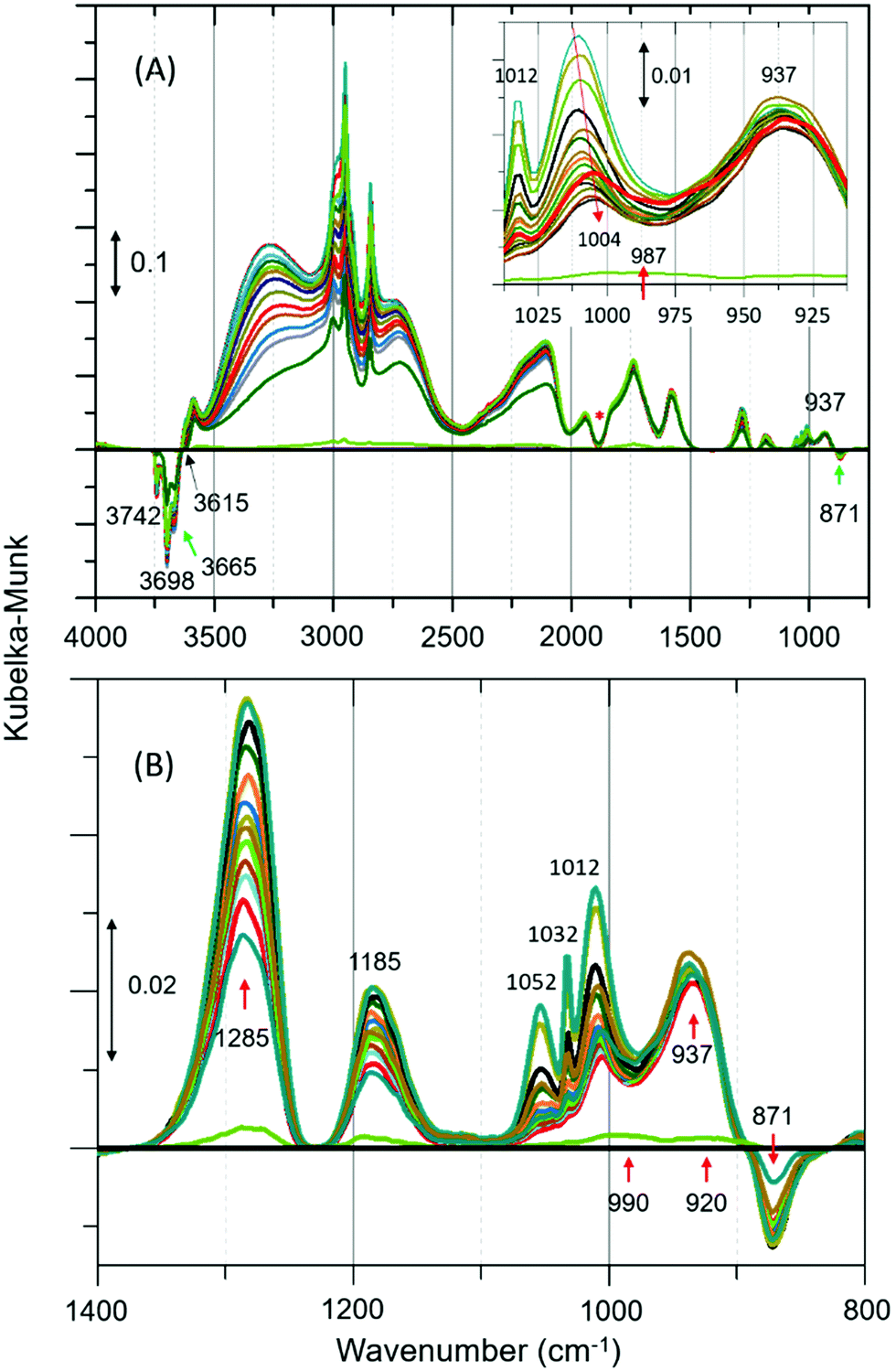 | ||
| Fig. 1 (A) Selected infrared difference spectra of zeolite ZSM-5 with a methanol pulse of 7 molecules per Brønsted acidic site at room temperature (RT) and a magnified region between 1050 and 800 is shown in the inset.‡ (B) Magnified region of Fig. 1A. Artefact arising from the subtraction process is indicated with asterisk (Fig. 1A).19 The complete data set is presented in Fig. S2 of ESI.† The spectral resolution is around 20 s. | ||
However, the P–Q–R bands disappear within the first five minutes of the reaction (as expected under He flow) and a new band attributable to C–O stretch of the hydrogen bonded methanol appeared at 1004 cm−1 as evident from the red shift of the band position from 1012 to 1004 cm−1 (see Fig. 1A inset). The band at 1004 cm−1 extends well below 975 cm−1 enveloping the band at 990 cm−1. Likewise, the band at 1285 cm−1 could be a combination of different species. For example (see Fig. S3, ESI†), the C–H bending mode of either methanol or methoxy species can contribute to the spectrum between 1400 and 1250 cm−1, in addition to a potential contribution from the zeolite framework (see Fig. S1, ESI†). Thus, the assignment of the band at 1285 cm−1 should be treated with caution. However, the assignment of band at 1185 cm−1 is straightforward (mainly to methyl rock of methoxy species) as there is neither gas phase methanol (see Fig. S3, ESI†) nor C–H bending modes of methoxy/methanol contributions. This is further corroborated by the fact that the band is symmetric (FWHM of ≈40 cm−1) and its relative intensity is similar to the corresponding C–O stretch at 937 cm−1, which is opposite to the band at 1285 cm−1 (FWHM of ≈80 cm−1). The latter shows a sharp dip in intensity towards lower and a long extended tail towards higher wavenumbers. Also, a potential contribution to 1185 cm−1 from the zeolite framework that might arise on subtraction is unlikely as there are no significant framework bands below 1200 cm−1 (see Fig. S1, ESI†).
The complexity of the low frequency methoxy bands requires us to seek additional evidence from the C–H stretching region. The signature C–H stretching modes of the methoxy species at 2980 and 2863 cm−1 (Fig. 2A)9 are obscured by an intense triplet arising from protonated hydrogen bonded methanol species.12,19,20 Thus, the difference spectra derived from the earliest measurement and different stages of the reaction are compared in the inset of Fig. 2A. It is clear that the bands assignable to methoxy emerge at 2980 and ≈2875 cm−1, and 2967 and 2865 cm−1,4,9 which imply the occurrence of at least two types of methoxy species. The band at ≈2875 cm−1 corresponds to symmetric C–H stretch enveloped by a broad feature between 2850–2880. In addition, all other features attributable to hydrogen bonded methanol are present (Fig. 1 and 2A). Clearly both methoxy and hydrogen bonded species immobilize methanol in ZSM-5 pores (between RT and 50 °C). The immobility of methanol in the pores unambiguously explains the quasi-elastic neutron scattering (QENS) data.8,22–25
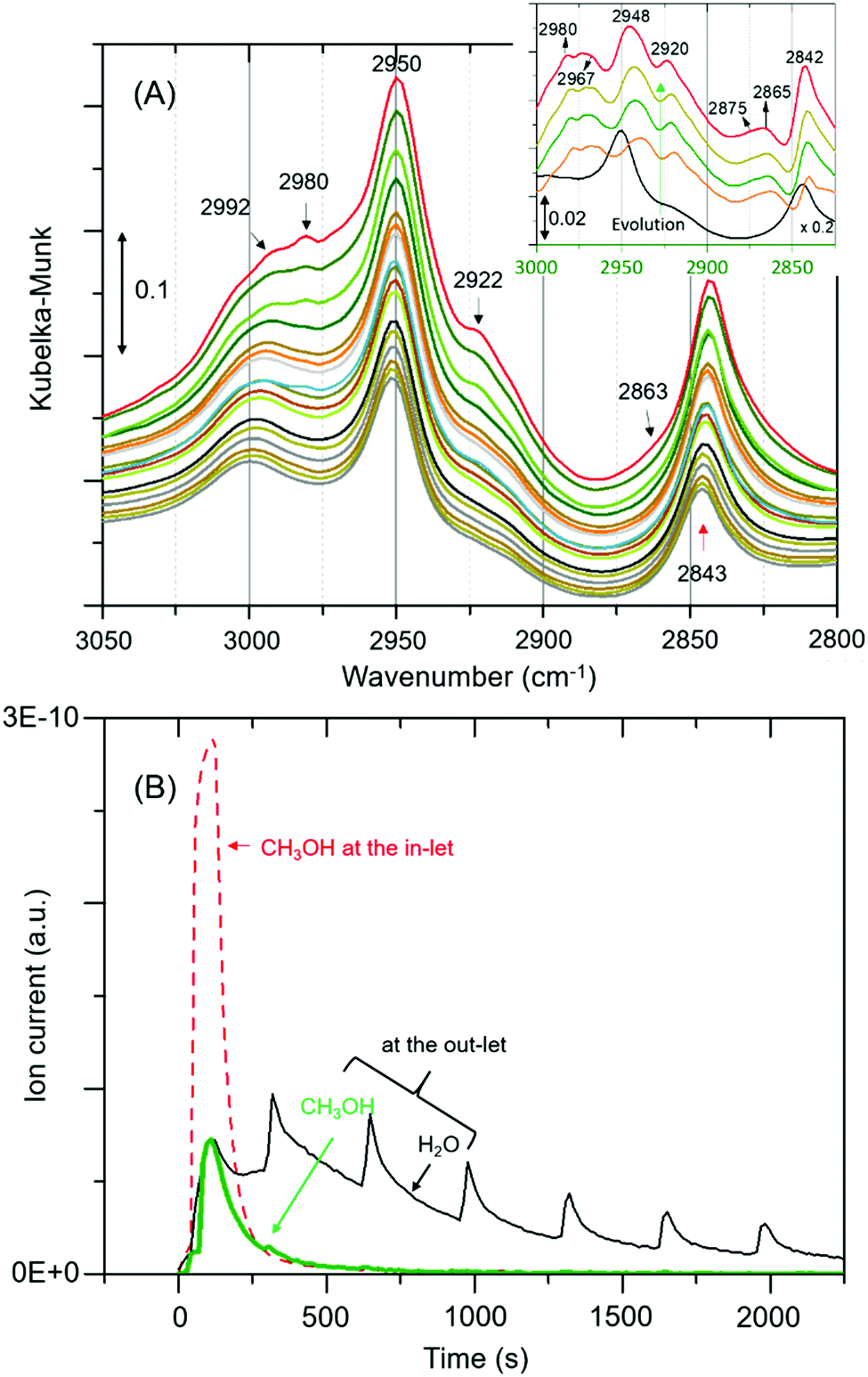 | ||
| Fig. 2 (A) Magnified infrared spectra of Fig. 1A. Inset compares difference spectra of the earliest measurement and different stages of the reaction. (B) MS data, including the initial concentration of methanol (broken red line for reference).§ The repetitive water desorption peaks are in tandem with the heating tape (of the exhaust pipe) fluctuation (±20 °C) cycle. As reaction proceeds water formation decreases due to a single methanol pulse that accounts for variations in water desorption peaks. | ||
In line with the formation of methoxy species and protonated hydrogen bonded methanol, consumption of different hydroxyls is evident from negative bands above 3600 cm−1 (Fig. 1A and Fig. S4, ESI†). Bands at 3615 and 3742 cm−1 are attributed to consumption of Brønsted acidic sites and silanol groups, respectively (Table S1, ESI†). The consumption of silanol groups is consistent with the negative band at 871 cm−1 and with C–H stretching bands at 2967 and 2865 cm−1.9 The corresponding methyl rock band of the species falls at around 1150 cm−1, which is overlapped by the broad band at 1185. The consumption of Brønsted acidic hydroxyls is consistent with methoxy species reflected by bands at 937, 1185, 2875 and 2980 cm−1. These results confirm the occurrence of at least two types of methoxy species in ZSM-5, in line with earlier IR and INS studies.9,20 A similar observation is also reported by NMR and, significantly these methoxy species on ZSM-5 are not completely eroded upon hydrolysis with water at RT unlike on Y and SAPO-34,10 implying the unique intrinsic nature of active sites in ZSM-5. In the present study, the band at 3665 cm−1 is assigned to hydrolysed extra-framework Al (Al–OH).19,20 Although the assignment of 3698 cm−1 band is not straight forward, it is safe to suggest the consumption of different hydroxyl groups (Table S1, ESI†). The strength of acidity of these hydroxyls may decrease from 3600 to 3742 cm−1.
The formation of methoxy species is corroborated with MS data in Fig. 2B that show continuous evolution of water molecules during the reaction under He flow, indicating that the rate of reaction (eqn (1)) is very slow, as expected, under these reaction conditions. We note that the evolution of water demonstrates the occurrence of methoxylation at RT, and rules out displacement of any physisorbed water from the zeolite pores that would rather result in a fast and single step process, matching with the methanol MS profile. Overall around 80% methanol is converted as evident from the amount of methanol detected at the cell outlet.
The occurrence of room temperature methoxylation is further supported by analysing the methyl rock band at 1185 cm−1 and the water formation as a function of time in Fig. 3. The band at 1185 cm−1 grows rather rapidly and thereafter decreases gradually suggesting the conversion of methoxy species into other products. Given the reaction temperature, the gradual decrease in intensity of the band can be attributed to partial hydrolysis of methoxy species due to significant amounts of water formed during the methoxylation reaction.10
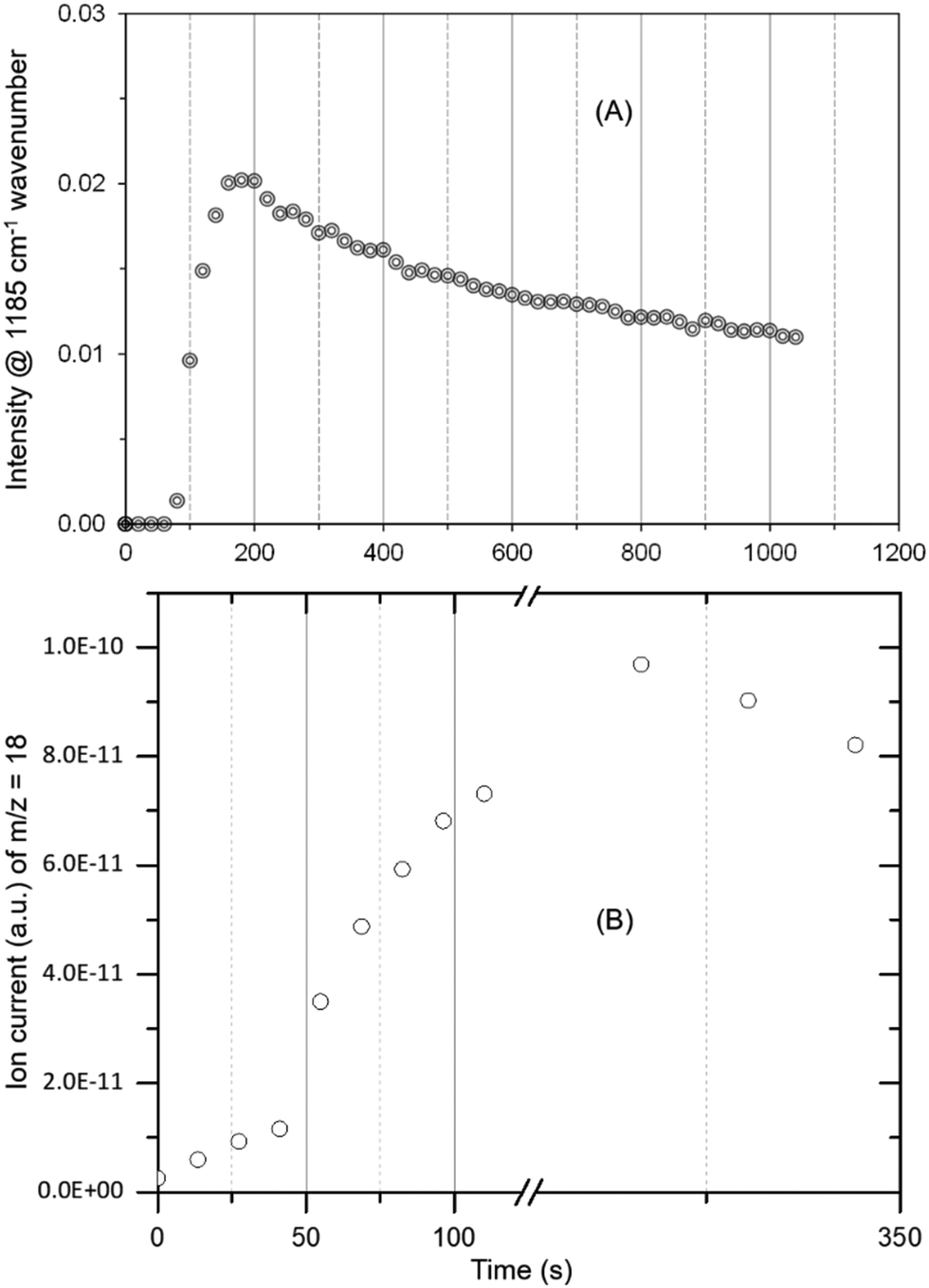 | ||
| Fig. 3 The evolution of methoxy species reflected by the band at 1185 cm−1 (A) and water formation (B) as a function of time. The peak maximum is slightly delayed for MS data (B) as compared to DRIFTS (A) due to hampered water desorption in the exhaust pipe (refer Fig. 2) and more than one methoxy contribution to water formation. The latter could also explain slight differences in the evolution of methoxy and water profiles. | ||
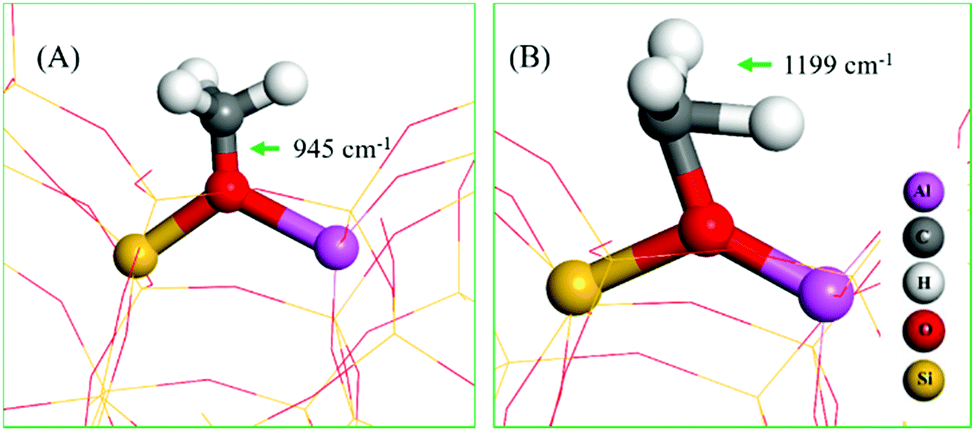
The assignment of vibrational frequencies is verified by DFT calculations using optimized structures of ZSM-5 with and without adsorbed species (Table 1). The vibrational frequencies of methoxy or hydrogen bonded species are affected by the presence of a second molecule such as water or methanol molecule in the unit cell. A methoxy species at the Brønsted acidic site is calculated to have a C–O stretch at 945 cm−1 (structure A in Table 1) and a pair of methyl rock bands at 1138 and 1199 cm−1 (the latter is represented by structure B in Table 1). These methoxy bands are affected upon co-adsorption of either a methanol or water molecule in the unit cell.
| Structure | Vibrational frequencies wavenumbers (cm−1) | Vibrational mode | Ref. |
|---|---|---|---|
| a Interaction of methanol with methoxy. b Interaction of water with methoxy. | |||
| A | 945 | ν CO (Si/Al–OCH3) | 14 |
| 927 | ν CO OCH3⋯CH3OHa (or H2O)b | This work | |
| B | 1138/1199 | ρ CH3 OCH3 | This work |
| 1144/1193 | ρ CH3 OCH3⋯CH3OHa | ||
| 1138/1176 | ρ CH3 OCH3⋯H2Ob | ||
|
|||
A degree of fluctuations in calculated vibrational frequencies of the same species as a function of local environment of the zeolite unit cell imply the sensitivity and complexity of the IR bands. In line with this observation, C–O stretch and methyl rock of methoxy species in DRIFTS appeared between 920–950 cm−1, and 1130–1185 cm−1, respectively. Both experimental and calculated vibrational frequencies of gas phase methanol fall at higher wavenumbers as compared to than that of adsorbed species, making our assignments of the surface adsorbed species plausible.
To summarise, we have found clear and unambiguous evidence that with a saturation level of methanol loading in H-ZSM-5 pores, methoxylation takes place under ambient conditions along with a proportion of remaining hydrogen bonded species with protonated geometry, rationalising the apparent contradictions between earlier IR and neutron scattering data.
The UK Catalysis Hub is thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/I038748/1, EP/I019693/1, EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). Johnson Matthey plc is thanked for the provision of the ZSM5. Dr A. J. O’Malley and Dr S. F. Parker are thanked for fruitful discussion.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249 CrossRef CAS.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329 CrossRef CAS.
- V. van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell, C. Richard and A. Catlow, Chem. Soc. Rev., 2015, 44, 7044 RSC.
- U. Olsbey, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155 RSC.
- F. Haase and J. Sauer, J. Phys. Chem., 1991, 98, 3083 CrossRef.
- C. M. Zicovich-Wilson, P. Viruela and A. Corma, J. Phys. Chem., 1995, 99, 13224 CrossRef CAS.
- R. Hunter and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1985, 644 Search PubMed.
- A. J. O’Malley, S. F. Parker, A. Chutia, M. R. Farrow, I. Silverwood, V. G. Sakai, C. Richard and A. Catlow, J. Chem. Soc., Chem. Commun., 2016, 52, 2897 RSC.
- T. R. Forester, S. T. Wong and R. F. Howe, J. Chem. Soc., Chem. Commun., 1986, 1611 RSC; T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 1987, 109, 5076 CrossRef CAS.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260 CrossRef CAS PubMed.
- J. Van der Mynsbrugge, S. L. C. Moors, K. De Wispelaere and V. Van Speybroeck, ChemCatChem, 2014, 6, 1906 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Spano and F. Geobaldo, J. Chem. Soc., Faraday Trans., 1996, 92(23), 4863 RSC.
- P. E. Sinclair and C. R. A. Catlow, J. Chem. Soc., Faraday Trans., 1996, 92(12), 2099 RSC.
- P. E. Sinclair, C. Richard and A. Catlow, J. Chem. Soc., Faraday Trans., 1997, 93(2), 333 RSC.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2006, 45, 1714 CrossRef CAS PubMed.
- S. R. Blaszkowski and R. A. van Santen, J. Phys. Chem. B, 1997, 101, 2292 CrossRef CAS.
- E. Sandre, M. C. Payne and J. D. Gale, J. Chem. Soc., Chem. Commun., 1998, 2445 RSC.
- M. C. Payne, M. Hytha, I. Stich, J. D. Gale and K. Terakura, Microporous Mesoporous Mater., 2001, 48, 375 CrossRef CAS.
- B.-T. L. Bleken, L. Mino, F. Giordanino, P. Beato, S. Svelle, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2013, 15, 13363 RSC.
- Suwardiyanto, R. F. Howe, E. K. Gibson, C. Richard A. Catlow, A. Hameed, J. McGregor, P. Collier, S. F. Parker and D. Lennon, J. Chem. Soc. Faraday Discuss., 2017, 197, 447 RSC.
- M. El-Roz, P. Bazin, M. Daturi and F. T-Starzyk, Phys. Chem. Chem. Phys., 2015, 17, 11277 RSC.
- S. K. Matam, A. J. O’Malley, C. Richard A. Catlow, Suwardiyanto, P. Collier, A. P. Hawkins, A. Zachariou, D. Lennon, I. Silverwood, S. F. Parker and R. F. Howe, Catal. Sci. Technol., 2018, 8, 3304 RSC.
- H. Jobic, A. Renouprez, M. Bec and C. Poinsignon, J. Phys. Chem., 1986, 90, 1059 CrossRef CAS.
- N. M. Gupta, D. Kumar, V. Kamble, S. Mitra, R. Mukhopadhyay and V. Kartha, J. Phys. Chem. B, 2006, 110, 4815 CrossRef CAS PubMed.
- S. Mitra, V. S. Kamble, A. K. Tripathi, N. M. Gupta and R. Mukhopadhyay, Pramana – J. Phys., 2004, 63, 443 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DRIFTS spectra of the dehydrated H-ZSM-5. See DOI: 10.1039/c8cc07444e |
| ‡ The zeolite ZSM-5 (Si/Al ≈ 30) was obtained from Johnson Matthey and details are reported elsewhere.20,22 Prior to experiments, the zeolite was dehydrated under He flow (100 ml min−1) at 500 °C for few hours and cooled to room temperature (RT). Methanol pulse (of 60 s) experiment was conducted under the same He flow at RT with a pulse of 7 molecules per Brønsted acidic site that corresponds to the saturation level of loading under flow conditions.8,22 After a methanol pulse, the evolution of surface adsorbed species was monitored by DRIFTS, and reaction cell outlet by MS, for around 30 min under the same He flow. |
| § The observed DRIFTS bands were verified by simulations. For this purpose, Density Functional Theory with a dispersion correction (DFT+D) was employed via the Vienna Ab-initio Simulation Package (VASP).26–28 The projector augmented wave (PAW) method and a plane-wave cut-off energy of 450 eV was used with single k-point at the gamma point. The converged bulk energies were within 10−4 eV. The convergence criterion of 0.02 eV Å−1 was used for the forces to optimise the structures. The Perdew–Burke–Ernzerhof (PBE) version of the generalized gradient approximation (GGA) was used for energy calculations and geometry optimizations.29 PBE exchange–correlation functional was used with Grimme's D2 correction scheme for dispersive interactions to allow the study of hydrogen bonding.30 The vibrational frequencies were calculated by finite differences to determine the second derivatives. A single unit cell of the MFI structure was used which has dimensions of 20.35 Å × 20.22 Å × 13.6 Å containing 95 Si atoms, 1 Al atom and 192 O atoms. |
| This journal is © The Royal Society of Chemistry 2018 |