
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrawing on biology to inspire molecular design: a redox-responsive MRI probe based on Gd(III)-nicotinamide†
Michael
Harris
a,
Jacek L.
Kolanowski
 b,
Edward S.
O’Neill
b,
Céline
Henoumont
c,
Sophie
Laurent
cd,
Tatjana N.
Parac-Vogt
a and
Elizabeth J.
New
*b
b,
Edward S.
O’Neill
b,
Céline
Henoumont
c,
Sophie
Laurent
cd,
Tatjana N.
Parac-Vogt
a and
Elizabeth J.
New
*b
aDepartment of Chemistry, KU Leuven, Celestijnlaan 200F, Heverlee 3001, Belgium
bSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: elizabeth.new@sydney.edu.au
cGeneral, Organic and Biomedical Chemistry Unit, NMR and Molecular Imaging Laboratory, University of Mons, Mons 7000, Belgium
dCenter for Microscopy and Molecular Imaging (CMMI), 8 rue Adrienne Boland, Gosselies 6041, Belgium
First published on 2nd November 2018
Abstract
A novel, reversible redox-active MRI probe, GdNR1, has been developed for the study of redox changes associated with diseased states. This system exhibits switching in relaxivity upon reduction and oxidation of the appended nicotinimidium. Relaxivity studies and cyclic voltammetry confirmed the impressive reversibility of this system, at a biologically-relevant reduction potential. A 2.5-fold increase in relaxivity was observed upon reduction of the complex, which corresponds to a change in the number of inner-sphere water molecules, as confirmed by luminescence lifetimes of the Eu(III) analogue and NMRD studies. This is the first example of a redox-responsive MRI probe utilising the biologically-inspired nicotinimidium redox switch. In the future this strategy could enable the non-invasive identification of hypoxic tissue and related cardiovascular disease.
The non-ionizing, non-invasive and unlimited tissue penetration of magnetic resonance imaging (MRI) offers an exceptional opportunity to visualize biological events with probes that modulate the MRI signal upon a signaling event.1 Numerous complexes have been developed for chemical stimuli such as enzymatic activity,2,3 Ca2+, Cu2+, Zn2+,4,5 or neurotransmitters,6 or characteristics of the chemical environment such as pH,7 redox state,8 or temperature.9 Currently reported approaches to achieving redox-responsive MRI systems include using redox-active metals such as cobalt,10 copper,11 and manganese,12 and europium,13 self-assembly triggered ligands,14 or appended functional groups such as quinols,15 thiols16 and merocyanine/spyrooxazine.17
Local changes in biological redox potential play an important role in signal transduction, influencing many cellular functions. The uncontrolled production of pro-oxidants in oxidative stress18,19 is increasingly being understood to play a role in various pathologies, including Alzheimer's disease, cardiovascular disease, obesity, diabetes and arthritis.20–23 On the other hand, hypoxia occurs when blood supply to tissues is restricted in a variety of pathologies including solid tumors and atherosclerosis. We are particularly interested in the preparation of reversible contrast agents that can sense dynamic changes in biological redox environments,10,24 and here sought to develop a Gd-based reversible redox sensor.
In this study we report Gd-nicotinamide redox sensor 1 (GdNR1), a Gd(III) contrast agent with a redox-sensitive switch that operates at a physiologically-relevant reduction potential (Scheme 1). We chose a nicotinamide-based redox sensor as this moiety is used in the redox-active NAD/NADH cofactor in biological systems.
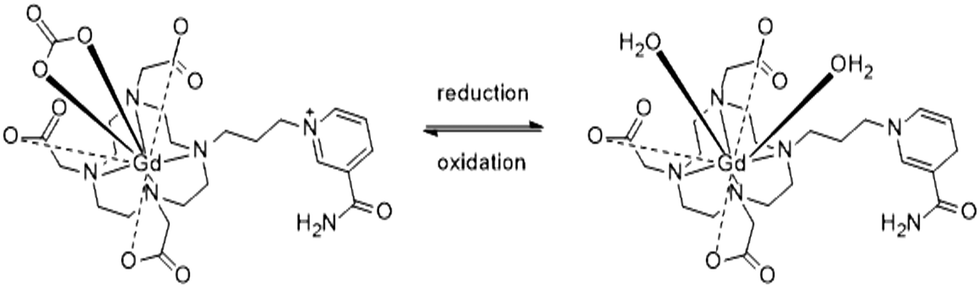 | ||
| Scheme 1 GdNR1 in oxidized (left) and reduced (right) forms. | ||
DO3A derivatives are commonly synthesized starting from DO3A-OtBu, but due to challenges in purification of the ligand, a less-commonly utilized approach was employed to synthesize GdNR1, enabling a facile final purification step (Scheme S1, ESI†). After alkylating nicotinamide with 1,3-dibromopropane and isolating the product by precipitation in acetonitrile, this salt was used directly in a one-pot, two-step synthesis with cyclen (2 eq.) followed by addition of excess tBu bromoacetate. This yielded a mixture of our desired, charged target material and tetra-substituted cyclen (DOTA-OtBu), which could be separated using aqueous/organic liquid–liquid extraction. TFA deprotection of the tBu ester groups and a Cl−-exchange column gave the free ligand with a single counter-ion. Lanthanide(III) complexes were then prepared by complexation with lanthanide(III) chlorides in water.
Electrospray ionization mass spectrometry (ESI-MS) of the ligand was performed to confirm the nature of reduced and oxidized species, with studies carried out before deprotection of the tertiary butyl ester groups due to very poor ionization of the reduced-neutral free ligand or complex. The charged nicotinimidium appeared at 677.4 (+ve m/z) and the reduced ligand was found as a sodium adduct (+ve m/z 701.4) (Fig. S9 and S10, ESI†). This single proton difference confirms the nicotinimidium-to-dihydronicotinamide reduction. 1H and 13C NMR characterization of the analogous diamagnetic La(III) complex showed the loss of the aromatic peaks and appearance of the expected reduced nicotinamide peaks upon reduction of the system (Fig. S11–S14, ESI†).
Cyclic voltammetry (CV) of the complex revealed well-separated redox peaks consistent with the two electron reduction of the nicotinimidium ring to dihydronicotinamide (Fig. 1). The electrochemical reversibility of the system was confirmed over 12 cycles, highlighting the advantages of our system over previously-reported contrast agents that exhibit only quasi-reversibility.25
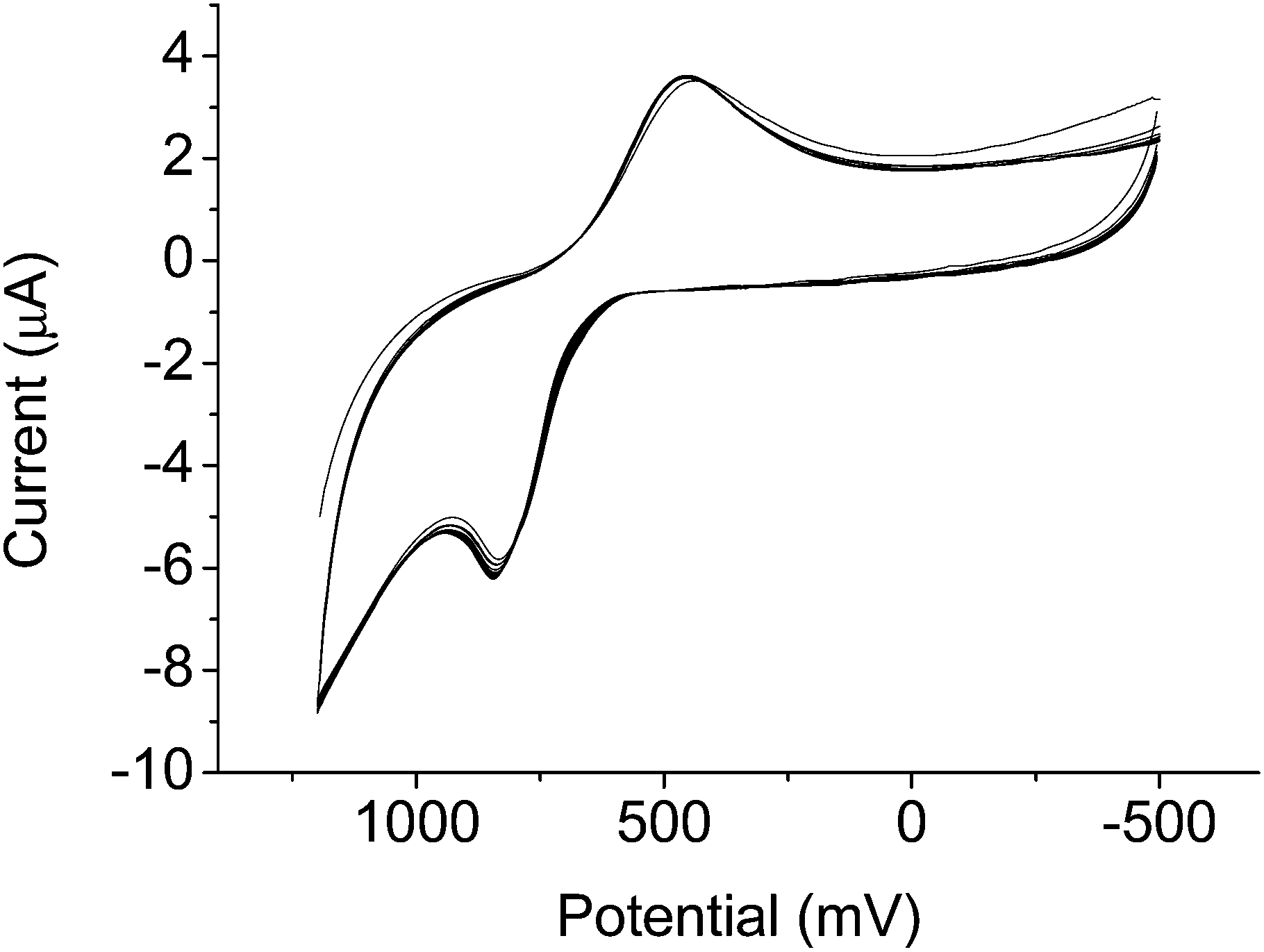 | ||
| Fig. 1 Cyclic voltammogram of GdNR1 in water containing 0.1 M KCl as supporting electrolyte for 12 cycles. CV recorded using glassy C as working electrode, Ag+/AgCl as reference electrode, and Pt wire as counter electrode. Scan rate 100 mV min−1. | ||
To investigate redox-induced changes in the hydration state of the complex, a luminescent Eu(III) analogue was synthesized and the luminescence lifetimes measured in H2O and D2O with addition of reductant, oxidant and bicarbonate. The hydration number (q) of Eu(III) complexes can be estimated (±0.1 to 0.3) using the empirical formula:26
| qEu(H2O) = 1.2(Δkobs − 0.25 − 1.2qNH − 0.075qCONH), |
This analysis confirmed a decrease in the water coordination number from 1.9 to 0.3 upon oxidation (in non-degassed MES buffer, pH 7) (Table 1). This corresponds to a change from 2 bound water molecules in the reduced form to 0. We speculated that binding of a counteranion, promoted by the positively charged oxidized nicotinimidium, was responsible for occluding water binding. In non-degassed solutions and biological systems alike, bicarbonate is the major coordinating anion, with biological bicarbonate concentrations on the order of 20 mM, so we hypothesised that this ion binds to the oxidized form, as has previously been observed for responsive lanthanide complexes.27 Addition of HCO3− (10 eq.) to the buffered sample showed no change, and when this sample was oxidized, the q value decreased in the same manner. In comparison, in samples degassed by boiling and purging with argon to remove bicarbonate, the oxidized complex showed q = 2.1, confirming our hypothesis that bicarbonate binding is responsible for this large change in hydration state. HEPES and MES buffers are reported to be mild reductants which can be used for synthesis of gold nanoparticles from AuCl4.28 This, together with a positive reduction peak observed in the CV (450 mV, Fig. 1) of GdNR1 could result in a partial reduction of the complex upon dissolution in the buffers, and subsequently higher r1 and q values observed by the luminescence lifetimes and NMRD measurements in buffered solution.
| τ H2O /ms | τ D2O /ms | qH2O | |
|---|---|---|---|
| a Average of 3 measurements that vary by less 0.01 ms. b Reduced by buffer. c Oxidised by addition of 10 eq. H2O2. d Native form in pure water. | |||
| EuNR1-reducedb (MES, non-degassed) | 0.32 | 0.80 | 1.9 |
| EuNR1-oxidizedc (MES, non-degassed) | 0.44 | 0.55 | 0.3 |
| EuNR1-reducedb + HCO3− (10 eq.) (MES, non-degassed) | 0.34 | 0.95 | 1.9 |
| EuNR1-oxidizedc + HCO3 (10 eq.) (MES, non-degassed) | 0.42 | 0.56 | 0.4 |
| EuNR1-oxidizedd (H2O & D2O degassed) | 0.26 | 0.57 | 2.1 |
Nuclear magnetic resonance dispersion (NMRD) was used to study the relaxivity of the system in the oxidized and reduced forms in buffered non-degassed water. The r1 values of the native reduced complex in HEPES and MES buffers were 6.9 and 7.5 s−1 mM−1 respectively (Fig. 2 and Table S1, ESI†). Addition of the reductant sodium dithionite to GdNR1 in buffer showed no significant change to the NMRD profile (Fig. 2). H2O2 was then used to oxidize the complex in buffered solution and the r1 was observed to decrease to 4.6 s−1 mM−1 at 60 MHz and 37 °C for both buffers.
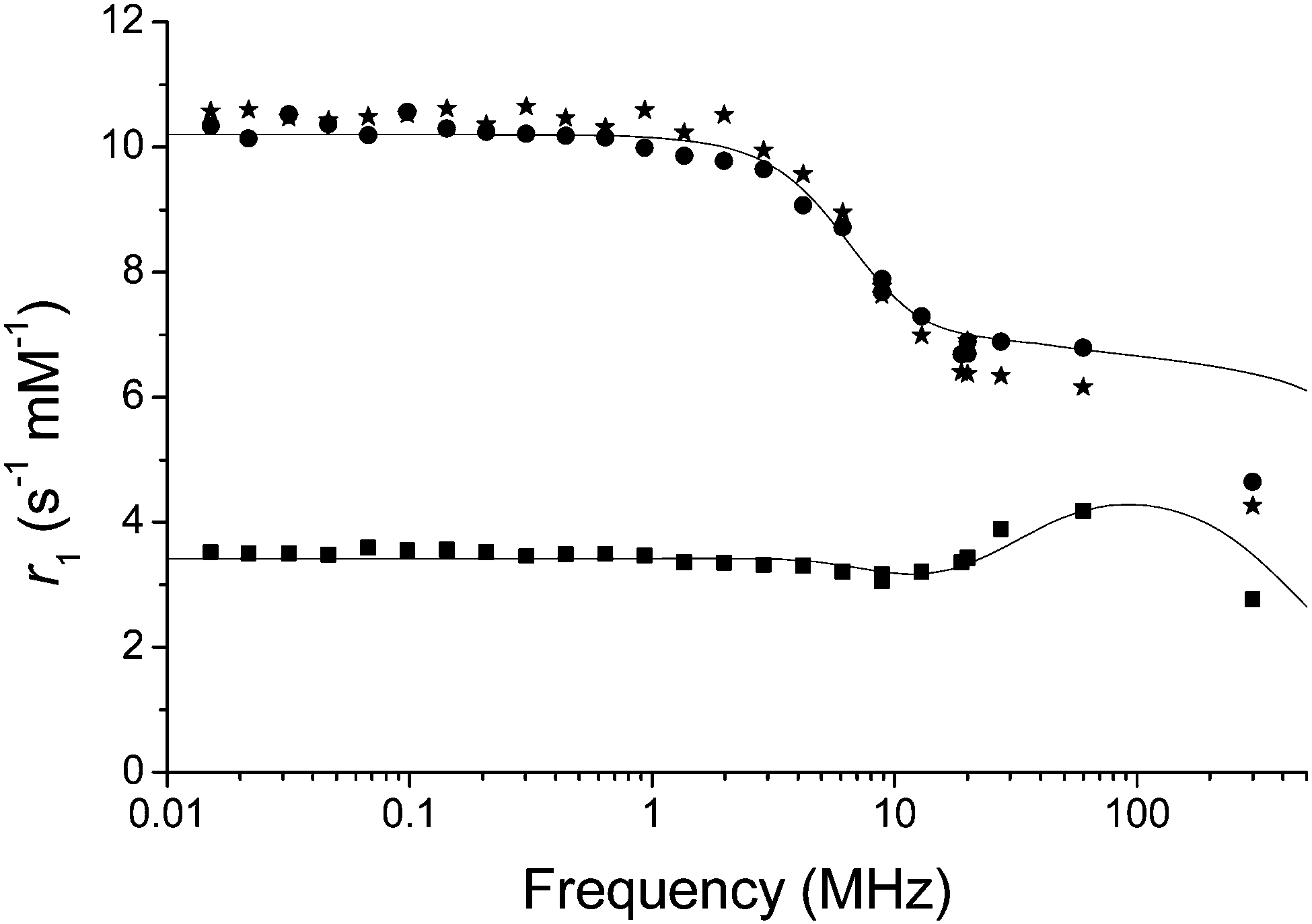 | ||
| Fig. 2 Proton NMRD profiles of GdNR1 in MES (20 mM, pH 7). Circles: GdNR1 dissolved directly in buffer, non-degassed, squares: addition of H2O2 (10 eq.) and stars: addition of sodium dithionite (10 eq.). Solid lines represent fitted data. | ||
The NMRD curve at low field confirmed the three-fold change in relaxivity from oxidized to reduced forms (Fig. 2). To further rule out any role for H2O2 other than as an oxidant, we repeated the experiment with DOTAREM (Gd-DOTA) in clinical preparation, and observed no difference in the presence of excess H2O2 (Fig. S16, ESI†).
The system could be reversibly switched between oxidized and reduced forms, although repeated cycling was limited by the buffering capacity due to the acidity of both H2O2 and sodium dithionite (Fig. 3A). The synthesized version of the probe bearing reduced nicotinamide showed a relaxation rate of 6.9 s−1 mM−1 at 60 MHz and 37 °C. Upon oxidation with H2O2, the relaxation rate decreased to 3.7 s−1 mM−1. This clear change in relaxation rate with oxidation and reduction could also be observed in phantom images of tubes containing oxidized and reduced GdNR1 (Fig. 3B).
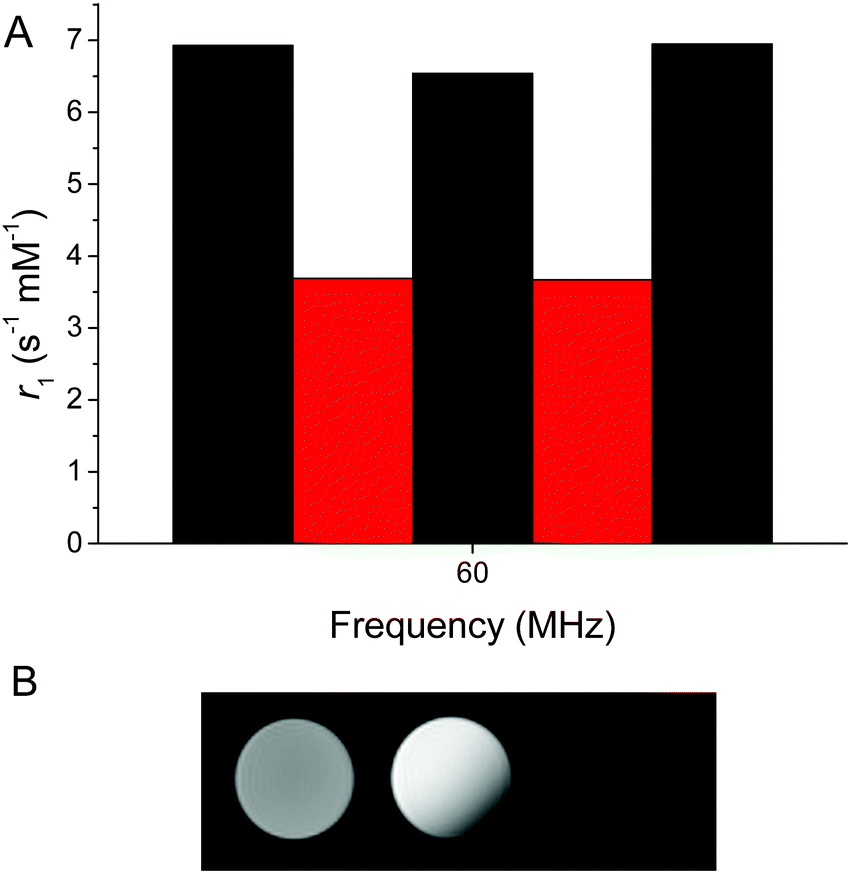 | ||
| Fig. 3 (A) Relaxivity measured at 60 MHz and 37 °C with alternating addition of H2O2 (10 eq. oxidant) or sodium dithionite (10 eq. reductant) (GdNR1 2 mM, MES 100 mM, bicarbonate 2 mM, pH 7). (B) Phantom MRI images of GdNR1 (1 mM) at 300 MHz (left oxidized using H2O2, middle reduced and right blank) (non-degassed water, HEPES 100 mM, pH 7.4). | ||
Consistent with our studies of the Eu complex, the r1 of the oxidized form was observed to be 7.6 s−1 mM−1 at 60 MHz and 37 °C when the water was degassed (boiling and argon purging) and the relaxivity did not change significantly upon addition of either oxidant (H2O2) or reductant (sodium dithionite). In contrast, in the presence of bicarbonate, the r1 decreased more than double on oxidation (Fig. 4 inset). Upon reduction with sodium dithionite in the presence of bicarbonate, the r1 value recovers to near that of the sample in high-purity degassed water of 7.1 s−1 mM−1 at 60 MHz and 37 °C. This is consistent with our hypothesis from the Eu lifetime studies that the changing charge of the complex from +1 to 0 modulates the binding and release of bicarbonate. This in turn affects water accessibility to the Gd(III) paramagnetic centre, one of the main parameters determining the relaxivity.
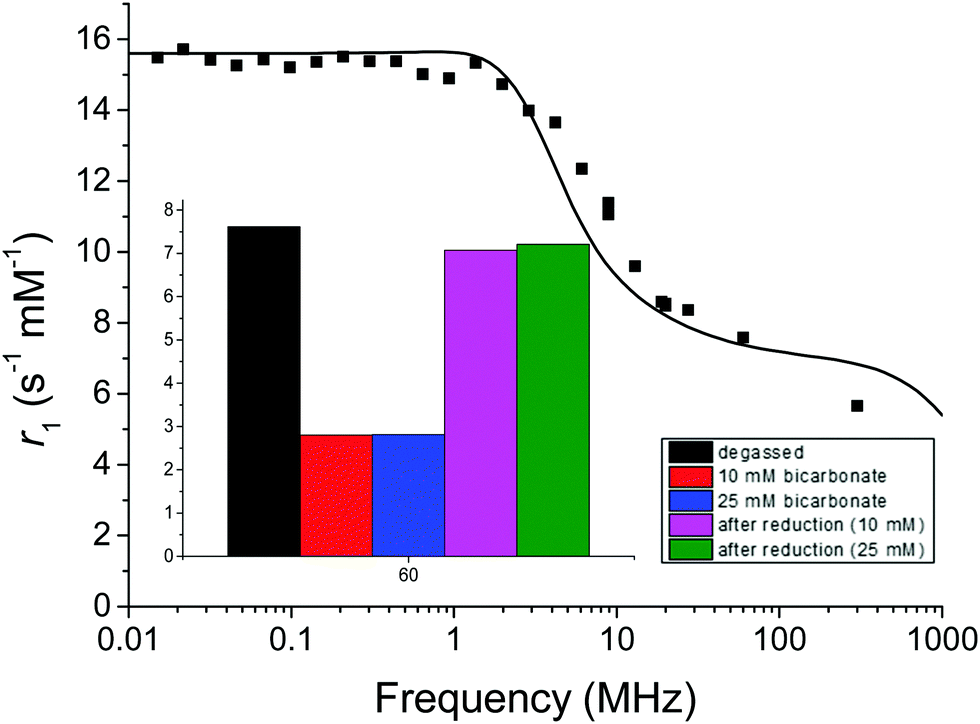 | ||
| Fig. 4 Proton NMRD profile of GdNR1 in high-purity degassed water at 37 °C. Solid line represents the fitted data. Inset shows relaxivity at 60 MHz for GdNR1 in high-purity degassed water, 10 mM and 25 mM bicarbonate spiked solutions (MES pH 7), and addition of sodium dithionite (10 eq.) to bicarbonate spiked solutions. | ||
Investigating the role of competing species in the relaxation process, we measured the changes in relaxivity upon the addition of citrate, lactate, HSA and in F12 (+10% FBS) cell culture medium (GdNR1 1 mM). Citrate (0.13 mM), lactate (2.3 mM) and cell culture medium did not disturb the relaxivity switching (Fig. S17, ESI†). In 4% HSA solution, relaxivity increased indicating non-covalent binding of GdNR1 to the protein associated with an increased rotational correlation time; however, the switching of the system was not impeded (Fig. S17, ESI†). Lowe et al. previously observed this phenomenon in DO3A derived complexes, where water coordination decreased up to five-fold upon binding of endogenous anions such as bicarbonate and relaxivity dramatically increased with HSA.29
In HEPES and MES the oxidized complex showed an increase in relaxivity in the NMRD curve from 20 to 60 MHz, characteristic of aggregation.30–35 Dynamic light scattering confirmed the presence of ∼100 nm aggregates in the MES buffered solution with H2O2 added (Fig. S19, ESI†). However, this aggregation was not visibly observed in the presence of HSA or in cell culture media, so it is likely that under these conditions the complex has sufficient solubility to overcome this problem.
Fitting of proton NMRD profiles to Solomon-Bloembergen-Morgan theory (ESI† text, Fig. 2 and 4) was performed to evaluate the effect of hydration number and other parameters (especially τR) on relaxivity, using parameters previously described for Gd(III) complexes (Table S2, ESI†).22,23 As expected for a small DO3A derived complex the rotational correlation time (τR) is short. Fitting the NMRD data from Fig. 4 before oxidation shows a very similar result to that observed for GdNR1 in high-purity water. Upon oxidation of GdNR1 with H2O2, the shape of the NMRD profile changed dramatically with a peak in relaxivity occurring at higher Larmor frequency (100 MHz). Such a profile is typical of a slow tumbling system, and the slow tumbling implies that aggregation has occurred. However, the relaxivity is much lower than expected for a slow tumbling Gd-complex with inner-sphere water ligand(s),36–39 and this low relaxivity implies that the hydration number is decreased and likely q is close to 0. We then investigated the effects of pH on relaxivity. r1 is constant over pH 3–6, decreases by 25% at pH 7, and further at pH 9 (Fig. S18, ESI†). This relaxivity decrease between pH 6 and 7 correlates with the first pKa of carbonic acid, at ∼6.4, suggesting that it may be result from formation of bicarbonate from water-dissolved CO2 from air and its subsequent coordination to the Gd centre. At pH 9, the relaxivity drops to ∼4 s−1 mM−1, higher than the relaxivity of ∼2.5 s−1 mM−1 (Fig. 4 inset) measured for GdNR1 in pure water spiked with bicarbonate (pH 8–9), indicating that the effects of the environment on relaxivity of the complex are a combination of bicarbonate-related as well as other pH-related mechanisms, as described previously for Gd-based complexes.29,40
In summary, we report the first example of a reversible redox-responsive MRI probe based on a Gd(III)-DO3A nicotinamide chelate, and provide a detailed insight into its mechanism and performance. Using a novel synthetic approach, it was possible to produce the final chelate in a few facile steps. The system shows response to sodium dithionite and H2O2. There is a large signal enhancement upon reduction of the nicotinimidium in the presence of bicarbonate. Luminescence lifetimes of the Eu(III) analogue revealed a change in binding of inner-sphere water molecules upon oxidation and reduction events. Bicarbonate is endogenous to blood serum and would be available to function with this MRI probe for signal suppression of the oxidised form. GdNR1 represents a useful prototype for future contrast agents; for example, by tuning the redox potential, or further conjugation to useful functionalities through the amide of the nicotinamide. In considering future biological application of this probe, we have confirmed its non-toxicity in cultured cells at a concentration of 0.1 mM even after 72 h incubation (Fig. S20, ESI†). The potential applications for this complex could include imaging of hypoxic tissue and dynamic monitoring of reducing biological environments.
This work was supported by the Research Foundation-Flanders (FWO Flanders, MH), a Westpac Research Fellowship (EJN), Australian Postgraduate Award (ESO), and the Australian Research Council (DP150100649). We acknowledge Dr Nick Proschogo and the Mass Spectrometry Facility at The University of Sydney. High-resolution mass spectrometry at KU Leuven was supported by the Hercules Foundation of the Flemish Government (grant 20100225-7). We thank Professor David Parker for the helpful discussion of results and Mr Marcus Graziotto for assistance with cytotoxicity studies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Angelovski, Angew. Chem., 2016, 55, 7038–7046 CrossRef CAS PubMed.
- F. Touti, P. Maurin and J. Hasserodt, Angew. Chem., Int. Ed., 2013, 52, 4654–4658 CrossRef CAS PubMed.
- C. Gondrand, F. Touti, E. Godart, Y. Berezhanskyy, E. Jeanneau, P. Maurin and J. Hasserodt, Eur. J. Inorg. Chem., 2015, 1376–1382 CrossRef CAS.
- E. L. Que and C. J. Chang, Chem. Soc. Rev., 2010, 39, 51–60 RSC.
- C. Shen and E. J. New, Curr. Opin. Chem. Biol., 2013, 17, 158–166 CrossRef CAS PubMed.
- G. Angelovski and E. Tóth, Chem. Soc. Rev., 2017, 46, 324–336 RSC.
- L. Q. Chen and M. D. Pagel, Adv. Radiol., 2015, 2015, 206405 Search PubMed.
- Q. N. Do, J. S. Ratnakar, Z. Kovács and A. D. Sherry, ChemMedChem, 2014, 9, 1116–1129 CrossRef CAS PubMed.
- P. K. Senanayake, N. J. Rogers, K. N. A. Finney, P. Harvey, A. M. Funk, J. I. Wilson, D. O'Hogain, R. Maxwell, D. Parker and A. M. Blamire, Magn. Reson. Med., 2017, 77, 1307–1317 CrossRef PubMed.
- E. S. O’Neill, A. Kaur, D. P. Bishop, D. Shishmarev, P. W. Kuchel, S. M. Grieve, G. A. Figtree, A. K. Renfrew, P. D. Bonnitcha and E. J. New, Inorg. Chem., 2017, 56, 9860–9868 CrossRef PubMed.
- D. Xie, T. L. King, A. Banerjee, V. Kohli and E. L. Que, J. Am. Chem. Soc., 2016, 138, 2937–2940 CrossRef CAS PubMed.
- G. S. Loving, S. Mukherjee and P. Caravan, J. Am. Chem. Soc., 2013, 135, 4620–4623 CrossRef CAS PubMed.
- L. A. Basal and M. J. Allen, Front Chem., 2018, 6, 1–12 CrossRef PubMed.
- D. Ye, P. Pandit, P. Kempen, J. Lin, L. Xiong, R. Sinclair, B. Rutt and J. Rao, Bioconjugate Chem., 2014, 25, 1526–1536 CrossRef CAS PubMed.
- M. Yu, M. B. Ward, A. Franke, S. L. Ambrose, Z. L. Whaley, T. M. Bradford, J. D. Gorden, R. J. Beyers, R. C. Cattley, I. Ivanović-Burmazović, D. D. Schwartz and C. R. Goldsmith, Inorg. Chem., 2017, 56, 2812–2826 CrossRef CAS PubMed.
- B. Jagadish, G. P. Guntle, D. Zhao, V. Gokhale, T. J. Ozumerzifon, A. M. Ahad, E. A. Mash and N. Raghunand, J. Med. Chem., 2012, 55, 10378–10386 CrossRef CAS PubMed.
- C. Tu, R. Nagao and A. Y. Louie, Angew. Chem., Int. Ed. Engl., 2009, 48, 6547–6551 CrossRef CAS PubMed.
- H. Sies, Exp. Physiol., 1997, 82, 291–295 CrossRef CAS PubMed.
- T. C. Jorgenson, W. Zhong and T. D. Oberley, Cancer Res., 2013, 73, 6118–6123 CrossRef CAS PubMed.
- H. K. Vincent and A. G. Taylor, Int. J. Obes., 2006, 30, 400–418 CrossRef CAS PubMed.
- R. Sultana and D. A. Butterfield, J. Alzheimer's Dis., 2010, 19, 341–353 Search PubMed.
- J. W. Baynes, Diabetes, 1991, 40, 405–412 CrossRef CAS PubMed.
- Y.-R. Chen and J. L. Zweier, Circ. Res., 2014, 114, 524–537 CrossRef CAS PubMed.
- E. S. O'Neill, J. L. Kolanowski, G. H. Yin, K. M. Broadhouse, S. M. Grieve, A. K. Renfrew, P. D. Bonnitchab and E. J. New, RSC Adv., 2016, 6, 30021–30027 RSC.
- S. J. Ratnakar, S. Viswanathan, Z. Kovacs, A. K. Jindal, K. N. Green and A. D. Sherry, J. Am. Chem. Soc., 2012, 134, 5798–5800 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. d. Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- M. Giardiello, M. P. Lowe and M. Botta, Chem. Commun., 2007, 4044–4046 RSC.
- S. R. Ahmed, S. Oh, R. Baba, H. Zhou, S. Hwang, J. Lee and E. Y. Park, Nanoscale Res. Lett., 2016, 11 Search PubMed.
- M. P. Lowe, D. Parker, O. Reany, S. Aime, M. Botta, G. Castellano, E. Gianolio and R. Pagliarin, J. Am. Chem. Soc., 2001, 123, 7601–7609 CrossRef CAS PubMed.
- E. Debroye, G. Dehaen, S. V. Eliseeva, S. Laurent, L. V. Elst, R. N. Muller, K. Binnemans and T. N. Parac-Vogt, Dalton Trans., 2012, 41, 10549–10556 RSC.
- E. Debroye, S. V. Eliseeva, S. Laurent, L. V. Elst, S. Petoud, R. N. Muller and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2013, 2629–2639 CrossRef CAS.
- M. Harris, S. Carron, L. V. Elst, S. Laurent, R. N. Muller and T. N. Parac-Vogt, Chem. Commun., 2015, 51, 2984–2986 RSC.
- M. Harris, S. Carron, L. V. Elst and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2015, 4572–4578 CrossRef CAS.
- M. Harris, L. V. Elst, S. Laurent and T. N. Parac-Vogt, Dalton Trans., 2016, 45, 4791–4801 RSC.
- M. Harris, H. D. Keersmaecker, L. V. Elst, E. Debroye, Y. Fujita, H. Mizuno and T. N. Parac-Vogt, Chem. Commun., 2016, 52, 13385 RSC.
- E. Debroye, S. V. Eliseeva, S. Laurent, L. V. Elst, R. N. Muller and T. N. Parac-Vogt, Dalton Trans., 2014, 43, 3589–3600 RSC.
- F. Carniato, L. Tei, J. Martinelli and M. Botta, Eur. J. Inorg. Chem., 2018, 2363–2368 CrossRef CAS.
- Z. Wang, F. Carniato, Y. Xie, Y. Huang, Y. Li, S. He, N. Zang, J. D. Rinehart, M. Botta and N. C. Gianneschi, Small, 2017, 13, 1701830 CrossRef PubMed.
- M. Filippi, D. Remotti, M. Botta, E. Terreno and L. Tei, Chem. Commun., 2015, 51, 17455 RSC.
- J. Hall, R. Häner, S. Aime, M. Botta, S. Faulkner, D. Parker and A. S. d. Sousa, New J. Chem., 1998, 627–631 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation and further NMRD details. See DOI: 10.1039/c8cc07092j |
| This journal is © The Royal Society of Chemistry 2018 |