
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTelechelic polymers from reversible-deactivation radical polymerization for biomedical applications
Daniele
Vinciguerra
 ,
Johanna
Tran
and
Julien
Nicolas
*
,
Johanna
Tran
and
Julien
Nicolas
*
Institut Galien Paris-Sud, UMR CNRS 8612, Univ Paris-Sud, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296 Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Tel: +33 1 46 83 58 53
First published on 6th December 2017
Abstract
The main strategies for the design of telechelic polymers synthesized by reversible-activation radical polymerization for biomedical applications are covered spanning from drug delivery and targeting to theranostics and sensing.
 Daniele Vinciguerra | Daniele Vinciguerra obtained in 2014 his Master's Degree in Pharmaceutical Chemistry and Technology from the University of Turin (Italy) with a final internship in the group of Prof. Marco Lucio Lolli. After another internship in the research group of Prof. Cristina Prandi in the Department of Chemistry, University of Turin (Italy), he was awarded with a Marie Skolodowska-Curie actions PhD fellowship from the Marie Curie ITN European NABBA project, under the supervision of Dr Julien Nicolas and Prof. Patrick Couvreur at Institut Galien Paris-Sud (Univ. Paris-Sud) in Châtenay-Malabry (France). He is currently working on the synthesis and the biological evaluation of polymer prodrug nanocarriers for biomedical applications. |
 Johanna Tran | Johanna Tran obtained in 2015 an MSc with honors in Molecular Chemistry from Pierre et Marie Curie University and the Ecole Normale Supérieure (ENS Paris) Diploma with a major in Chemistry, Paris (France). During this period, she had the opportunity to do an internship in the groups of Prof. Peter J. Sadler and Dr Nicolas Barry at Warwick University where she worked on the encapsulation of Ru- and Os-based complexes into polymers. She then, joined Prof. Patrick Couvreur's group at Paris-Sud University to pursue a PhD degree under the supervision of Dr Julien Nicolas. Her work focuses on the synthesis of functional polyesters made by radical polymerization for biomedical applications. |
 Julien Nicolas | Julien Nicolas completed his PhD in 2005 under the supervision of Prof. Bernadette Charleux at the University Pierre and Marie Curie in Paris (France), where he studied nitroxide-mediated polymerization. He then joined the group of Prof. David M. Haddleton at the University of Warwick (UK), as a postdoctoral researcher in the field of polymer–protein bioconjugates. In 2007, he obtained a CNRS researcher position at Institut Galien Paris-Sud (Univ. Paris-Sud) in Châtenay-Malabry (France) and got promoted to CNRS research Director in 2016. His current research activities lie in advanced macromolecular synthesis and in the design of innovative polymer-based nanomedicines. He is a (co)author of more than 85 peer review articles in international journals, 7 patents and 13 book chapters. He serves as an Associate Editor for Chemistry of Materials (ACS) and is part of the Editorial Advisory Board of Polymer Chemistry (RSC). He received the 2016 SCF/GFP award and the 2017 Polymer Chemistry Lectureship award. |
Introduction
Functional polymers are of paramount importance in materials sciences to conceive macromolecular architectures with advanced properties.1–3 According to IUPAC, they are defined as polymers that bear “specified chemical groups” or polymers that have “specified physical, chemical, biological, pharmacological, or other uses which depend on specific chemical groups”.4 They include polymers with self-assembling, selective binding/coupling or catalytic abilities and are used in various applications such as nanomedicine,5,6 organic electronic devices,7 solar energy conversion and storage,8 or smart materials (e.g., self-healing polymers).9Among the different classes of functional polymers, end-functional polymers possess important benefits as their structures are crucial for the design of many industrially relevant materials (e.g., block copolymers, network, elastomers, macromonomers, surfactants) or strong value-added polymers (e.g., bioconjugates, nanoparticles, organic electronics).10 Telechelic polymers are end-functional polymers bearing reactive end-groups at both chain-ends.11 They can be either homotelechelic (same functionality at both chain-ends) or heterotelechelic (different chain-ends), also called heterobifunctional polymers (Fig. 1a). A convenient way to prepare telechelic polymers relies on the use of reversible-deactivation radical polymerization (RDRP), such as nitroxide-mediated radical polymerization (NMP),12 atom-transfer radical polymerization (ATRP)13 or reversible addition–fragmentation chain transfer (RAFT) polymerization.14 Not only do these polymerization techniques allow well-defined polymers to be obtained, but they also exhibit high α,ω chain-end fidelity enabling facile and efficient α,ω functionalization.12,15,16
 | ||
| Fig. 1 (a) Schematic representations of homotelechelic (left) and heterotelechelic (right) polymers. (b) Main biologically relevant entities currently used for the design of telechelic polymers from RDRP techniques for biomedical applications: drugs, proteins and oligonucleotides (drug delivery), fluorescent probes (tracing/imaging), gold nanoparticles/surfaces (biosensing, hyperthermia) and iron oxide nanoparticles (imaging, magnetic guidance). | ||
In the biomedical field, telechelic polymers are particularly interesting as they offer an opportunity to embed two different biologically active entities at the chain-ends, such as drugs, ligands, imaging agents or functional sites for binding or advanced properties (Fig. 1b). This Feature Article will cover the main strategies for the design and use of telechelic polymers from RDRP (both organic and organic/inorganic systems) for biomedical applications, spanning from drug delivery to targeting, imaging/sensing and other applications.
1. Organic systems
Telechelic polymers end-functionalized with organic (macro)molecules allow a broad range of different polymers to be synthesized for application in drug delivery, protein bioconjugation, imaging and sensing.1.1 Drug delivery
The design of drug-functionalized polymers (often termed polymer prodrug) is of crucial importance for drug delivery as they allow the limitations associated with traditional drug-loaded polymer nanocarriers (e.g., the “burst release”, the difficulty to encapsulate poorly soluble drugs and the poor drug loadings) to be circumvented, or at least alleviated.17 The development of polymer prodrugs represents a relatively new research field that has gained popularity in recent times, mostly in the field of cancer therapy.17,18 Among the different approaches to prepare polymer prodrug nanocarriers by RDRP, the most popular are the “grafting to” and “grafting from” approaches that consist in functionalizing, with the drug of interest, a preformed polymer or monomer, respectively. As for the emerging “grafting through” strategy (also called “drug-initiated”),19 it relies on the controlled growth of a short polymer chain from a drug, used as an initiator to position the drug at one of the polymer chain-end, generally on the α-position (Fig. 2).20–24 Exploiting the presence of RDRP moieties at the other extremity (i.e., nitroxide for NMP, halide atom for ATRP and chain transfer agent for RAFT), providing that the living chain fraction is high, can allow another molecule of interest to be positioned for biomedical applications. | ||
| Fig. 2 “Drug-initiated” synthesis of drug–polymer conjugates by RDRP methods. The drug is first functionalized by an RDRP moiety (alkoxyamine for NMP or chain transfer agent for RAFT polymerization), followed by the controlled growth of a polymer chain to yield a drug–polymer prodrug that can be formulated into the corresponding drug–polymer prodrug nanoassemblies. Adapted with permission from ref. 19. | ||
To combine therapeutic and targeting purposes, heterobifunctional polymers have been conceived to display a drug and a targeting ligand. Among the different ligands available for targeting purposes,25 antibodies are perhaps the most reliable ones as they are very specific regarding their target with high interaction affinities and have specific binding sites in many diseases, including cancer.26
For instance, a heterotelechelic polyacrylamide (PAAm) bearing biotin and doxorubicin (Dox) at each chain-end was synthesized from a biotin-functionalized chain transfer agent (CTA) to perform RAFT polymerization (Fig. 3).27 Dox was further linked at the ω-chain-end via thiol-maleimide chemistry. The polymer backbone also contained pendant metal-chelating moieties (MCP) functionalized with diethylenetriaminepentaacetic acid (DTPA) to chelate 111In, an Auger electron emitting radionuclide to cause lethal DNA double strand breaks. By exploiting the strong non-covalent bond between streptavidin (SAv) and biotin (this interaction is the strongest known non-covalent bond with KD ∼ 10−15 M, allowing the formation of stable complexes),28 trastuzumab Fab (tmFab) fragments were then covalently linked to SAv prior its linkage to the biotinylated polymer for the targeting of cancer cells via the HER2 receptor. The efficiency of targeting was assessed by surface plasmon resonance, and confocal microscopy revealed the uptake of the functionalized polymer by SK-BR-3 breast cancer cells.
 | ||
| Fig. 3 Structure of heterotelechelic α-biotin, ω-doxorubicin PAAm for further binding with the streptavidin–trastuzumab Fab fragment and targeting of cancer cells. Adapted with permission from ref. 27. | ||
Even though antibodies are highly specific ligands, other molecules could advantageously be used for targeting purposes,25 such as small peptides deriving from the RGD sequence that can specifically bind to an overexpressed receptor at the surface of tumour cells. They are less bulky, less fragile and less expensive than antibodies, and similar strategies could be developed to position RDG sequences to the polymer chain-end.
Following a similar concept to that for the construction of α-biotin, ω-Dox PAAm, a drug and a fluorescent dye were positioned at each polymer chain-end to combine therapeutic and imaging abilities.29 This was achieved by combining the “drug-initiated” synthesis of an α-drug polymer conjugate and its post-functionalization to link the fluorescent dye at the ω-extremity. In brief, paclitaxel (Ptx) was first conjugated to a RAFT agent and a short, well-defined poly(N,N-dimethylacrylamide) (PDMA) chain was subsequently grown from it. By taking advantage of the end-terminal thiol obtained after the removal of the RAFT agent, the ω-chain-end of the polymer was subsequently functionalized with a rhodamine derivative through maleimide chemistry to yield Ptx–PolyDMA–Rho conjugates (Fig. 4).
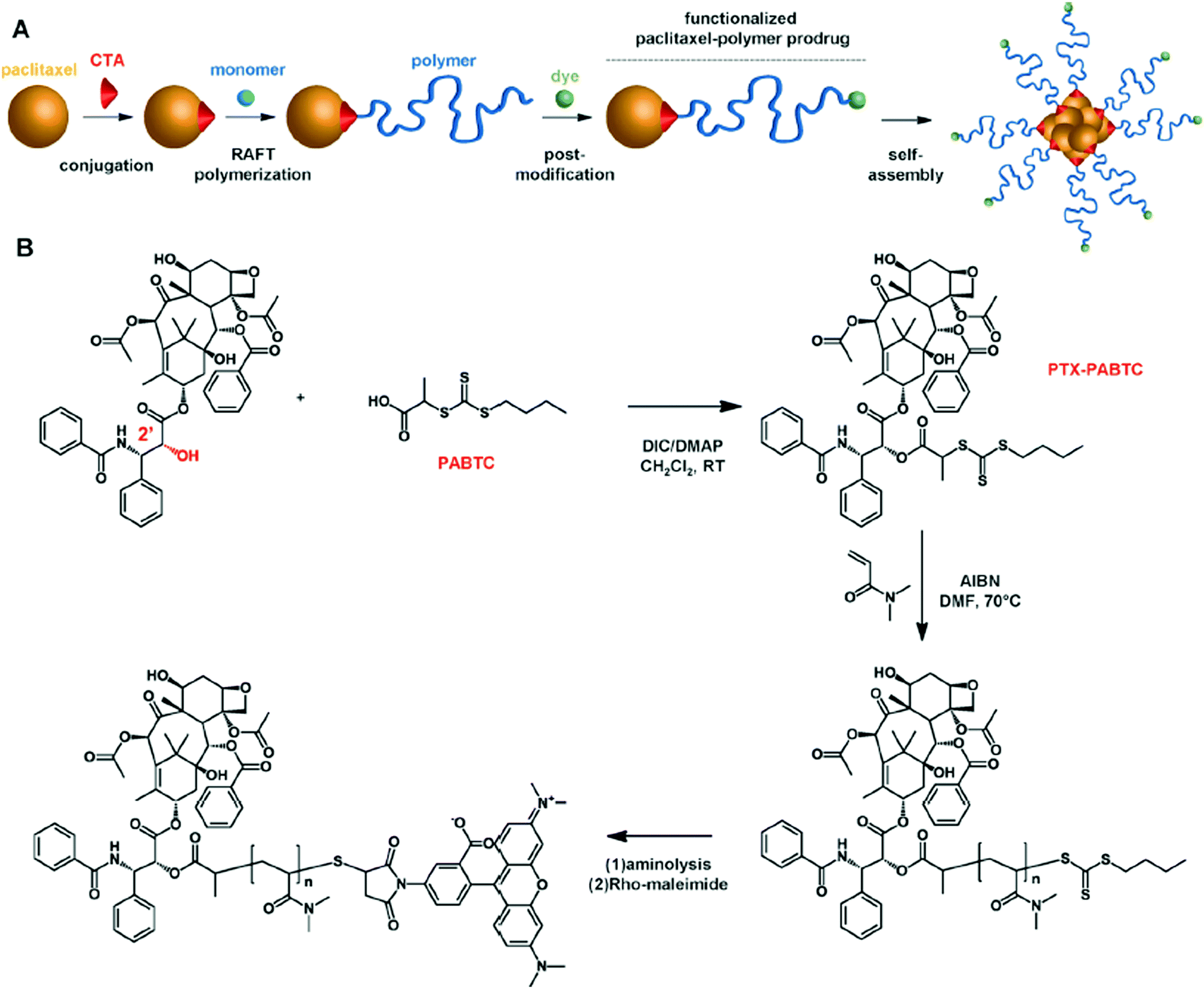 | ||
| Fig. 4 (A) Synthesis of heterotelechelic α-Ptx, ω-rhodamine PDMA by RAFT polymerization from a Ptx-RAFT agent followed by a post-functionalization to link the fluorescent dye at the ω-extremity and nanoprecipitation to yield the corresponding fluorescent polymer prodrug nanoparticles. (B) Synthetic strategy for the synthesis of heterotelechelic α-Ptx, ω-rhodamine PDMA. Adapted with permission from ref. 29. | ||
The fluorescent polymer prodrugs were formulated into nanoparticles and gave high anticancer activity in vitro on SKOV-3 human ovarian cancer cells, close to that of the parent drug as shown by MTT assays. Interestingly, they exhibited similar in vitro efficacy to genexol-PM and abraxane. On the other hand, the rhodamine moiety enabled cell internalisation experiments.
Interestingly, Matyjaszewski and co-workers showed the preparation of telechelic di-biotin polymers by ATRP from a difunctional ATRP initiator.30 One can imagine the use of such a heterobifunctional polymer as an intermediate block builder to link two different complexes through biotin/SAv binding.
Another utility of telechelic polymers is to design high molecular weight, segmentable polymers for drug delivery applications, to enhanced accumulation in the tumour tissue by the enhanced permeability and retention effect. To this aim, homotelechelic poly[N-(2-hydroxypropyl) methacrylamide] (PHPMA) chains were prepared by RAFT polymerization from a difunctional CTA and connected together by means of difunctional GFLG sequences via copper(I)-catalysed azide–alkyne Huisgen cycloaddition (CuAAC),31 thiol–divinylsulfone32 or thiol–maleimide coupling (Fig. 5).32
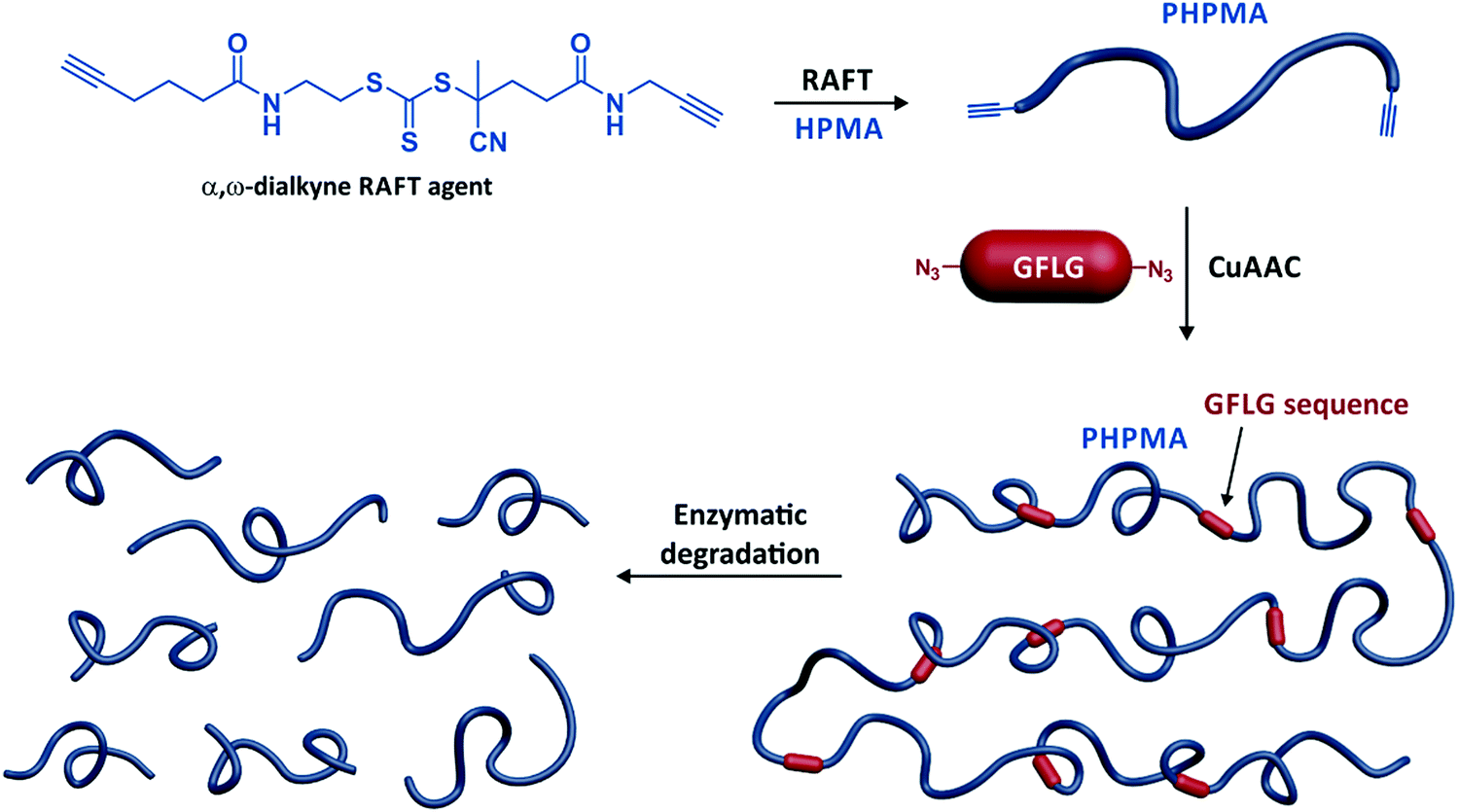 | ||
| Fig. 5 Synthesis of multisegmented PHPMA by a combination of RAFT polymerization of HPMA from a α,ω-dialkyne trithiocarbonate chain transfer agent, followed by CuAAC between the resulting α,ω-dialkyne PHPMA and α,ω-diazide GFLG peptide sequence. Adapted with permission from ref. 33. | ||
This approach was further extended to multi-segmented polymer prodrugs by copolymerizing HPMA and drug-bearing methacrylamide derivatives based on gemcitabine (Gem),34 Ptx,35 or both for combination therapy.36 The in vivo activity was compared to that of low molecular weight polymer counterparts and free drugs, showing major advantages in the use of the high molecular weight polymer.37 Using the same approach, another combination involving Gem and DACH platinum (mP-GEM and mP-DACH Pt, respectively), was prepared and tested on A2780 human ovarian cancer cells. The best synergistic effect was found when mP-GEM was administered for 24 h followed by mP-DACH Pt for 48 h.38
1.2 Imaging and sensing
Small fluorochromes are commonly used as model molecules to prepare functionalized polymers, usually to prove the easiness or the efficacy of functionalization. Model heterotelechelic polymers bearing two different dyes can easily be prepared by ATRP39 or RAFT40 by taking benefit from efficient chain-end functionalization. The latter was also selected to synthesize homotelechelic polymer carrying pyrene41 or naphthalene.42Such polymers bearing two different dyes could find applications in imaging/sensing such as FRET (Fluorescence Resonance Energy Transfer),43 bioimaging, theranostics, or simply for tracing purposes, for instance by confocal microscopy or other optical imaging techniques. FRET refers to the distant-dependant energy transfer that can occur between two different chromophores; a donor and an acceptor. This may be of great importance for polymer physics allowing polymer conformation, end-to-end distance or end group interactions to be measured.44,45
RDRP techniques are well-suited to design materials for FRET studies because of the narrow dispersity of the resulting polymer (the polymer chains have nearly the same length and thus end-to-end distances are rather similar from one chain to another) and the easy functionalization of both chain-ends. For instance, ATRP46 was employed to synthesize α,ω-dye-functionalized polymers bearing anthryl and carbazolyl groups (Fig. 6). They were prepared from anthryl-functionalized ATRP initiators followed by substitution of the bromine group by an azide moiety and subsequent CuAAC with carbazolyl alkyne.
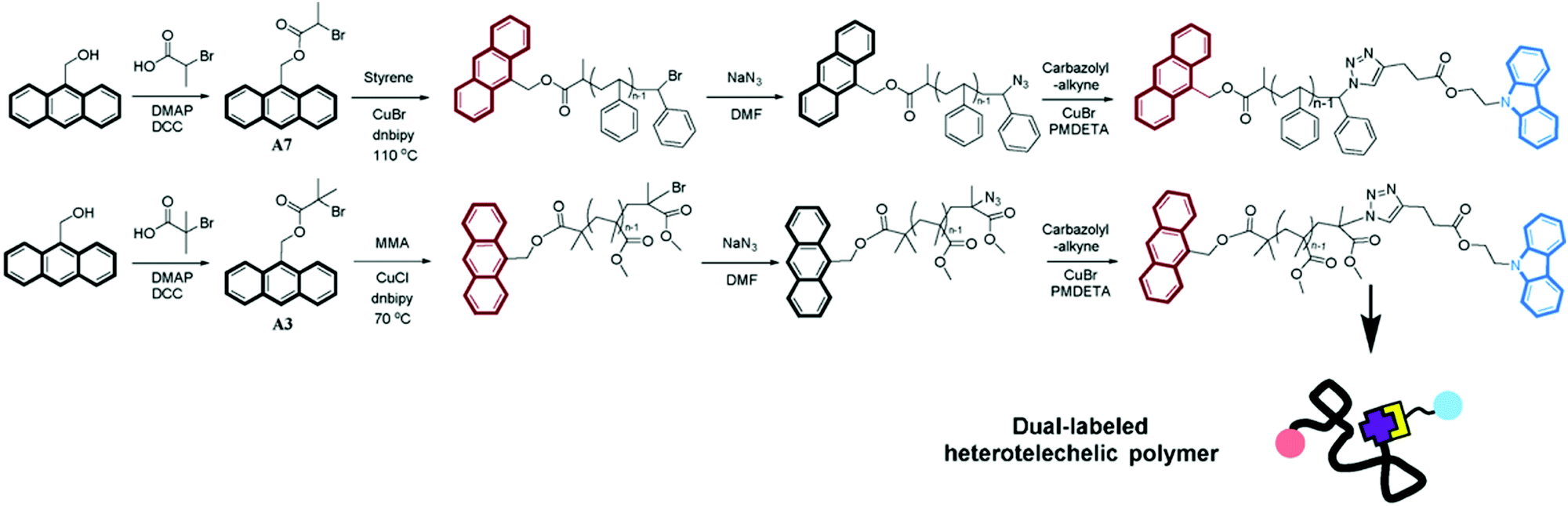 | ||
| Fig. 6 Synthetic routes for α,ω-dye-functionalized PS and PMMA bearing anthryl and carbazolyl groups as FRET pairs. Anthryl moieties are first derivatized with ATRP initiating groups for the subsequent ATRP of styrene or methyl methacrylate, followed by chain end azidation and CuAAC with carbazolyl-alkyne. Adapted with permission from ref. 46. | ||
A similar approach was reported on oregon green cadaverin and texas red as FRET pairs for the conception of α,ω-dye-functionalized polymers.47 Diethylene glycol methacrylate (DEGMA) was polymerized by RAFT from a pentafluorophenyl (PFP) activated ester RAFT agent followed by: (i) amination of PFP with Oregon Green Cadaverin to the α position and (ii) aminolysis of the RAFT end group by excess of n-propylamine in the presence of texas red-2-sulfonamidoethyl methanethiosulfonate to create a disulfide bond connecting the dye (Fig. 7). Assuming a Förster-type process, an average end-to-end distance of 4.5 nm was calculated, in reasonable agreement with data obtained from light scattering.
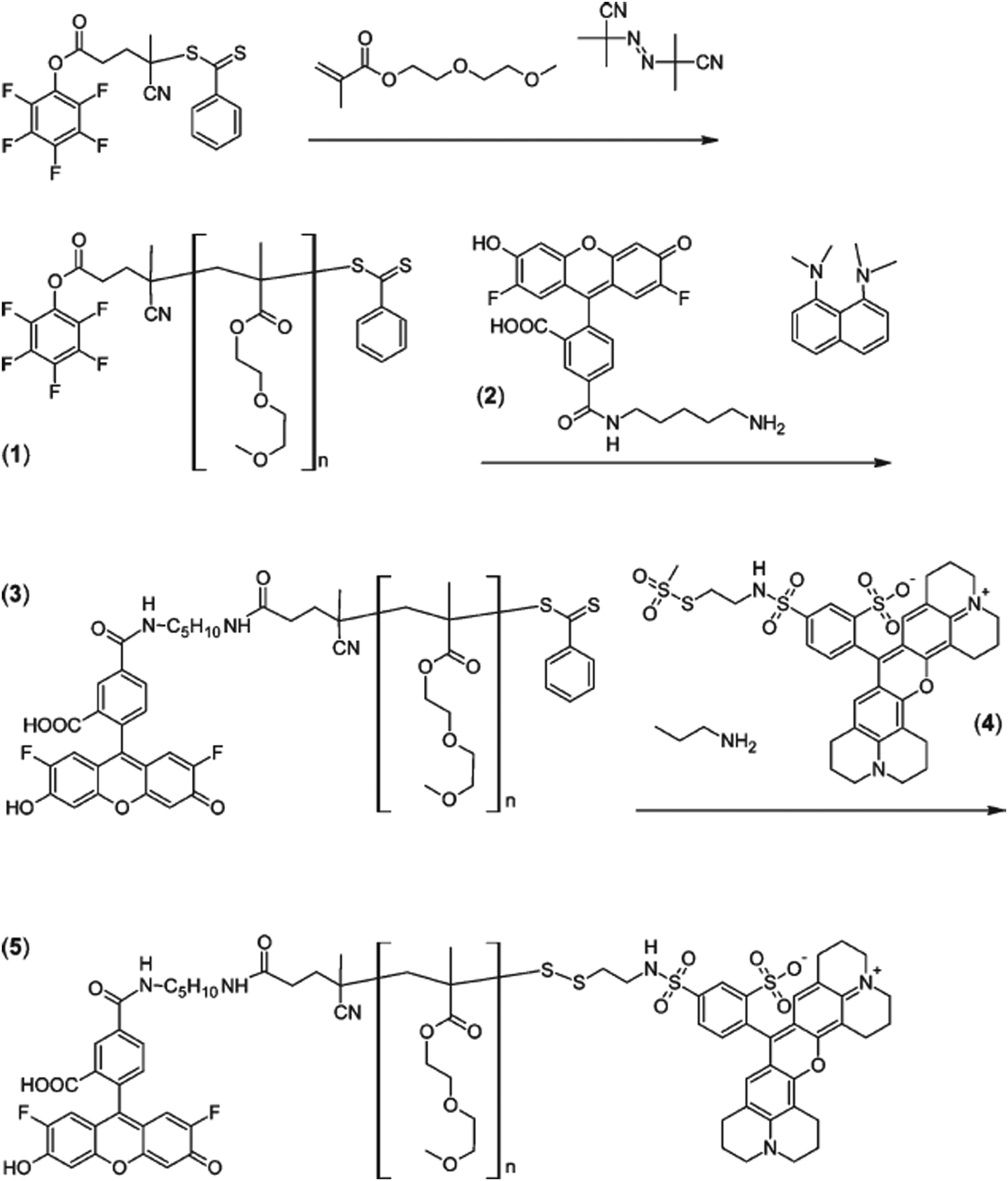 | ||
| Fig. 7 RAFT synthesis of PDEGMA (1) from pentafluorophenyl activated ester RAFT followed by successive attachment of oregon green cadaverin (2) by amidation on the α position (3) and texas red-2-sulfonamidoethyl methanethiosulfonate (4) by disulfide bond formation after aminolysis of the RAFT end group (5). Adapted with permission from ref. 47. | ||
1.3 Peptide/protein conjugation
Therapeutic proteins are used for a broad range of pathologies with different purposes, such as increasing the amount of lacking natural proteins, upregulating existing metabolic pathways, inhibiting molecules or organisms or as targeting agents in tumour therapy.48 Unfortunately, some limitations are still present: proteins are rapidly cleared from the circulatory system by the reticuloendothelial system or metabolized by peptidases or proteases leading to a loss of biological activity.The construction of polymer–protein bioconjugates can help overcome these problems. The well-known “PEGylation” technique, that consists in the attachment of a poly(ethylene glycol) (PEG) chain to a protein/peptide, brings several advantages: it increases bioavailability and plasma half-lives, it enhances biocompatibility and it decreases immunogenicity, reduces proteolysis and enhances solubility and stability.49,50 In the past few decades, RDRP techniques offered the possibility to use more complex and tailor-made polymers with advanced properties to synthesize polymer–protein/peptide bioconjugates.51–53
The introduction of a second biological moiety at the other extremity of a polymer–protein/peptide bioconjugate can be of great interest to confer additional beneficial properties. Amino acids or small peptides are commonly used as model molecules, leading to interesting proofs of concept. For instance, a heterotelechelic α-aldehyde, ω-thiol polyvinylpyrrolidone (PVP) synthesized by RAFT polymerization was functionalized with glycine-D,L-serine and phenyl acrylate, respectively.54 Also, by starting from difunctional trithiocarbonate RAFT agents bearing aminooxy groups or pyridyl disulfide (PDS) groups (Fig. 8a and b), corresponding homotelechelic poly[oligo(ethylene glycol)methyl ether acrylate] (POEGA) polymers were successfully prepared (Fig. 8c and d). They were then post-functionalized at both chain-ends with either glutathione (GSH) or benzyl mercaptan.55
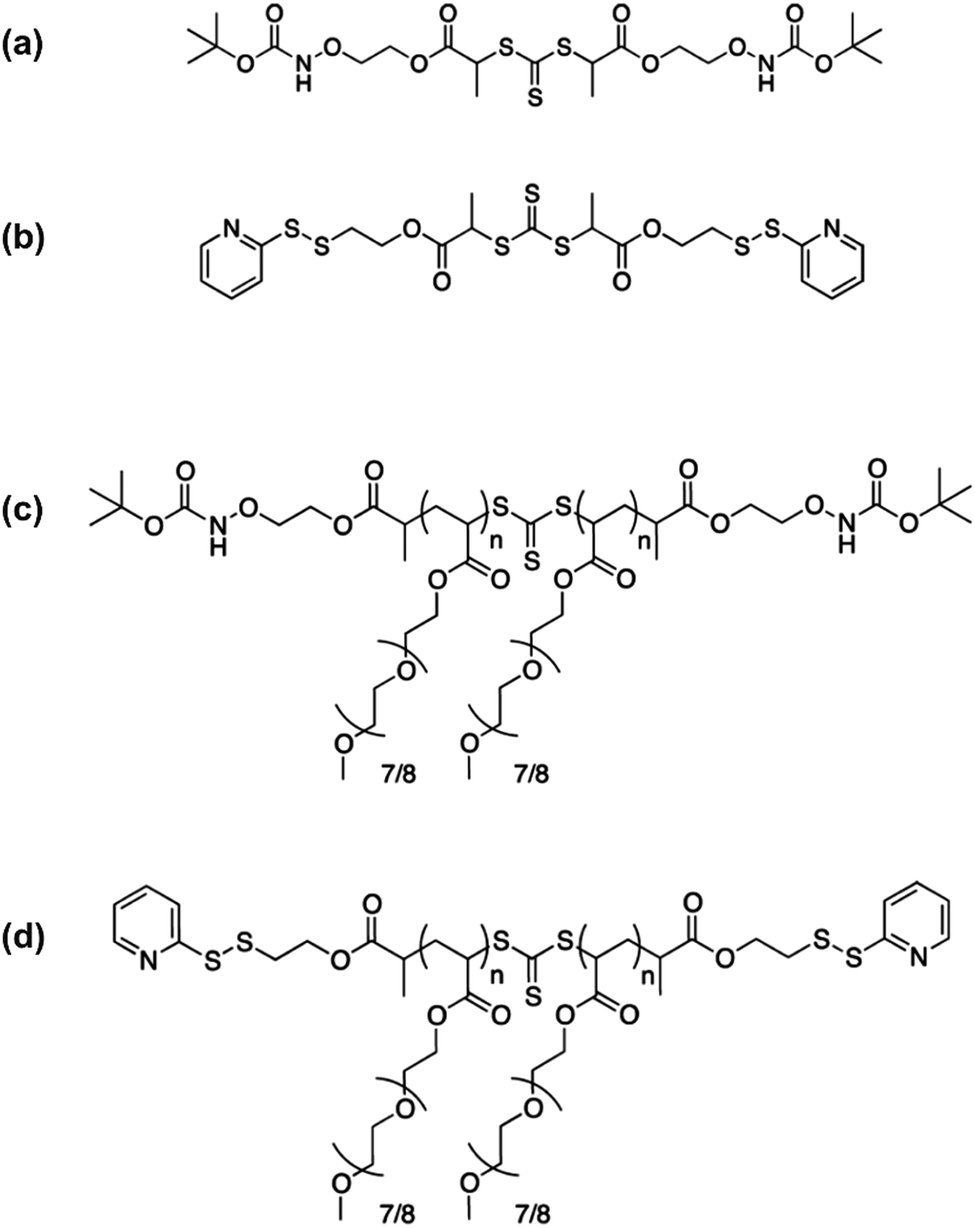 | ||
| Fig. 8 Structure of difunctional, symmetric (a) N-Boc-aminooxy trithiocarbonate, (b) pyridyl disulfide trithiocarbonate, (c) telechelic N-Boc-aminooxy POEGA and (d) telechelic pyridyl disulfide POEGA. Adapted with permission from ref. 55. | ||
Synthesis of a telechelic thiol-reactive polymer and its subsequent conjugation to N-acetyl-L-cysteine methyl ester as a thiol-containing model molecule was also reported.56 ATRP of styrene was initiated from a protected maleimide ATRP initiator followed by atom-transfer radical coupling to yield the corresponding homotelechelic polymer. The amino acid was then linked through maleimide chemistry to both chain-ends (Fig. 9). Noteworthy is the fact that this strategy could be easily applied to other polymers and thiol-containing biomolecules that would be relevant for biomedical applications.
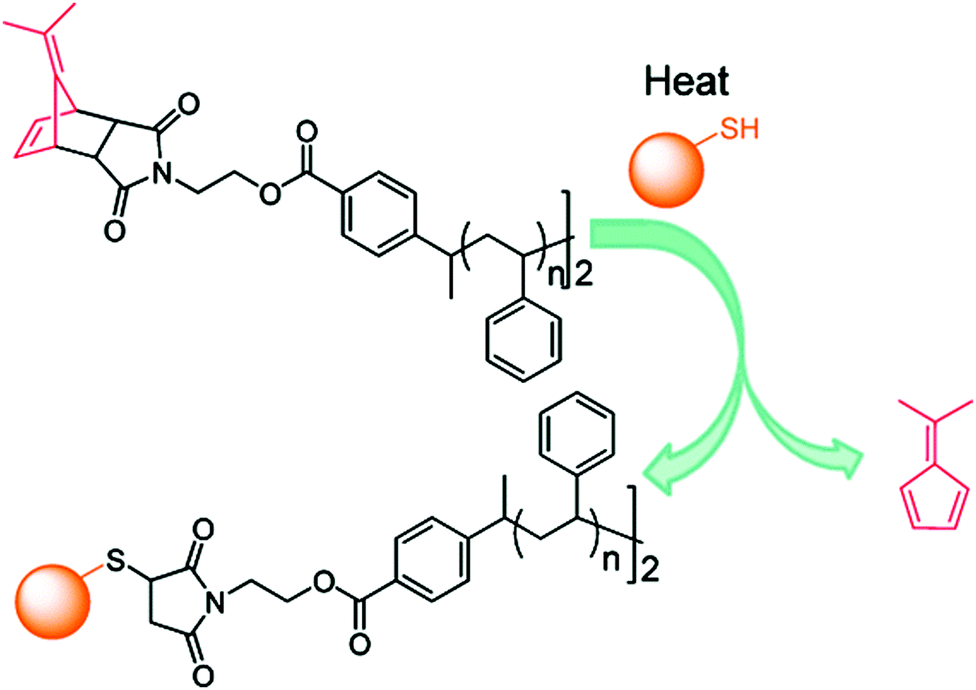 | ||
| Fig. 9 Synthesis of a telechelic thiol-reactive polymer by ATRP and subsequent atom-transfer radical coupling, followed by conjugation to N-acetyl-L-cysteine methyl ester. Adapted with permission from ref. 56. | ||
Bovine serum albumin (BSA) and lysozyme are commonly used as model proteins because of the easiness of covalent linkage offered by the presence of a free cysteine residue (Cys34) or several lysine residues, respectively. Different approaches were employed to synthesize heterotelechelic polymers bearing BSA on one chain-end (via thiol/maleimide linkage) and another biomolecule at the other extremity. For instance, an alkyne–biotin was clicked through CuAAC to RAFT-constructed α-azide, ω-PDS heterobifunctional poly(N-isopropylacrylamide) (PNIPAAm) before subsequent coupling to BSA to form a disulfide linkage and to avidin (Av) via its binding to the biotinylated end group.57 Similarly, BSA and SAv were linked to a heterobifunctional α-biotin, ω-maleimide PNIPAAm made by RAFT, through its maleimide and biotin end groups, respectively (Fig. 10).58
 | ||
| Fig. 10 Synthesis of heterotelechetic polymer–protein bioconjugates by RAFT polymerization of NIPAAm from a biotinylated chain transfer agent, followed by end-functionalization with a maleimide group and subsequent coupling with BSA via thioether bond formation and SAv via binding with biotin. Adapted with permission from ref. 53. | ||
In another study, heterobifunctional polymers bearing BSA on one side and another molecule on the other were prepared.59 A BSA macro-RAFT agent bearing a PDS group was prepared from the reaction between BSA and a PDS-difunctionalized CTA, leaving one PDS group intact because of steric hindrance. This was followed by RAFT polymerization of OEGA or HPMA leading to BSA–POEGA and BSA–PHPMA bioconjugates in situ. Then thiol-functionalized rhodamine B and thiocholesterol were linked through PDS linkage to form the corresponding heterobifunctional polymers.
Strategies to position the same protein at both chain-ends were also reported. For instance, linear homotelechelic PNIPAAm bearing on the α and ω positions a mutant V131C T4 lysozyme (T4L), that contains one free cysteine, was successfully prepared either via maleimide linkage (Fig. 11a)60 or via a new tetrazine linker (Fig. 11b) which improved the coupling yields.61 The synthetic route was based on a bis-carboxylic acid RAFT agent that was reacted, after polymerization, with either a furan-protected maleimide alcohol or (4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanol to install at both chain-ends maleimide or tetrazine moieties, respectively (Fig. 11c). This approach was further extended to four-armed star-PNIPAAm with T4L at each polymer end.62
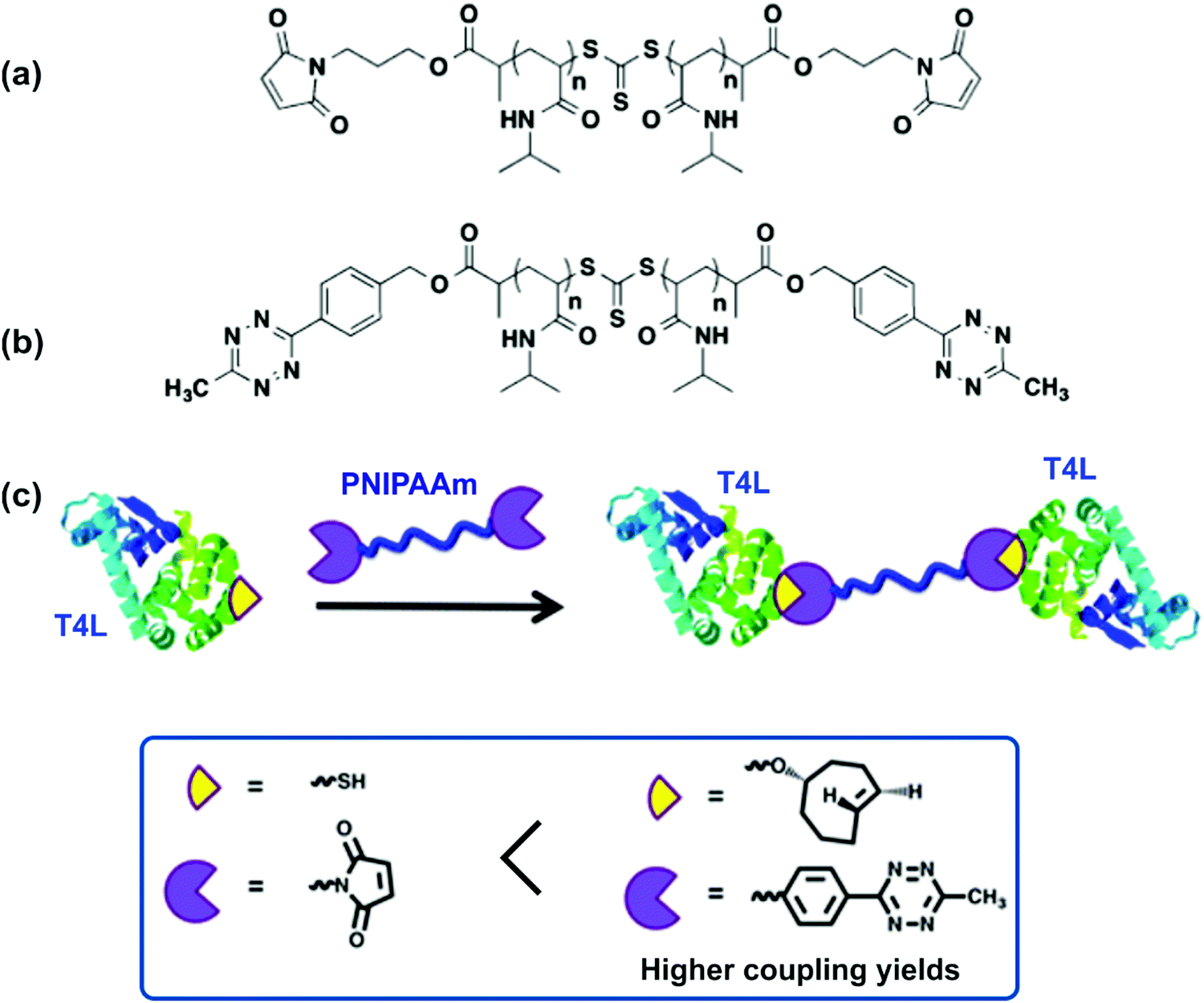 | ||
| Fig. 11 Structure of homotelechelic PNIPAAm bearing (a) maleimide or (b) tetrazine moieties, and (c) homodimerization of T4L with homotelechelic PNIPAAm bearing either maleimide or tetrazine functional groups. Adapted with permission from ref. 61. | ||
More sophistication can be implemented by conferring reversible linkages in the structure of dual-functionalized bioconjugates. A heterotelechelic α-biotin, ω-maleimide PNIPAAm containing a cleavable disulfide bond between biotin and the polymer was synthesized by RAFT polymerization and used to reversibly modify surfaces with proteins.63 T4L was linked to the maleimide group and the conjugate was anchored to an Av-coated surface through biotin/Av linkage. The conjugate was released from the surfaces under mild reducing conditions demonstrating the reversibility (Fig. 12).
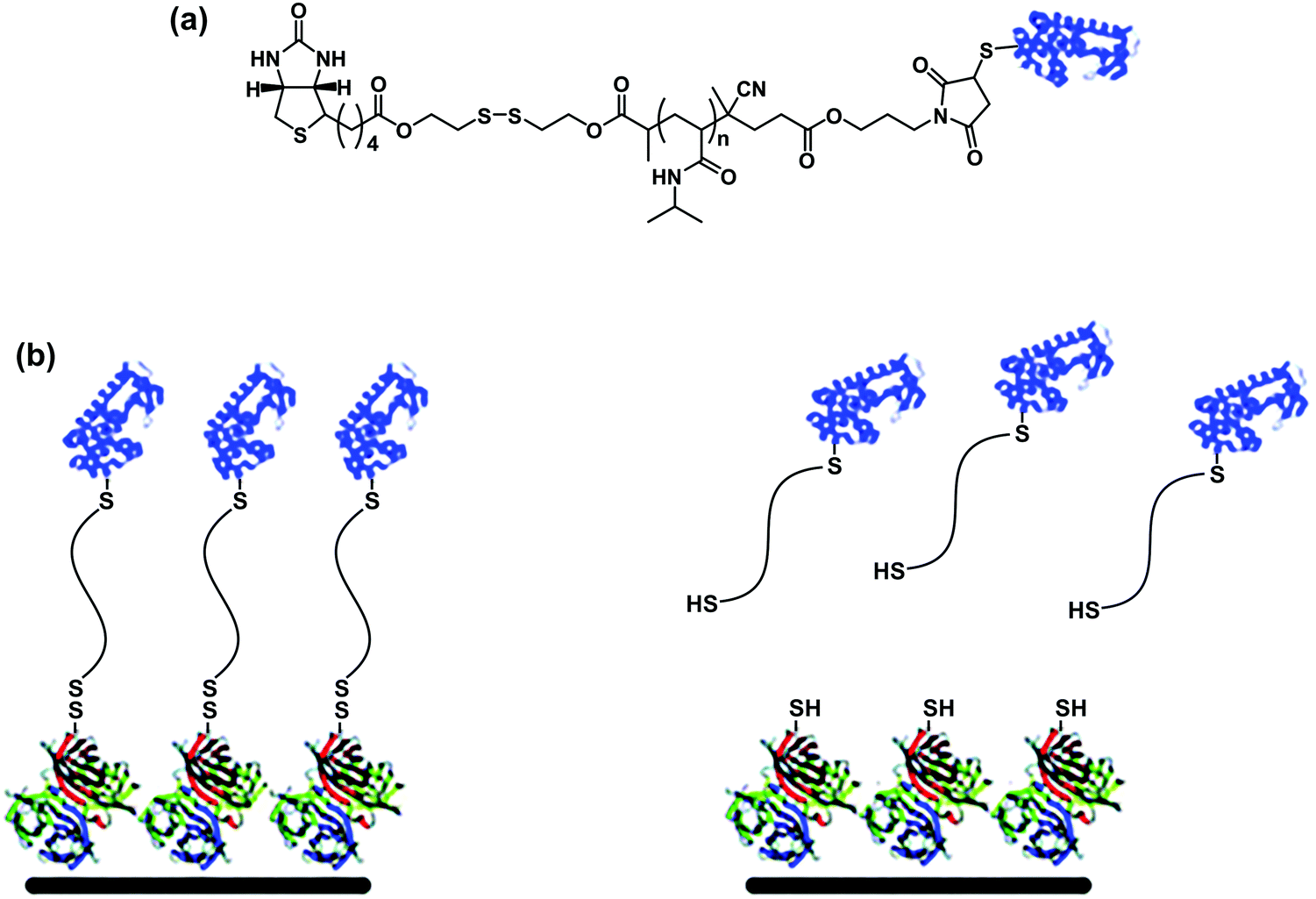 | ||
| Fig. 12 (a) Structure of heterotelechelic α-biotin, ω-maleimide PNIPAAm containing a cleavable disulfide bond between the biotin and the polymer, after conjugation of BSA via thiol/maleimide linkage. (b) Immobilization of the conjugate via the biotin–SAv interaction and release from the surface by reduction of the disulfide bound. Adapted with permission from ref. 63. | ||
Non-covalent interactions other than biotin/SAv (or biotin/Av or biotin/neutravidin (NAv)) have been reported for the design of biorelated telechelic polymers. For instance, Theato and co-workers built on their previous study47 where a pentafluorophenyl (PFP) activated ester RAFT agent was used to prepare heterotelechelic polymers bearing two different dyes, to report a heterotelechelic biofunctionalized polymer via hormone thyroxine-prealbumin linkage (Fig. 13).64 A PFP-poly[di(ethylene glycol)methyl ether methacrylate] (PFP-PDEGMA) was reacted with the hormone thyroxine (T4) followed by end-capping with biotinylated methane thiosulfonate during aminolysis of the RAFT end group. T4 and biotin end groups specifically bonded to prealbumin and SAv, respectively. Subsequent immobilization onto a self-assembled monolayer of T4-disulfide surface was performed with the polymer connecting the two different proteins with its end groups.
 | ||
| Fig. 13 Synthesis of α-hormone thyroxine (T4), ω-S-S-biotin PDEGMA and subsequent binding with prealbumin and SAv, respectively, followed by surface immobilization. Adapted with permission from ref. 64. | ||
The non-covalent interaction between GSH and glutathione-S-transferase (GST) was also employed for the synthesis of heterotelechelic polymers.65 Synthesis of heterotelechelic α-GSH, ω-biotin PNIPAAm was performed by RAFT, followed by coupling with GST and SAv in solution. α-GSH, ω-biotin PNIPAAm was further shown to separately bind GST-tagged Rac1, a potential cancer marker, and biotin-tagged BSA.
1.4 Oligonucleotide bioconjugation
Even though not many examples of dual-functionalization are present in the literature, nucleic acids (e.g., DNA, RNA, or small interfering RNA (siRNA)) are another class of macromolecules that gain great benefit from their conjugation to synthetic polymers, helping to resolve issues such as serum stability, low blood circulation time and lack of targeting.66A convenient strategy to bind them at a polymer chain-end is to react a thiol-terminated nucleotide with a PDS end group.67 For instance, a PDS end-functionalized polymer-dendritic carbohydrate scaffold was obtained by RAFT polymerization of HPMA from a RAFT agent bearing 2-mercaptothiazolidine active ester function. The resulting polymer was further reacted with dendritic-mannose G2Man on the ω-end group, followed by aminolysis to install a PDS group on the α position. Conjugation to a short interfering RNA then led to the heterobifunctional conjugate (Fig. 14). This PDS end-functionalized PHPMA-dendritic carbohydrate scaffold is believed to be a versatile precursor for bioconjugations, as the synthetic procedure can easily accommodate a range of sugar functionalities.
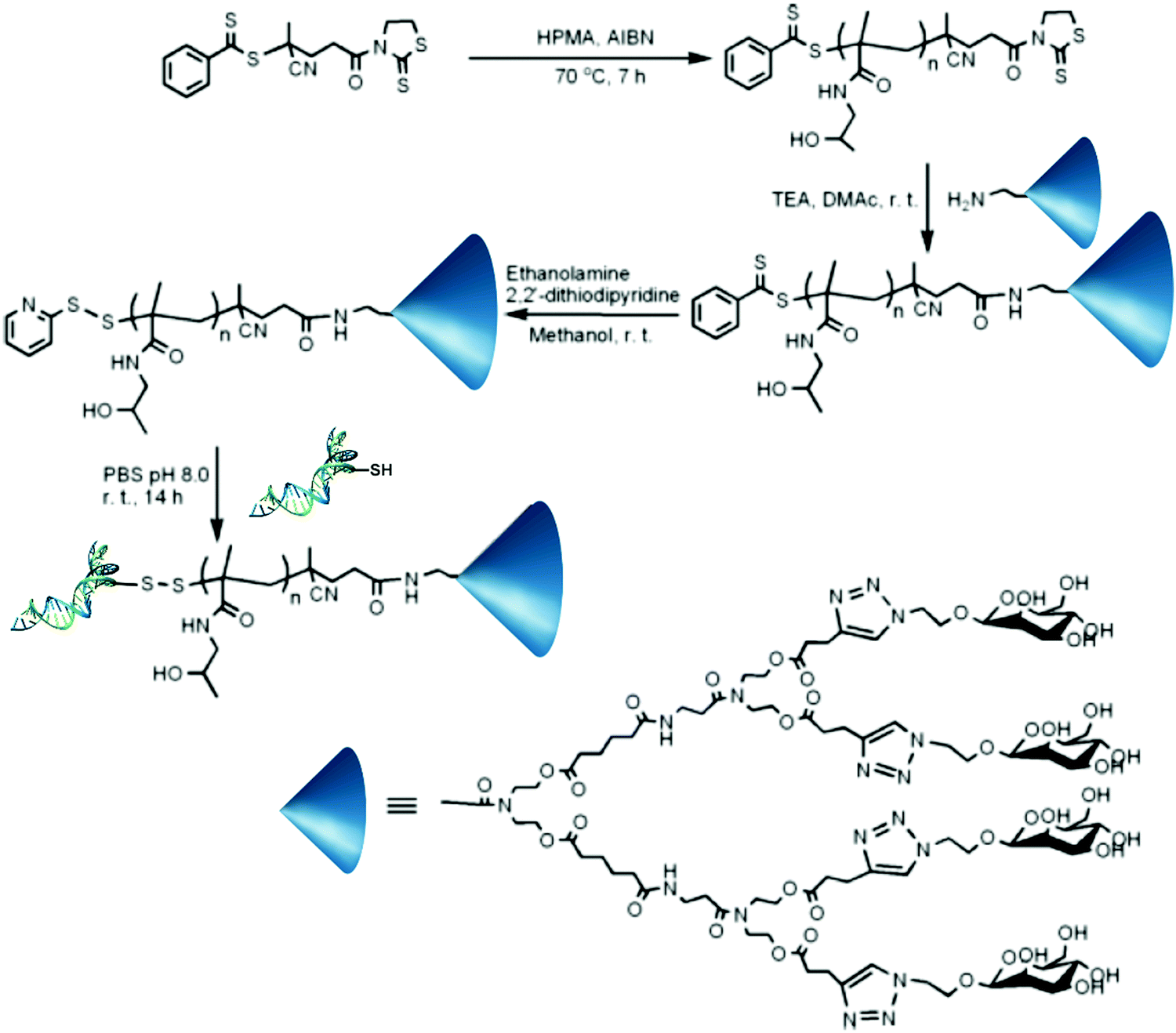 | ||
| Fig. 14 Synthesis of heterobifunctional siRNA–PHPMA–dendritic mannose biohybrid conjugates by RAFT polymerization of HPMA from a chain transfer agent bearing a 2-mercaptothiazolidine active ester group followed by coupling with dendritic-mannose G2Man on the ω chain-end and with a short, thiolated interfering RNA on the α position via disulphide bond formation after the reaction with a PDS moiety. Adapted with permission from ref. 67. | ||
2. Organic/inorganic hybrid systems
The synthesis of organic/inorganic hybrid systems can give access to materials with interesting features because of the nature and unique properties of inorganic entities (e.g., optical, electronic, physical). The use of RDRP techniques enables the synthesis of a broad range of well-defined functional polymers that can be attached to inorganic materials including particles, flat surfaces or rods. In this area, the design of telechelic polymers is of crucial importance as it allows to incorporate other biorelevant (macro)molecules for advanced applications. Below are reported representative strategies for the synthesis of organic/inorganic hybrid systems based on preformed telechelic polymers. Given the field of surface modification is extremely broad and rich, especially from surface-initiated RDRP strategies, the readers interested in this field are referred to the following comprehensive reviews.68–702.1 Gold
Gold nanoparticles (AuNPs) are the subject of an incredible amount of work because of their magnetic, optic and electrical properties. It explains why they are widely investigated in bionanotechnology and biomedicine, among many other fields, especially as a biosensor material.71–73 Moreover, given their biocompatibility and stability, AuNPs can be used as a nanocarrier for nucleic acids or drugs for instance.74–76 Grafting a polymer at the surface of AuNPs comes with great advantages: (i) it ensures the colloidal stabilization of the system and can improve biocompatibility; (ii) it can tune the size of the particles, which plays a significant role in the modulation of optical, electric and magnetic properties and (iii) it opens the door to further conjugation with biorelevant entities.Given the most common functionalization strategy involves thiol chemistry, this makes RAFT polymerization the technique of choice to link RDRP polymers at the surface of AuNPs.77–81 It is explained by the easy reduction of the Z group of the RAFT agent to disclose a thiol end group which displays high affinity for gold surfaces. Reduction can also be carried out in situ during AuNP formation. This approach has naturally been extended to gold surfaces82,83 or gold nanorods (AuNRs),84,85 in a rather straightforward manner.
Gibson and co-workers reported on poly(hydroxyethyl acrylamide) (HEA),86 PVP87 and POEGA87 synthesised by RAFT and further grafted at the surface of AuNPs via the ω-thiol function after removal of the CTA's Z group. By taking advantage of the carboxylic acid from the CTA's R group, 2-deoxy-2-amino glucose (GlcNH2) or 2-deoxy-2-amino-galactose (GalNH2) were coupled to the α-terminus of the polymer to generate glycosylated nanoparticle AuNPs. No significant binding to ovine red blood cells, nor haemolysis was noticed, suggesting that they are compatible with blood. This work is intended to guide the development of new nanoparticles for drug delivery, imaging or biosensing, with enhanced specificity, affinity, and stability.
Interestingly, heterotelechelic α-amino, ω-thiol PNIPAAm prepared by RAFT polymerization was used as “polymeric glue” to interlink two types of gold building blocks: AuNPs and AuNRs, via a two-step ligand exchange approach. It created a satellite-like structure that shows unique thermo-responsive optical properties (Fig. 15).84 Given these unique properties, this nanomaterial assembly could be of interest in the field of biomedicine and biosensing.
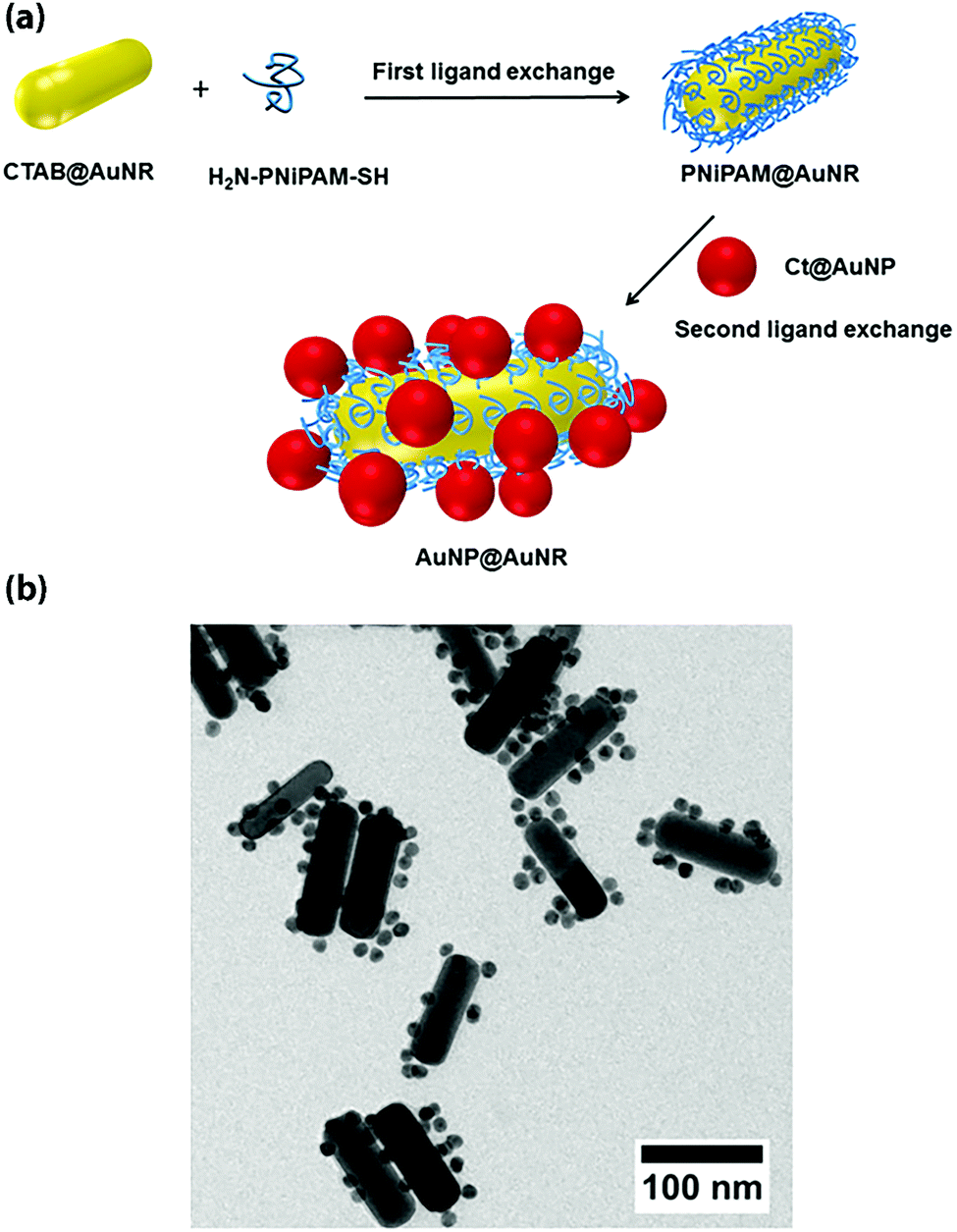 | ||
| Fig. 15 (a) Linkage of heterotelechelic α-amino, ω-thiol PNIPAAm at the surface of AuNRs and further immobilization of AuNPs. (b) Transmission electron microscopy of AuNP@AuNR assembly. Adapted with permission from ref. 84. | ||
Heterotelechelic polymers are naturally of great interest to immobilize proteins at the surface of gold nanoparticles for potential applications in drug delivery or biosensing. For instance, NIPAAm was polymerized by RAFT from 2-dodecylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (DMP) as a RAFT agent.88 After aminolysis to recover ω-thiol end groups and conjugation of biotin to the α-carboxylic acid group, a α-biotin, ω-thiol PNIPAAm was obtained. Its anchoring at the surface of AuNPs enabled their homogeneous coating and further surface functionalization with avidin mediated by biotin/Av binding. Similarly, Narain and co-workers immobilized heterotelechelic α-biotin, ω-thiol glycopolymers and PNIPAAm at the surface of AuNPs.79,80 Their incubation with Av showed the accessibility of the biotin ligands for conjugation and the Av-conjugated glyconanoparticles were found to selectively immobilize lectins.
Flat gold surfaces are also of major interest for several biorelevant applications, such as biosensing, chip-based bioassays, or cell adhesion studies.89–91 Many different biorelevant molecules can be grafted by means of telechelic polymers used as a bridge between them and the molecule to be linked. For instance, a RAFT-synthesized aminooxy end-functionalized PNIPAAm was either used for direct conjugation to BSA or was immobilized on a gold surface after reduction of the trithiocarbonate end-group (Fig. 16).82 The resulting PNIPPAm surface was then incubated with an aldehyde-modified heparin to yield the polysaccharide-functionalized surface. This technique could be applied to the stabilization and storage of growth factors on surfaces or used to coat medical devices where the anticoagulant activity of heparin is needed such as for renal dialysis instruments.
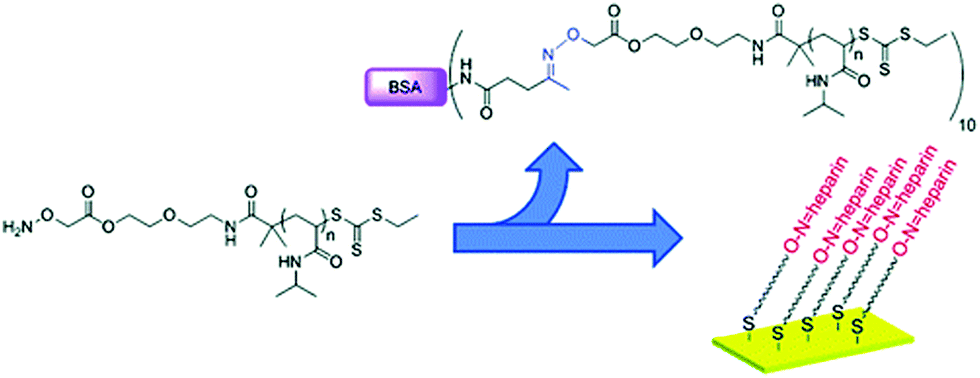 | ||
| Fig. 16 Structure of α-aminooxy end-functionalized PNIPAAm prepared by RAFT used either for the synthesis of BSA-based bioconjugates or for the grafting to flat gold surfaces via the ω-thiol function, followed by heparin attachment via the α-aminooxy end group. Adapted with permission from ref. 82. | ||
In the context of biosensing and bioimaging applications, Theato, Stenzel and co-workers reported on a general approach towards difunctionalized RAFT polymers bearing a fluorescent dye on the α-position and an anchoring group (S–S–CH3) on the ω-position, for the surface decoration of AuNPs or quantum dots (QDs).92 For AuNPs, FRET caused a fluorescence quenching of the dye, while QDs led to fluorescence enhancement by FRET from the QD to the tethered dyes (Fig. 17). This synthetic route, and especially the use of an RDRP technique, allows hybrid materials with a tuneable distance between the inorganic species and the dyes to be prepared.
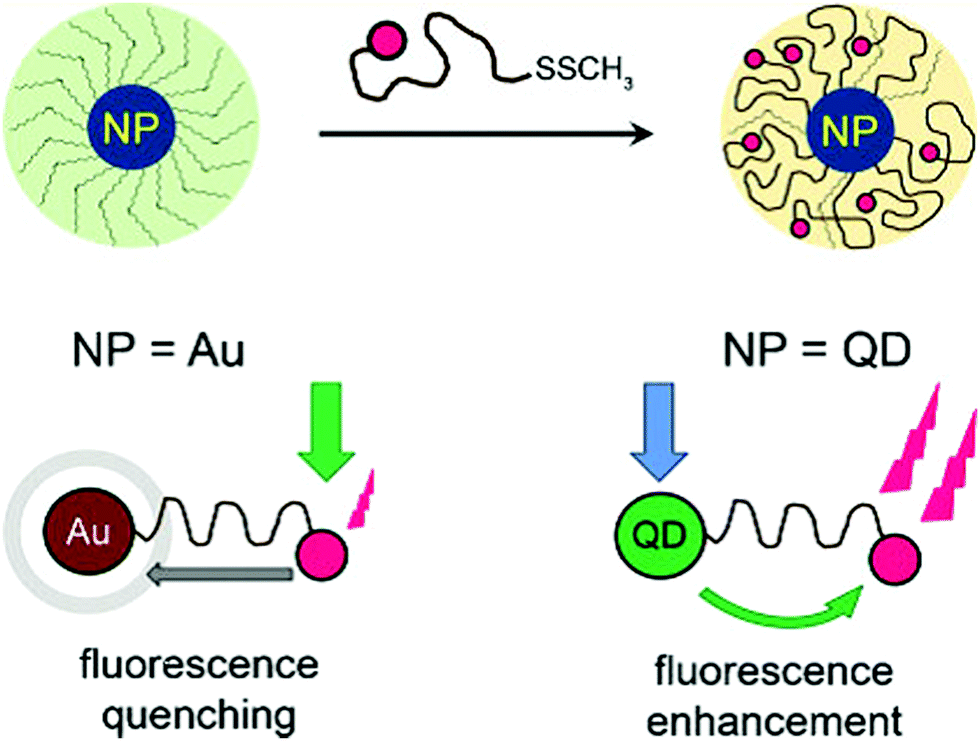 | ||
| Fig. 17 Au nanoparticle or QD decoration by a heterotelechelic α-dye, ω-methyl disulfide poly(methyl methacrylate) and effect of the nearby Au core (fluorescence quenching) or QD (fluorescence enhancement) on the emission of the dye. Adapted with permission from ref. 92. | ||
An additional benefit of RGD-based sequences lies in the fact that they correspond to the minimal adhesive domain of fibronectin and therefore allow control over cell adhesion.93 To finely tune cell adhesion, a targeted GGRGDS-functionalized PNIPAAm was grafted on a gold surface after reduction of a ω-trithiocarbonate moiety. When mouse fibroblasts (L929), which are adhesive cells,83 were cultured on the surface, a faster adhesion on the functionalized surface was observed compared to that on the unmodified PNIPAAm coating.
2.2 Iron oxide
Beyond small fluorochromes and QDs, other imaging agents can be used. Iron oxide nanoparticles (IONPs) are involved in a broad range of applications, such as immunoassay, detoxification of biological fluids, drug delivery, tissue repair and as contrast agents for magnetic resonance imaging (MRI).94–96 Surface functionalization of IONPs can be easily performed from functional polymers embedding suitable binding motifs.For example, a heterotelechelic α-phosphonic acid, ω-dithiopyridine POEGA obtained by RAFT was grafted to IONPs via its α-phosphonic acid moiety, allowing for IONP stabilization in different media (e.g., water, PBS).97 The resulting POEGA-stabilized IONPs were then successfully functionalized with two different peptides after modification of the ω-trithiocarbonate into an ethylpyridyl disulfide end-group: reduced glutathione as a model peptide and an NGR motif as a tumour targeting peptide (Fig. 18).
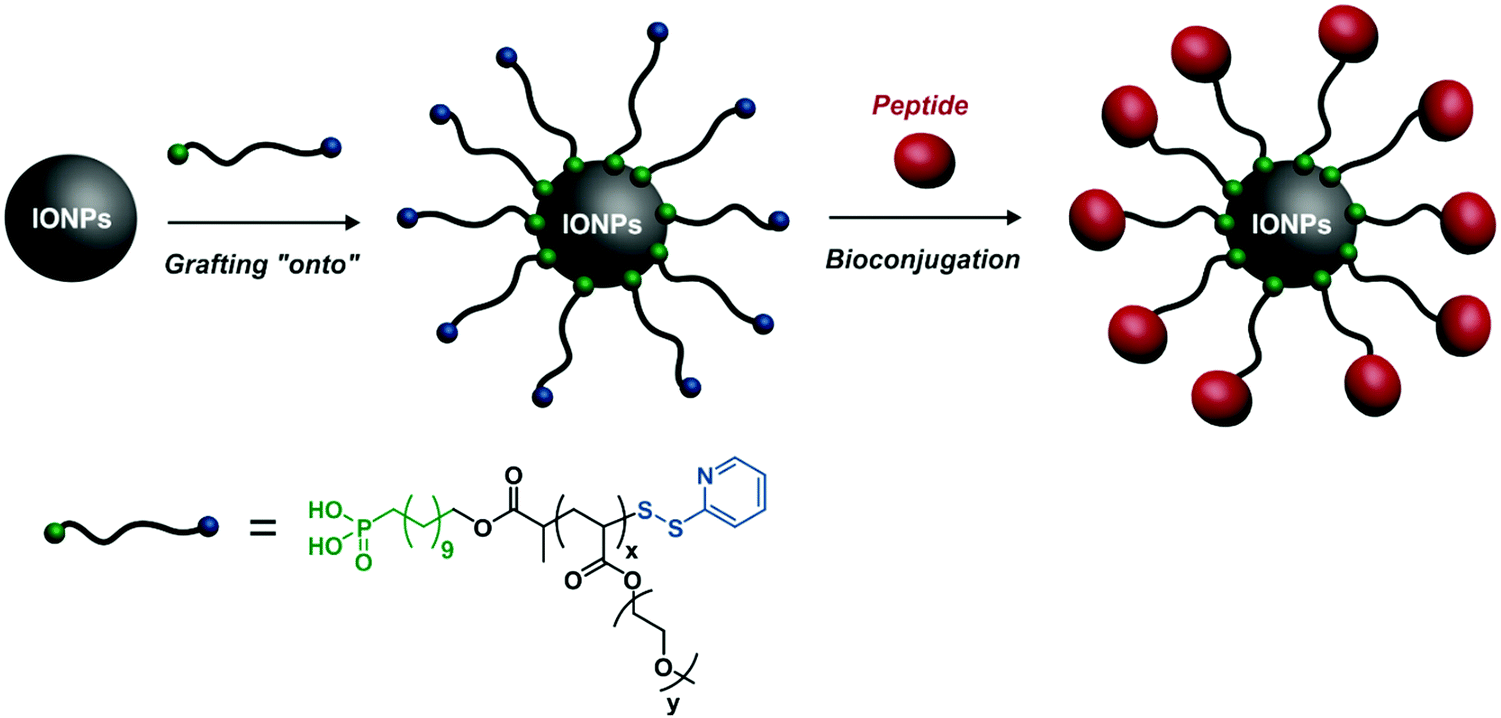 | ||
| Fig. 18 Grafting of heterotelechelic α-phosphonic acid, ω-dithiopyridine POEGA onto IONPs and further surface coupling via the ω-dithiopyridine end group with glutathione as a model peptide or the NGR motif for targeting purposes. Adapted with permission from ref. 97. | ||
Conclusions
The aim of this Feature article was to cover the recent achievements in the design of telechelic polymers by RDRP techniques for biomedical applications. In light of this survey, it appears that these well-defined functional materials present unique features that could be of great value in many several key bio-related areas including drug delivery, targeting and imaging/sensing. Enabling molecular weight control and precise positioning of biologically active moieties at both polymer chain-ends gave access to valuable tools or building blocks that could serve in tomorrow's biomedical applications.The different examples also shed light on the robustness and a kind of supremacy of the RAFT technique over other RDRP methods to achieve telechelic structures for biomedical applications, owing to facile chain-end functionalization via removal of the RAFT agent, the broad applicability of thiol chemistry for bioconjugation and easy access to a broad range of functional RAFT agents.
One could however regret the too few examples in the field of drug delivery from small molecules, whereas telechelic polymer–proteins/peptides have been extensively investigated. To achieve such a goal, the “drug-initiated” method appears key as it inherently leads to conjugates with one drug molecule linked at the α-position. Efficient ω-chain-end modification by drugs, biologically active ligands or imaging agents should result in a broad range of heterotelechelic polymer prodrugs that could find use in combinatorial therapy, targeted drug delivery or theranostics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Some results described in this review were obtained thanks to a Marie Skłodowska Curie Intra-European Fellowship from the European Commission (No. 623501) and to the French National Research Agency (ANR-11-JS08-0005). The PhD fellowships of DV and JT are supported by the European Union's Horizon 2020 research and innovation programme under Marie Skłodowska Curie grant agreement no. 642028 (NABBA) and by the French National Research Agency (ANR-15-CE08-0019), respectively. CNRS and Université Paris-Sud are also acknowledged for financial support.References
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed.
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- http://https://goldbook.iupac.org/html/F/FT07174.html .
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- F. Lo Verso and C. N. Likos, Polymer, 2008, 49, 1425–1434 CrossRef CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455–567 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS PubMed.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef CAS PubMed.
- Y. Bao, T. Boissenot, E. Guégain, D. Desmaële, S. Mura, P. Couvreur and J. Nicolas, Chem. Mater., 2016, 28, 6266–6275 CrossRef CAS.
- Y. Bao and J. Nicolas, Polym. Chem., 2017, 8, 5174 RSC.
- Y. Bao, V. Nicolas and J. Nicolas, Chem. Commun., 2017, 53, 4489–4492 RSC.
- S. Harrisson, J. Nicolas, A. Maksimenko, D. T. Bui, J. Mougin and P. Couvreur, Angew. Chem., Int. Ed., 2013, 52, 1678–1682 CrossRef CAS PubMed.
- A. Maksimenko, D. T. Bui, D. Desmaële, P. Couvreur and J. Nicolas, Chem. Mater., 2014, 26, 3606–3609 CrossRef CAS.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- P. Carter, Nat. Rev. Cancer, 2001, 1, 118 CrossRef CAS PubMed.
- P. Liu, Z. Cai, J. W. Kang, A. J. Boyle, J. Adams, Y. Lu, G. Ngo Ndjock Mbong, S. Sidhu, R. M. Reilly and M. A. Winnik, Biomacromolecules, 2014, 15, 715–725 CrossRef CAS PubMed.
- E. P. Diamandis and T. K. Christopoulos, Clin. Chem., 1991, 37, 625–636 CAS.
- B. Louage, L. Nuhn, M. D. Risseeuw, N. Vanparijs, R. De Coen, I. Karalic, S. Van Calenbergh and B. G. De Geest, Angew. Chem., Int. Ed., 2016, 55, 11791–11796 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh, H. Gao, S. A. Bencherif, F. Perineau, A. K. Bohaty, J. O. Hollinger and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 2179–2193 CrossRef CAS.
- K. Luo, J. Yang, P. Kopeckova and J. Kopecek, Macromolecules, 2011, 44, 2481–2488 CrossRef CAS PubMed.
- H. Pan, M. Sima, S. C. Miller, P. Kopeckova, J. Yang and J. Kopecek, Biomaterials, 2013, 34, 6528–6538 CrossRef CAS PubMed.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- J. Yang, K. Luo, H. Pan, P. Kopeckova and J. Kopecek, React. Funct. Polym., 2011, 71, 294–302 CrossRef CAS PubMed.
- R. Zhang, K. Luo, J. Yang, M. Sima, Y. Sun, M. M. Janat-Amsbury and J. Kopecek, J. Controlled Release, 2013, 166, 66–74 CrossRef CAS PubMed.
- N. Larson, J. Yang, A. Ray, D. L. Cheney, H. Ghandehari and J. Kopecek, Int. J. Pharm., 2013, 454, 435–443 CrossRef CAS PubMed.
- R. Zhang, J. Yang, M. Sima, Y. Zhou and J. Kopecek, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12181–12186 CrossRef CAS PubMed.
- A. Duangjai, K. Luo, Y. Zhou, J. Yang and J. Kopecek, Eur. J. Pharm. Biopharm., 2014, 87, 187–196 CrossRef CAS PubMed.
- I. Singh, Z. Zarafshani, F. Heaney and J.-F. Lutz, Polym. Chem., 2011, 2, 372–375 RSC.
- Z. An, W. Tang, M. Wu, Z. Jiao and G. D. Stucky, Chem. Commun., 2008, 6501–6503 RSC.
- F. Segui, X.-P. Qiu and F. M. Winnik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 314–326 CrossRef CAS.
- J. Fu, Z. Cheng, N. Zhou, J. Zhu, W. Zhang and X. Zhu, e-Polym., 2009, 9, 206–216 Search PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- G. D. Scholes, Annu. Rev. Phys. Chem., 2003, 54, 57–87 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387 CrossRef CAS PubMed.
- Y. Sha, Y. Xu, D. Qi, Y. Wan, L. Li, H. Li, X. Wang, G. Xue and D. Zhou, Macromolecules, 2016, 49, 8274–8281 CrossRef CAS.
- P. J. Roth, M. Haase, T. Basche, P. Theato and R. Zentel, Macromolecules, 2010, 43, 895–902 CrossRef CAS.
- D. S. Dimitrov, Ther. Proteins, 2012, 1–26 CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS PubMed.
- M. Roberts, M. Bentley and J. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- E. M. Pelegri-O’Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- P. W. Reader, R. Pfukwa, S. Jokonya, G. E. Arnott and B. Klumperman, Polym. Chem., 2016, 7, 6450–6456 RSC.
- G. N. Grover, J. Lee, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2012, 45, 4858–4965 CrossRef PubMed.
- Z. P. Tolstyka, J. T. Kopping and H. D. Maynard, Macromolecules, 2008, 41, 599–606 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360–2367 CrossRef CAS PubMed.
- J. Liu, H. Liu, V. Bulmus, L. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399–1405 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. Ogorzalek Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148–2150 RSC.
- M. M. Lorenzo, C. G. Decker, M. U. Kahveci, S. J. Paluck and H. D. Maynard, Macromolecules, 2016, 49, 30–37 CrossRef CAS PubMed.
- L. Tao, C. S. Kaddis, R. R. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028–8033 CrossRef CAS PubMed.
- K. L. Heredia, L. Tao, G. N. Grover and H. D. Maynard, J. Polym. Sci., Part A: Polym. Chem., 2010, 1, 168–170 CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS PubMed.
- X. Huang, C. Boyer, T. P. Davis and V. Bulmus, Polym. Chem., 2011, 2, 1505 RSC.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581 CrossRef CAS PubMed.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- X. Zhang, Cell Biochem. Biophys., 2015, 72, 771–775 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- R. K. DeLong, C. M. Reynolds, Y. Malcolm, A. Schaeffer, T. Severs and A. Wanekaya, Nanotechnol., Sci. Appl., 2010, 3, 53 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169–183 CrossRef CAS PubMed.
- S. G. Spain, L. Albertin and N. R. Cameron, Chem. Commun., 2006, 4198–4200 RSC.
- A. Housni, H. Cai, S. Liu, S. H. Pun and R. Narain, Langmuir, 2007, 23, 5056–5061 CrossRef CAS PubMed.
- R. Narain, A. Housni, G. Gody, P. Boullanger, M.-T. Charreyre and T. Delair, Langmuir, 2007, 23, 12835–12841 CrossRef CAS PubMed.
- X. Jiang, A. Housni, G. Gody, P. Boullanger, M.-T. Charreyre, T. Delair and R. Narain, Bioconjugate Chem., 2010, 21, 521–530 CrossRef CAS PubMed.
- A. L. Parry, N. A. Clemson, J. Ellis, S. S. Bernhard, B. G. Davis and N. R. Cameron, J. Am. Chem. Soc., 2013, 135, 9362–9365 CrossRef CAS PubMed.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650–7656 CrossRef CAS PubMed.
- J. Hentschel, K. Bleek, O. Ernst, J.-F. Lutz and H. G. Börner, Macromolecules, 2008, 41, 1073–1075 CrossRef CAS.
- Z. Fan, M. Tebbe, A. Fery, S. Agarwal and A. Greiner, Part. Part. Syst. Charact., 2016, 33, 698–702 CrossRef CAS.
- J. Lu, W. Zhang, S.-J. Richards, M. I. Gibson and G. Chen, Polym. Chem., 2014, 5, 2326–2332 RSC.
- L. E. Wilkins, D. J. Phillips, R. C. Deller, G. L. Davies and M. I. Gibson, Carbohydr. Res., 2015, 405, 47–54 CrossRef CAS PubMed.
- N. Sze Ieong, C. I. Biggs, M. Walker and M. I. Gibson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1200–1208 CrossRef.
- A. Aqil, H. Qiu, J.-F. Greisch, R. Jérôme, E. De Pauw and C. Jérôme, Polymer, 2008, 49, 1145–1153 CrossRef CAS.
- D. Grieshaber, R. MacKenzie, J. Vörös and E. Reimhult, Sensors, 2008, 8, 1400 CrossRef CAS PubMed.
- H. Y. Song, T. I. Wong, A. Sadovoy, L. Wu, P. Bai, J. Deng, S. Guo, Y. Wang, W. Knoll and X. Zhou, Lab Chip, 2015, 15, 253–263 RSC.
- S.-H. Yoon and M. R. K. Mofrad, Biomaterials, 2011, 32, 7286–7296 CrossRef CAS PubMed.
- P. J. Roth, K. S. Kim, S. H. Bae, B. H. Sohn, P. Theato and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1274–1278 CrossRef CAS PubMed.
- E. Ruoslahti and M. Pierschbacher, Science, 1987, 238, 491–497 CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111–123 RSC.
| This journal is © The Royal Society of Chemistry 2018 |