
Supramolecular polymeric biomaterials
Joseph L.
Mann†
a,
Anthony C.
Yu†
a,
Gillie
Agmon
b and
Eric A.
Appel
 *ab
*ab
aDepartment of Materials Science and Engineering, Stanford University, 476 Lomita Mall, Stanford, CA 94305, USA. E-mail: eappel@stanford.edu
bDepartment of Bioengineering, Stanford University, 476 Lomita Mall, Stanford, CA 94305, USA
First published on 2nd November 2017
Abstract
Polymeric chains crosslinked through supramolecular interactions—directional and reversible non-covalent interactions—compose an emerging class of modular and tunable biomaterials. The choice of chemical moiety utilized in the crosslink affords different thermodynamic and kinetic parameters of association, which in turn illustrate the connectivity and dynamics of the system. These parameters, coupled with the choice of polymeric architecture, can then be engineered to control environmental responsiveness, viscoelasticity, and cargo diffusion profiles, yielding advanced biomaterials which demonstrate rapid shear-thinning, self-healing, and extended release. In this review we examine the relationship between supramolecular crosslink chemistry and biomedically relevant macroscopic properties. We then describe how these properties are currently leveraged in the development of materials for drug delivery, immunology, regenerative medicine, and 3D-bioprinting (253 references).
 Joseph L. Mann | Joseph Mann is a PhD student in the Materials Science & Engineering department at Stanford University with a soft spot in his heart for controlled radical polymerization. He is currently funded by a Department of Defense Graduate Fellowship and is interested in the controlled synthesis of topologically diverse polymeric backbones for materials that interface with proteins. More specifically, he is exploring the rational design of soluble excipients, thermo-responsive artificial chaperones, and injectable supramolecular hydrogels. |
 Anthony C. Yu | Anthony Yu is a PhD student in the Materials Science & Engineering department at Stanford University and is currently funded by a Kodak Graduate Fellowship. He is interested in developing a fundamental understanding of dynamically cross-linked polymeric materials in order to engineer practical materials solutions to biomedical challenges. In particular, he is exploring the structure–property relations that control injectability and solute release kinetics of supramolecular hydrogels and how the relations can be leveraged for rational design. Currently, Anthony is designing new injectable materials for controlled drug delivery and sprayable materials for fire retardant encapsulation and retention. |
 Gillie Agmon | Gillie Agmon is working toward her PhD in bioengineering at Stanford University and is currently funded by an NSF Graduate Fellowship. She is fascinated by the prospect of using bioengineering as a platform for translating groundbreaking discoveries from biology and engineering into novel medical solutions. Gillie is working towards improving vaccine efficacy by probing the effects of sustained release of antigen and adjuvants on the immune response. She uses an injectable hydrogel platform and advanced immunology assays to study the humoral and cell mediated immune responses. Simultaneously, she is also developing novel materials platforms for sustained drug delivery. |
 Eric A. Appel | Eric Appel is an Assistant Professor of Materials Science & Engineering at Stanford University. He received his BS + MS (2008) in Chemistry and Polymer Science from Cal Poly San Luis Obispo, and PhD (2013) in Chemistry from Cambridge University. For his PhD work he received a Graduate Student Award from the Materials Research Society and a Jon Weaver PhD Prize from Macro Group UK. He was awarded a Ruth L. Kirschstein National Research Service Award and a Wellcome Trust Fellowship to pursue postdoctoral research with Robert Langer at MIT. In 2016, he moved to Stanford to establish his independent research efforts in the design, synthesis, characterization and application of supramolecular biomaterials for a number of biomedical applications. |
1 Introduction
It is a privilege to live in a time where the global life expectancy continues to increase. Over the past 30 years the fraction of humans aged 65+ has increased over 140%; multiple countries contain over a fifth of their population in this bracket! However, as the human body ages it becomes more fragile. A rising global life expectancy has been met with increased incidences of cancer and degenerative diseases. We are now left with the challenge of an aging population seeking improved quality of life. Addressing this problem, the healthcare community has looked towards smarter and more creative medical solutions through the development of novel therapeutic agents and strategies, or innovative delivery of existing therapeutics. Common to both approaches is an increasing reliance on creative and adaptable biomaterials.Biomaterials—substances in contact with tissues or biological fluids that are not food or drugs1,2—have evolved from crude wooden prostheses dating back millennia to advanced metals, ceramics, and polymers to augment, repair, or replace diseased and damaged tissue.3 These materials have played a pivotal and transformational role in orthopedic joint replacement, vascular grafting, plastic surgery, dental augmentation and bone fusion,4,5 and their usage has been explored in the field of regenerative medicine.6–8 While traditional materials make great structural analogues for native bone and tissue, they have trouble recapitulating the dynamics and complexity of the native biological environment.
New biomaterials for novel delivery and therapeutic design must then have marked and intentional differences from what is currently available. Addressing delivery, materials should be easily processed, administered, and able to deliver a wide range of therapeutics with controlled release profiles over various timescales. For addressing novel regenerative therapeutics, we need biomaterials that demonstrate physical and chemical cues in a biologically relevant manner. A significant body of research over recent decades has shed light on the utility of hydrogels for this very matter.9–13
Hydrogels are 3D networks of crosslinked macromolecules that can entrap substantial amounts of water through surface tension and capillary forces. They may be categorized into chemical and physical networks. Chemically crosslinked hydrogels, described as polymeric chains interconnected by permanent non-reversible bonds, have been fabricated through a wide range of chemistries.14–20 Chemical crosslinking can be easily tuned to alter the mechanical properties of the final material and has been commonly employed when tough and stable hydrogels are required.21 However, due to the covalent nature of the crosslink, these hydrogels are often brittle, lack transparency, and cannot self-heal after network failure.22 Moreover, the use of metal catalysts, photoinitiators, and UV-light coupled with incomplete conversion of reactive functional groups limits biocompatibility of these systems.23 Further, a poorly defined structure with network defects and large equilibrium swelling volumes hamper in vivo material performance. In contrast, physical hydrogels rely on the formation of transient crosslinks between polymer chains. Because gelation is driven by molecular self-assembly, it avoids the deleterious implications of chemical crosslinks (brittleness, swelling, etc.) at the price of mechanically weaker systems. Moreover, physical crosslinks afford tailored viscoelasticity, an important property for biological recapitulation and injection.24,25
Physical crosslinking is made available through the utilization of supramolecular chemistry—chemistry beyond the molecule.26–31 These crosslinks generally fall into two broad classes: (1) entanglements between one-dimensional fibrous assemblies of supramolecular monomers27 and (2) supramolecular crosslinks between polymeric precursors.32 The former approach has been famously employed with self-assembling peptide amphiphiles33,34 and functionalized amphiphilic benzene-tricarboxamides (BTA).35 Many excellent reviews have been written to describe these low molecular weight gelators (LMWG), so they will not be covered here.27,28,36
Employing supramolecular crosslinks between polymeric chains effectively couples the beneficial properties of polymeric based hydrogels with the diversity and orthogonality of supramolecular chemistry.31,32 Typical supramolecular crosslinks employable in aqueous media include host–guest, ionic, metal–ligand, shielded hydrogen bond, and protein based interactions. Similar to chemical crosslinking, altering the density of crosslinks directly affects the mechanical properties of resulting hydrogel. Dissimilar, however, is the transient nature of the crosslink; both the rate and degree of association can be controlled through the judicious selection of the crosslinking moiety. These parameters have direct implications in the mechanical and viscoelastic response of the material.
Due to the modularity of crosslinks, facile gelation mechanism, beneficial mechanical properties, and outstanding ability to replicate biological viscoelasticity, supramolecular polymeric hydrogels are increasingly employed as advanced biomaterials for applications that range from sustained therapeutic delivery to 3D-bioprinting for regenerative medicine.31,32 We wrote this review to highlight emerging biological applications of supramolecular polymers and specify why their chemical properties make them great candidates for clinical translation. More directly, this review intends to illustrate the toolbox of supramolecular chemistries for researchers trying to address current biomedical challenges while highlighting emerging biomedical applications for researchers working on developing new supramolecular interactions (Fig. 1). We intend to do this by first creating a design space of supramolecular polymeric hydrogels. We will describe the chemical moieties of crosslinks and their respective kinetic and thermodynamic parameters. We will then describe the macroscopic mechanical properties these interactions afford and how they can be controlled. After introducing the design space, we will review how the chemistries and mechanical properties of these materials are leveraged in emerging biomedical applications.
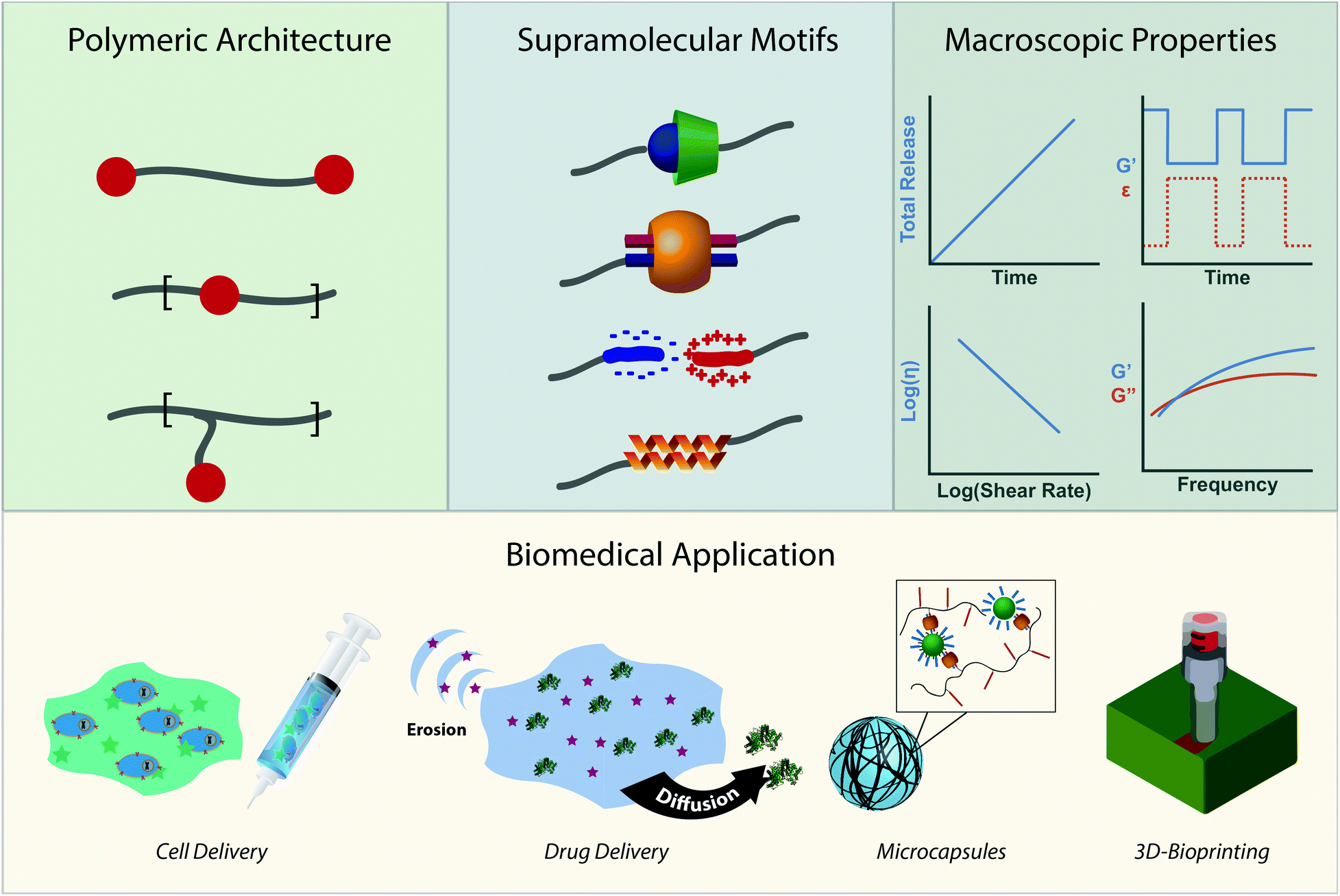 | ||
| Fig. 1 When designing novel supramolecular polymeric biomaterials, researchers must first choose a polymeric architecture. Illustrated in descending order are a telechelic design, network embedding, and grafting from a linear chain. In this section, the red dot represents the supramolecular motif, chosen for its kinetic and thermodynamic properties. Among the many interactions available, we have illustrated, in a descending list, cyclodextrin, CB[8] ternary, ionic, and protein based coiled-coil interactions. The judicious choice of the architecture and motif affords very specific and unique material properties, such as (left to right) extended release, self-healing, shear-thinning, and frequency-dependent mechanical properties. These considerations make up the toolbox for supramolecular polymeric biomaterial design. Control over the structural architecture and chemical motifs enable fine control over the macroscopic properties, allowing for systematic development of advanced biomaterials. Displayed above are but a few of the applications discussed in this review: cellular injection, cargo delivery on different timescales, microcapsules, and 3D-bioprinting. | ||
2 Chemistry of the crosslink
The past two decades have seen an explosion of research employing specific, directional, non-covalent moieties, including hydrogen bonding, metal–ligand coordination, host–guest complexation, and ionic interactions. The non-covalent interactions are realized through different binding mechanisms with corresponding associative equilibrium constants (Keq) and associative (ka) and dissociative (kd) rate constants. These thermodynamic and kinetic parameters offer insight into the binding mechanisms and provide a handle for the judicious choice of a supramolecular system for a biomaterial. Generally, Keq, a thermodynamic constant, will relate the degree of association in a system. The kinetic parameters, ka and kd, describe the binding dynamics of the system. With only these thermodynamic and kinetic parameters, one can begin to envision the degree of connectivity with an understanding of how transient that connectivity is. This connectivity, however, only comprises part of a physical hydrogel's complexity. Beyond the transient nature of the crosslinks, polymer molecular weight, crosslink density, chain rigidity, and polymer concentration must be considered when designing a supramolecular biomaterial. The upcoming section will provide a brief background and discussion on thermodynamic and kinetic parameters, followed by highlights of the currently employed supramolecular interactions and the design handle one has over their thermodynamic and kinetic properties.2.1 Visualizing thermodynamics and kinetics
When looking at the reaction and free energy diagram of the supramolecular interaction (Fig. 2), we can visualize both the thermodynamic and kinetic parameters. The equilibrium constant is directly related to the relative free energy levels of the unbound and bound supramolecular complex and is described through eqn (1). As the bound complex becomes more energetically favorable, the degree of association increases. Indeed, the degree of association is proportional to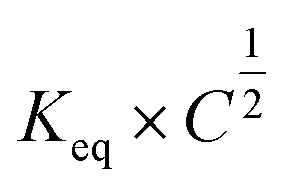 .32 Hydrogel formation occurs only when the degree of association has reached a critical ratio so that a contiguous network forms. Fig. 2 also illustrates that the supramolecular complex spends time as an active (bound) and inactive (unbound) crosslink. The rates of interchange between these states is a direct relation of the forward and reverse activation energy (Eaa, Ead). The Arrhenius relationship, shown in eqn (2), describes the temperature dependence of the reaction rate, where A represents the pre-exponential factor and R is the universal gas constant. The associative and dissociative rate constant, respectively, are directly related to the change in free energy between the transition state and the unbound and bound complex. It is not uncommon for the energetic barriers of supramolecular association to approach or fall below values of RT. In these cases, ka is limited by the rate of transport through the medium, and the process is described as diffusion-limited.
.32 Hydrogel formation occurs only when the degree of association has reached a critical ratio so that a contiguous network forms. Fig. 2 also illustrates that the supramolecular complex spends time as an active (bound) and inactive (unbound) crosslink. The rates of interchange between these states is a direct relation of the forward and reverse activation energy (Eaa, Ead). The Arrhenius relationship, shown in eqn (2), describes the temperature dependence of the reaction rate, where A represents the pre-exponential factor and R is the universal gas constant. The associative and dissociative rate constant, respectively, are directly related to the change in free energy between the transition state and the unbound and bound complex. It is not uncommon for the energetic barriers of supramolecular association to approach or fall below values of RT. In these cases, ka is limited by the rate of transport through the medium, and the process is described as diffusion-limited.ΔG° = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Keq) ln(Keq) | (1) |
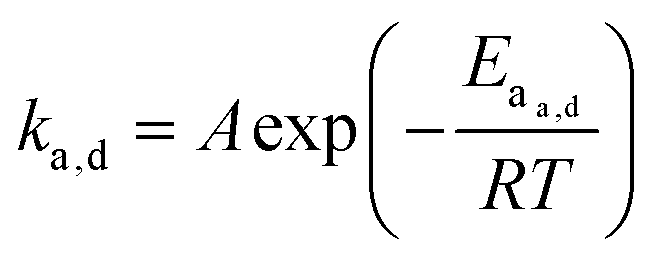 | (2) |
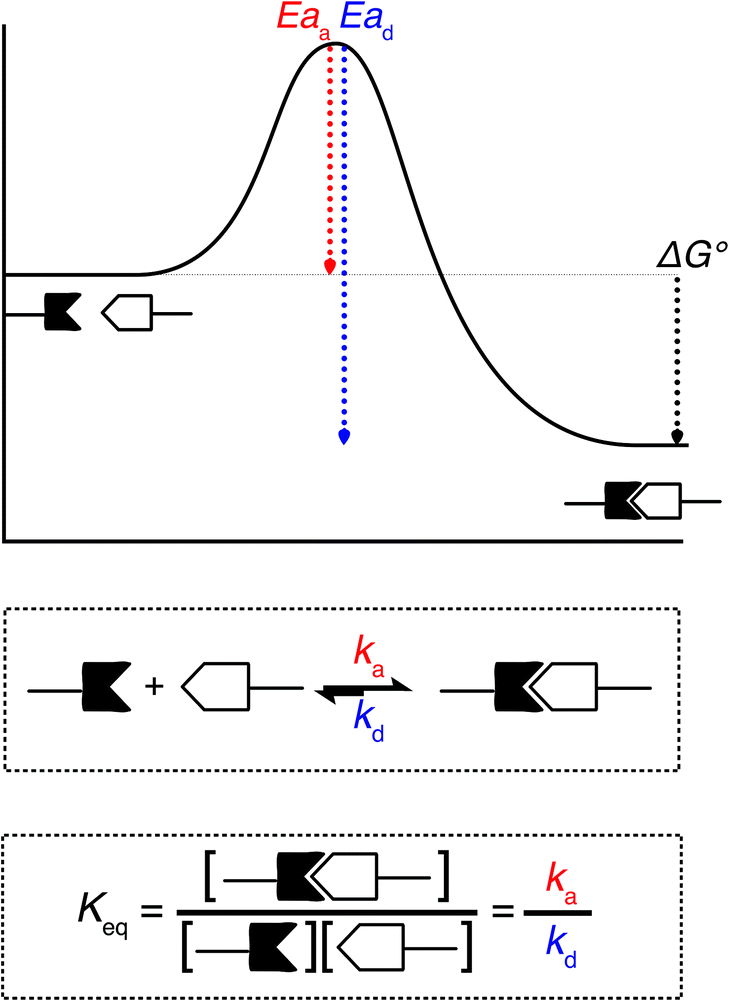 | ||
| Fig. 2 A visualization of the thermodynamics and kinetics of supramolecular association. (a) Reaction coordinate diagram of a host–guest interaction illustrating the activation energy and free energy of association; factors that are correlated to the values ka, kd, and Keq. (b) Equation illustrating the kinetic parameters of association. (c) Equation illustrating the thermodynamic parameters of association. | ||
These parameters are implicitly tied to the performance and functionality of the biomaterial. An in-depth understanding of their behavior allows for the judicious selection of a supramolecular moiety when trying to design a new system. However it is important to understand that choice of polymeric architecture can drastically effect the process of association between chains. A separate thermodynamic treatment employing statistical mechanics can be used to describe a global energetic minimum for interchain connectivity. When Ead is large (such as with certain metal–ligand interactions), associating chains may initially crosslink into kinetically trapped states that do not have enough thermal energy to reach a global energetic minimum. If Ead is neither excessively large or small, associating polymeric chains can dissociate and will diffuse through the network, a phenomenon observed in model protein hydrogels.37 This enables the transition from an initial kinetically impeded state of connectivity towards a global energetic minimum and would physically manifest in morphological and rheological changes over time. In the proceeding section we will discuss and review different examples of supramolecular interactions and available polymeric architectures, and begin to relate them back to their fundamental kinetic and thermodynamic parameters.
2.2 The chemical toolbox of the supramolecular crosslink
Supramolecular polymeric hydrogels have been prepared using a wide variety of supramolecular motifs, including host–guest complexation, H-bonding, metal–ligand coordination, electrostatic, and protein based interactions. The interactions most suitable for use in aqueous media are described below. Briefly, more attention in the literature has been given to the thermodynamics and kinetics of macrocyclic host–guest interactions. As such, the chemistry of host–guest interactions will be discussed within the lens of their thermodynamic and kinetic parameters. The following sections on hydrogen bonding, metal–ligand coordination, electrostatic and protein based interactions will describe trends in polymeric architecture and molecular interaction and how modulation of both parameters affects the process of association.2.2.1.1 Cyclodextrin. Cyclodextrins are cyclic oligosaccharides in which D-glucose units are coupled through α-1,4-glycosidic linkages.38 α-, β-, and γ-CDs, composed respectively of 6, 7, and 8 D-glucose repeat units, are most commonly encountered and generally act as hosts to hydrophobic guests.39 As different CDs contains different sized cavities, they bind different types of guests. Most commonly, α-CD will bind linear alkyl chains while β-CD binds “spherical” diamondoid guests like adamantine and its derivatives.39 Their 3D structure can be represented as a truncated cone with a solubilizing hydrophilic surface composed of hydroxyl groups and a more hydrophobic interior. While hydrophobic and van der Waals interactions between the host and guest in the inner cavity provide the driving force for host–guest interactions, hydroxyl groups on the surface promote self-association mediated by hydrogen bonding.
From a high-level description, these respective interactions can be leveraged to form hydrogels in two ways. The first method takes advantage of the host–guest interaction as a dynamic crosslink; CD and respective guests are grafted onto a polymer backbone (Fig. 3a). In this usage, hydrogels will form in systems where a polymer is grafted with both guest and CD,40–42 polymers grafted with either CD or guest are allowed to mix,43–47 or linear polymers grafted with a guest are mixed with a CD dimer.48,49 The host–guest dynamic crosslink can also be employed in hybrid hydrogels, where a linear polymer functionalized with either CD or a guest is mixed with a nanoparticle50 or vesicle51 that has been functionalized with its compliment (Fig. 3b). In a recent study employing a mixture of polymers grafted with either CD or guest, Burdick demonstrates microstructural evolution of a homogenous hydrogel into porous structures.47 This is indicative of an initial formation of a kinetically impeded state (homogenous mixture) which subsequent rearranges to a more energetically favorable conformation (porous structure). Implementation of these different strategies, coupled with control over polymer size distribution and topology, may affect the tendency to form kinetically impeded states, however this remains a largely unexplored area of research.
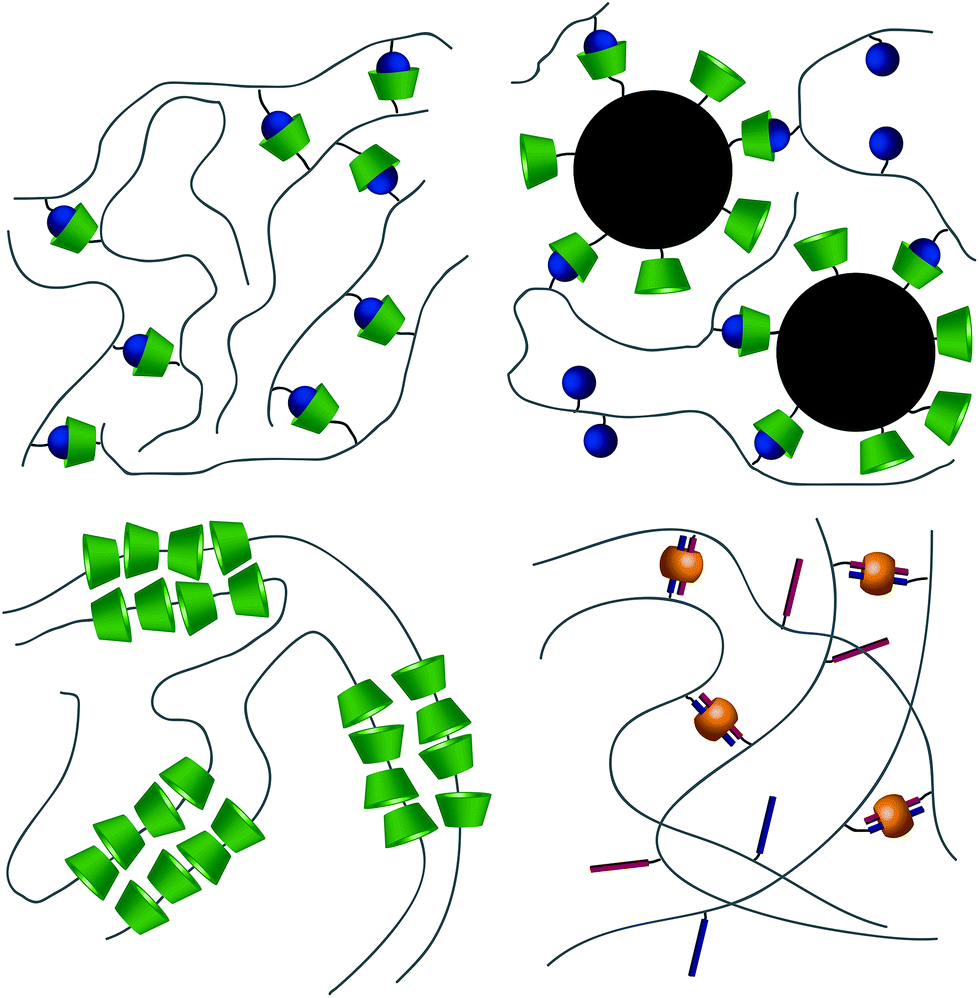 | ||
| Fig. 3 Schematic illustration of the different ways that hydrogels can be formed through host–guest interactions. (a) A typical host–guest interaction, with hosts grafted to one polymer and guests grafted to another. While CD is depicted here it could just as easily be CB[n] (n = 6, 7). (b) A host grafted onto a nanoparticle, with guests grafted onto polymers. (c) CDs that thread polymeric backbones form poly(pseudo)rotoxanes and that have dynamic crosslinks between their surfaces (d) The CB[8] specific ternary interaction where CB[8] can act as a host for two similar or different guests. | ||
The second method takes advantage of CD's ability to thread linear polymers, forming poly(pseudo)rotoxanes.52,53 When implemented into poly(pseudo)rotoxanes, CDs have been demonstrated to thread nonfunctional PEG,54,55 poly(propylene-glycol),56 aliphatic polyesters,57,58 poly(ε-lysine),59 and poly(vinyl alcohol)s60 in aqueous solvents (Fig. 3c). While this threading alone does not lead to the formation of hydrogels, the threaded CDs self-associate through hydrogen bonding interactions, forming the physical crosslinks between the linear chains.61
2.2.1.2 Cucurbit[n]uril. Cucubit[n]urils (CB[n], n = 5–8, 19) are macrocyclic oligomers containing repeat units of glycoluril connected through methylene bridges. CB[n]'s visual structure can be represented by barrels of increasing radius.62 CB[n]'s chemical structure is understood as a hydrophobic cavity surrounded by polar carbonyl functional units at either portal. Much like CDs, CB[n]s bind guest through hydrophobic and van der Waals interactions in the hydrophobic cavity. However, unlike CDs, CB[n]s interactions are aided by the release of “high-energy” water molecules trapped in the hydrophobic cavity.63,64 Further, CB[n]s have a tendency to bind metal or organic cations through ion–dipole interactions on the carbonyl portal.
Increasing repeat number (n) increases the CB[n]'s portal and cavity size, altering what type of molecule the CB[n] host tends to bind with. CB[5], the smallest CB[n], can encapsulate a variety of gas molecules as well as small solvents, but will not be discussed further as this plays little relevance in the creation of supramolecular biomaterials.65 Of greater relevance, CB[6] tends to bind neutral and positively charged organic guests, CB[7] binds larger amphiphilic guests, and CB[8] binds positively charged and large organic guests.66 Moreover, CB[8] is unique in the fact that it can bind two guests simultaneously, forming ternary complexes.67,68 When guests differ, CB[8] will bind the electron deficient guest before the electron rich guest.
CB[n]s have been utilized to form physically crosslinked hydrogels in two ways. Similar to CDs, CB[n]s are appended to a polymer while guest molecules are appended to a complimentary polymer. Subsequent mixing yields robust supramolecular crosslinks and hydrogel formation (Fig. 3a).69–71 Common guests for CB[6] hosts are diamines, such as 1,6-diaminohexane (DAH). In the second system, CB[8] is titrated into a solution of guest molecules grafted to polymers, creating ternary inclusion complex crosslinks with either the same72–74 or different guests (Fig. 3d).75–79 Common guests include amino acids, viologen (MV, first guest), and 2-napthoxy (Np, second guest).
2.2.1.3 Kinetics and thermodynamics. Due to the shared host–guest nature of CDs and CBs, discussing their kinetics and thermodynamics in tandem affords insights into their similarities and differences and helps motivate a choice of motif for future applications. One thing that becomes immediately apparent is that CB-guest binding is much stronger than CD-guest binding, often with Keqs that are many orders of magnitude larger. The thermodynamic equilibrium constant (Keq) for CD range between 101–5 M−1 with the popular β-CD@Adamantane motif at 104 M−1 and a α-CD@C12 at 103 M−1.39 Equilibrium rate constants between CB[n]s of different sizes vary more than constants of different sized CDs. CB[6] binds guests with equilibrium constants as high as 1012 M−1,80 but is centered around 105 for a variety of alkyl ammonium salts.66 CB[7] includes the strongest association interactions and contain equilibrium constants as high as 1017 M−1,81 but are typically between 105–9 M−1.66 CB[8] presents a slightly more convoluted case because there are parameters needed for the first guest, heterodimer, and homodimer inclusion interactions. In the interest of materials properties, we will look at overall equilibrium constants. In the kinetic section, we will explore dissociation constants of the second guest. Typically, both first guest and second guest Keq values fall between 104–6 M−1 resulting in overall bond equilibrium constants of 1010–13 M−2.66
While many thermodynamic parameters have been reported for CB and CD based host–guest systems, quantitative determination of association and dissociation rate constants have proved challenging due to short relaxation times (<1 s) of these supramolecular complexes that demand high time resolution.82 Common to both CD and CB (second guest), if the fit is right, ka approaches the diffusion limit (within 1–2 orders of magnitude).77,83–88 However, the ka will lower as the host or guest is attached to a slower diffusing (larger) backbone, when there is a rate limiting shape reorganization between the host, or as recently demonstrated, when the pressure is lowered or the viscosity is increased.88 While viscosity and pressure affects CB[8]'s ka they leave kd seemingly unchanged (resulting in Keq alteration). In the same study, they demonstrate that when ka was near the diffusion limited state, only kd (and thus Keq) was affected by different guest molecules, agreeing with previous discussions of CD.87 Together, the data argues that the host affects association, while the guest controls dissociation. It is imperative to remember, however, that association in hydrogel networks happens in a range of viscosities and on large backbones, resulting in equilibrium and rate constants that may vary from those of stop-flow experiments. While CB and CD based systems both share control over their association and dissociation rate constants, when the ka is rate limiting, the kd is inherently tied to Keq. Thus CB moieties that have large Keqs will have larger dissociation constants than CD moieties. For CB[8], however, the relevant kd value is tied to the Keq of the second guest, which is similar or marginally larger than the Keq of CDs.
A singular hydrogen bond pair renders a relatively weak interaction. However, due to their specificity and directionality, one can greatly increase the association constant through multivalent interactions. Seminal work by Meijer demonstrated that both the ordering of donor (D) and acceptor (A) units (e.g. DAAD/ADDA vs. ADAD/DADA) and the number of adjacent hydrogen bonding actors affected the relative association constants between complimentary interacting motifs.89 Generally, the relative strength can be thought of as an interplay between attractive and repulsive secondary interactions, leaving DAAD/ADDA pairs with higher Keq values than ADAD/DADA pairs.
The equilibrium constant of the hydrogen bonding interaction is strongly solvent dependent.89,90 Polar solvent molecules, such as water, compete for binding sites and impede the ability of complementary units to associate. To incorporate hydrogen bonding motifs into biological materials the hydrogen bonds must then be shielded by a hydrophobic spacer. The most common structural design involves poly ethylene glycol (PEG) surrounded by a hydrophobic spacer with telechelic hydrogen bonding units. This design has been used with the ureido-pyrimidinone (UPy),91,92 and benzene tricarboxamide (BTA)35 hydrogen bonding motif. In this motif, the supramolecular polymers assemble into long 1D fibers of the hydrogen bonding units (Fig. 4a). The hydrophillic chains can either attach two hydrogen bonding units on the same fiber, or on different fibers, actings as a crosslink between the fibrous units. A different strategy embeds hydrogen bonding units surrounded with hydrophobic spacers directly into the polymeric backbone (Fig. 4b). Instead of creating interconnected 1D fibers, dimeric hydrogen bonding units act as dynamic crosslinks. This strategy has been employed with UPy93 and urea94,95 hydrogen bonding units. Instead of embedding the hydrogen bonding unit into the polymeric backbone, Upy groups have also been grafted onto polymeric backbones with a C6 hydrophobic spacer.96–98
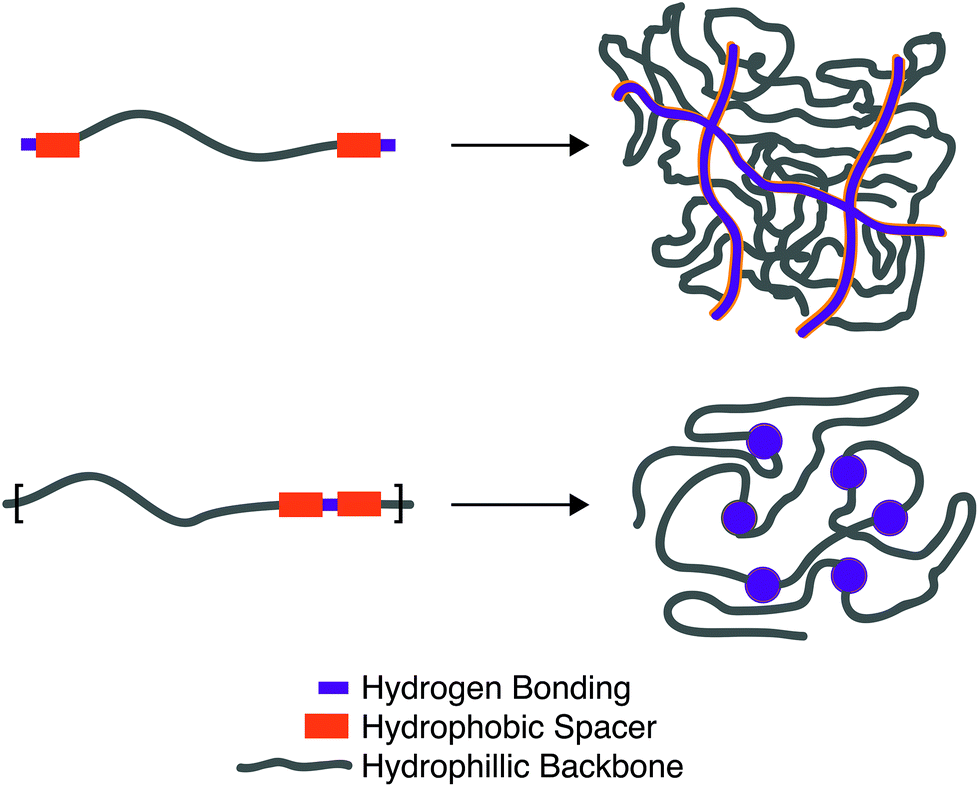 | ||
| Fig. 4 Schematic illustration of the two most common forms of hydrogels formed through hydrogen bonding interactions. (a) A telechelic water soluble unit forms 1D fibers that bundle and strengthen the hydrogel. (b) Hydrogen bonding units shielded by hydrophobic spacers on a water soluble backbone dimerize to form dynamic crosslinks. | ||
Looking towards the polymeric design space there are three main components: (1) choice of the hydrogen bonding units, (2) choice and length of the hydrogen bonding spacer, (3) length of the hydrophillic chain. Choice of the hydrogen bonding unit will ultimately afford a different Keq, ka, and kd, affecting the mechanical properties of the material. The length of the hydrogen spacer will dictate both the degree of crystallinity of the hydrogel and the degree of shielding from protic solvents. The length of the hydrophillic spacer will both describe the flexibility of chains and dictate the ratio of a polymer self-associating (loop) or associating with a different polymeric backbone (crosslink).
Perhaps the most anomalous biological example of the metal–ligand interaction is found on intertidal coastal surfaces, where mussels attach themselves through byssal threads terminated in adhesive plaque.99 While the feat of underwater adhesion is outstanding by itself, the unique self-healing properties of the byssal threads have directed a field of mussel-inspired metal–ligand hydrogels. Synthetically, this design motif is incorporated into polymeric hydrogels by crosslinking telechelic catechol or histidine units on multi-arm PEG with a variety of transition metal cations, notably Fe3+, In3+, Al3+, V3+ (catechol) Zn2+, Cu2+, Co2+, and Ni2+ (histidine). Transition metals have different characteristic relaxation times with their respective ligand; judicious mixtures of ligands and metals afford predictable control over timescales of energy dissipation in bulk materials.100,101
Fe3+ catechol interactions have stability and strength approaching covalent bonds; measured equilibrium constants approach 1040. The mechanical properties of these hydrogel are strongly dependent on the coordination ratio of catechol:Fe3+, a function of hydrogel pH. Optimal mechanical properties are realized at pH 12 (coordination ratio of 3:1, Fig. 5a), however hydrogel formation is still realized at a pH of 8 (coordination ratio 2:1, Fig. 5b).102 Implementing different transition metals103 or catechol motifs104 afford different coordination ratios at a given pH, leading to more robust and translatable synthetic protocols. Histidine-transition metal hydrogels are often facilitated at neutral pH (coordination ratio 2:1). It has been noted that increasing the coordination ratio by lowering the relative concentration of transition metal imparts more rigidity on the network.105
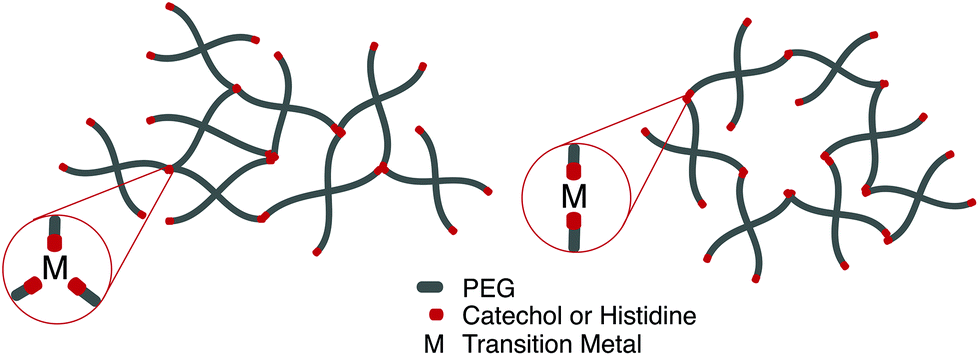 | ||
| Fig. 5 Depending on the choice of chelating unit (catechol, histidine), transition metal, and pH, hydrogels with coordination ratio (a) 3:1 and (b) 2:1 will form. | ||
While strong, mussel-inspired hydrogels are often brittle with little elastic recovery force, an attempt to address this issue involves altering the transition metal bound to a catechol (vanadium and aluminum).103 This tends to alter the coordination number at a given pH, affording control over the degree of elastic and viscous character but not necessarily overcoming the flaws of a multi-arm PEG system. Alternatively, Hawker demonstrated that interpenetrating networks could be formed between Fe-catechol crosslinked PEG and loosely crosslinked acrylamide.106 This dynamic crosslink provides stiffness and energy dissipation at strain rates above the characteristic relaxation times and minimal reinforcement at slow rates. Redesigning the molecular architecture of the catechol crosslink, Ma and coworkers chelated iron with methacrylamide functionalized catechols and polymerized acrylamide, rendering an acrylamide network with Fe-catechol crosslinks.107 These hydrogels are scalable and maintain demonstrable extensibility, however the synthetic process incorporates a non-insignificant amount of DMSO in the hydrogel.
Alternate examples of metal–ligand supramolecular crosslinks include the appendage of ligands to linear polymer backbones with titration of a transition metal crosslink. Polyacrylic acid homo- and copolymers have been used with Fe3+ to fabricate hydrogels with high toughness and photo-redox responsive sol–gel transitions.108,109
In general, tethered 2D nanosheets form strong, high water content (97%) and elastic hydrogels.110,111 Clay nanosheets are implemented and functionalized with a surfactant, impeding aggregation and increasing anionic surface area. These nanosheets are then mixed with PEG containing dendritic,110 or linear111 telechelic guanadinium (cationic) groups. The telechelic PEG functions as a supramolecular tether; mechanical properties of the gel are affected by its macromolecular architecture. As the ionic interactions are multivalent, gels become stronger with increasing length of the charge section (or increasing dendrimer generation). Moreover, increasing the length of the PEG spacer (to an extent) increases the probability that the PEG tethers two nanosheets (increasing the modulus) rather than forming a closed loop with itself (lowering the modulus). ABA triblock copolymers represent a strong class of medium water content (85–90%) hydrogels with mechanical responsiveness to salt content, stoichiometry of charge, pKa of ionic moiety and pH (depending on the ionic A block used). In this polymer architecture, A represents ionic charge (anionic or cationic) while B represents a neutral PEG midblock. This class of material is further separated into two groups: ABA triblock copolymers that are mixed with an oppositely charged homopolymer (C)112 and ABA triblock copolymers that are mixed with an oppositely charged triblock copolymer (CBC).113,114 While the ABA-C system form a network of interconnected polyelectrolyte micelles stabilized by a corona of neutral PEG, the ABA–CBC tend to form ordered body center cubic arrays of complex coacervates (polymer-rich regions formed via liquid–liquid phase separation) tethered by PEG chains (Fig. 6). ABA–CBC systems tend to be stronger and more ordered than ABA-C systems because they have an increased number of PEG bridges between domains. Both structures tend towards amorphous behavior and display weaker mechanical properties as either polymer concentration decreases or salt concentration increases.
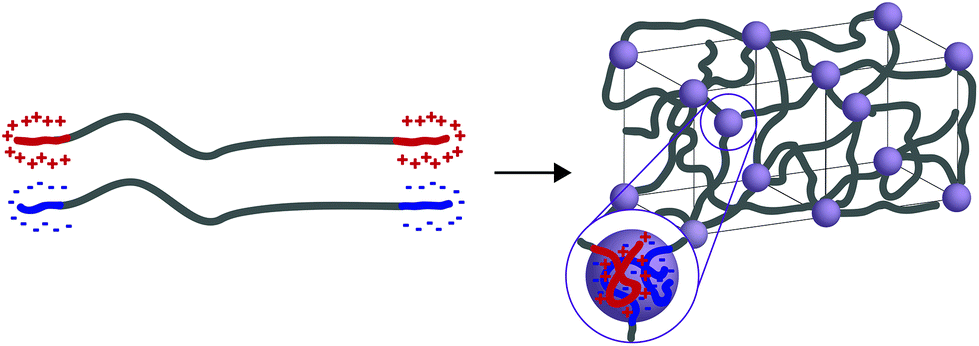 | ||
| Fig. 6 When ABA and CBC triblock copolymers (A and C have opposite charges) are mixed they form ordered body center cubic (BCC) arrays of complex coacervates tethered by PEG chains. | ||
The polyampholyte motif consists of polymers containing both cationic and ionic monomers and represents a class of very tough, modular, lower water content (50–70%) hydrogels intended for use as structural biomaterials.115 The strength of the electrostatic interaction is proportional to the multivalency of the interaction. By employing a random polymerization, segments of the polymer backbone will contain alternating cationic and anionic domains and repeating cationic (and anionic) domains. The alternating domains will have interactions with lower multivalency, thus acting as weak crosslinks, while the repeating domains will have high multivalency, acting as strong crosslinks. This is akin to utilizing a weak and strong transition metals in metal–ligand interactions; the mixture of strong and weak interactions allows for different and distinct timescales for energy dissipation, imparting strength, self-healing, and toughness into the materials. This hydrogel becomes weaker by employing either a lower concentration, or utilizing more hydrophilic monomers. While not explored in this setting, one could imagine that employing monomers with strategically chosen reactivity ratios could affect the timescales of viscoelasticity in the material.
Strategies to use proteins and peptides either involve a pure multiblock recombinant protein, or the grafting of proteins and peptides to synthetic backbones. Common strategies include appending a histidine tag (his6) to the peptide with a synthetic polymer backbone that chelates Ni2+, thiol–ene chemistry with a cysteine on the protein and a double bond on the backbone, and substitution reactions with lysine residues on the protein and activated carboxylic acids on the polymer backbone.116
The leucine zipper, an example of a recombinant protein, is a helical peptide with a heptad periodicity represented as (abcdefg)n.117 The a and d residues are generally hydrophobic and frequently leucine, while e and g are charged. The a and d residues land on the same helical face and dimerize with other leucine zippers through hydrophobic interactions. Through recombinant engineering, the leucine zipper motif is incorporated into supramolecular hydrogels through the synthesis of an ABA triblock polypeptide, in which A groups represent the leucine zipper and B groups are a flexible water soluble peptide domain.118 The self-assembly of these triblock polypeptides are illustrated in Fig. 7. Flexibility in n,119 the number of heptad repeats, and subtle changes in heptad amino acids120 can change the equilibrium number of associating helixes (i.e. dimer vs. trimer vs. tetramer) and thermal stability of those associations. Gel stability and strength can be increased by reducing the number of primary loops (vide infra), a larger problem with ABA coiled-coil hydrogels. This can be overcome by increasing the B block length121 or by imagining a novel ABC triblock copolypeptide, where A and C blocks can only, respectively, form coiled-coil interactions with A and C blocks.122 A simpler usage of the leucine zipper coil functionalizes a water-soluble polymer with the helical polypeptide, a technique that promotes gelation with >99% water content.123 While these hydrogels tend to be weaker, they demonstrate incredible environmental responsiveness that can be modulated by clever choices of amino acid residues; their mechanical properties are generally affected by pH, temperature, and salt concentration.
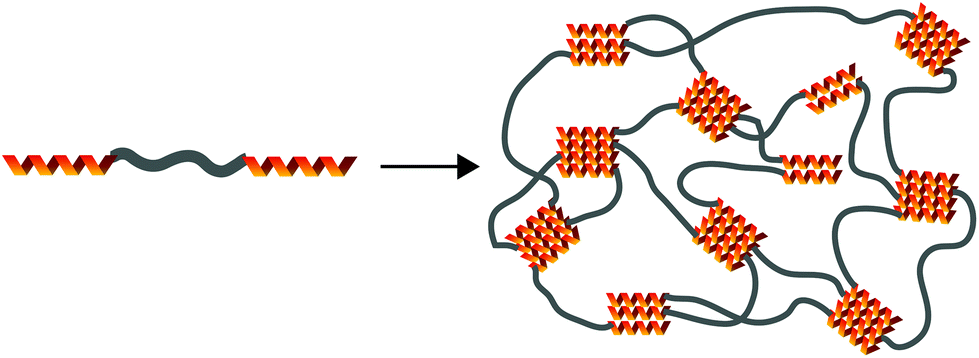 | ||
| Fig. 7 Demonstration of leucine zipper hydrogels forming from an ABA triblock copolypeptide. Here, the equilibrium number of associating helixes is 3, although this can be changed by altering the amino acid of the heptad or the number of heptads in the A block. | ||
The coiled-coil motif has also been incorporated into supramolecular hydrogels through use in multiblock (AB)n polypeptides, where A is the coiled-coil block and B is a flexible polyelectrolyte block.37,124 Like the telechelic leucine zipper, the synthetic design space includes the number of heptad repeat units in A, heptad amino acid choice, and length of the flexible polyelectrolyte block. Introduction of cysteine residues on the N and C terminal of the multiblock polypeptide affords oxidative chain extension and introduces linear entanglements into the hydrogel, affording viscoelastic responses on different timescales.124 Appending a thermoresponsive polymer, such as N-isopropylacrylamide (NIPAM), to the N and C terminal of the multiblock polypeptide introduces a secondary network reinforcement at temperatures above the lower critical solution temperature (LCST).125–127 This technique affords network strengthening after injection into physiologically relevant environments and has also been employed with associating WW and proline rich hydrogels developed by Heilshorn.128–130
The use of proteins as supramolecular crosslinks also allows for environmental response to other proteins or ligands. In a landmark study, Uragami grafted an antigen and a complimentary antibody to separate acrylamide backbones which, upon mixing, association into hydrogels.131 Grafting the antigen onto the acrylamide backbone results in slight denaturation and a lower Keq (108 M−1) and faster kd (104 s−1) than the free antigen with the antibody.132 Because free antigen binds more competitively than grafted antigen, the hydrogel reversibly swells in the presence of free antigen, releasing molecular cargo. Weber was able to form antibiotic responsive hydrogels by grafting recombinantly engineered bacterial gyrase subunit B (GyrB) to linear polyacrylamide with a histidine tag.133 Coumermycin, an antibiotic ligand, serves as a crosslink between two GyrB units. Addition of a competitively binding antibiotic that only associates with a single GyrB results in hydrogel degradation.
In addition to environmental responsiveness, appropriate use of protein–polysaccharide interactions can better mimic the interactions of proteins and glycosaminoglycans in the extracellular matrix (ECM).134 In this regard, the Kiick group has functionalized 4-arm PEG polymers with telechelic low molecular weight heparin (LMWH) or heparin interacting proteins (HIP). Weak hydrogels (97.5% water) form upon mixture of the PEG-LMWH and PEG-HIP, which have equilibrium association constants on the order of 106 M−1 and dissociation rate constants on the order of 103 s−1.135,136 Mechanical properties can be controlled through changing concentration of the molar ratio of LMWH:HIP. Weak hydrogels can also be formed through the interactions of 4-arm PEG-LMWH and heparin-binding growth factors (VEGF).137 This strategy could be used to employ hydrogels with targeted degradation under the presence of VEGF receptors.
While peptide and protein based hydrogels afford incredible environmental responsiveness, peptide interactions can also be used to demonstrate strain resilient, shear-thinning hydrogels with tunable mechanical moduli. Burdick cleverly utilized the dimerization domain of a protein kinase with the anchoring domain of the kinase anchoring protein to create a “dock-and-lock” hydrogel that formed with simple mixing. The dimerization domain formed the A block of an ABA triblock polypeptide that would self-associate (“dock”). A multi-arm PEG conjugated with the anchoring domain on the ends of the arms subsequently reinforced the dimerization (“lock”).138 Through altering the molar ratio of docking and locking groups, the number of PEG arms, and the concentration of polymer, the mechanical properties and degradation rates of the hydrogel could be controlled while demonstrating resistance to yielding at strain rates of 400%.
3 Macroscopic mechanical properties
Self-assembly using the preceding non-covalent chemistries is the primary mechanism for crosslinking of hydrogels and has been leveraged to impart dynamic and transient mechanical properties to biomaterials. These properties include the ability to flow under an applied shear strain (shear-thinning) and to reform the mechanical properties of the network once that strain is released (self-healing). Furthermore, they can be finely engineered for a specific application by carefully choosing appropriate supramolecular interacting chemistries with the appropriate interaction thermodynamics (Keq) and kinetics (ka, kd). Naturally, these interactions will also govern the overall strength, stiffness, and elasticity of the material as well as the final microstructure. While each of these considerations provides an additional variable to tune, the increased complexity also allows for many creative materials strategies with precisely designed macroscopic properties. For example, due to the dynamism of supramolecular interactions, hydrogels made with supramolecular crosslinks typically will not swell when submerged in water—the stresses are dissipated through rearrangement of the non-covalent network. In the following sections, we attempt to highlight how these chemistries have been used to control gel strength, shear-thinning, self-healing, and solute release kinetics.3.1 Viscoelastic materials
Supramolecular biomaterials are often designed to mimic the mechanical properties of biological materials. From a structural standpoint, biological materials are commonly formed from self-assembling, non-covalent interactions between small molecules or polymers. These biomaterials exhibit viscous and elastic behaviors and are therefore classified as viscoelastic materials. Due to these characteristics, engineered biomaterials seeking to mimic biological materials must match the elastic behavior as well as the viscous behavior of the material to impart time-dependent mechanical properties.142While there are many useful in-depth discussions on viscoelasticity,142–145 the development of models that correlate bulk rheological properties to supramolecular structure remains a complex challenge and active area of research. The following discussion presents the basic rheological grammar commonly used in characterizing and designing supramolecular biomaterials.
Oscillatory rheometry is a powerful way to probe the molecular origins of a material's viscoelasticity and also the primary way of quantifying the elastic and viscous properties. In oscillatory rheometry, a sinusoidal shear strain is applied to the material, while the corresponding stresses are monitored. An ideal elastic material will have a stress response exactly in-phase with the applied strain (δ = 0), while an ideal viscous material will be exactly out of phase 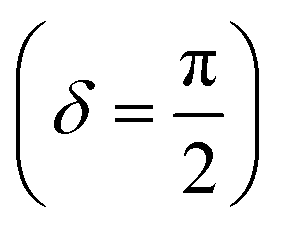 . For viscoelastic materials, the response lays somewhere in between
. For viscoelastic materials, the response lays somewhere in between  and researchers typically report the storage modulus (G′), the loss modulus (G′′), and the ratio between G′′ and G′ (tan(δ)).144
and researchers typically report the storage modulus (G′), the loss modulus (G′′), and the ratio between G′′ and G′ (tan(δ)).144
While mathematically G′ and G′′ represent moduli multiplied by the in-phase or out-of-phase factor of the stress–strain response, they can be loosely thought of as the amount of stored energy (G′) and the amount of dissipated energy (G′′) by the material. Tan(δ) represents the ratio between G′′ and G′ and is often treated as the relative elasticity of the material. As a result, G′ and tan(δ) are commonly reported together in order to summarize the material elasticity, while providing information on how fluid-like the material is. These values are obtained through common rheological measurements like frequency sweeps (Fig. 8b and d), or strain sweeps. Frequency sweeps are one of the most common measurements reported for characterizing viscoelastic materials and Fig. 8d illustrates an example plot labelled with a crossover point and a plateau modulus. Notably, the viscoelastic moduli exhibit frequency dependent values that are related to the transient crosslinking interactions. In particular, the crossover point indicates the timescale of viscoelastic network relaxation while the plateau modulus represents a pseudo-equilibrium of the crosslinked network. Both of these parameters are directly influenced by the network's degree of crosslinking and the Keq of the crosslink interactions. As the degree of crosslinking is increased, the crossover point will shift towards lower frequencies, until there is no visible crossover point in the frequency range tested. A widely used method of qualifying a material as a gel is to demonstrate no crossover point in a standard frequency range (0.1–100 rad s−1). When the Keq of the crosslinks approach covalent-like values, the plateau modulus will increase and extend towards lower frequencies, approaching a frequency sweep profile of an elastic material (Fig. 8b). A rich understanding of how microstructures are represented by these oscillatory measurements is invaluable when characterizing and engineering new viscoelastic materials.
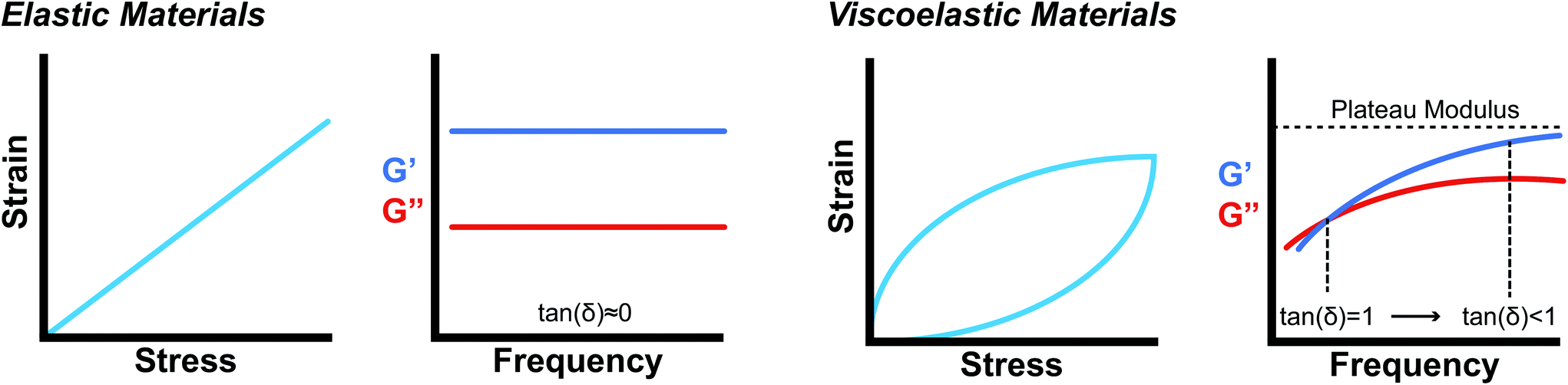 | ||
| Fig. 8 (a., c) Comparison of ideally elastic materials with viscoelastic materials. While elastic materials have a linear strain response to stress, viscoelastic materials exhibit a hysteresis loop, where energy is being lost during the cycle. (b) and (d) represent typical log–log plots of frequency sweeps for elastic materials and viscoelastic materials over a standard experimental range (0.1–100 rad s−1), respectively. Viscoelastic materials exhibit a frequency dependence on the modulus and a crossover point where tan(δ) = 1 that represents the timescale for network relaxation. | ||
While oscillatory rheometry is capable of examining a wide range of moduli and time scales typically desired for biomaterials, the development of microrheology has enabled local probing measurements on heterogeneous samples, smaller sample volumes over significantly larger frequency ranges, and the possibility of high throughput screening.146 These methods include atomic force microscopy,147 optical interferometry,143 video-particle tracking,148–151 and magnetic twisting cytometry.152 However, current microrheological techniques also suffer from several challenges, including the limitation that the materials must be partially transparent and a significant amount of computational analysis must be done to extract data. Furthermore, while microrheology excels at analyzing low viscosity samples, it has trouble with very stiff materials that limit movement of probe particles.
Using these characterization techniques, many different strategies have been employed to develop supramolecular materials with robust elastic properties. Poly(pseudo)rotoxane gels formed with α-CD and PEG chains achieved a G′ of 500 Pa, tan(δ) = 0.02 with linear viscoelastic responses to strains upwards of 300%.153 These gels utilized host–guest interactions between the α-CD and bis(2,4-dinitrophenyl)-capped PEG chains to form topological restrictions that acted as the crosslinkers. Additionally, double network hydrogels composed of interpenetrating polymer networks achieve high moduli through the combined elastic contributions from both networks. Commonly, the two networks exhibit drastically different mechanical properties, where one network might act as a sacrificial dynamic network to dissipate stress and increase stretchability of the material. Ping Gong and coworkers used bacterial cellulose (BC) and gelatin to form a double network gel with a tensile modulus of 2.9 MPa. The BC provided a hydrogen bonding network between cellulose fibers, while the gelatin provided a strong covalently bonded network. The supramolecular BC network itself showed weak mechanical properties, while the gelatin on its own was brittle. The formation of a double network allowed the final material to be strong, while maintaining stretchability.154
Supramolecular motifs also allow for independent modulation of network strength and network stretchability. Alginate hydrogels can be crosslinked by covalent and ionic interactions. Mooney demonstrated that the incorporation of ionic crosslinking allows for independent tuning of hydrogel modulus and toughness (energy stored before fracture). While increasing covalent crosslink density increased the modulus, it led to brittle, non-stretchable gels. Yet, by increasing ionic crosslink density, the resultant gels had higher moduli and increased stretchability.155
Whereas consideration of interaction thermodynamics provides intuition on how strong each crosslink is in the network, the examples above utilized the dynamic property of the bonds to impart additional mechanical properties. This concept will be explored further in discussions about shear-thinning and self-healing. Moreover, beyond the chemistry of the non-covalent interactions, the examples illustrate how creatively combining the interactions can significantly influence the final modulus and relative elasticity.
3.2 Self-healing and shear-thinning
Injectable therapeutics and drug delivery strategies rely on biomaterials that can be easily pushed through a syringe or catheter. To allow for injection, these materials are designed to decrease in viscosity under an applied shear force (shear-thinning). For drug delivery materials, they must also rapidly reform their initial structure with minimal changes to mechanical properties when that stress is released (self-healing) in order to reduce burst release of cargo. Materials formed with supramolecular interactions offer both shear-thinning and self-healing properties by nature of the dynamic non-covalent bonds that act as the primary driving force for self-assembly. As a result, many developing biomaterials utilize supramolecular interactions to maintain injectability.In the majority of cases, shear-thinning behavior has been fundamentally characterized as either a shear-induced splitting of non-covalent interactions, or a localized region of high strain rate affecting flow properties.156 The degree to which a material shear-thins and self-heals is a direct consequence of the polymer architecture and bond association and dissociation kinetics. Consequently, a comprehensive understanding of the supramolecular chemistry is essential towards imbuing materials with these properties. It should be noted that covalently crosslinked materials can also exhibit shear-thinning properties through dynamic covalent bonds that are thermodynamically controlled rather than the traditional kinetically controlled bonds.157 Yet, in covalently crosslinked materials, as well as most non-covalently crosslinked materials, the viscosity differential upon shearing is limited to 1–3 orders of magnitude. In both covalent and non-covalently crosslinked materials, controlling these properties requires understanding the chemistry and kinetics of the intramolecular bonds, as well as the mechanism of bond dissociation and association. The following discussions will focus on supramolecular materials.
While there is no single unifying parameter than can be used to compare between shear-thinning abilities across different mechanisms, chemistries, and materials platforms, the relaxation time of the network is often used as a point of reference. This relaxation time encompasses the dynamics of network rearrangement and can provide insight into the material's mechanical behavior regardless of the specific chemistry used. Craig and coworkers expanded on this idea and identified an untangled, a semi-dilute, and an entangled regime that all have competing relaxation mechanisms and lifetimes that determine the final shear-thinning abilities.158,159 Furthermore, it has been demonstrated that materials can shear-thin or shear-thicken depending on the kinetics of bond dissociation. Faster bond dissociation was attributed to the supramolecular bonds rapidly associating and dissociating, which led to network disentanglement. Slower bond dissociation led to intrachain crosslinks being converted into interchain crosslinks upon shearing, resulting in an increase in entanglement.77
Despite the lack of a unifying parameter, the rheological experiments used to measure self-healing and shear-thinning are well defined. Typically, demonstration of self-healing is done using either a step-strain experiment, where high and low strains are alternately applied and the moduli are measured in oscillatory mode, or a step-shear experiment, where a high and low shear rate are alternately applied and the viscosity is measured in flow mode. These experiments probe the ability for the material to recover its original structure after network scission and characterize how quickly the network heals. While step-strain experiments probe the elastic network by measuring the shear moduli, step-shear experiments monitor the shear viscosity of the bulk material. Although both experiments can characterize the time-scale of self-healing, a step-strain experiment is insufficient when characterizing a material for injectability, a process that is fundamentally flow-based. Moreover, data points are often “lost” between the strain transitions due to the time restraints of oscillatory mode (data must be averaged over several cycles) and it is harder to relate modulus and shear strain to a material passing through a needle. On the other hand, step-shear experiments measure the viscosity based on a varying shear rate—native parameters when considering injectability. Therefore, step-shear experiments are better for characterizing injectability, while step-strain experiments provide information on the network structure and elasticity under high shear strains.
Shear-thinning is fully characterized using a steady-shear experiment, where a constantly increasing flow rate is applied at a fixed rate and the viscosity is measured. In simple terms, the viscosity (η) can often be experimentally related to the applied shear rate (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ) through a power law relationship (3).
) through a power law relationship (3).
η = k![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/char_e0a2.gif) −a −a | (3) |
This relationship provides insight into the degree of shear-thinning through the power law index (a). A value of a = 0 represents a Newtonian fluid, such as water or honey, that does not shear-thin. As the value of a increases, the degree of shear-thinning increases. This feature is particularly important when considering the stress required to inject a supramolecular hydrogel. Fig. 9a illustrates the calculated pressure (P) required to push 1 mL of material through a 1 mL syringe in 10 seconds. As a general trend, more pressure is required to push material out of a higher gauge needle (lower needle diameter). The magnitude of this relationship 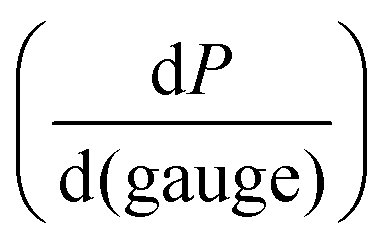 is dependent on the power law index, where materials with higher power law indices exhibit less dependence on needle gauge (Fig. 9b). This lack of dependence is desirable as higher gauge needles afford less invasive procedures. Newtonian materials like honey may be injected through lower gauge needles, but becomes increasingly more difficult to push through higher gauge needles as indicated by the dotted line (the maximum achievable pressure applied to a 1 mL syringe by 98% of subjects from ref. 164). In contrast, MV/Np@CB[8] and PNP hydrogels exhibit significant shear-thinning, with minimal pressure dependency on needle gauge (Fig. 9a). Notably, for a 31 gauge needle typically used to inject insulin, MV/Np@CB[8] hydrogels approach the injectability of blood, while exhibiting moduli ranging up to several hundreds of pascals under working conditions.78 It is important to point out that for injectable applications, both step-shear and steady-shear experiments are crucial to demonstrate appropriate injectability properties. In a steady-shear experiment, low shear rate data provides insight into the material viscosity in operando and high shear rate data characterizes injection viscosity (Fig. 9d). The corresponding log–log plot is used to determine the power law index, allowing for comparison of shear-thinning ability between materials. Studies of shear-thinning materials regularly only perform a step-strain experiment, which lacks information about how easily the material would actually flow through a needle and diminishes the ability to truly judge or engineer injectability.
is dependent on the power law index, where materials with higher power law indices exhibit less dependence on needle gauge (Fig. 9b). This lack of dependence is desirable as higher gauge needles afford less invasive procedures. Newtonian materials like honey may be injected through lower gauge needles, but becomes increasingly more difficult to push through higher gauge needles as indicated by the dotted line (the maximum achievable pressure applied to a 1 mL syringe by 98% of subjects from ref. 164). In contrast, MV/Np@CB[8] and PNP hydrogels exhibit significant shear-thinning, with minimal pressure dependency on needle gauge (Fig. 9a). Notably, for a 31 gauge needle typically used to inject insulin, MV/Np@CB[8] hydrogels approach the injectability of blood, while exhibiting moduli ranging up to several hundreds of pascals under working conditions.78 It is important to point out that for injectable applications, both step-shear and steady-shear experiments are crucial to demonstrate appropriate injectability properties. In a steady-shear experiment, low shear rate data provides insight into the material viscosity in operando and high shear rate data characterizes injection viscosity (Fig. 9d). The corresponding log–log plot is used to determine the power law index, allowing for comparison of shear-thinning ability between materials. Studies of shear-thinning materials regularly only perform a step-strain experiment, which lacks information about how easily the material would actually flow through a needle and diminishes the ability to truly judge or engineer injectability.
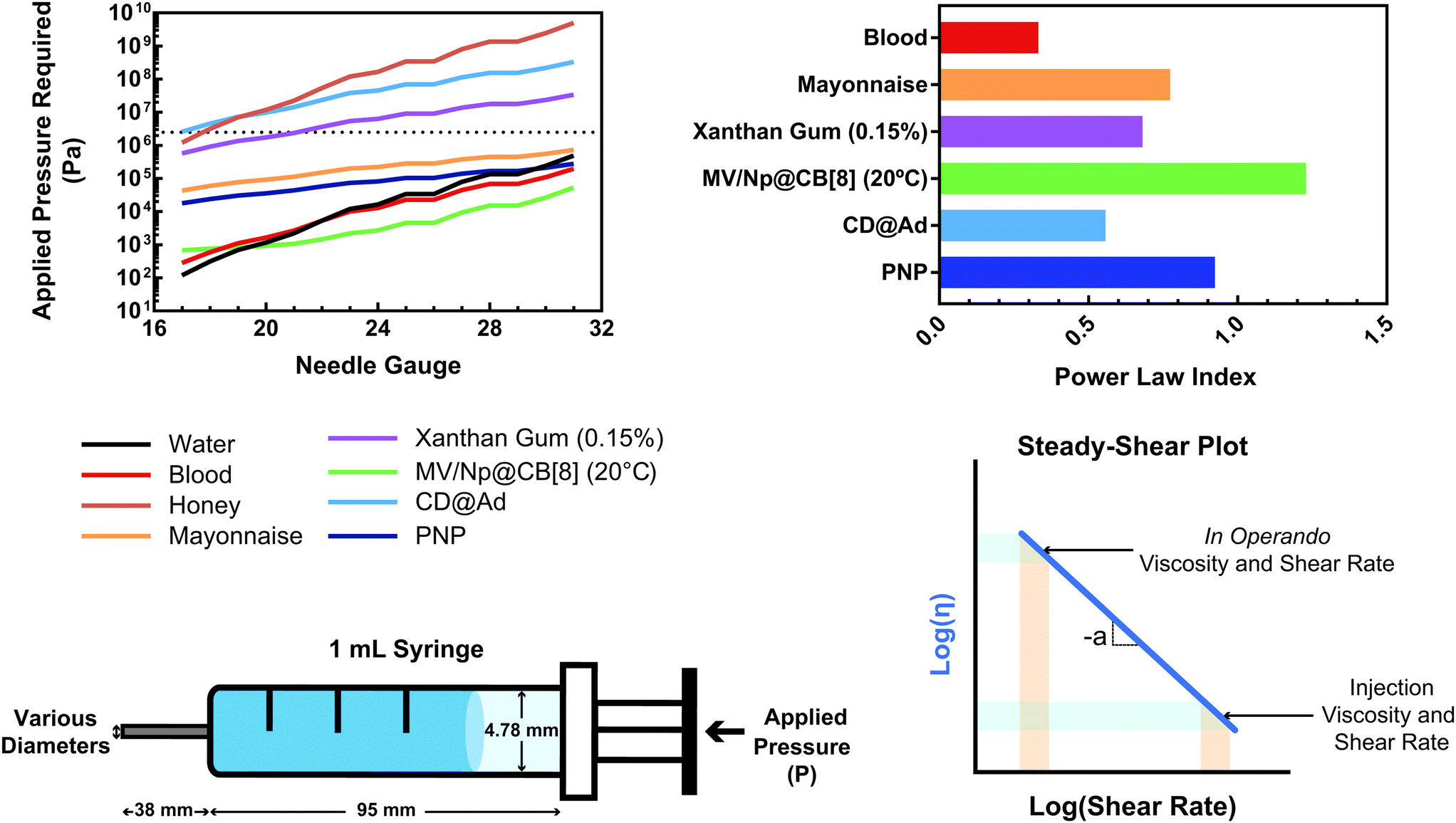 | ||
| Fig. 9 (a) The calculated pressure required to push 1 mL of material [water, blood,160 honey, mayonnaise,161 xanthan gum (0.15%),162 HEC-Np + PVA@CB[8] (0.1%),78 Ad-MeHA/CD-MeHA (10%),163 and HPMC-C12/PEG-PLA PNPs139] through a 1 mL syringe over 10 seconds. The dotted line represents the average pressure subjects were able to apply to a 1 mL syringe from ref. 164. Plateaus in the plot result from gauges that have the same inner diameter. (b) A plot of the power law index (a) of the shear-thinning materials pushed through the syringe. All data were obtained at 25 °C unless otherwise stated. (c) Calculations were done assuming a horizontal needle exhibiting lamellar flow and pressure loss from the syringe and needle walls. (d) Example of a steady-shear plot, where viscosity is plotted vs. shear rate in a log–log graph. Viscosities at low shear rates are indicative of in operando viscosities, while high shear rate viscosities exemplify injection conditions. The power law index values were calculated from the referenced data from viscosity points around 10 s−1. | ||
The physical chemistries behind disentanglement and dissociation processes have been extensively studied and exploited through using various bonding chemistries and bonding motifs.165 For supramolecular biomaterials, shear-thinning materials have been loosely categorized under three groups: (1) protein-based hydrogels, (2) hydrogel nanocomposites, and (3) host–guest based hydrogels.166
Protein–protein interactions used to develop shear-thinning and self-healing hydrogels leverage hydrogen bonding between specific β-sheet structures to provide the non-covalent, dynamic interactions. Specifically, intracellular WW domains bind with Pro-Pro-X-Tyr motifs with Keq values as high as 106 M−1. These components self-assemble to crosslink the network (G′ ∼ 50 Pa) and self-heal within 5–30 minutes depending on the strength of the interacting peptide pairs.128,167 As mentioned before, leucine zipper motifs have been utilized for making shear-thinning hydrogels. In these motifs, the bundle formation of helical structures can be disrupted at high or low pH, which allows for stimulus controlled shear-thinning and self-healing of the structure.168
Hydrogels that form through interactions with nanoparticles or nanodisks provide another set of shear-thinning hydrogels, though these interactions are not strictly supramolecular due to the lack of directionality. In these materials, the primary network structure is usually adsorbed onto the surface of the nanoparticle or nanodisk through hydrophobic interactions, hydrogen bonding, and ionic interactions. PEG based gels have been created through physically crosslinking the PEG chains with silicate nanodisk surfaces. It is imperative in these structures that each polymer chain interconnects several nanodisks or nanoparticles. Schmidt and coworkers demonstrate this requirement by combining LAPONITE® clay nanodisks with PEG chains of different molecular weight. Using small angle neutron scattering (SANS) and rheology, they were able to show that the onset of shear-thinning decreases as the PEG chain molecular weight is decreased, since the chains lose their ability to effectively interconnect multiple disks.169 While clay nanodisk based nanocomposites utilize ionic interactions, purely hydrophobic interactions can also be used to create shear-thinning, self-healing gels. Diblock copolymers of PEG-b-poly(lactic acid) (PEG-PLA) can be nanoprecipitated to form polymer nanoparticles and act as crosslinks in a cellulosic network. Previously, Langer and Appel showed that hydroxypropylmethyl cellulose (HPMC) can interact with PEG-PLA polymer nanoparticles to form hydrogels. To strengthen this interaction while maintaining injectability, pendant alkyl groups could be added to the HPMC backbone.139
Host–guest interactions also provide facile methods to tune the shear-thinning and self-healing abilities of hydrogels. Burdick and coworkers have extensively studied how to leverage host–guest interactions between β-CD and adamantane to form injectable hydrogels. In particular, they designed hyaluronic acid hydrogels with β-CD and adamantane pendant groups to drive supramolecular self-assembly of the hydrogel. The combination of HA with CD was chosen due to both being abundantly used in biomedical applications and the modularity of the interaction. By changing the concentration of β-CD and adamantane modified HA, the authors were able to tune the shear-thinning and self-healing properties as well as overall gel modulus.43,170,171 These CD based polymeric materials have continued to be explored for 3-D printing of biomaterials due to their injectability.163 Similarly, CB based complexes have also been used as supramolecular crosslinks. In 2010 Scherman reported the first example of a hydrogel based on CB[8] host–guest inclusion complexes. These host–guest interactions involved two hosts, forming a ternary complex that had independent kinetics for each host. The pore size, mechanical properties, and thermal reversibility of these materials as well as their shear-thinning, self-healing properties could be modulated through tuning the concentration of CB[8] present.77
3.3 Cargo release kinetics
Supramolecular polymeric biomaterials have increasingly been developed for drug delivery applications. One of the main advantages of using supramolecular hydrogels for release is the ability to finely control the kinetics of cargo release. When designing a new material for a specific desired kinetic profile, it is important to consider the drug-network interactions and the overall network stability over time.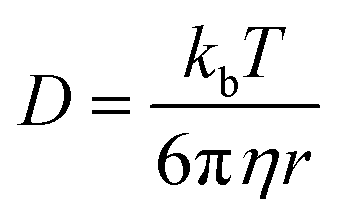 .173 Once the mesh size is close to the solute size, solute diffusion and determination of the diffusivity becomes more complex. If the mesh size is smaller than the cargo, no diffusion occurs. However, supramolecular networks are composed of dynamic crosslinks that serve as molecular gates for the meshes (Fig. 10a). The rate at which this gate opens and closes is dependent on the ka and kd of the supramolecular interaction (a function of
.173 Once the mesh size is close to the solute size, solute diffusion and determination of the diffusivity becomes more complex. If the mesh size is smaller than the cargo, no diffusion occurs. However, supramolecular networks are composed of dynamic crosslinks that serve as molecular gates for the meshes (Fig. 10a). The rate at which this gate opens and closes is dependent on the ka and kd of the supramolecular interaction (a function of 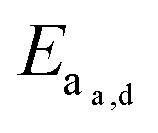 ; eqn (1)). This is to say that a material with supramolecular crosslinks, where ka and kd can be controlled independently of Keq, can be engineered to demonstrate different release profiles (Fig. 10b).174
; eqn (1)). This is to say that a material with supramolecular crosslinks, where ka and kd can be controlled independently of Keq, can be engineered to demonstrate different release profiles (Fig. 10b).174
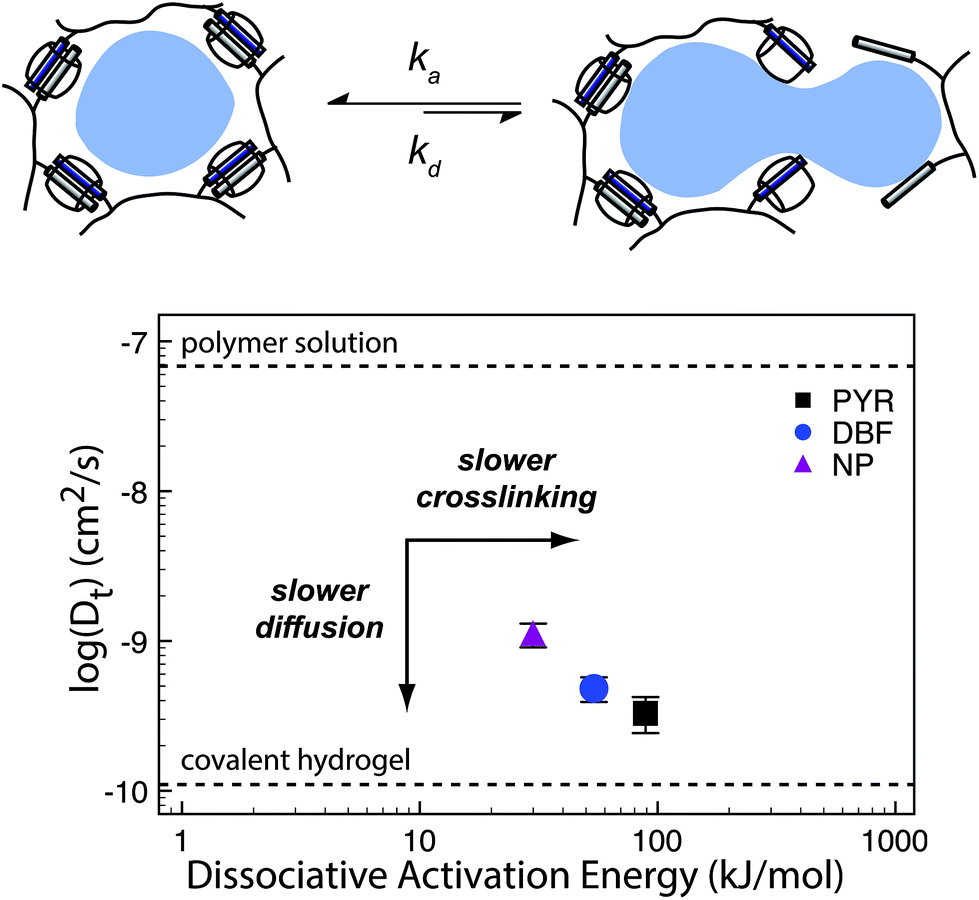 | ||
| Fig. 10 (a) Supramolecular hydrogels that use non-covalent crosslinks, such as the CB[8] ternary complexes shown above, exhibit time-dependent mesh sizes that depend on the kinetics of the crosslink interactions. These hydrogels can exhibit identical crosslink thermodynamics, but varying kinetics due to the different guest moieties. (b) A supramolecular interaction can have different dissociation kinetics, but similar binding thermodynamics. Using the example of hydrogels prepared with CB[8] ternary complex crosslinks with data adapted from ref. 175 we demonstrate that different diffusion profiles for the same cargo are achieved for materials of similar moduli due to varying guest moieties (PYR = pyrone (slow); DBF = dibenzofuran (medium); Np = naphthalene (fast)). Moreover, scattering experiments illustrated that these gels were isostructural. | ||
Scherman exploited this phenomena using CB[8] based hydrogels with different second guest moieties. These second guest moieties displayed a range of ka and kd values, while exhibiting identical Keq values. Interestingly, this preparation leads to a set of gels that have the same plateau G′ due to the identical crosslink Keq, but different release kinetics depending on the rate of the crosslink kinetics (ka and kd). This phenomenon is due to faster kinetics resulting in larger effective mesh sizes and therefore faster release of cargo.175 These results illustrate an important distinction between interaction strength and interaction kinetics, and how they must be independently considered when engineering complex properties like cargo diffusion. While reports of G′ and tan(δ) can provide insight into the overall elasticity of a material, it is not sufficient to draw conclusions about release kinetics solely from those values. These complex relations have not been explored thoroughly and further research would offer more insight into engineering solute diffusion in a number of important areas.
A different strategy to tune the diffusion of the solutes involves deliberately engineering solute-network interactions. This method provides an additional interaction to impede solute diffusion kinetics and can involve ionic interactions, hydrogen bonding, and hydrophobic interactions, to name a few. Guldberg and coworkers utilized negatively charged alginate hydrogels to delivery cationic, heparin binding growth factors. The cargo release was primarily controlled by hydrogel degradation and the protein-network interaction provided additional stability to the protein encapsulated in the gel.176,177 Using a CD/Ad host–guest based HA hydrogel, Burdick and coworkers demonstrated control over solute release kinetics by modifying the CD:Ad pendant group stoichiometry.178 While CD typically acts as a host for Ad, when CD is in excess it will be available to reversibly trap solute molecules in its hydrophobic cavity as well. Tuning the CD:Ad ratio as well as the solute affinity to CD allowed for fine control over release kinetics.
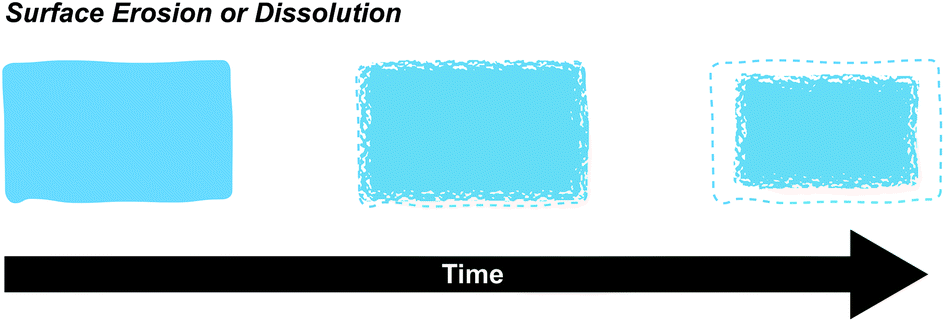 | ||
| Fig. 11 Surface degradation (erosion or dissolution) occurs when chemical bonds or supramolecular interactions at the surface are broken. Surface erosion occurs when chemical bonds are broken, usually through a hydrolysis reaction, while surface dissolution occurs when non-covalently crosslinked polymer chains detach from the surface. Surface degradation kinetics are directly proportional to the surface area and arise when the rate of degradation at the surface exceeds bulk erosion. | ||
Surface based degradation is often desired in supramolecular polymeric material systems and is a direct result of the Keq and kinetic rate constants, as well as the polymer molecular weight, degree of connectivity, and open surface area. In the β-CD-Ad-HA gel system described previously, the hydrogels exhibited an initial burst release of bovine serum albumin (BSA) due to residual BSA on the surface, followed by a nearly linear release rate. These release rates were tunable by varying the initial amount of BSA. The combination of a shear-thinning, self-healing material with surface dominated erosion kinetics makes these gels excellent candidates for extended drug delivery (vide infra).43 Previously discussed cyclodextrin poly(pseudo)rotoxane motifs have also been shown to form hydrogels with surface dominated release kinetics. In these studies, the authors loaded PEG-α-CD poly(pseudo)rotoxane hydrogels with BSA and lysozyme and measured kinetics of release while keeping track of the exposed surface area. The final results were best fit to zero order release kinetics, insinuating surface dominated kinetics.55
4 Biomaterial applications
Given the toolbox of chemical binding moieties and design criteria for macroscopic mechanical properties, one can begin to engineer new biomaterials for several biomedical applications. In the following sections, we will describe how researchers have used these tools to design biomaterials for drug delivery, immunology, regenerative medicine, and 3D-bioprinting.4.1 Drug delivery
Drug delivery using polymers began in the 1960s, when researchers began to explore biocompatible materials to manipulate the pharmacokinetics and pharmacodynamics of a delivered drug.179 Since then, the field has progressed in producing a variety of strategies for “smart” delivery, where the materials allow the drug to overcome certain biological barriers for delivery. These advances were largely motivated by improving efficacy of the delivered drugs, requiring a lower dosage and therefore cost of therapy, and increasing patient compliance. Currently, there is a focus on creating advanced delivery systems, which involve additional functionality to delivery systems. These functionalities include extended cargo release and stimuli responsiveness, which both fall under the category of controlled drug delivery.Supramolecular polymeric biomaterials provide significant advantages towards realizing advanced delivery systems. The main criteria required for effective controlled release systems include localization of the payload, stable therapeutic release of cargo, no toxicity arising from the delivery system, stability of the encapsulated drug, amenability to stimuli responsive release, and a non-harmful fate for the material at end-of-use. In light of these criteria, supramolecular hydrogels and microcapsules utilizing supramolecular motifs are excellent candidates as a general platforms for engineering delivery materials.
When applying supramolecular materials for drug delivery, localization of the payload is contingent on how the material is delivered to the site of interest. This most often translates to designing hydrogels that are injectable, or using microcapsules. Injectability allows the material to be administered relatively easily and implies fast shear-thinning and self-healing kinetics in the case of hydrogels. Fast self-healing kinetics reduce burst release, allowing more of the payload to be released at a kinetic rate governed by the engineered microstructure and transient behavior of the material. Stable therapeutic release of cargo is related to the overall release kinetics and whether the kinetic profile is consistantly at therapeutic levels. Toxicity can be reduced by choosing components of the material that are highly biocompatible, such as those “generally recognized as safe” by the Food and Drug Administration (GRAS-listed). Due to the modularity of supramolecular interactions, there is a significant amount of flexibility in choosing components that can result in the same final macroscopic properties, while maintaining minimal toxicity. Furthermore, as demonstrated in a previous section, the modulus and dynamic properties can be tuned through increasing the amount of supramolecular interactions, or devising ways to tailor the thermodynamics/kinetics of these interactions (vide supra). This flexibility allows for materials to have moduli and viscoelastic properties most compatible with the surrounding biological interface. To address drug stability in the material, strategies have utilized non-covalent drug-network interactions that can protect the drug from being exposed to enzymes and maintain a specific conformation. Stimulated release is also a property that is amenable to supramolecular polymeric systems that use pH, temperature, ionic, or other external stimuli to change the release behavior (vide supra).
Host–guest interactions provide a facile way of independently controlling thermodynamics and kinetics of the interactions. As described above, Burdick has developed a β-CD-Ad-HA system that forms shear-thinning, self-healing hydrogels with tunable strength. In particular, they designed hyaluronic acid hydrogels with β-CD and adamantane pendant groups to drive supramolecular self-assembly of the hydrogels.170,171,178,182 By changing the concentration of β-CD and adamantane modified HA, the authors were able to tune the shear-thinning, self-healing properties as well as the overall gel strength. These initial studies also demonstrated the ability to tune the release of BSA to beyond 60 days. Specifically, by changing the hydrogels from a 2.5 wt% to 10 wt% gel, the time it took to achieve 90% cumulative release doubled.43
Using this same system, Burdick and coworkers also demonstrated modular tuning of release.178 The authors were able to tune the cumulative release profile through either varying the total polymer wt%, the Ad:CD ratio, or the affinity of the peptide cargo. All release profiles achieved sustained release of the two peptides for up to three weeks, while exhibiting different controllable parameters that can be used to tailor the material for a wide range of payloads. This work culminated in tuning the HA-CD gels to deliver two clinical therapeutics of different affinities with CD for two weeks. Collectively, these results illustrate how hydrogels dynamically crosslinked with CD complexes can be used to create injectable materials capable of releasing peptide drugs over weeks, while also remaining versatile as a materials platform to be tuned for a wide range of cargo.178,183
In another example of hydrogels based on host–guest interactions, Scherman and coworkers developed a system based on CB[8] self-assembly with two different guests.77 The authors modified cellulose derivatives with either viologen or naphthoy derivatives and demonstrated that the presence of CB[8] induced gelation due to the formation of ternary complexes. By tuning the crosslink density, the authors were able to control pore sizes, mechanical strength, and drug release kinetics.75,76 Extremely sustained release of BSA was observed over 5 months from a hydrogel containing only 1.5 wt% of polymer. Furthermore, the bioactivity of the proteins were maintained for over a month, demonstrating the potential for using these hydrogels for long-term controlled release.78,79
Kim has also shown hydrogel formation using CB[6] and alkylamonium guests derived from 1,6-diaminohexane (DAH) and spermine (SPM).69 These gels were made by grafting (allyloxy)12CB[6] to a thiol-functionalized HA through a thiol–ene reaction. By mixing the CB[6] containing polymer with DAH-containing HA, a hydrogel was produced after several minutes. These gels exhibited a storage modulus as high as 3.4 kPa for a hydrogel comprised of 2 wt% polymer. Moreover, cytocompatibility studies demonstrated high cell viability, enzymatic degradability, and negligible cytotoxicity of the hydrogels. When injected under the skin of mice, the hydrogel formed within a few minutes and was stable for longer than two weeks. More recently, the authors have been investigating the same CB[6] based platform for long term delivery of engineered mesenchymal stem cells (eMSCs).71 In this work the authors used CB[6]-HA combined with DAH-HA and drug conjugated HA. The eMSCs produced enhanced green fluorescence protein in mice for more than 60 days, demonstrating the hydrogel's feasibility in providing long term cell therapies. This work exemplifies how the modularity of supramolecular chemistries enable the researchers to finely tailor their materials platform for cell and protein delivery. This flexibility has allowed for supramolecular materials to solve many challenges in biomedical fields such as immunology and regenerative medicine (vide infra).
Protein interactions have also been employed to assemble gels for drug delivery. Kiick illustrated this strategy by using heparin-modified PEG copolymers to drive assembly when introduced to proteins or peptides.134 Star PEG polymers terminated with heparin-binding peptides (HBP) were mixed with heparin-modified star PEG polymers to form robust gels with G′ ∼ 200 Pa (tan(δ) ∼ 0.05) over 0.1–100 s−1. Contrary to the previous drug delivery systems, the solute release kinetics were mediated by matrix erosion as the star PEG polymers eroded off the matrix. By changing the ratio of heparin containing PEG to PEG-HBP, they were able to obtain a wide range of cumulative release profiles over a two week period. Notably, the modulus and relative elasticity of these heparinized hydrogels can be easily tuned by modulating the heparin:HBP ratio. This tunability in conjunction with heparin's biological versatility has allowed heparin based assembly motifs to be used for a variety of different drug delivery strategies.184
While not strictly based on directional supramolecular interactions, nanocomposites formed from polymer–nanoparticle (PNP) interactions have also been explored for their potential for injectable, controlled release platforms. Langer developed shear-thinning injectable hydrogels that exploit dynamic non-covalent crosslinks between polymers and nanoparticles.185 These PNP gels were made of hydrophobically modified (hydroxypropyl)methyl cellulose (HPMC-Cx) that acted as the main network component and nanoparticles composed of poly(ethylene glycol)-block-poly(lactic acid) (PEG-b-PLA). When mixed, the HPMC-Cx would rapidly and selectively adsorb onto the nanoparticles, forming the physically and dynamically crosslinked gel. Using this materials platform, they demonstrated hierarchical drug release by encapsulating cargo inside the nanoparticles in addition to the bulk of the PNP gel.139 This co-loading allowed for control over two different mechanisms of drug release—Fickian diffusion from the bulk and erosive release from the nanoparticles. Lastly, they demonstrated in vivo functional validation by simultaneously delivering a model hydrophobic cargo and dye labeled BSA.
Cyclodextrins have typically been used for capsule formation due to their GRAS-listed status, industrial scale production, and biocompatibility.39,191,192 Captisol uses β-CD derivatives to solubilize a number of different drugs and Stella and coworkers have demonstrated that a variety of charged drugs can be encapsulated by using different β-CD derivatives.193 Extending LBL assembly technologies for construction of CD multilayers,194 Auzély-Velty and coworkers developed hollow capsules using HA-β-CD to deliver paclitaxel, an anti-cancer drug.195 β-CD formed host–guest complexes with the hydrophobic paclitaxel, greatly enhancing water solubility of paclitaxel, which is a major problem for clinical administration. Release of paclitaxel is controlled by the Keq of the host–guest complex and the authors demonstrated controlled release kinetics, while maintaining efficacy towards killing breast cancer cells. Moreover, the ability for β-CD to pre-complex with paclitaxel circumvents reduced encapsulation efficiency typically seen in capsule formation using LBL techniques, where cargo is loaded after fabrication of the capsule. Beyond CD, calix[4]arenes have also been explored since the 1990s for their use in capsule formation and molecular recognition. These capsules involve the formation of dimeric architectures with two calix[4]arenes encapsulating various small molecule guests and hope to overcome the low Keq values and relatively poor selectivity of CD complexes.196 Some examples of improving calix[4]arene capsule solubility involve attaching large benzylic groups to the lower rim, or alkyl substituted aromatics along the urea groups of the upper rim. Liu and coworkers demonstrated the formation of supramolecular capsules based on p-sulfonatocalix[4]arene and asymmetric viologen host–guest complexes to release doxorubicin.197 Notably, these capsules were shown to be stimuli responsive to temperature, redox, and host–guest inclusion. This multi-responsiveness is exemplary of the supramolecular host–guest interactions. Due to the enthalpy-driven viologen-calix[4]arene interaction, the interactions are weakened with increasing temperature. Furthermore, redox responsiveness was included by deliberately using a viologen guest, which are known to transform into radical cations or neutral molecules via redox reactions. This study not only demonstrates the use of calix[4]arenes in designing capsules for drug encapuslation and delivery, but also exemplifies how the inherent modularity of host–guest interactions enable novel and creative materials.197
CB-based complexes have also been used to make capsules and provide additional tunability. While CB complexes have relatively slow guest dissociation kinetics and more rigid structures than other hosts, CB[5] and CB[7] have relatively high water solubilities of 20–30 mM in neutral water and have demonstrated suitable biocompatibility.198–200 Accordingly, CB[7] has been widely studied for encapsulating drugs, while maintaining bioactivity.201–204 Isaacs and coworkers demonstrated that acyclic CB-based containers increased the solubility of ten different insoluble drugs by several orders of magnitude. Furthermore, the CB materials exhibited low in vitro toxicity in the presence of human liver, kidney, and monocyte cells, and also increased concentrations of paclitaxel encapsulated in the capsules.205 In addition to CB[5] and CB[7], there has been considerable interest in using CB[8] for capsule formation due to the ability for CB[8] to form higher stoichiometric complexes.206–209 Abell and coworkers used CB[8] to form 1:1:1 ternary complexes through multiple supramolecular interactions between an electron-deficient methyl viologen (MV) and an electron-rich naphthol (Np) derivative (Fig. 12). By leveraging the ternary structure formation of CB[8] host–guest interactions, the authors were able to “handcuff” viologen modified gold nanoparticles to a polymer network containing naphthol pendant groups to form controlled dispersions of NPsin the network. Furthermore, the highly stable and strong 1:1:1 ternary complexes allowed the microcapsules to be stable at 100 °C and lower pressures (20 Pa). Similarly to the use of viologens in multifunctional theranostic particles (vide supra), the use of MV allowed for stimuli responsive degradation of the capsules by reduction of the MV group. The use of microfluidics allowed the authors to obtain monodisperse, high throughput, one-step fabrication of these CB[8] microcapsules, demonstrating the efficiency and scalability of the process (Fig. 12).207
 | ||
| Fig. 12 CB[8] ternary complexes were used to form microcapsules using microfluidics processing. The MV functionalized Au NPs were mixed with a copolymer functionalized with Np pendant groups and a CB[8] solution for efficient and facile formation of microcapsules. Microfluidics processing allowed for formation of monodisperse capsules with diameters of 59.6 ± 0.8 μm. | ||
As a whole, these examples illustrate how supramolecular chemistries can improve current microcapsule designs. In particular, the innate modularity and tunability of host–guest interactions enables many different functionalities that are amenable to many current and future microcapsule designs.
4.2 Immunology
Over the last 30 years our understanding of the immune system has increased tremendously, enabling new approaches for engineering immune control. Biomaterials are a powerful tool for designing new therapeutic strategies to modulate the immune system. Below we outline the few examples of immune modulating platforms that exploit the supramolecular systems that have been described in this review. To elucidate the potential for growth in the use of supramolecular polymeric biomaterials for immune applications, we will also provide examples that utilize the highly specific, directional, and strong supramolecular interactions in materials platforms not explored previously in this review. These materials have been used in many areas of this field such as vaccine design, cancer immunotherapies, and general immunomodulation. The research applications that follow indicate the great potential for the use of supramolecular materials for understanding and influencing the immune system.Supramolecular materials held together by host–guest chemistries have been used to provide controlled release of cytokines and therapeutics for immunomodulation. Physically crosslinked hydrogels are desirable materials for cargo delivery because they are both easily loaded and self-heal after injection. Heilshorn and Soranno take advantage of these properties when creating an injectable hyaluronic acid hydrogel grafted with adamantane and CD to deliver interleukin-10 (IL-10) and anti-transforming growth factor β into chronic kidney disease affected kidneys. This treatment successfully produced local delivery depots that reduced macrophage infiltration in a kidney disease model, though the combined delivery of these molecules did not improve long-term fibrosis.210 Similarly, Burdick employed the “dock-and-lock” hydrogels (vide supra) for the delivery of IL-10 to treat chronic kidney disease. The gels delivered IL-10 locally for up to 30 days leading to a decrease in fibrosis in mice.211 Host–guest interactions are also valuable for their modularity. A study used CB[8] to create multipartite vaccines, circumventing challenging synthetic protocols associated with covalently bonding conjugate vaccines. The components of their model antitumor vaccine (tumor antigen, T-helper cell epitope, and TLR2 ligand) were functionalized with either a first or second guest and mixed with CB[8] to create the linked “supramolecular” vaccines. This supramolecular approach to producing conjugate vaccines provides previously unattainable adaptability and ease of synthesis.212
Hydrophobic and entropically driven interactions are also useful supramolecular motifs for immunomodulating applications. Kiyono adapted a cholesteryl-functionalized pullulan (CHP) for intranasal vaccination. CHP self-assembles into physically crosslinked nanogels in water through hydrophobic interactions and traps cargo as it assembles. A similar CHP nanogel successfully improved humoral and T cell responses to the NY-ESO-1 human tumor antigen in a clinical study.213,214 Kiyono incorporated cationic functionality on the cholesteryl bearing pullulan to improve nasal uptake of a subunit fragment of Clostridium botulinum type-A neurotoxin from the CHP nanogels. This vaccine improved systemic and mucosal immune responses in mice and successfully avoided central nervous system accumulation of the antigen.215 A different study developed a self-assembling nanogel from pyridine functionalized poly(hydroxyethyl methacrylate) (pHEMA-pyridine). The polymer, dispersed when unperturbed, self-assembled into nanogels in the presence of protein due to noncovalent interactions. The study demonstrated that these self-assembled nanogels released protein cargo intracellularly in dendritic cells and led to the activation of CD8+ T cells. They describe a new platform for delivery of protein antigens for achieving cell mediated immune responses.216
Targeted drug delivery to the lymph nodes (LNs), tumor sites, or other tissues of interest remains an important goal of immunoengineering. A particularly innovative application for self-assembling polymer micelles involved the design of amphiphilic polymers with a retinoic acid hydrophobic region in the core and a cationic polyethylenimine hydrophilic outer region. After the polymers self-assembled into micelles, the cationic charges were shielded with hyaluronic acid (HA), limiting the toxicity. After the particles passively diffused to tumor sites, the high expression of hyaluronidase in tumor microenvironments degraded the HA coating, unshielding the cationic charges and leading to tumor specific cell necrosis. This necrosis activated an immune response against the tumor, therefore achieving an immune modulation effect.217 Targeting LNs can also be particularly valuable when designing vaccines in order to maximize the interaction necessary for immune cell activation. Irvine and collaborators were inspired by the supramolecular interactions that allow commonly used LN tracers to bind to serum albumin in order to “hitch-hike” to the LNs. They created polymers with three components; an albumin binding domain, PEG, and the peptide antigen. These polymers formed micelles that were able to bind to albumin and be transported into the LNs in mice. This strategy increased LN accumulation of their model vaccine and led to a 30-fold increase T cell priming.218
Rational peptide design enables immunoengineers to create peptide based vaccines that self-assemble like LMWGs, affording unique characteristics for improved immune responses. One approach for preparing supramolecular peptide vaccines employs the well-known β-sheet motif to create highly modular nanofibers and cylindrical micelles.219 By conjugating immunological epitopes to the self-assembling β-sheets, engineers have precise control over the epitope content, dose, and the multivalency of their vaccine. Chong emphasized the utility of precise epitope display while conjugating a CD4+ T cell epitope and B cell epitope to their supramolecular peptide platform in specific ratios. These ratios affected the immune phenotype, favoring T follicular helper, Th1, or Th2 responses.220 These technologies provide a strategy to improve the immunogenicity of subunit vaccines without introducing inflammatory adjuvants. Another way to design peptide vaccines requires conjugating the peptides to a lipid tail that will self-assemble into micelles. It is predicted that these micelles provide high local concentrations of the peptide antigen, protect the peptide from degradation, and increase antigen uptake by dendritic cells. A study showing the preliminary efficacy of peptide micelles for protecting against tumors used micelles with an ovalbumin peptide as a prophylactic tumor vaccine. The mice were inoculated with ovalbumin expressing lymphoma cells after receiving three doses of the vaccine. The peptide micelle treatment stimulated a CD8+ T cell response against the tumor challenge and improved the protection from the tumor compared to using a traditional adjuvant system.221
The above examples provide a snapshot into the potential impact of supramolecular materials in the design of immune modulating therapeutics. The materials can be used alone or as delivery vehicles to produce state-of-the-art therapies. The relatively few studies that utilize the supramolecular systems we focused on in this review demonstrate that this field is still in its infancy. The examples of additional supramolecular materials used for immune applications reveal the advantages of these interactions in the context of modulating the immune system, and show that there are many directions to explore in developing new biomaterials for this field. For immunoengineering applications, supramolecular material features that have been most impactful are their modularity, injectability, loading efficiencies, and stimuli responsiveness. The design parameters are endless in this space; we believe there is significant room for innovation in both prophylactic and therapeutic immune modulating therapies by employing supramolecular polymeric biomaterials.
4.3 Regenerative medicine
In a succinct editorial, Mason and Dunnill describe regenerative medicine as a tool to replace or regenerate human cells, tissues, or organs, restoring or establishing normal functions.222 Translationally minded research in the field parses this out into the combinatorial delivery of soluble factors, cells, and scaffolds. The design and employment of novel biomaterials requires intimate knowledge of how cells interact with their physical environment (mechanotransduction) and the temporal presentation of soluble factors.223There are many excellent reviews describing the intricacies of mechanotransduction; we will only briefly discuss material handles on cellular fate.224–226 It has been demonstrated that stem cells are specifically responsive to their ability to generate traction forces through integrin mediating binding with their microenvironment. This can be modulated mechanically through changes in both matrix elasticity227 and visoelastic relaxation timescales.228 Moreover, changes in dimensionality229—culturing cells above (2D) or within (3D) a matrix—and presentation of the ligand—density and spatial clustering230—affect how cells pull on their matrix and can have profound effects on cell morphology and fate. Like the viscoelastic compliance, the degradability of matrices also change the temporal interaction of cellular materials with their ligands.231 Similarly, there are a collection of great reviews on cellular interaction with soluble factors for regenerative medicine.232–234 In regards to soluble factor delivery, it is becoming more apparent that spatiotemporal control of multiple agents over extended timeframes will be required to affect cellular fate and regenerative processes.235 In addition to these environmental factors, it is important to understand how these materials will be delivered into the body. While surgical implantation relaxes material property constraints, the use of non-invasive protocols through injection provide a more translational alternative.
Supramolecular polymeric materials make a great platform for addressing these needs due to their facile modulation and dynamic nature. The modulus of hydrogels can be affected by changing polymer concentration, backbone architecture, degree of crosslinking, and moiety of the crosslink. The timescale of viscoelasticity can be modulated by selecting and combining crosslinks with different dissociation rate constants. Moreover, it has been demonstrated that precursors to the hydrogels are non-toxic to cells, allowing for high viability encapsulation into 3D networks. Because these materials are synthetic, it is simple to incorporate biological functionality (degradable motifs and RGD adhesion domains) through polymeric backbone modification or by incorporation into the actual backbone through recombinant engineering. Due to the dynamic nature of the crosslinks, engineered materials are shear-thinning, and can be either injected directly or through a catheter. The following section will describe the initial efforts mainly clustered into the following two areas: using supramolecular polymeric hydrogel to (1) deliver soluble factors and to (2) create a 3D microenvironment suitable for injection.
Within the scope of encapsulation, Erythroprotein (EPO), a hormone that can prevent apoptosis while enhancing neovascularization for myocardial infarctions (MI), was delivered via injection in an α-CD PEG-PCL-PEG hydrogel.236 While sustained release of EPO led to improvement of cardiac function in a MI rat model, the authors argue that its localization into an injected hydrogel reduced systemic off-target effects, such as polycythaemia. A telechelic UPy system has also been employed to deliver soluble factors through a catheter for MI, and deliver 2 growth factors, HGF and IGF.237,238 A porcine study demonstrated improved cardiac function and cardiomyocyte generation over a bolus injection of the respective growth factors. Rather than being employed as cargo, growth factors, such as VEGF, can be employed as the actual physical crosslink of a material due to their affinity with LMWH.137 Using VEGF as a physical crosslink resulted in triggered degradation of the material in the presence of VEGF receptors, found on cells, opening the ability to design materials for regenerative medicine with cellular mediated release profiles.
Using α-CD poly(pseudo)rotoxane hydrogel networks, mesenchymal stem cells (MSC) were injected into rats and a rabbit MI model.239,240 While it seems important to note that in the rabbit model the components were mixed during the injection process using a Duploject applicator, both demonstrated beneficial cell morphology, limited material cytotoxicity, and better regenerative profiles than with simple bolus injection.
Kim and coworkers have initiated studies employing CB[6] and guests grafted onto polymeric backbones for cellular delivery through in situ gelation.69–71 When cells are mixed with a hydrogel precursor solution (either CB[6]-grafted or guest-grafted polymer) and injected simultaneously with their precursor counterpart, a hydrogel network results in situ; this strategy is used to deliver a 3D cellular microenvironment in animal models. Because the supramolecular hydrogel is synthetically created, chemical motifs can be introduced to release soluble factors to the cells in the engineered microenvironment. Tethering a steroid that promotes chondrogenesis to the polymeric backbone via a hydrolytically cleavable ester while physically encapsulating a growth factor afforded independent control of two soluble factors over radically different timescales.70 Creative design with the modularity of a supramolecular system allowed control over release and hydrogel mechanics, promoting chondrogenesis through injection into a mouse model. Moreover, a very similar material was used to inject engineered MSCs in a cancer model (Fig. 13).71 This system released two small molecules, dexamethasone and retanoic acid, that acted as cues for transgene expression of the engineered MSCs. To achieve different release profiles, the cues for transgene expression were directly grafted to the polymeric backbone or appended to a soluble CB[6] (letting the cue-CB[6] attach to a free guest on the polymer backbone). The release of the two agents was then mediated by hydrolysis or the dissociation kinetics of the CB[6] guest pair. The use of this supramolecular hydrogel afforded rapid in situ gelation while demonstrating effective transgene expression of engineered MSCs for 5 weeks and increased cellular viability over Matrigel, a traditionally used extracellular matrix for in vitro cell culture. The use of this system retarded tumor growth and increased average lifespan over the delivery of engineered MSCs in Matrigel. While these supramolecular motifs afforded synthetic control of release kinetics and hydrogel viscoelasticity, they ultimately were not able to demonstrate the direct injection of a 3D microenvironment; cells in a hydrogel environment formed before injection. To accomplish this, researchers must look to employ material systems with larger power law indexes (larger values of a) (Fig. 9a and b).
 | ||
| Fig. 13 The usage of post-modified CB[6] based hydrogels to deliver engineered mesenchymal stem cells (eMSC). (a) General scheme: eMSCs are mixed with CB[6] (host) functionalized HA and diaminohexane (DAH, guest) and retinoic acid (RA, transgene expression agent) to form a hydrogel with excess guest molecules. The excess guest molecules then react with addition of dexamethasone (D) functionalized CB[6]. (b) Real time bioimaging of enhanced green fluorescent protein (EGFP) expression in eMSCs in (i) the hydrogel system, (ii) matrigel, and (iii) serum free media to 50 days. (c) Degradation profile of hydrogel samples in mice over 3 months. The post-modification of D-CB[6] shows no negative effects on degradation profile. Figure reproduced with permission from from ref. 71, Wiley. | ||
Using associating protein hydrogels to accomplish this, Heilshorn employed the WW and proline rich domains for stem cell encapsulation and delivery.128,241 Specifically, the use of recombinant proteins afforded direct insertion of cell adhesion motifs into the polymer backbone, enabling encapsulation of a wide range of cell types. As is, this engineered system increased cellular retention over both alginate and collagen post-injection in a mouse model. Incorporating a polymeric segment with a LCST below body temperature provided a thermally induced second gelation, increasing the modulus of the material and overall cellular retention post injection.129
4.4 3D-Bioprinting
The field of regenerative engineering aims to replace or regenerate human cells, tissues, or organs, restoring or establishing normal functions. Complex spatio-temporal control in the fabrication process of materials for regenerative engineering is necessary to replicate the complexities of biological tissue and is only afforded through 3D-bioprinting; precise layer-by-layer depositions of biological materials, biochemical cues, and living cells.242 3D-bioprinting is a complex and multidisciplinary problem that involves advances in imaging, biological design, materials selection, cell selection, printer selection, and application engineering. While these have been reviewed elsewhere,242–244 we will briefly discuss printer technology, material requirements, and demonstrate the benefits of supramolecular crosslinks in advanced bio-inks.The most common 3D-bioprinting machines utilize either laser assisted, inkjet, or microextrusion printing technologies.245 Laser assisted bioprinting involves focusing laser pulses onto an absorbing layer of a material, generating a high-pressure bubble that directs cell-containing materials to a collector. Inkjet printers use thermal or acoustic forces to eject drops of liquid onto a substrate. Microextrusion printers deposit continuous beads of material through a nozzle with pneumatic or mechanical (piston or screw) force. The benefits afforded by dynamic crosslinks are most closely associated with aiding printability in microextrusion printing because it requires printing a viscous material through narrow nozzles under force. This is not say that other printing methods should not employ supramolecular bio-inks. For example, an inkjet bioprinter containing two cartridges of liquid polymer with separate but complimentary supramolecular crosslink components could print supramolecular materials. However, the benefits of using supramolecular materials for this application are no different than the benefits of using supramolecular interactions for traditional regenerative engineering approaches. For this reason, we will continue by describe the benefits of dynamic crosslinks in extrusion based systems.
The spatio-temporal control afforded by 3D-bioprinting requires that the material demonstrate printability, a novel material property similar to injectability.246 In addition to being biocompatible, having controlled degradation kinetics and mechanical properties, and having dynamic biomimetic materials properties, bio-inks must also be suitable to be extruded from a nozzle under pressure and form defined printed architectures at high resolution while maintaining high cell viability.
While readily available biomaterials have been explored for printing, they are met with many problems. Agarose, methylcellulose, gelatin, and collagen possess gel transition temperatures and must be printed at high temperatures potentially dangerous to cells.247 Further, they form weakly bound structures that often need a secondary crosslinking step. While use of alginate forms robust hydrogels, it is printed as a viscous fluid in which cells experience both sedimentation which can lead to printer clogging and substantial shear forces during extrusion.248,249
To address issues of printability of alginate materials, Heilshorn employed tailored supramolecular interactions on the alginate backbone.250 Alginate was functionalized with a proline rich domain and mixed with alginate functionalized with a WW domain, forming weak (20 Pa), shear-thinning hydrogels. This pre-gel was printed into a bath of CaCl2, forming a robust alginate hydrogel (4 kPa) whose material properties largely matched those of crosslinked alginate without peptide crosslinks. By printing alginate as a weak gel rather than a viscous liquid, they demonstrated homogeneity of cells in the cartridge, increased cell membrane integrity during printing, and >90% viability of hASCs and 3T3s a week after printing. While originally demonstrated with complimentary peptide sequences, alginate printability ostensibly would increase with other, more modular supramolecular crosslinks, offering the ability to correlate macroscopic pre-gel properties with cell printability.
Similarly, Burdick modified hyaluronic acid (HA), a utilized bioink, with either β-CD or Ad and printed the hydrogel into a support gel of near identical material—a unique printing strategy afforded by the shear-thinning of the extruded gel and the rapid self-healing of both the extruded gel and the displaced support network (Fig. 14).251 Appending methacrylate units to the HA polymers in the cartridge or HA support gel gives the ability to reinforce the printed architecture with a photo-crosslinking step and subsequently wash away the support. This strategy led to a complex 3D architecture unobtainable without printing into a medium. The washing process, however, involves disrupting all (print and support) host–guest interactions, leaving only the covalent methacrylate crosslinks in the printed structure. In a similar fashion, methacrylate units can be appended to only the support supramolecular gel. Here the support is reinforced and the printed structure is removed through flow applied at the inlet and outlet of the channel by the printer, leaving a support gel with supramolecular crosslinks and well defined channels. Excitingly, the printer could pattern MSCs in the extruded gel, demonstrating >90% viability immediately after printing and >80% viability after 3 days in culture. While printing into a support gel had many advantages, it ultimately could not produce a printed design with physical crosslinks removed from the support. To address this, Burdick explored printing HA appended with methacrylate and either CD or Ad motifs directly onto a substrate with a subsequent photo-crosslinking step.163 Here they explore how changes in crosslink density, crosslink nature (supramolecular vs. chemical), and curing method affect the printability and mechanical properties of the printed gel. While they could functionalize printed scaffolds with RGD and seed the scaffolds with 3T3 fibroblasts, direct printing of cells from the printer was not demonstrated.
 | ||
| Fig. 14 Burdick's process of 3D printing into a support gel. (a) Illustration of the injection process where the injected gel (red) contains the same CD host–guest chemistry as the support gel (green). (b) Printing different resolutions of rhodamine (red) labeled gel into a fluorescein (green) labeled support gel through a 20 (i), 27 (II), and 34 (iii) gauge needle (d) Confocal image of two different inks (rhodamine and fluorescein) printed in different geometries. (d) Printing of mesenchymal stem cells (green) into a support gel containing 3T3 fibroblasts. Scalebars in (b): 100 μm, (c), (d): 200 μm. Figure reproduced with permission from from ref. 251, Wiley. | ||
Rather than modifying an already used bioink, Duncan and collaborators created a new ink backbone with appended dynamic biological crosslinks.252 Here they separated two components, a polypeptide with a grafted nucleic acid sequence (A) complimentary to the sticky ends of a DNA crosslinker (B), into separate printer valves. Strong, optically transparent hydrogels formed in seconds upon co-release of bioink A and B through the printer microvalves. Further, because these hydrogels are composed of peptides with DNA crosslinks they demonstrate biodegradability in the presence of specific nucleases and proteases. Because the dissociation rate constant is much smaller for multivalent, complimentary hydrogen bonding, similar levels of shear-thinning are not recognized for this system as they are for Burdick's, removing the possibility of directly printing gels. This limits the resolution of the system to that of the droplet, 500 μm (as opposed to 35 μm for printing into a support gel and 100 μm for freestanding CD@Ad gels). They demonstrated the printability of AtT-20 cells at exceptional viability (>98%) but did not demonstrate the printing of cell lines more relevant to regenerative medicine applications.
Future development in this field will benefit from utilizing supramolecular crosslinks to tease out structure property relationships between bio-ink and printability. Scalable, tailorable new bioinks should be created that are shear-thinning and maintain high cell viability. With this knowledge, hydrogel properties can be specifically engineered for use in a 3D printer for applications in regenerative medicine.
5 Conclusions
This review illustrates the design toolbox for supramolecular polymeric hydrogels—water soluble polymeric chains that assemble into contiguous networks through physical crosslinks (host–guest, hydrogen bonding, metal–ligand, electrostatic, and protein based interactions). Different from conventional crosslinking, supramolecular crosslinks exploit specific, directional, tunable, and reversible non-covalent interactions. Accordingly, supramolecular polymeric hydrogels exhibit unique and desirable properties not found in covalent systems. Through careful choice of crosslinking motif and polymeric architecture, novel biomaterials can be systematically built to have engineered viscoelasticity and release profiles, with shear-thinning and rapid self-healing properties.Supramolecular polymeric hydrogels are increasingly used as novel biomaterials because their macroscopic material properties can be precisely engineered. The specific, directional, tunable, and reversible nature of a supramolecular interaction facilitates creative solutions to complicated immunological problems. Controlled viscoelasticity enables rheological properties similar to in vivo biological tissue, making these materials excellent candidates for controlled microenvironments. Shear-thinning and rapid self-healing allow mechanically strong gels to flow through high gauge needles during injection and then reform in situ with minimal cargo loss. Physical crosslinks unperturbed by traditional network swelling afford controlled and extended release through kinetically controlled mesh dynamics. The controlled viscoelasticity, shear-thinning, and self-healing properties enable pneumatic and mechanical printers to extrude well defined cellular microenvironments with incredible precision.
While many studies have developed the synthetic toolbox for designing supramolecular polymeric biomaterials, exploration of the relationship between crosslinking chemistries and material properties has only just begun. The ability to precisely correlate, model, and engineer the relationship between network architecture and supramolecular kinetics and thermodynamics to defined rheological and mechanical properties is presented as a future goal. Work towards this understanding will enable facile implementation of these chemistries into the biomedical community.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Hellman Faculty Scholars fund and a Research Starter Grant from the PhRMA Foundation. J. L. M. thanks the Department of Defense for the National Defense Science and Engineering Graduate Fellowship. A. C. Y. is grateful for the Eastman Kodak Fellowship. G. A. thanks the National Science Foundation for the Graduate Research Fellowship.References
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- N. A. Peppas and R. Langer, Science, 1994, 263, 1715–1720 CAS.
- N. Huebsch and D. J. Mooney, Nature, 2009, 462, 426–432 CrossRef CAS PubMed.
- K. S. Katti, Colloids Surf., B, 2004, 39, 133–142 CrossRef CAS PubMed.
- M. Navarro, A. Michiardi, O. Castaño and J. A. Planell, J. R. Soc., Interface, 2008, 5, 1137–1158 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- M. J. Webber, O. F. Khan, S. A. Sydlik, B. C. Tang and R. Langer, Ann. Biomed. Eng., 2015, 43, 641–656 CrossRef PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- J. Kopeček, Biomaterials, 2007, 28, 5185–5192 CrossRef PubMed.
- C. P. Pathak, A. S. Sawhney and J. A. Hubbell, J. Am. Chem. Soc., 1992, 114, 8311–8312 CrossRef CAS.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 34, 4312–4314 RSC.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS PubMed.
- R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- C. D. Pritchard, T. M. O'Shea, D. J. Siegwart, E. Calo, D. G. Anderson, F. M. Reynolds, J. A. Thomas, J. R. Slotkin, E. J. Woodard and R. Langer, Biomaterials, 2011, 32, 587–597 CrossRef CAS PubMed.
- H. Zhang, A. Qadeer, D. Mynarcik and W. Chen, Biomaterials, 2011, 32, 890–898 CrossRef CAS PubMed.
- A. B. W. Brochu, S. L. Craig and W. M. Reichert, J. Biomed. Mater. Res., Part A, 2011, 96, 492–506 CrossRef PubMed.
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Macromolecules, 1993, 26, 581–587 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- M. P. Lutolf, Nat. Mater., 2009, 8, 451–453 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed.
- C. M. A. Leenders, T. Mes, M. B. Baker, M. M. E. Koenigs, P. Besenius, A. R. A. Palmans and E. W. Meijer, Mater. Horiz., 2014, 1, 116–120 RSC.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- S. Tang, M. Wang and B. D. Olsen, J. Am. Chem. Soc., 2015, 137, 3946–3957 CrossRef CAS PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- T. Kakuta, Y. Takashima and A. Harada, Macromolecules, 2013, 46, 4575–4579 CrossRef CAS.
- Y. Takashima, Y. Sawa, K. Iwaso, M. Nakahata, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 3254–3261 CrossRef CAS.
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125–4134 CrossRef CAS PubMed.
- C. B. Rodell, R. J. Wade, B. P. Purcell, N. N. Dusaj and J. A. Burdick, ACS Biomater. Sci. Eng., 2015, 1, 277–286 CrossRef CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- C. B. Rodell, C. B. Highley, M. H. Chen, N. N. Dusaj, C. Wang, L. Han and J. A. Burdick, Soft Matter, 2016, 12, 7839–7847 RSC.
- O. Kretschmann, S. W. Choi, M. Miyauchi, I. Tomatsu, A. Harada and H. Ritter, Angew. Chem., Int. Ed., 2006, 45, 4361–4365 CrossRef PubMed.
- Y. Guan, H.-B. Zhao, L.-X. Yu, S.-C. Chen and Y.-Z. Wang, RSC Adv., 2014, 4, 4955 RSC.
- M. Guo, M. Jiang, S. Pispas, W. Yu and C. Zhou, Macromolecules, 2008, 41, 9744–9749 CrossRef CAS.
- S. Himmelein, V. Lewe, M. C. A. Stuart and B. J. Ravoo, Chem. Sci., 2014, 5, 1054–1058 RSC.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Macromolecules, 1993, 26, 5698–5703 CrossRef CAS.
- J. Li, X. Ni and K. W. Leong, J. Biomed. Mater. Res., Part A, 2003, 65, 196–202 CrossRef PubMed.
- P. L. Chee, A. Prasad, X. Fang, C. Owh, V. J. J. Yeo and X. J. Loh, Mater. Sci. Eng., C, 2014, 39, 6–12 CrossRef CAS PubMed.
- J. Li, X. Ni and K. Leong, Angew. Chem., Int. Ed., 2003, 42, 69–72 CrossRef CAS PubMed.
- X. Li, J. Li and K. W. Leong, Macromolecules, 2003, 36, 1209–1214 CrossRef CAS.
- K. L. Liu, S. H. Goh and J. Li, Macromolecules, 2008, 41, 6027–6034 CrossRef CAS.
- H. S. Choi, K. Yamamoto, T. Ooya and N. Yui, ChemPhysChem, 2005, 6, 1081–1086 CrossRef CAS PubMed.
- R. Hernández, M. Rusa, C. C. Rusa, D. López, C. Mijangos and A. E. Tonelli, Macromolecules, 2004, 37, 9620–9625 CrossRef.
- J. Li, in Cyclodextrin Inclusion Polymers Forming Hydrogels, ed. G. Wenz, Springer, Berlin Heidelberg, 2009, pp. 175–203 Search PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323 CrossRef CAS PubMed.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed.
- Y. Miyahara, K. Abe and T. Inazu, Angew. Chem., Int. Ed., 2002, 41, 3020–3023 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- J. W. Lee, K. Kim, S. Choi, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Chem. Commun., 2002, 2692–2693 RSC.
- K. M. Park, J. A. Yang, H. Jung, J. Yeom, J. S. Park, K. H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS PubMed.
- H. Jung, J. S. Park, J. Yeom, N. Selvapalam, K. M. Park, K. Oh, J. A. Yang, K. H. Park, S. K. Hahn and K. Kim, Biomacromolecules, 2014, 15, 707–714 CrossRef CAS PubMed.
- J. Yeom, S. J. Kim, H. Jung, H. Namkoong, J. Yang, B. W. Hwang, K. Oh, K. Kim, Y. C. Sung and S. K. Hahn, Adv. Healthcare Mater., 2015, 4, 237–244 CrossRef CAS PubMed.
- M. J. Rowland, M. Atgie, D. Hoogland and O. A. Scherman, Biomacromolecules, 2015, 16, 2436–2443 CrossRef CAS PubMed.
- M. J. Rowland, E. A. Appel, R. J. Coulston and O. A. Scherman, J. Mater. Chem. B, 2013, 1, 2904–2910 RSC.
- C. Li, M. J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman and D. Liu, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS PubMed.
- E. A. Appel, R. A. Forster, M. J. Rowland and O. A. Scherman, Biomaterials, 2014, 35, 9897–9903 CrossRef CAS PubMed.
- E. A. Appel, R. A. Forster, A. Koutsioubas, C. Toprakcioglu and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 10038–10043 CrossRef CAS PubMed.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, C. A. Dreiss and O. A. Scherman, Biomaterials, 2012, 33, 4646–4652 CrossRef CAS PubMed.
- Y. Kim, H. Kim, Y. H. Ko, N. Selvapalam, M. V. Rekharsky, Y. Inoue and K. Kim, Chem. – Eur. J., 2009, 15, 6143–6151 CrossRef CAS PubMed.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- J. Zhang, H. Li, L. Sun and C. Wang, in Determination of the Kinetic Rate Constant of Cyclodextrin Supramolecular Systems by High-Performance Affinity Chromatography, ed. S. Reichelt, Springer, New York, New York, NY, 2015, pp. 309–319 Search PubMed.
- M. Christoff, L. Okano and C. Bohne, J. Photochem. Photobiol., A, 2000, 134, 169–176 CrossRef CAS.
- W. Al-Soufi, B. Reija, M. Novo, S. Felekyan, R. Kühnemuth and C. A. M. Seidel, J. Am. Chem. Soc., 2005, 127, 8775–8784 CrossRef CAS PubMed.
- W. Al-Soufi, B. Reija, S. Felekyan, C. A. M. Seidel and M. Novo, ChemPhysChem, 2008, 9, 1819–1827 CrossRef CAS PubMed.
- W. M. Nau and X. Zhang, J. Am. Chem. Soc., 1999, 121, 8022–8032 CrossRef CAS.
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed.
- E. A. Appel, F. Biedermann, D. Hoogland, J. del Barrio, M. D. Driscoll, S. Hay, D. J. Wales and O. A. Scherman, J. Am. Chem. Soc., 2017, 139(37), 12985–12993 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- R. E. Kieltyka, A. C. H. Pape, L. Albertazzi, Y. Nakano, M. M. C. Bastings, I. K. Voets, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- Y. Cui, M. Tan, A. Zhu and M. Guo, J. Mater. Chem. B, 2015, 3, 2834–2841 RSC.
- Y. Cui, M. Tan, A. Zhu and M. Guo, J. Mater. Chem. B, 2014, 2, 2978–2982 RSC.
- S. Hou, X. Wang, S. Park, X. Jin and P. X. Ma, Adv. Healthcare Mater., 2015, 4, 1491–1495 CrossRef CAS PubMed.
- G. Zhang, T. Ngai, Y. Deng and C. Wang, Macromol. Chem. Phys., 2016, 217, 2172–2181 CrossRef CAS.
- Y. Wu, L. Wang, X. Zhao, S. Hou, B. Guo and P. X. Ma, Biomaterials, 2016, 104, 18–31 CrossRef CAS PubMed.
- J. Waite, Int. J. Adhes. Adhes., 1987, 7, 9–14 CrossRef CAS.
- D. E. Fullenkamp, L. He, D. G. Barrett, W. R. Burghardt and P. B. Messersmith, Macromolecules, 2013, 46, 1167–1174 CrossRef CAS PubMed.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. DiMarco, L. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC.
- M. S. Menyo, C. J. Hawker and J. H. Waite, Soft Matter, 2013, 9, 10314–10323 RSC.
- S. C. Grindy, M. Lenz and N. Holten-Andersen, Macromolecules, 2016, 49, 8306–8312 CrossRef CAS.
- M. S. Menyo, C. J. Hawker and J. H. Waite, ACS Macro Lett., 2015, 4, 1200–1204 CrossRef CAS PubMed.
- S. Hou and P. X. Ma, Chem. Mater., 2015, 27, 7627–7635 CrossRef CAS PubMed.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- F. Peng, G. Li, X. Liu, S. Wu and Z. Tong, J. Am. Chem. Soc., 2008, 130, 16166–16167 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- S. Tamesue, M. Ohtani, K. Yamada, Y. Ishida, J. M. Spruell, N. A. Lynd, C. J. Hawker and T. Aida, J. Am. Chem. Soc., 2013, 135, 15650–15655 CrossRef CAS PubMed.
- M. Lemmers, J. Sprakel, I. Voets, J. v. d. Gucht and M. C. Stuart, Angew. Chem., Int. Ed., 2010, 49, 708–711 CrossRef CAS PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- D. V. Krogstad, N. A. Lynd, S.-H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- S. A. Fisher, A. E. Baker and M. S. Shoichet, J. Am. Chem. Soc., 2017, 139, 7416–7427 CrossRef CAS PubMed.
- W. H. Landschulz, P. F. Johnson and S. L. McKnight, Science, 1988, 240, 1759–1764 CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS PubMed.
- C. Xu and J. Kopeček, Pharm. Res., 2008, 25, 674–682 CrossRef CAS PubMed.
- C. Xu, V. Breedveld and J. Kopeček, Biomacromolecules, 2005, 6, 1739–1749 CrossRef CAS PubMed.
- W. Shen, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2007, 40, 689–692 CrossRef CAS.
- W. Shen, K. Zhang, J. A. Kornfield and D. A. Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS PubMed.
- J. Yang, C. Xu, C. Wang and J. Kopeček, Biomacromolecules, 2006, 7, 1187–1195 CrossRef CAS PubMed.
- S. Tang, M. J. Glassman, S. Li, S. Socrate and B. D. Olsen, Macromolecules, 2014, 47, 791–799 CrossRef CAS PubMed.
- M. J. Glassman, J. Chan and B. D. Olsen, Adv. Funct. Mater., 2013, 23, 1182–1193 CrossRef CAS PubMed.
- M. J. Glassman and B. D. Olsen, Soft Matter, 2013, 9, 6814–6823 RSC.
- M. J. Glassman and B. D. Olsen, Macromolecules, 2015, 48, 1832–1842 CrossRef CAS.
- C. T. S. Wong Po Foo, J. S. Lee, W. Mulyasasmita, A. Parisi-Amon and S. C. Heilshorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22067–22072 CrossRef PubMed.
- L. Cai, R. E. Dewi and S. C. Heilshorn, Adv. Funct. Mater., 2015, 25, 1344–1351 CrossRef CAS PubMed.
- L. Cai, R. E. Dewi, A. B. Goldstone, J. E. Cohen, A. N. Steele, Y. J. Woo and S. C. Heilshorn, Adv. Healthcare Mater., 2016, 5, 2758–2764 CrossRef CAS PubMed.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- T. Miyata, N. Asami and T. Uragami, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2144–2157 CrossRef CAS.
- M. Ehrbar, R. Schoenmakers, E. H. Christen, M. Fussenegger and W. Weber, Nat. Mater., 2008, 7, 800–804 CrossRef CAS PubMed.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- N. Yamaguchi, B.-S. Chae, L. Zhang, K. L. Kiick and E. M. Furst, Biomacromolecules, 2005, 6, 1931–1940 CrossRef CAS PubMed.
- N. Yamaguchi and K. L. Kiick, Biomacromolecules, 2005, 6, 1921–1930 CrossRef CAS PubMed.
- N. Yamaguchi, L. Zhang, B.-S. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040–3041 CrossRef CAS PubMed.
- H. D. Lu, M. B. Charati, I. L. Kim and J. A. Burdick, Biomaterials, 2012, 33, 2145–2153 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS PubMed.
- S. Rose, A. Prevoteau, P. Elziere, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382–385 CrossRef CAS PubMed.
- A. Parisi-Amon, D. D. Lo, D. T. Montoro, R. E. Dew, M. T. Longaker and S. C. Heilshorn, ACS Biomater. Sci. Eng., 2017, 3, 750–756 CrossRef CAS.
- M. Rubinstein and R. H. Colby, in Polymer Physics, Oxford University Press, Oxford, New York, 2003 Search PubMed.
- Q. Wen, A. Basu, P. A. Janmey and A. G. Yodh, Soft Matter, 2012, 8, 8039–8049 RSC.
- R. B. Bird, in Dynamics of Polymeric Liquids, Wiley, New York, 1987 Search PubMed.
- D. L. Chen, P. F. Yang and Y. S. Lai, Microelectron. Reliab., 2012, 52, 541–558 CrossRef CAS.
- J. Vermant and M. J. Solomon, J. Phys.: Condens. Matter, 2005, 17, R187–R216 CrossRef CAS.
- P. Cicuta and A. M. Donald, Soft Matter, 2007, 3, 1449–1455 RSC.
- T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 1995, 75, 2770–2773 CrossRef CAS PubMed.
- S. Yamada, D. Wirtz and S. C. Kuo, Biophys. J., 2000, 78, 1736–1747 CrossRef CAS PubMed.
- T. H. Larsen and E. M. Furst, Phys. Rev. Lett., 2008, 100, 146001–146004 CrossRef PubMed.
- K. Schultz, K. Kiick and E. Furst, AIP Conf. Proc., 2008, 1027, 1096 CrossRef CAS.
- M. Puig-de Morales, M. Grabulosa, J. Alcaraz, J. Mullol, G. N. Maksym, J. J. Fredberg and D. Navajas, J. Appl. Physiol., 2001, 91, 1152–1159 CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- H. J. Kong, E. Wong and D. J. Mooney, Macromolecules, 2003, 36, 4582–4588 CrossRef CAS.
- E. Buhler, S. J. Candau, E. Kolomiets and J. M. Lehn, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 061804 CrossRef PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- D. Xu, J. L. Hawk, D. M. Loveless, S. L. Jeon and S. L. Craig, Macromolecules, 2010, 43, 3556–3565 CrossRef CAS PubMed.
- T. Indei, T. Koga and F. Tanaka, Macromol. Rapid Commun., 2005, 26, 701–706 CrossRef CAS.
- E. M. Cherry and J. K. Eaton, Phys. Fluids, 2013, 25, 073104–073119 CrossRef.
- D. Izidoro, M. R. Sierakowski, N. Waszczynskyj, C. W. I. Haminiuk and A. D. Scheer, Int. J. Food Eng., 2007, 3, 17 Search PubMed.
- L. Zhong, M. Oostrom, M. J. Truex, V. R. Vermeul and J. E. Szecsody, J. Hazard. Mater., 2012, 244, 160–170 Search PubMed.
- L. Ouyang, C. B. Highley, C. B. Rodell, W. Sun and J. A. Burdick, ACS Biomater. Sci. Eng., 2016, 2, 1743–1751 CrossRef CAS.
- W. A. P. Hayward, L. J. Haseler, L. G. Kettwich, A. A. Michael, W. L. Sibbitt and A. D. Bankhurst, Scand. J. Rheumatol., 2011, 40, 379–382 CrossRef CAS PubMed.
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- V. Kanelis, D. Rotin and J. D. Forman-Kay, Nat. Struct. Biol., 2001, 8, 407–412 CrossRef CAS PubMed.
- B. D. Olsen, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2010, 43, 9094–9099 CrossRef CAS PubMed.
- E. Loizou, P. Butler, L. Porcar and G. Schmidt, Macromolecules, 2006, 39, 1614–1619 CrossRef CAS.
- C. B. Rodell, J. W. MacArthur, S. M. Dorsey, R. J. Wade, L. L. Wang, Y. J. Woo and J. A. Burdick, Adv. Funct. Mater., 2015, 25, 636–644 CrossRef CAS PubMed.
- C. B. Rodell, N. N. Dusaj, C. B. Highley and J. A. Burdick, Adv. Mater., 2016, 28, 8419–8424 CrossRef CAS PubMed.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed.
- M. E. Young, P. A. Carroad and R. L. Bell, Biotechnol. Bioeng., 1980, 22, 947–955 CrossRef CAS.
- R. Mateen and T. Hoare, J. Mater. Chem. B, 2014, 2, 5157–5167 RSC.
- E. A. Appel, R. A. Forster, M. J. Rowland and O. A. Scherman, Biomaterials, 2014, 35, 9897–9903 CrossRef CAS PubMed.
- Y. M. Kolambkar, K. M. Dupont, J. D. Boerckel, N. Huebsch, D. J. Mooney, D. W. Hutmacher and R. E. Guldberg, Biomaterials, 2011, 32, 65–74 CrossRef CAS PubMed.
- E. A. Silva and D. J. Mooney, J. Thromb. Haemostasis, 2007, 5, 590–598 CrossRef CAS PubMed.
- J. E. Mealy, C. B. Rodell and J. A. Burdick, J. Mater. Chem. B, 2015, 3, 8010–8019 RSC.
- H. Rosen and T. Abribat, Nat. Rev. Drug Discovery, 2005, 4, 381–385 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS.
- J. Su, B. H. Hu, W. L. Lowe, D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 308–314 CrossRef CAS PubMed.
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Bioconjugate Chem., 2015, 26, 2279–2289 CrossRef CAS PubMed.
- L. L. Wang, J. N. Sloand, A. C. Gaffey, C. M. Venkataraman, Z. Wang, A. Trubelja, D. A. Hammer, P. Atluri and J. A. Burdick, Biomacromolecules, 2017, 18, 77–86 CrossRef CAS PubMed.
- Y. K. Liang and K. L. Kiick, Acta Biomater., 2014, 10, 1588–1600 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, J. M. Greer, O. S. Fenton, K. Kreuels, D. G. Anderson and R. Langer, ACS Macro Lett., 2015, 4, 848–852 CrossRef CAS.
- C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS PubMed.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- R. C. Luo, S. S. Venkatraman and B. Neu, Biomacromolecules, 2013, 14, 2262–2271 CrossRef CAS PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210–211, 831–835 CrossRef CAS.
- T. Boudou, T. Crouzier, K. F. Ren, G. Blin and C. Picart, Adv. Mater., 2010, 22, 441–467 CrossRef CAS PubMed.
- R. A. Rajewski and V. J. Stella, J. Pharm. Sci., 1996, 85, 1142–1169 CrossRef CAS PubMed.
- L. Szente and J. Szejtli, Adv. Drug Delivery Rev., 1999, 36, 17–28 CrossRef CAS PubMed.
- K. Okimoto, R. A. Rajewski, K. Uekama, J. A. Jona and V. J. Stella, Pharm. Res., 1996, 13, 256–264 CrossRef CAS.
- R. C. Smith, M. Riollano, A. Leung and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 8974–8977 CrossRef CAS PubMed.
- J. Jing, A. Szarpak-Jankowska, R. Guillot, I. Pignot-Paintrand, C. Picart and R. Auzély-Velty, Chem. Mater., 2013, 25, 3867–3873 CrossRef CAS.
- V. Bohmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
- K. Wang, D. S. Guo, X. Wang and Y. Liu, ACS Nano, 2011, 5, 2880–2894 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 73, 4347 Search PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- Y. J. Jeon, S. Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122–2125 CAS.
- N. Dong, S. F. Xue, Q. J. Zhu, Z. Tao, Y. Zhao and L. X. Yang, Supramol. Chem., 2008, 20, 663–671 CrossRef.
- A. L. Koner, I. Ghosh, N. Saleh and W. M. Nau, Can. J. Chem., 2011, 89, 139–147 CrossRef CAS.
- Y. J. Zhao, D. P. Buck, D. L. Morris, M. H. Pourgholami, A. I. Day and J. G. Collins, Org. Biomol. Chem., 2008, 6, 4509–4515 CAS.
- S. M. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed.
- J. Mohanty, S. D. Choudhury, H. P. Upadhyaya, A. C. Bhasikuttan and H. Pal, Chem. – Eur. J., 2009, 15, 5215–5219 CrossRef CAS PubMed.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690–694 CrossRef CAS PubMed.
- X. Xu, E. A. Appel, X. Liu, R. M. Parker, O. A. Scherman and C. Abell, Biomacromolecules, 2015, 16, 2743–2749 CrossRef CAS PubMed.
- Z. Y. Yu, J. Zhang, R. J. Coulston, R. M. Parker, F. Biedermann, X. Liu, O. A. Scherman and C. Abell, Chem. Sci., 2015, 6, 4929–4933 RSC.
- C. B. Rodell, R. Rai, S. Faubel, J. A. Burdick and D. E. Soranno, J. Controlled Release, 2015, 206, 131–139 CrossRef CAS PubMed.
- D. E. Soranno, H. D. Lu, H. M. Weber, R. Rai and J. A. Burdick, J. Biomed. Mater. Res., Part A, 2014, 102, 2173–2180 CrossRef PubMed.
- Y. Gao, Z. Y. Sun, Z. H. Huang, P. G. Chen, Y. X. Chen, Y. F. Zhao and Y. M. Li, Chem. – Eur. J., 2014, 20, 13541–13546 CrossRef CAS PubMed.
- R. Kawabata, H. Wada, M. Isobe, T. Saika, S. Sato, A. Uenaka, H. Miyata, T. Yasuda, Y. Doki, Y. Noguchi, H. Kumon, K. Tsuji, K. Iwatsuki, H. Shiku, G. Ritter, R. Murphy, E. Hoffman, L. J. Old, M. Monden and E. Nakayama, Int. J. Cancer, 2007, 120, 2178–2184 CrossRef CAS PubMed.
- A. Uenaka, H. Wada, M. Isobe, T. Saika, K. Tsuji, E. Sato, S. Sato, Y. Noguchi, R. Kawabata, T. Yasuda, Y. Doki, H. Kumon, K. Iwatsuki, H. Shiku, M. Monden, A. A. Jungbluth, G. Ritter, R. Murphy, E. Hoffman, L. J. Old and E. Nakayama, Cancer Immun., 2007, 7, 9 Search PubMed.
- T. Nochi, Y. Yuki, H. Takahashi, S. Sawada, M. Mejima, T. Kohda, N. Harada, I. G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS PubMed.
- A. Purwada, Y. F. Tian, W. Huang, K. M. Rohrbach, S. Deol, A. August and A. Singh, Adv. Healthcare Mater., 2016, 5, 1413–1419 CrossRef CAS PubMed.
- H. Yim, W. Park, D. Kim, T. M. Fahmy and K. Na, Biomaterials, 2014, 35, 9912–9919 CrossRef CAS PubMed.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- Y. Wen and J. H. Collier, Curr. Opin. Immunol., 2015, 35, 73–79 CrossRef CAS PubMed.
- R. R. Pompano, J. Chen, E. A. Verbus, H. Han, A. Fridman, T. McNeely, J. H. Collier and A. S. Chong, Adv. Healthcare Mater., 2014, 3, 1898–1908 CrossRef CAS PubMed.
- M. Black, A. Trent, Y. Kostenko, J. S. Lee, C. Olive and M. Tirrell, Adv. Mater., 2012, 24, 3845–3849 CrossRef CAS PubMed.
- C. Mason and P. Dunnill, Regener. Med., 2008, 3, 1–5 CrossRef PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- D. E. Ingber, FASEB J., 2006, 20, 811–827 CrossRef CAS PubMed.
- N. Wang, J. D. Tytell and D. E. Ingber, Nat. Rev. Mol. Cell Biol., 2009, 10, 75–82 CrossRef CAS PubMed.
- M. A. Schwartz, Cold Spring Harbor Perspect. Biol., 2010, 2, 1–9 Search PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Nat. Commun., 2015, 6, 6364 CrossRef PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- K. Lee, E. A. Silva and D. J. Mooney, J. R. Soc., Interface, 2011, 8, 153–170 CrossRef CAS PubMed.
- R. R. Chen and D. J. Mooney, Pharm. Res., 2003, 20, 1103–1112 CrossRef CAS.
- Y. Tabata, Tissue Eng., 2003, 9(Suppl 1), S5–S15 CrossRef CAS PubMed.
- F. M. Chen, M. Zhang and Z. F. Wu, Biomaterials, 2010, 31, 6279–6308 CrossRef CAS PubMed.
- T. Wang, X.-J. Jiang, T. Lin, S. Ren, X.-Y. Li, X.-Z. Zhang and Q.-z. Tang, Biomaterials, 2009, 30, 4161–4167 CrossRef CAS PubMed.
- S. Koudstaal, M. M. C. Bastings, D. A. M. Feyen, C. D. Waring, F. J. Van Slochteren, P. Y. W. Dankers, D. Torella, J. P. G. Sluijter, B. Nadal-Ginard, P. A. Doevendans, G. M. Ellison and S. A. J. Chamuleau, J. Cardiovasc. Transl. Res., 2014, 7, 232–241 CrossRef PubMed.
- M. M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2014, 3, 70–78 CrossRef CAS PubMed.
- T. Wang, X.-J. Jiang, Q.-Z. Tang, X.-Y. Li, T. Lin, D.-Q. Wu, X.-Z. Zhang and E. Okello, Acta Biomater., 2009, 5, 2939–2944 CrossRef CAS PubMed.
- D.-Q. Wu, T. Wang, B. Lu, X.-D. Xu, S.-X. Cheng, X.-J. Jiang, X.-Z. Zhang and R.-X. Zhuo, Langmuir, 2008, 24, 10306–10312 CrossRef CAS PubMed.
- A. Parisi-Amon, W. Mulyasasmita, C. Chung and S. C. Heilshorn, Adv. Healthcare Mater., 2013, 2, 428–432 CrossRef CAS PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- T. Jungst, W. Smolan, K. Schacht, T. Scheibel and J. Groll, Chem. Rev., 2016, 116, 1496–1539 CrossRef CAS PubMed.
- C. Mandrycky, Z. Wang, K. Kim and D. H. Kim, Biotechnol. Adv., 2016, 34, 422–434 CrossRef CAS PubMed.
- I. T. Ozbolat, K. K. Moncal and H. Gudapati, Addit. Manuf., 2017, 13, 179–200 CrossRef CAS.
- D. Chimene, K. K. Lennox, R. R. Kaunas and A. K. Gaharwar, Ann. Biomed. Eng., 2016, 44, 2090–2102 CrossRef PubMed.
- D. M. Kirchmajer, R. Gorkin III and M. in het Panhuis, J. Mater. Chem. B, 2015, 3, 4105–4117 RSC.
- W. Zhu, X. Ma, M. Gou, D. Mei, K. Zhang and S. Chen, Curr. Opin. Biotechnol., 2016, 40, 103–112 CrossRef CAS PubMed.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2012, 18, 806–815 CrossRef CAS PubMed.
- K. Dubbin, Y. Hori, K. K. Lewis and S. C. Heilshorn, Adv. Healthcare Mater., 2016, 5, 2488–2492 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- C. Li, A. Faulkner-Jones, A. R. Dun, J. Jin, P. Chen, Y. Xing, Z. Yang, Z. Li, W. Shu, D. Liu and R. R. Duncan, Angew. Chem., Int. Ed., 2015, 54, 3957–3961 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |