DOI:
10.1039/C7BM00882A
(Review Article)
Biomater. Sci., 2018,
6, 38-59
Phenylalanine and derivatives as versatile low-molecular-weight gelators: design, structure and tailored function
Received
28th September 2017
, Accepted 3rd November 2017
First published on 3rd November 2017
Abstract
Phenylalanine (Phe) is an essential amino acid classified as neutral and nonpolar due to the hydrophobic nature of the benzyl side chain. In the field of materials science, the chemical modification of phenylalanine at C or N terminus has enabled to synthesize a large number of low-molecular-weight gelators over the past decade. Thus, many physical (or supramolecular) softgel materials have been fabricated by self-assembly of Phe-derived building blocks, which can be programmed with atomic level information and modification. The process of self-assembly and gelation must balance the parameters that influence the solubility as well as the contrasting forces that dictate epitaxial growth into entangled fibrillar aggregates. Gelator–gelator and solvent–gelator interactions are known to be highly important for the gelation process, and the non-covalent nature of these interactions provides physical gels with important properties such as reversible phase transitions and responsiveness towards external stimuli. Among other applications, these gels have been used for drug delivery, as extracellular matrix for tissue engineering, for oil spills recovery, removal of dyes, extraction of heavy metals or pollutants, and for the detection of explosives. In this tutorial review, we highlight the advances in the design, synthesis and applications of supramolecular gels made of Phe and derivatives.
 Tanmay Das | Tanmay Das obtained his MSc in Chemistry from Indian Institute of Technology Delhi in 2014. Then, he moved to Indian Institute of Science Education and Research Kolkata, where he is currently in the 3rd year of his PhD under supervision of Professor Debasish Haldar. His research focuses on the self-assembly behavior and stimuli-responsive properties of small peptides and peptidomimetics. |
 Marleen Häring | Marleen Häring obtained both BSc and the MSc degrees from the University of Regensburg (Germany). She completed her Master's thesis on biopolymer-based catalysis under the supervision of Prof. David Díaz Díaz at the University of Regensburg. Since 2015 she has been pursuing her PhD in Chemistry in the same group working on the development and applications of new chemical and physical gels. |
 Debasish Haldar | Debasish Haldar received his M.Sc. in Organic Chemistry from University of Calcutta in 1999. In 2005, he was awarded with the PhD degree from Jadavpur University. After post-doctoral research stays at University of Bordeaux, France; University of Manchester, U.K. and University of Würzburg, Germany, he joined Indian Institute of Science Education and Research Kolkata as Assistant Professor in August 2008. Currently, he is an Associate Professor of Organic Chemistry and his research interests range across synthesis of amino acids and analogues, peptides, peptidemimetics and their applications. Haldar is co-author of 80 research papers, four books and six book chapters. |
 David Díaz Díaz | David Díaz Díaz received his PhD in Chemistry from University of La Laguna (Spain). In 2002, he joined Prof. Finn's group as postdoc at TSRI (USA). Since 2006, he has held various positions in academia and industry (‘Ramón y Cajal’ Researcher, UAM, Spain, 2006; Sr Chemist, Dow Chemical, Switzerland, 2007; Tenured Scientist, CSIC, Spain, 2009; Alexander von Humboldt Experienced Researcher, Univ. Regensburg, Germany, 2010). In 2013, he was awarded with the prestigious DFG Heisenberg Professorship and appointed as Associate Professor at the University of Regensburg. He has received the Young Investigator Award from the Polymer Network Group and is the Editor-in-Chief of Gels. He has co-authored more than 160 publications and his main research interest focuses on the development of new functional soft materials. |
Introduction
Viscoelastic gel-based materials are highly important in everyday life. Natural hydrogels made from pectin, gelatine and agar are major ingredients in food products, whereas synthetic gels (including both organo- and hydrogels) such as lubricants, adhesives, soaps, cosmetics, medical implants and explosives play a key role in the development of our modern society.1 During the last few decades, supramolecular gels have gained immense attention due to their potential applications in various fields such as drug delivery,2–4 clinical therapies,5 catalysis,6 tissue engineering,7,8 biosensing,9 cell culture matrix,10 self-healing materials,11 antimicrobial materials,12 pollutant removal,13–15 sensing and soft lithography,16 microarray kit design,17 and templated nanomaterial syntheses.18 Gels have a continuous macroscopic structure and display a solid-like rheological behavior. In contrast to chemical gels, which are based on covalent bonds (usually cross-linked polymers unable to redissolve), physical (or supramolecular) gels are made of either low-molecular-weight (LMW) compounds or polymers, so-called gelators, through different non-covalent interactions such as hydrogen-bonding, π–π stacking, van der Waals, charge-transfer, donor–acceptor, dipole–dipole, and coordination interactions.19
The solid-like appearance of the gels is the result of the entrapment of the liquid, the major component, in the interstices of a solid 3D self-assembled matrix, usually through capillary forces. In the case of physical gels, the viscoelastic matrix is formed via entanglement of one dimensional (1D) supramolecular nanoscale or microscale structures, usually fibers, which is typically induced by cooling their hot isotropic solutions to room temperature.20–26
Over the last two decades such LMW gelators27–30 have turned out to be attractive alternative for polymeric systems to fabricate gels because of their unique properties, which are unattainable by polymeric gelators such as the above-mentioned reversible responsiveness to a variety of external stimuli.31,32 Nevertheless, despite the rapid increase in the number of LMW gelators observed during the last decade, it is still difficult to predict whether a particular compound will form gel or not. There are reports where only a simple modification in a peptide sequence can create a perfect gelator from a non-gelator substance.66 This difficulty stems from the complex 3D intermolecular nature of gels and the different mechanisms associated to the hierarchical supramolecular organization of LMW gelators.33–36
Peptides are manufactured from short chains of amino acid monomers and are among the most popular LMW gelating moieties reported in the literature,37–40 where the gelation process can be triggered using a variety of stimuli including light, pH, temperature, ionic strength and enzymes.41–48 In addition, supramolecular gels derived from peptide-based gelators have attracted significant attention due to their inherent biocompatibility, biodegradability, and ability to easily manipulate their chemical functionality and properties simply by changing their primary building blocks.49,50
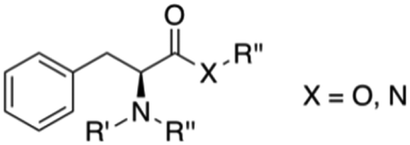
If we focus on the monomeric units of the peptides, phenylalanine (Phe) and its derivatives have been found to be also very effective gelators. Phe is an essential amino acid that is classified as neutral and non-polar due to the hydrophobic nature of the aromatic side chain. Both π–π interactions induced by the phenyl group and the hydrogen bonding characteristic of the amide group cooperate to bring about effective gelation in numerous solvents. During the last decade, the chemical modification of Phe at C or N terminus has allowed to develop a large number of LMW gelator with potential for different applications. This review summarizes the major achievements in the field of LMW gelators using Phe as the key moiety, in which modifications are limited either to the substitution at the amino group and/or the replacement of the carboxylic function by an amide unit (Fig. 1). Thus, gelators having substitution or modifications at the phenyl ring and Phe-containing peptide gelators are out of the scope of this review. The focus of this compilation lies on the design strategy used to prepare the building blocks and their self-assembly pattern.
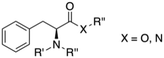 |
| | Fig. 1 General gelator structure related to Phe showing the positions of structural modifications covered in this review (i.e., R′, R′′, R′′′, X). | |
Modular architecture of Phe-based gelators
Gels made of unmodified Phe
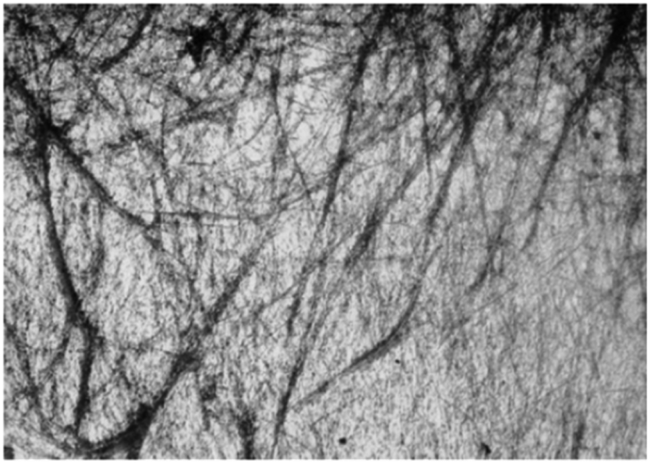
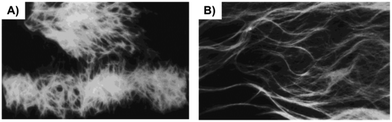
Out of 22 naturally occurring amino acids, Phe is the only one until now that has been proved to form supramolecular gels with a free N-terminus. Tverdislov and co-workers described the formation of strings in aqueous solution of Phe (10−1 M) (Fig. 2).51 Microscopic investigation revealed subsequent weaving of thin strings into thick ones with a clear correlation between the thickness of the strings and Phe concentration. This study indicated that biological fluids are the most likely anisometric fluids, which are built by associations and strings of many chiral molecules. In some cases, anisometric gels can be formed via associations of these chiral strings.
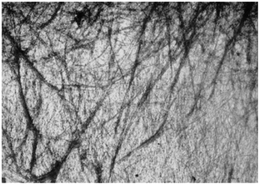 |
| | Fig. 2 Strings formed in aqueous solution of Phe, ×100. Reproduced with permission from ref. 51. Copyright 2012, Springer Science+Business Media, Inc. | |
Metallogels made of Phe
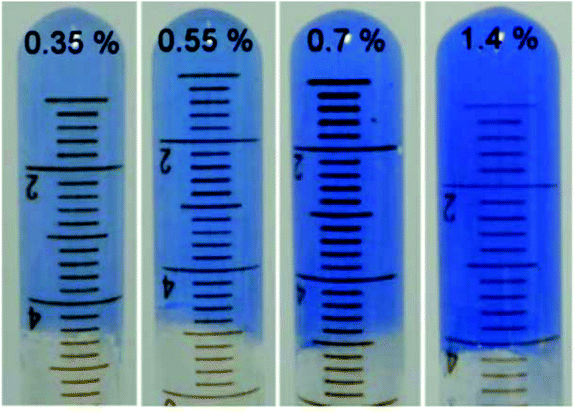
Metal–ligand coordination chemistry for direct assembly of small molecules constitutes also a valuable tool in materials synthesis.52 Thus, the incorporation of metal ions in gel-based materials, forming so-called metallogels, has gained increasing attention during the last decade.53 Within this context, Shen and co-workers reported the formation of metallogels by direct coordination of Cu(II) with unmodified Phe.54 In this particular case, Phe was found to be extraordinarily selective over other metal ions, and only in the presence of Cu(NO3)2 with a ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2 equivalents of NaOH, it allowed the in situ formation of blue translucent aqueous metallogels (Fig. 3). In addition, the authors demonstrated by circular dichroism (CD) spectroscopy that the chirality of Phe is able to modulate the formation of stable metallogels by using different enantiomeric excess (ee) of mixtures of L- and D- Phe. The Phe-Cu(II) system lost its gelation ability when ee% was lower than 30%. A semi-gel was obtained at 50% enantiomeric excess and a fully gel state was achieved at ee% higher than 80%. In case of DL-Phe-Cu(II) (ee% is equal to 0%) no gelation occurred even after increasing the content by 2%. These observations suggest that efficient intermolecular hydrogen bonding, hydrophobic and/or π–π stacking interactions, which are responsible for effective gelation might be weakened as ee% lowered. So this kind of gelation method can be used in chiral sensing as well as recognition applications. Furthermore, the study reveals that no gelation can be attained using other α-amino acids such as alanine, leucine, tryptophan and histidine, despite their potential coordination with Cu(II). These results suggest that the hydrophobic phenyl ring of Phe might play an important role in the gelation process.
:1 and 2 equivalents of NaOH, it allowed the in situ formation of blue translucent aqueous metallogels (Fig. 3). In addition, the authors demonstrated by circular dichroism (CD) spectroscopy that the chirality of Phe is able to modulate the formation of stable metallogels by using different enantiomeric excess (ee) of mixtures of L- and D- Phe. The Phe-Cu(II) system lost its gelation ability when ee% was lower than 30%. A semi-gel was obtained at 50% enantiomeric excess and a fully gel state was achieved at ee% higher than 80%. In case of DL-Phe-Cu(II) (ee% is equal to 0%) no gelation occurred even after increasing the content by 2%. These observations suggest that efficient intermolecular hydrogen bonding, hydrophobic and/or π–π stacking interactions, which are responsible for effective gelation might be weakened as ee% lowered. So this kind of gelation method can be used in chiral sensing as well as recognition applications. Furthermore, the study reveals that no gelation can be attained using other α-amino acids such as alanine, leucine, tryptophan and histidine, despite their potential coordination with Cu(II). These results suggest that the hydrophobic phenyl ring of Phe might play an important role in the gelation process.
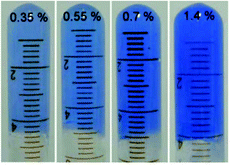 |
| | Fig. 3 Supramolecular metallogels obtained by varying L-Phe-Cu(II) content (wt%). Reproduced with permission from ref. 52. Copyright 2010, The Royal Society of Chemistry. | |
Gels made of Phe-amide derivatives
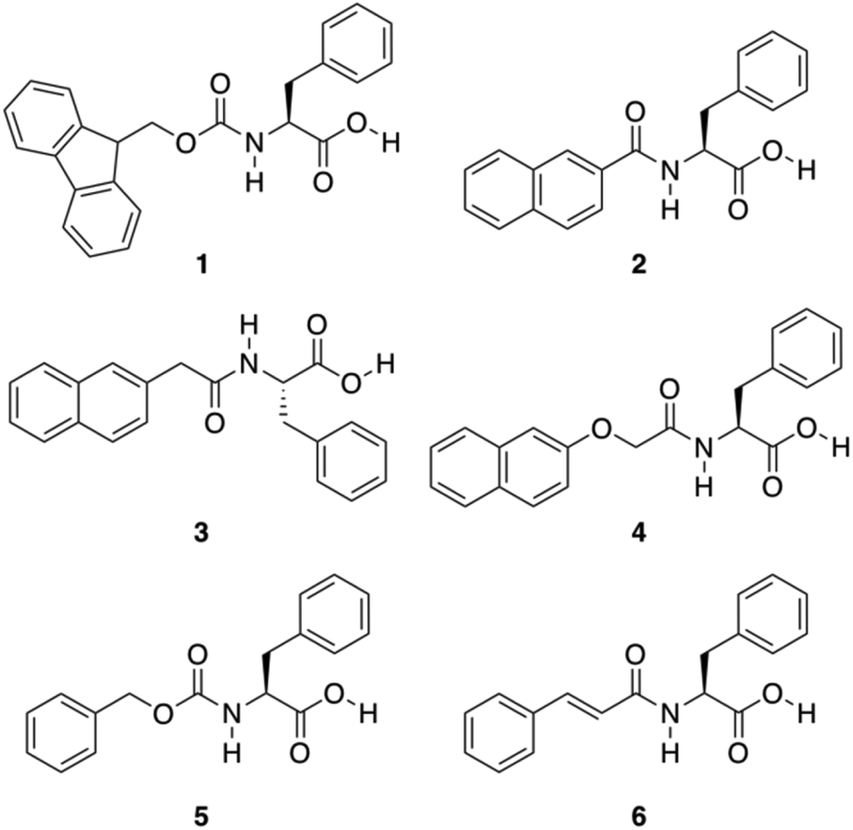
A great variety of gelators55–58 can be designed by introducing carbonyl amide groups into linker structures in order to provide hydrogen-bond formation sites and enhance their aggregation ability. Accordingly, Shi and co-workers evaluated the gelation ability of several compounds 1–6 where different aromatic moieties such as fluorenyl, naphthyl, naphthalenoxyl, and cinnamoyl groups were covalently attached to Phe via an amide linkage (Fig. 4).59 Besides the effect of the amide group in hydrogen-bonding, the influence of aromatic–aromatic interactions on the gelation ability was also investigated. Based on the emission spectra of the hydrogels, it was confirmed that aromatic–aromatic interactions increased in the gel state, which is supported by the fact that most of bands in the gel phase show red shift. The results revealed that these aromatic groups are able to induce the formation of well-defined cross-linking networks affording stable hydrogels. These gels showed critical gelation concentrations (CGCs) ranging from 0.3 to 1.0 wt%, and displayed pH and thermoreversibility. Interestingly, compounds 2 and 3 failed to form gels, which was atrributed to the conjugation between the carbonyl and the naphthyl groups, partially reducing the rotational freedom of the naphthyl group, which is essential for the self-assembly process. After screening several functional groups they came to the conclusion that the cinnamoyl group is the minimum structure motif to provide sufficient aromatic–aromatic interactions for a phenylalanine derivative to be a hydrogelator.
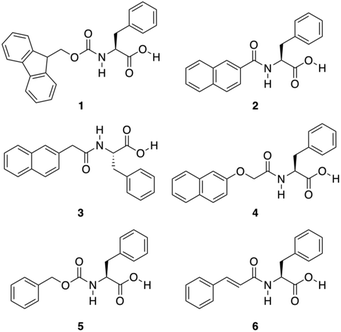 |
| | Fig. 4 Chemical structures of Phe derivatives 1–6. Adapted with permission from ref. 59. Copyright 2011, Beilstein-journals. | |
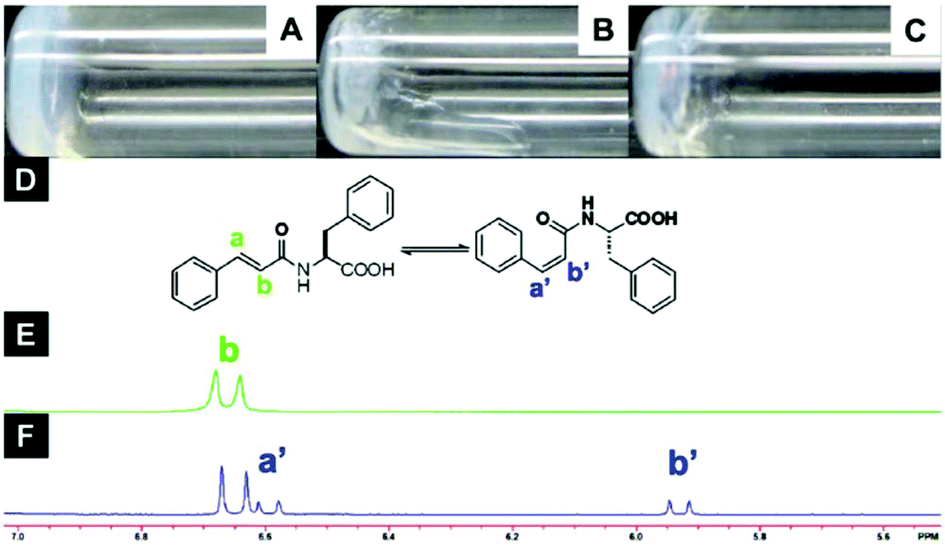
Furthermore, it is important to highlight that the highest tendency to aggregate is shown by 6, which also exhibits a photoinduced transition to cis isomer 7 upon UV irradiation with concurrent gel-to-sol transition (Fig. 5).
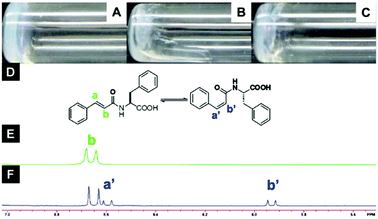 |
| | Fig. 5 Optical images of (A) gel made of 6 (1.5 wt%, pH = 4.6), (B) gel after UV irradiation (no aging), (C) gel after UV irradiation (aging 2 h), (D) structure of compound 6 in trans- and cis-isomers, (E) NMR spectra of compound 6 before and (F) after UV irradiation for 2 h. Adapted with permission from ref. 59. Copyright 2011, Beilstein-journals. | |
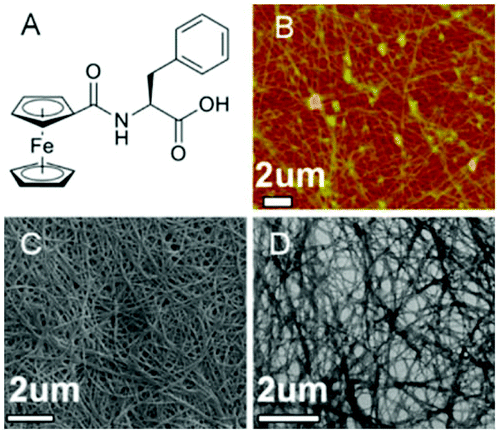
The inclusion of a ferrocenyl (Fc) residue attached by amide bond to Phe has been investigated in some studies. The synthesis of these new ferrocene derivatives is of considerable interest. It is expected that a change in the oxidation state of the central ion of the ferrocenyl residue would largely modulate the interaction of adjacent ferrocenyl moieties of these compounds, which would influence the intermolecular hydrogen bonding interaction and hence affect the supramolecular structures.60–62 Moreover, the ferrocene group is a possible building block in the design of potential new drugs due to its high stability, low toxicity, and bulkiness. Recently, Zhang and co-workers reported the discovery of ferrocenoyl Phe 8, an exceptionally small hydrogelator with a simple structure and potential responsive properties.63 This work was inspired by two previously reported organogelators: a redox-active ferrocenyl moiety, which is attached to one cholesteryl residue linked by different diamino units64 and an Fmoc-PhePhe.65 In general, the obtained hydrogels were very stable and achieved at very low CGC (between 2–3 g L−1). Morphological studies revealed the presence of a rare aggregation to well-organized fibrils of ferrocene attached Phe (Fig. 6). In order to evaluate the role that phenylalanine in the gelation process, they also synthesized the following series of Fc-amino acid conjugates: Fc-Tyrosine, Fc-Tryptophan, Fc-Histidine, Fc-Alanine, and Fc-Lysine. None of these compounds were observed to form hydrogels, which therefore suggests that the phenyl group of Fc-F plays a critically important role in gel formation. Moreover, additional assays also confirmed that the phenyl group of 8 plays a key role in the gel formation. Regarding the chirality, both levorotatory (L-) and dextrorotatory- (D-) Fc-F form stable hydrogels. However making hydrogels using a 1:1 mixture of L-Fc-F and D-Fc-F takes significantly longer time to self-assemble than the pure enantiomer alone. Furthermore, these gels showed responsiveness to pH changes, chemical and electrochemical redox, and the presence of a ligand receptor. The use of these hydrogels covers a wide spectrum of applications including electron transfer matrix for immobilized enzymes, drug carrier, catalyst support in chiral separation techniques, and gel electrolyte.
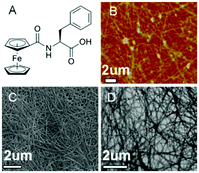 |
| | Fig. 6 (A) Ferrocene-appended Phe gelator 8. (B) AFM, (C) SEM, and (D) TEM images of the cryo-dried hydrogel, respectively. Adapted with permission from ref. 63. Copyright 2013, American Chemical Society. | |
The same research group reported the fabrication of well-organized hybrid aerogels of Fe3O4/N-doped graphene which were directly prepared from 8/GO supramolecular hydrogels via in situ hydrothermal treatments.6 Gelator 8 acted as Fe and N as precursor. They found that GO could trigger a sol-to-gel transition of gelator 8 significantly below the CGC, which indicated that GO acts as a positive co-gelator besides the solid matrix. GO nanosheets, with their abundant functional groups, stabilize supramolecular hydrogels and assist in a good distribution of Fe3O4 nanoparticles on ultrathin graphene films. Microscopical investigations were applied to get an insight into the structures and morphologies of hybrid 8/GO hydrogels. The electron microscopic results confirmed that the self-assembled fibrils of 8 are tightly bound onto the graphene sheets. The electrocatalytic properties of Fe3O4/N-GAs for ORR were studied using cyclic voltammetry, which showed high efficiency in catalyzing the electrochemical ORR compared to commercial Pt/C. In addition, the synthesized Fe3O4 nanoparticles were found to inhibit the self-aggregation among the graphene sheets during GO reduction process. It is also important to emphasize that these sheets contain many channels, which enhance charge mobility within the films.
Ferrocenoyl Phe 8 gelator has been found to be very effective also in other applications. For instance, a biosensor for glucose was developed by Yang and co-workers.9 Glucose oxidase (GOx) was immobilized into the hydrogel prepared from 8. For this purpose, a solution of GOx was mixed with the hydrogel, and dropped it on an electrode surface which was covered with a chitosan layer to stabilize it. The advantage of this arrangement is the presence of the ferrocene as artificial electron transfer mediator for GOx. Moreover, the authors investigated the performance of the proposed biosensor for the electrochemical detection of glucose. They found that their results were in agreement with the conventional colorimetric method in donated blood samples determined by a hospital. The favorable results were attributed to the biocompatible microenvironment provided by the hydrogel and the significant amount of ferrocene moieties.
Yang and co-workers also used the redox hydrogel prepared from 8 to design an electrochemical immunosensing platform for the label-free detection of the cancer biomarker prostate specific antigen (PSA).5 Here, the hydrogel modified electrode advantageously displays a pair of strong current peaks originated from the ferrocenyl moieties. The key mechanism is based on the interaction between the anti-PSA immobilized on the electrode and the PSA present in the sample. Thereby, this interaction allowed the formation of an immuno-complex on the electrode, which produced a decrease in the peak current allowing the quantitative detection of PSA. The current change was proportional to the concentration of PSA detected in the range from 1 pg mL−1 to 10 ng mL−1 with a detection limit of 0.5 pg mL−1.
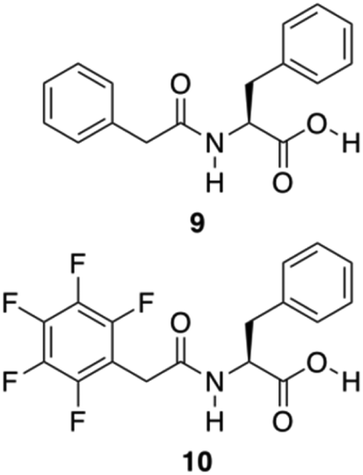
Further approaches have been developed to design Phe-amide based gelators. The phenyl/perfluorophenyl pair, a supramolecular synthon, has attracted great attention in designing structures for crystal engineering and for reaction control in solution.67 Hexafluorobenzene or benzene showed herringbone crystal packing,68,69 whereas an equimolar composition of hexafluorobenzene and benzene showed alternating and parallel packing.70 Even in the gas and liquid phases, the molecular planes between these pairs are almost parallel.71,72 Computational studies indicate that alternating and parallel packing of a hexafluorobenzene/benzene pair are due to an optimization of quadrupole–quadrupole, dispersion, and C–H⋯F–C interactions.73–75 A water mediated trimeric complex of hexafluorobenzene/water/benzene is also theoretically possible, and calculations determined that the water sandwiched between two aromatics has larger binding energy compared to the alternate and parallel dimer without water.76 Inspired by these observations, Lin and co-workers developed the phenyl/perfluorophenyl pair 9–10 for the formation of supramolecular hydrogels (Fig. 7).77
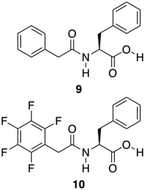 |
| | Fig. 7 Chemical structures of compounds 9 and 10. Adapted with permission from ref. 77. Copyright 2014, John Wiley and Sons. | |
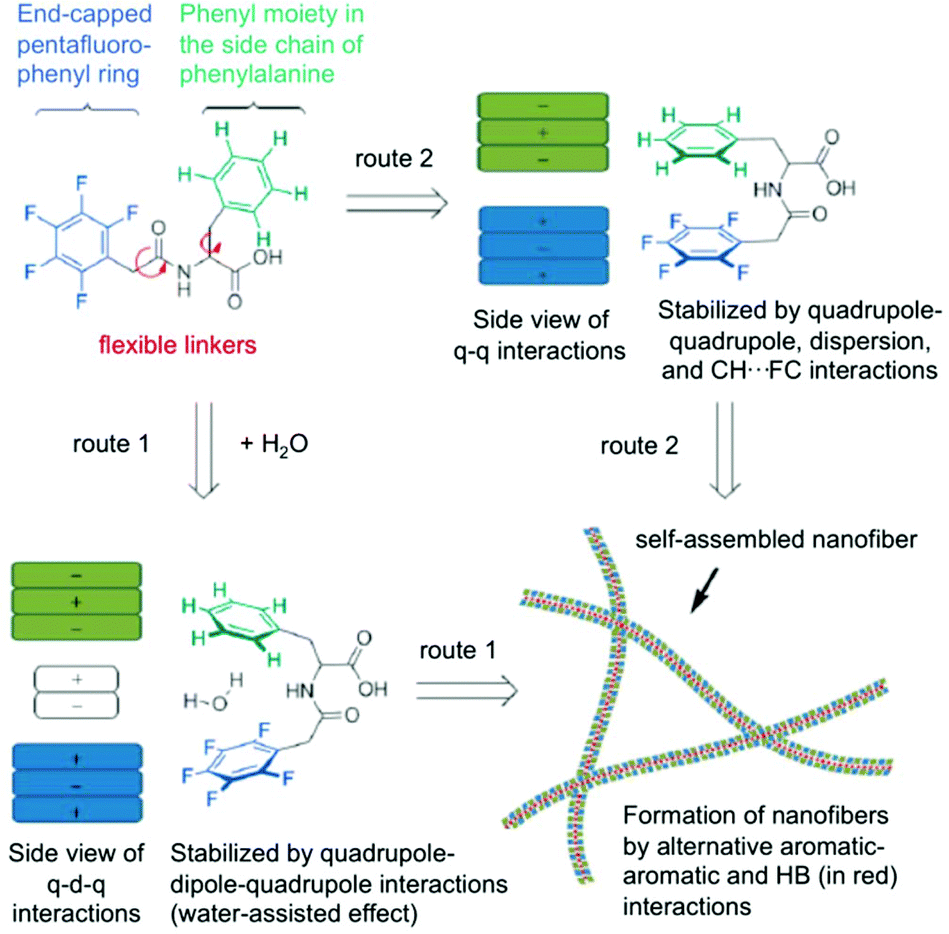
Here, two possible routes are proposed to form self-assembled nanofibers and hydrogels (Fig. 8). The first route contains an alternating packing of the phenyl 9/perfluorophenyl 10 pair by the quadrupole–dipole–quadrupole interaction where water is sandwiched between the two aromatic moieties. The second route includes an alternating arrangement of two aromatics resulting from the balance of quadrupole–quadrupole, dispersion and CH–FC interactions. Hydrogels could be prepared from these peptides by a sequential change in pH. The microscopic nanostructures were visualized from negatively stained TEM analysis, which revealed that the hydrogel consists a fibrous network. No gelation was achieved in the second route within the concentration range of 1–3 wt%. The dramatic difference in the hydrogel formation indicates a fluorous effect that is crucial on the self-assembly and hydrogelation. It is evident that the intramolecular effect of pentafluorobenzyl-L-Phe is responsible for the gel formation whereas the intermolecular interaction of the pentafluorobenzyl and phenyl groups, as the only difference between the two molecules is the position of the benzyl side chain of the amino acid, plays no crucial role in the assembly process. In conclusion, after combining all kinds of spectroscopic data, it can be deduced that a cooperative effect of aromatic–aromatic and hydrogen-bonding interactions are the main driving force for the formation of self-assembled nanofibers and hydrogels.
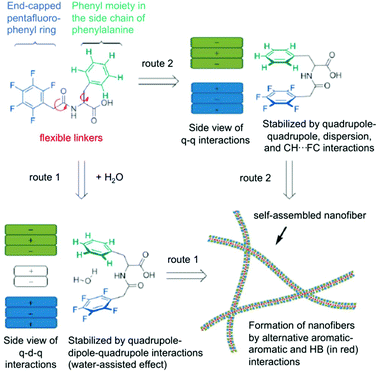 |
| | Fig. 8 Proposed routes for intramolecular interactions and intermolecular self-assembly of hydrogelators (q = quadrupole, d = dipole, HB = hydrogen bonding). Adapted with permission from ref. 77. Copyright 2014, John Wiley and Sons. | |
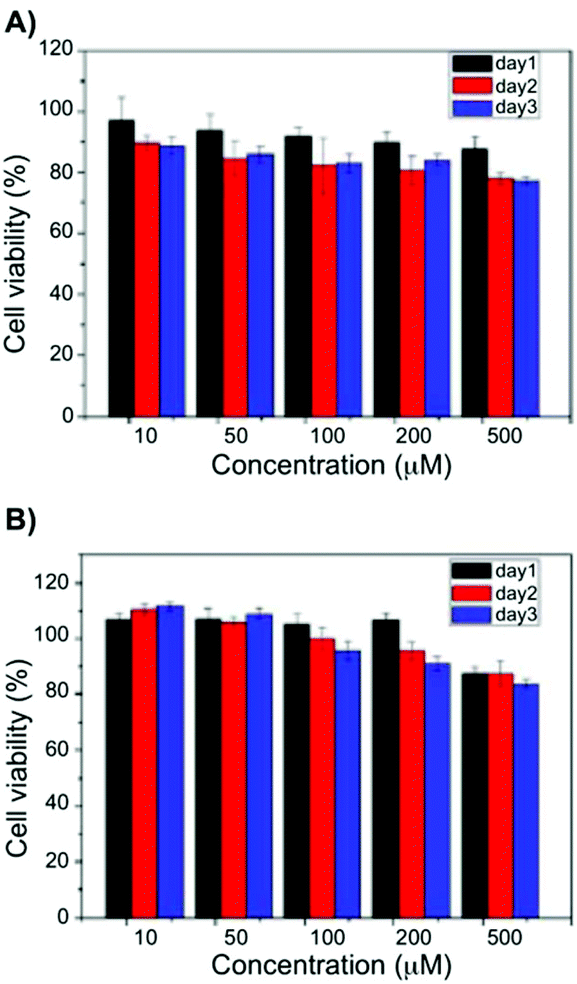
Later, the pentafluorobenzyl-Phe moiety was further applied in biomedical applications. In particular, the formation of self-assembled hydrogels at physiological pH and also various molar ratios of the hydrogelators based on pentafluorobenzyl-Phe and pentafluorobenzyl-diPhe was reported.78 At pH 7.4, self-supporting hydrogels were obtained via co-assembly in equimolar ratios. UV-Vis absorption spectra of the hydrogels showed absorption maximum at 260 nm due to the π–π* transitions of the typical aromatic rings. In case of pentafluorobenzyl-diPhe, an additional Phe is enhancing the π–π interactions in the gel, which explains band shift to 285 nm. In its gel state, a red shift in the emission spectra (ca. 360 nm) can be observed when compared to the solution phase (∼310 nm). Combining all the spectroscopic results of the hybrid gel, it can be confirmed that π–π interactions and hydrogen-bonding interactions are extremely important for the gel formation. Moreover, the authors found the hybrid hydrogel to be biocompatible by the results of cell survival ratio of CTX TNA2 and MCF-7 cells which makes it a potential candidate as scaffolding material for biomedical applications such as drug delivery and tissue engineering (Fig. 9).
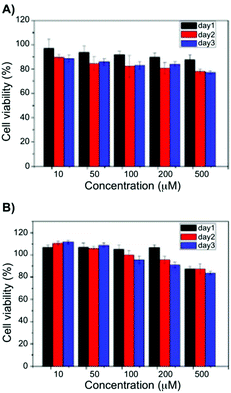 |
| | Fig. 9 Viability data of (A) CTX TNA2 and (B) MCF-7 cells incubated with 10–500 μM of a 1:1 blend hydrogel after 24, 48 and 7 h. Adapted with permission from ref. 78. Copyright 2015, The Royal Society of Chemistry. | |
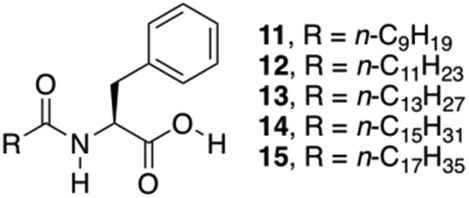
So far, several Phe derivatives with N-terminal protecting groups containing aromatic moieties have been reported. Huang and co-workers prepared a series of monochain L-Phe derivatives to check whether van der Waals interactions (via incorporation of aliphatic long chain in the N-terminal side) are equivalent to π–π interactions of the aromatic moieties during gelation.79 All these L-Phe derivatives do not display any gelation ability. However, in the presence of different aliphatic amines, L-Phe derivatives 11–15 (Fig. 10) underwent two-component gelation in various organic liquids. SEM images revealed that two-component organogels could self-assemble into fibrous or lamellar aggregates. FT-IR and 1H NMR results indicated that gel formation occurs through acid–base interactions, intermolecular hydrogen bonding of amide groups and van der Waals interactions of long alkyl chains. Hydrogen-bonding and acid–base interactions were found to be the key forces for the formation of self-assembled gels.
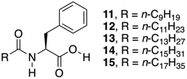 |
| | Fig. 10 Chemical structures of L-Phe derivatives 11–15. Adapted with permission from ref. 79. Copyright 2014, Elsevier B.V. | |
To learn more about the effect of larger π surfaces on the gelation of Phe, Banerjee and co-workers attached a γ-pyrene substituted butanoic acid at the N-terminal of Phe to evaluate the gelation ability of a pyrene conjugated Phe derivative 16.80 This gelator alone was able to form hydrogels in a wide range of basic pHs (7.46–14) showing CGC of just 0.037% at pH 7.46 in phosphate buffer solution. The appearance of those hydrogels were dependent on the pH. Specifically, transparent hydrogels were obtained in a pH range from 7.46 to 10.5, whereas translucent hydrogels were formed at more basic pHs. Most remarkably, when the pH was increased, a morphological change of the gel nanofibers from helical to tape-like morphology was observed under FE-SEM. They studied CD spectroscopy of the gelator at its minimum gelation concentration at different pH values. However, the ellipticity of the gel gradually diminished significantly with a slight blue shift with an increase in the pH value. At a higher range of basic pH (≥12.0), no apparent CD signal was observed. Diminishing of the CD signals at highly basic pH can be correlated with the pH dependent morphological transformation from helical chiral nanofiber to straight achiral gel nanofibers. This suggests that the orientation of gelator molecules in lower pH range (7.46–11.0) favors the formation of a chiral (helical) structure and the almost disappearance of the CD signal at higher pH (≥12.0) enables the non-helical achiral straight tape-like morphology. Besides, the increase in pH also promoted a thixotropic behavior, which was utilized to encapsulate vitamin B12 and anticancer drug doxorubicin. Remarkably, this system does not need heating–cooling cycles for gel formation which offers new opportunities for encapsulations, ensuring the non-decomposition of the encapsulated biomolecules.
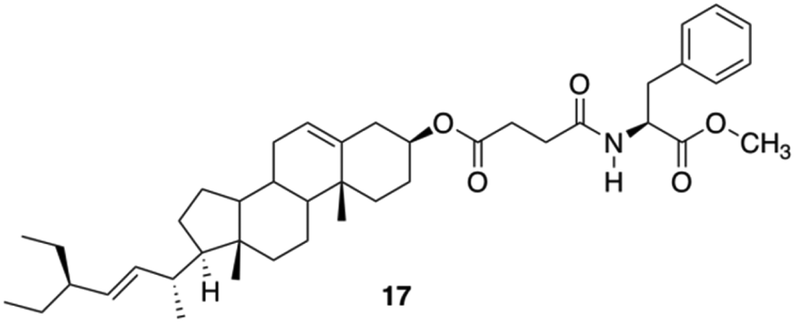
Due to the lipophilic nature of stigmasterols, Šusteková and co-workers considered their combination with an amino acid to generate a self-assembled system driven by hydrophobic interactions and hydrogen bonds. Thus, a library of conjugates of stigmasterol and L-Phe connected by ester and amide bonds through short-chained dicarboxylic acyl compounds was prepared.81 Among these compounds, only 17 (Fig. 11) allowed the formation of a stable gel in 1-octanol.
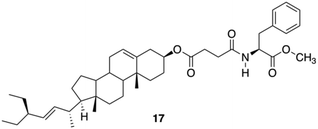 |
| | Fig. 11 Phe-based stigmasterol conjugate gelator 17. Adapted with permission from ref. 81. Copyright 2011, MDPI AG. | |
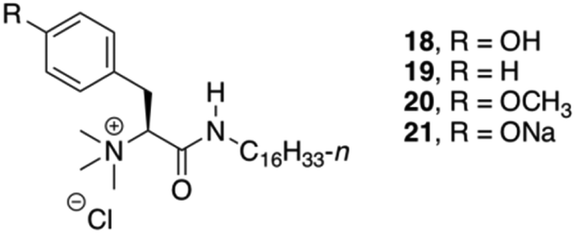
Das and co-workers studied the basic structural requirements to get amino acid-based amphiphilic pH responsive hydrogelators (Fig. 12).82 Therefore, 10 analogous amphiphiles based on L-Phe and L-tyrosine were tested toward their gelation abilities (structures of L-tyrosine derivatives are given in Fig. 12 only for comparison). The studies showed that L-tyrosine derivative 18 did not form any gel, whereas hydrogels could be achieved with L-Phe 19 in plain water at room temperature with an CGC of 5% w/v. Interestingly, 21 was able to form a hydrogel, which is sensitive to pH. CD spectroscopy suggested a highly ordered arrangement of chiral planes at the supramolecular level induced by the amino acid residues. The supramolecular chirality that emerged through non-covalent organized packing of molecular components in the process of self-aggregation was also investigated for 19 by temperature-dependent CD spectra. The intensity of the CD peak gradually decreased with increase in temperature as a result of gel to sol state transition due to the destruction of the 3D supramolecular arrangement. However, even at 90 °C (>Tgel), observed CD signal at sol state was probably due to the presence of small self-aggregates. Hence, the observed CD intensity at gel state is due to supramolecular aggregates not due to the molecular chirality.
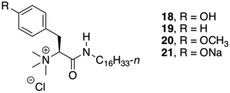 |
| | Fig. 12 Structures of quaternary gelators 18–21. Adapted with permission from ref. 82. Copyright 2008, American Chemical Society. | |
In addition, the newly discovered pH sensitivity of 21, a thermoreversible hydrogelator, was utilized to evaluate the controlled release of vitamin B12 and hemeprotein cytochrome c. The release of vitamin B12 and cytochrome c after 24 h, were 26% and 18%, respectively at pH 7.4. But after lowering the pH from 7.4 to 5.5/2.0, almost 95% of the entrapped vitamin B12 was released within 5–5.5 h due to the formation of insoluble precipitates of the gel structure of 21.
Gels made from Phe-diamide derivatives
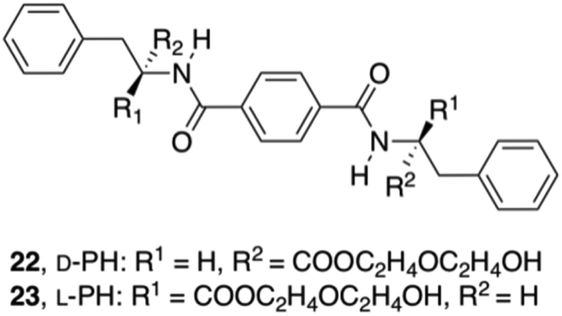
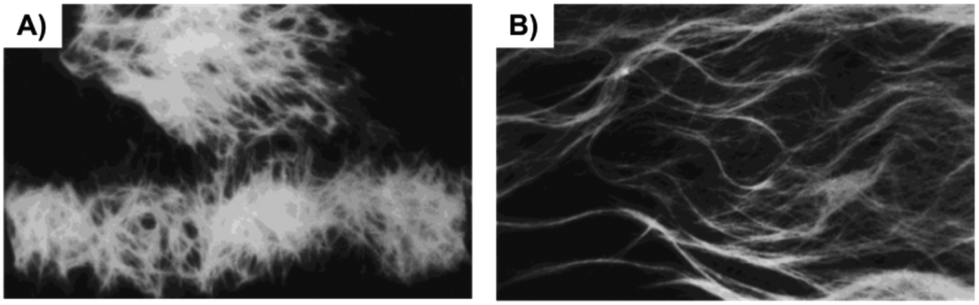
To produce more hydrogen bonding sites, Feng's group described the preparation of Phe-based building amide units at both ends. In particular, two enantiomers of a 1,4-benzenedicarboxamide Phe derivative 22–23 (Fig. 13) were investigated toward their gelation abilities.83 The effective concentration of the gelators both in methanol (solution state) and in phosphate-buffered saline (PBS; gel state) were kept constant. CD spectra demonstrated that the self-assembly of D/L-PH reached an equilibrium after about 1 h. All the results revealed that self-assembled aggregates or nanofibers are responsible for the chirality of the gels and not the individual gelator molecules. In addition, L-Phe, D-Phe, and r-Phe hydrogels with encapsulated NIH 3T3 cells and HUVECs were prepared in order to study how the chirality of the 3D environment influences cell adhesion and proliferation. In this study it was found that numerous HUVEC and NIH 3T3 cell clusters were evenly distributed in the L-Phe hydrogel after culture for 3 days. The clusters were formed as a result of cell proliferation rather than cell migration. Interestingly, NIH 3T3 cells developed the typical elongated polygonal cobblestone morphology throughout the L-Phe hydrogel, which indicated 3D proliferation behavior of the cells and a favorable interaction between the cells and L-type nanofibers. It was shown that cells cultured in 3D hydrogels constructed by D/L-Phe chiral nanofibers showed different adhesion and proliferation behavior. High cell growth and proliferation density were observed in L-Phe hydrogels, but not in D-Phe hydrogels. The chirality of nanofibrous structures has important role on cell adhesion and proliferation in the 3D ECM. These results are very useful for the development of a new generation of 3D matrices to control cell-adhesion and cell-proliferation density. They emphasize the key role of the chirality of nanostructures for materials design and offer a novel way to achieve high-density cell growth in the 3D ECM.
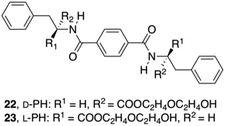 |
| | Fig. 13 Structures of diamide gelators 22 and 23. Adapted with permission from ref. 83. Copyright 2014, John Wiley and Sons. | |
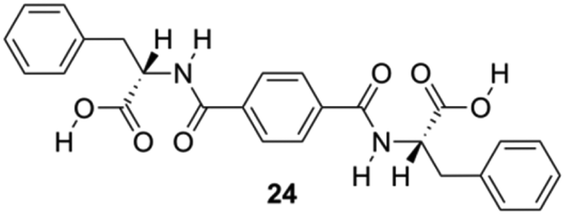
Feng and co-workers constructed a new type of redox-responsive supramolecular hydrogel by co-assembling 24 (Fig. 14) and disulfide-containing 4,4′-dipyridine disulphide (DPDS) to encapsulate dyes.84 The hydrogels were characterized by using 1H NMR, FT-IR and CD spectroscopy. These hydrogels can control the release of encapsulated dyes in response to intracellular reductive environments like tumor tissues, and hence may be used as drug carriers for controllable drug release for cancer therapy. Dithiothreitol (DTT) solution, which destroys the disulphide bonds in the gel network, was used to explore the responsive property of the hydrogels in reductive environments. A gradual disintegration of the hydrogels was observed upon DTT addition over a period of approximately 20 h while the composite hydrogel remained stable without DTT. In addition, the disintegration of hydrogels containing methyl violet (MW) as a model drug was studied to explore the possibility of using as redox-responsive hydrogel for controllable drug release. The release of MV was below 10% within 30 h in the absence of DTT, but higher release speeds from 3 to 10 h, and thus the total amount of MV was released to 80% in 30 h, which may be explained by assuming the hydrogel to suffer a severe damage after reacting for 3 h through thiol disulfide exchange reactions. The release kinetic process of MV dyes from 24-DPDS followed a pseudo first order model.
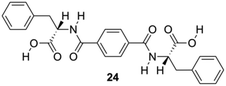 |
| | Fig. 14 Structure of diamide gelator 24. Adapted with permission from ref. 84. Copyright 2015, John Wiley and Sons. | |
In addition, Feng and co-workers reported the construction of a multiresponsive hydrogel co-assembled from 24 and bis(pyridinyl) derivative((E)-1,2-di(pyridin-4-yl)diazene) (AP), which shows sharp phase transitions in response to several different stimuli (i.e., temperature changes, photoirradiation, and pH changes).85 The incorporation of the azobenzene (Azo) group makes this material light-responsive, whereas the Phe and the pyridinyl groups in AP provide a dual pH response with the addition of both acid and base. In addition, the thermoresponsive property of the hydrogel was directly confirmed by the dissolution due to increasing temperature and gelation again upon cooling. Gelator 24-AP was found to be soluble in dimethyl sulfoxide (DMSO) and thermoreversible orange gels were formed quickly upon water addition. In addition, 24-AP formed a stable gel at pH 7. When the pH was changed to 4, the hydrogels turned into orange precipitates and at pH 9 yellow green solutions were obtained. Once the pH of the system was tailored to 2, rust red hydrogels were formed. However, 24 hydrogels can be also transformed into solutions at a higher pH value after adding base, whereas the gel-to-sol transformation cannot be triggered below pH 7. Upon UV (365 nm) irradiation, the stable orange gels gradually collapse causing a morphological change of the gel system. At the first 30 min of irradiation, degradation of the fibrous network were observed, whereas after exposure of UV light for 10 h, the whole gel was disassembled due the photoisomerization of AP. The authors showed that logic gates can be constructed tuning the stimuli concentrations of input signals. Such logic gates may promote the possibility of developing smart materials, such as gels that can be used as drug releaser in a quantitative manner with a fine control by external stimuli.
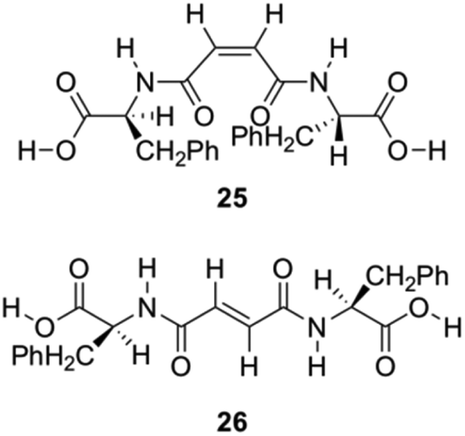
Zinic and his group described the preparation of bis(Phe) maleic acid amide 25 and fumaric acid amide 26 and showed that they were able to constitute an irreversible photoinduced gelation system in water. This system works based on photochemical isomerization of nongelling maleic acid amide 25, which gelates fumaric acid amide 26 (Fig. 15).86
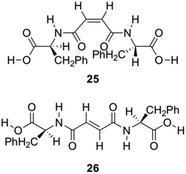 |
| | Fig. 15 Structures of gelators 25 and 26 for photoinduced hydrogelation. Adapted with permission from ref. 86. Copyright 2002, American Chemical Society. | |
An aqueous solution of compound 25 turned into gel almost instantaneously (<30 s) upon addition of trace amounts of bromine and irradiation with a 400 W high pressure Hg lamp (UV; λ > 330 nm; glass filter). The 1H NMR spectrum of the gel formed after UV shows that most of the gelator molecules are organized in a rigid network. TEM images of the hydrogels illustrated the conversion of microspheres into gel fibers indicating a configurational isomerization induced morphological transformation at the supramolecular level (Fig. 16).
 |
| | Fig. 16 TEM of hydrogel made of 26 (left, fiber diameters in the 10–20 nm range) and the light microscope image of the microspheres (right, diameters in the 2–10 μm range) formed by 25. Adapted with permission from ref. 86. Copyright 2002, American Chemical Society. | |
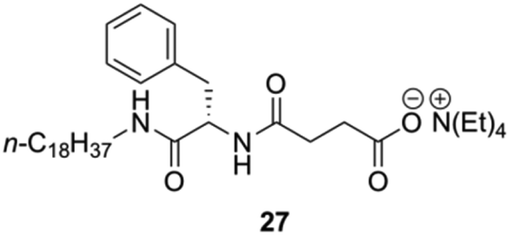
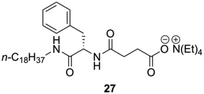
Yang and co-workers prepared a novel chiral hydrogelator 27 based on L-Phe and tetraethylammonium 3-{[(2R)-2-(octadecylamino)-3-phenylpropanoyl]} (Fig. 17).87 The formation mechanism of supramolecular hydrogels in presence of 27 was investigated by using FT-IR, FE-SEM, fluorescence spectroscopy and CD. Gelation experiments indicated that 27 could form gels with water and various aqueous media of different pH. The CGC in deionized water is 2 wt%, which reveals that only one molecule of 27 can immobilize about 1500 water molecules. Increasing the pH of aqueous media from pH 1 to pH 6 led to an increase of the CGC from 1.2 to 2 wt% as well as an increase in gelation time from about 10 to 25 min. This suggests that the gelation dynamics could be controlled by the ionic strength of the media (i.e., CGCs decrease with the increase of the ionic strength).
 |
| | Fig. 17 Structure of ammonium-containing hydrogelator 27. Adapted with permission from ref. 87. Copyright 2007, Elsevier B.V. | |
FE-SEM and polarized optical microscopy (POM) imaging gave insight into the pH dependent morphology of the hydrogels (Fig. 18). In deionized water, 27 self-assembled into fibril-like aggregates (Fig. 18a) whereas at pH 12 sheet-like aggregates (Fig. 18c) and at pH 2 spherical aggregates (Fig. 18b) were observed. For 27 aggregates, dendritic crystallites were obtained at pH 2 (Fig. 18e) and spheric crystallites at pH 12 (Fig. 18d) were observed under POM.
 |
| | Fig. 18 (A) FE-SEM images of dried hydrogels formed by 3.0 wt% of 27 in deionized water, (B) in pH 2, and (C) in pH 12 aqueous media. (D) and (E) POM images of hydrogels formed by 5 wt% of 27 in pH 12 and pH 2 aqueous media, respectively (magnification 200×). Adapted with permission from ref. 87. Copyright 2007, Elsevier B.V. | |
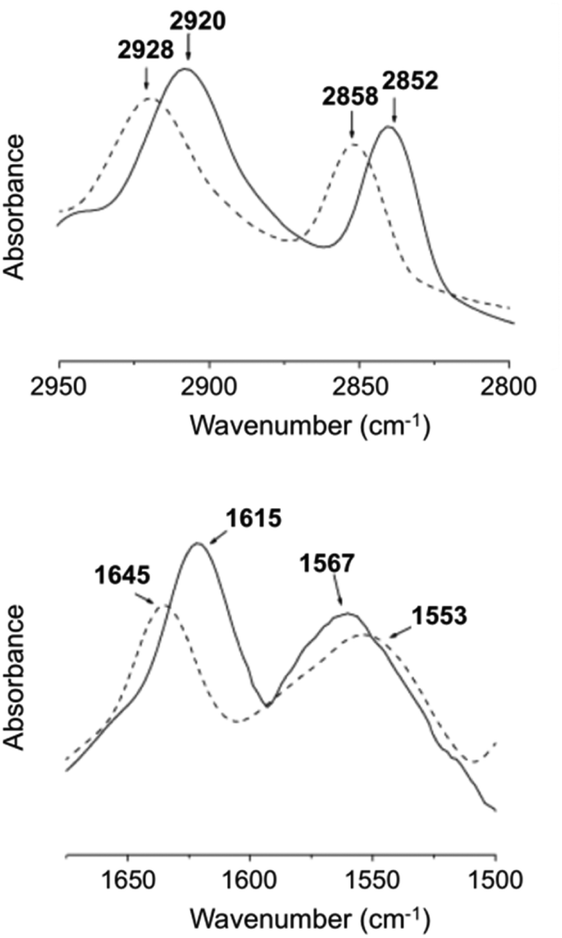
For FT-IR, D2O was used instead of H2O (Fig. 19), which confirmed that one of the main driving forces for 27 hydrogel formation is hydrogen bonding due the presence of intermolecular hydrogen bonded amide groups.
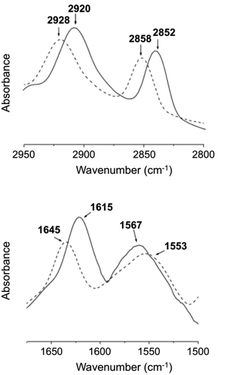 |
| | Fig. 19 FT-IR spectra of the D2O hydrogel (solid line) formed by 27 (3 wt%) and D2O solution (dashed line) of 27 (1 wt%) at 25 °C. Adapted with permission from ref. 87. Copyright 2007, Elsevier B.V. | |
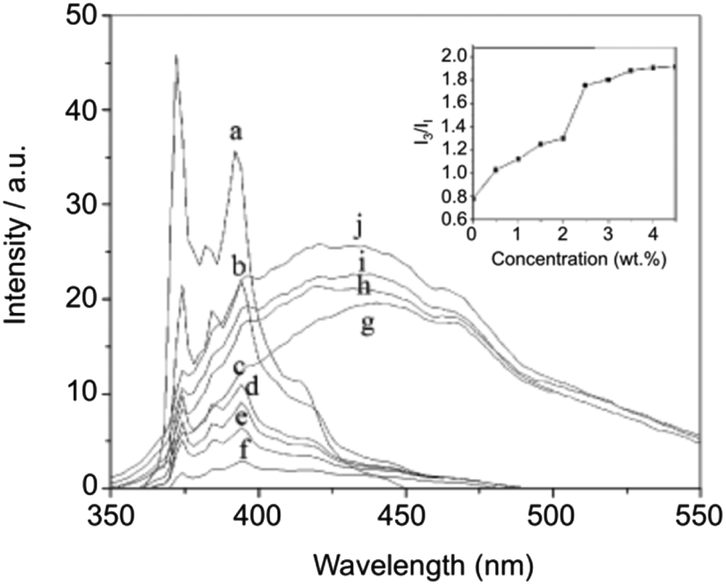
The hydrophobic interaction between long chain alkyl groups was characterized by fluorescence measurements in the presence of pyrene as a probe (Fig. 20). It was concluded that hydrophobic interaction between the alkyl groups of 27 is another driving force for the self-assembly of 27 in water.
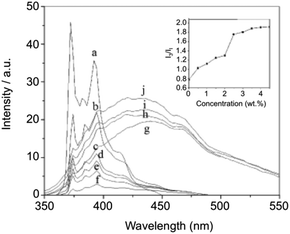 |
| | Fig. 20 Fluorescence spectra and relative intensities (I3/I1) of pyrene in aqueous solutions containing 0 (a), 0.5 (b), 1 (c), 1.5 (d), 2 (e), 2.5 (f), 3 (g), 3.5 (h), 4 (i), and 4.5 (j) wt% of 27 at 25 °C. Adapted with permission from ref. 87. Copyright 2007, Elsevier B.V. | |
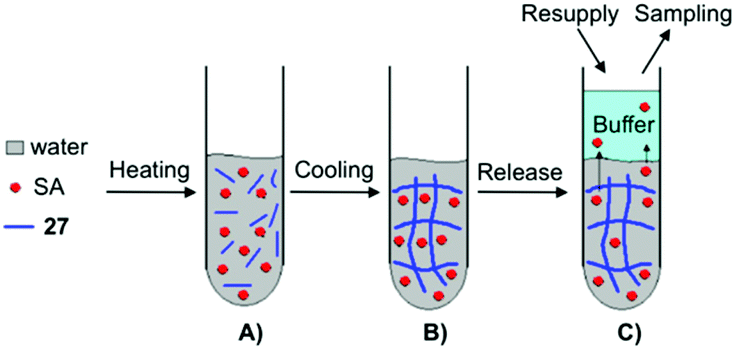
Yang and co-workers prepared supramolecular hydrogels through self-assembly of 27, which were used as drug carriers of salicylic acid (SA) (Fig. 21).88 The release behavior of SA from the supramolecular hydrogels was investigated by UV-Vis spectroscopy varying (i) the concentration of gelator, (ii) pH of the buffers used as receptors, (iii) the temperature during the release, and (iv) the concentration of SA. By changing the pH of the buffer solutions and the concentration of SA, the hydrogels displayed diverse phase status (i.e., from opaque to transparent gels). In addition, SA at (concentrations >50 mg L−1) worked as impurity within fibril-like aggregates, which partially disrupted the self-assembly of 27 resulting in a decrease of the Tgel values. Furthermore, the release ratio of SA from the hydrogels was decreased by increasing the concentration of 27, showing a clear pH dependency (4, 7.4 and 10) of the buffers used as receptors. Higher release ratios were obtained with the buffer at pH 4 in comparison to the pH 7.4 and pH 10 buffers. This suggests that SA, released from the hydrogel, favors an acidic receiving medium rather than an alkaline medium. Also the influence of the temperature on the release was evaluated which was higher at 37 °C than at 25 °C, showing that SA as an additive may participate in hydrogen-bonding that could become weaker with an increase of temperature. In all cases, the amount of SA released from the hydrogel was plotted against the square root of time, showing a good linear correlation in accordance with the Higuchi equation, which indicated that the release mechanism followed Fickian diffusion control.
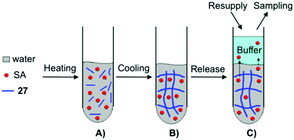 |
| | Fig. 21 Illustration of the formation of supramolecular hydrogel and SA released from the hydrogel. (A) Mixture of water, SA and gelator 27. (B) Formation of supramolecular hydrogel. (C) SA released from supramolecular hydrogel. Adapted with permission from ref. 88. Copyright 2008, Elsevier B.V. | |
Yang and co-workers synthesized and characterized an amphiphilic hydrogelator based on L-Phe, N-octadecyl-Nα-[6-(pyridinium-1-yl)hexanoyl]-L-phenylalaninamide bromide 28 (Fig. 22).89 The formation mechanism of supramolecular hydrogels in the presence of NP18PB was investigated by using 1H-NMR, polarized optical microscopy (POM), field emission scanning electron microscopy (FE-SEM), fluorescence, X-ray diffraction (XRD), and CD. The authors proved using 1H NMR that intermolecular H-bonding interaction is one of the main driving forces for the self-assembly of NP18PB molecules. In addition, fluorescence studies indicated that also hydrophobic interactions play a crucial role on the self-assembly of NP18PB into nanofibers. The observed supramolecular chirality of the gel of 28 emerged through non-covalent organized packing of molecular components. This was supported by a variable temperature CD study of NP18PB at 1.2 wt%. The intensity of the CD peak at 220–225 nm gradually diminished with an increase in temperature from 25 °C to 80 °C as a result of the gel to solution state transition. This leads to the destruction of the three-dimensional aggregate structure. The results indicated that the chirality of the gelator is responsible for the formation of the self-assembled aggregates and supramolecular hydrogel. Finally, the X-ray diffraction patterns of the xerogel of NP18PB prepared from H2O show periodical reflection peaks indicating that NP18PB assembles into a lamellar organization.
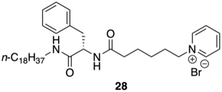 |
| | Fig. 22 Structure of pyridinium-containing gelator 28. Adapted with permission from ref. 89. Copyright 2010, John Wiley and Sons. | |
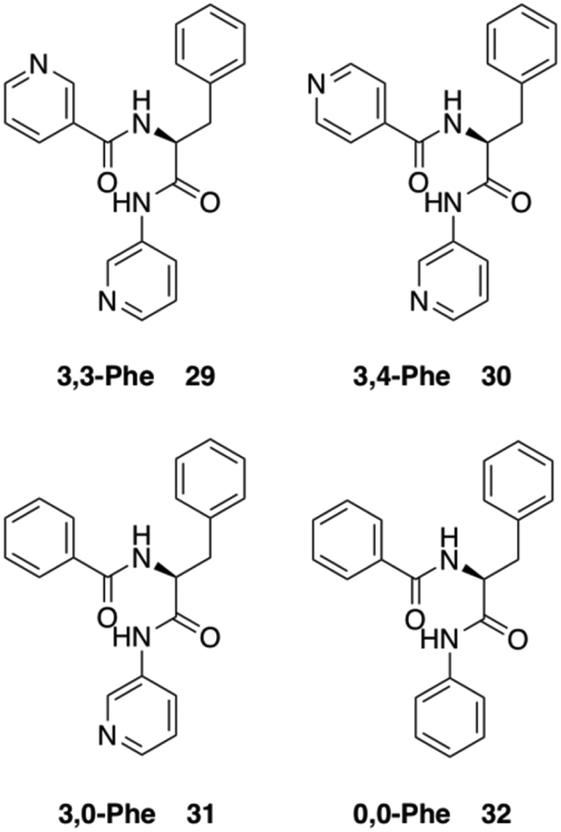
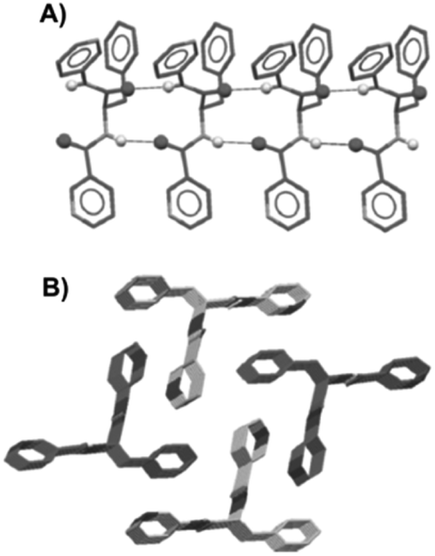
Dastidar and co-workers synthesized potential low-molecular-weight gelators based on bis-amides of L-alanine and L-phenyl alanine combined with various pyridyl and phenyl moieties as terminal groups (Fig. 23).90 The results showed that seven out of eight bisamides were able to form gels in both organic and aqueous solvents. Differential scanning calorimetry (DSC) experiments confirmed the thermoreversible nature of these gels, which were stable for months at ambient temperature. The role of the pyridyl group in gelation was explained by pH-dependent gelation experiments. Interestingly, 3,3-Phe did not undergo gelation in 1,2-dichlorobenzene below pH 3, which indicates the crucial role of neutral pyridyl moieties in gelation to provide additional H-acceptor sites to build an extended supramolecular network. In the single-crystal structure of 3,3-Phe, both 1D and 2D hydrogen bond networks were found to be present, apparently playing an important role in the gelation process (Fig. 24). Moreover, temperature-variable 1H NMR experiments gave insights into the presence of either π–π or C–H⋯π interactions during gelation. SEM and TEM imaging of the corresponding xerogels showed the presence of highly aligned (for 3,0-Ala, 0,0-Ala, and 3,0-Phe) or entangled (for 3,4-Phe) fibers, whereas 3,3-Phe displayed both fibers and colloidal particles. The gel made of 3,3-Phe 29 in 1,2-dichlorobenzene showed excellent shape-sustaining, load-bearing and self-healing ability, which are still considered to be uncommon properties for supramolecular gels (Fig. 25).
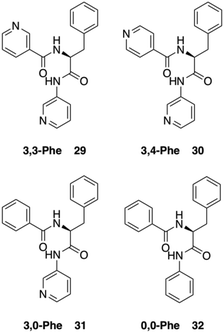 |
| | Fig. 23 Structures of bis-amide derivatives 29–32 of L-Phe. Adapted with permission from ref. 90. Copyright 2014, John Wiley and Sons. | |
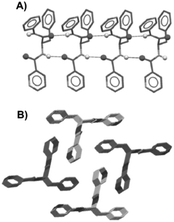 |
| | Fig. 24 Crystal structure of gelator 29; 1D hydrogen bond network through N_H⋯O interactions (A) and overall parallel packing (B). All hydrogen atoms (except N_H) were omitted for clarity. Adapted with permission from ref. 90. Copyright 2014, John Wiley and Sons. | |
 |
| | Fig. 25 (A) Free-standing and (B) load-bearing DCB gel made of 29 (1 wt%). (C) Gel slice with the abbreviation _IACS_ carved. (D) Self-healing of several gel blocks made of 29. Adapted with permission from ref. 90. Copyright 2014, John Wiley and Sons. | |
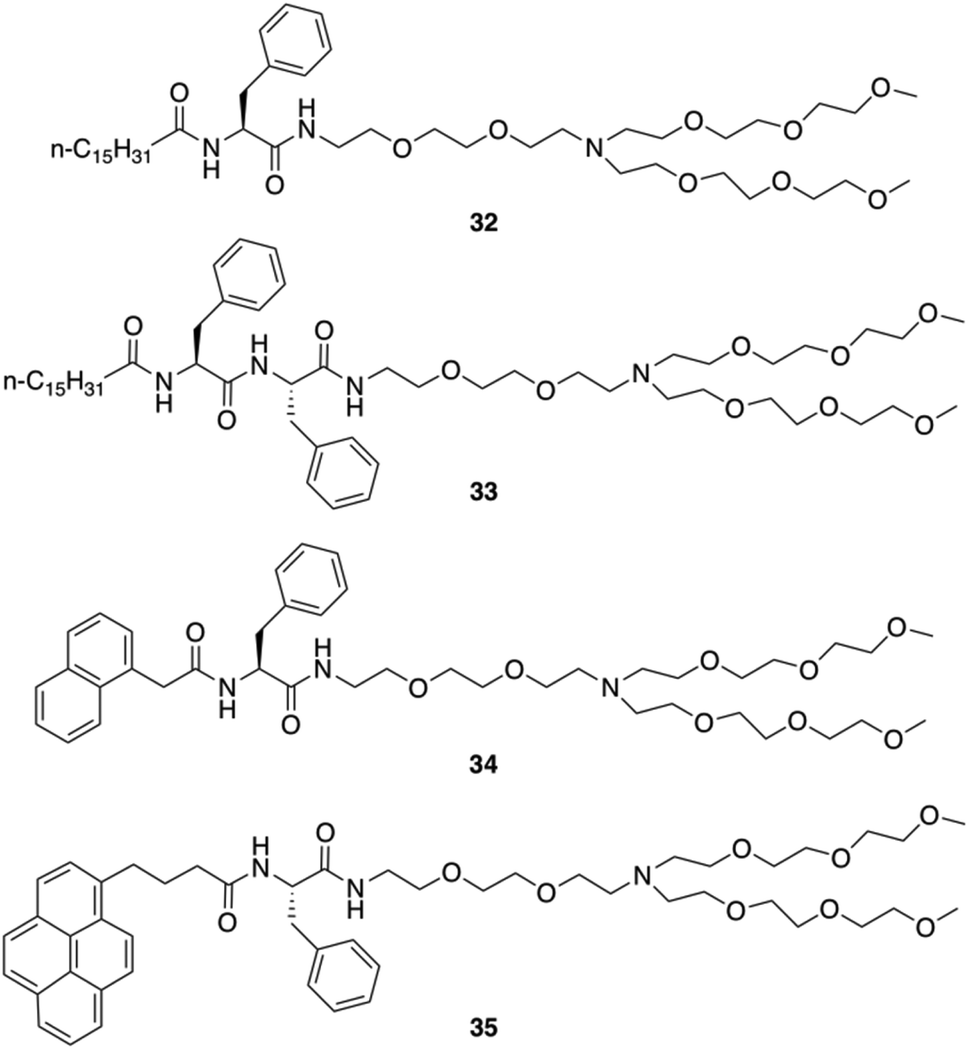
Neutral hydrogelators composed of hydrophobic terminal-group-modulated L-Phe with triethylene glycol monomethyl ether (TEG) tagged hydrophilic moiety were also designed by Das and co-workers (Fig. 26).91 Those hydrogels can contain nanomaterials which improve the mechanical stiffness and can be useful in the area of supercapacitors, nanoelectronics, photovoltaic devices, chemical sensors, biomedicine, and more. The amphiphiles 32, 34 and 35 self-assembled into hydrogels in water and aqueous buffer solutions of varying pH ranging from 3.0–7.0. Amphiphile 33 was found to self-aggregate to form a gel in a DMSO/water mixture (1:3, v/v). Hydrogelators 32, 34 and 35 were used to include various carbon nanomaterials (CNMs) within the hydrogel matrix due to their high hydrogelation efficiency in water. Two types of CNMs for integration were used: a 1D allotrope of carbon [single-walled pristine carbon nanotubes (SWNTs)] and a 2D allotrope of carbon [graphene oxide (GO)]. The studies showed that the long chain (C-16)-containing gelator 32 efficiently disperses, and includes 1D allotropes of carbon and single-walled carbon nanotubes (SWNTs) within the gel matrix. Notably, the pyrene-containing gelator 35 integrated both 1D and 2D allotropes of carbon (i.e., SWNTs and GO) in its SAFIN. TEM images of native hydrogels 32, 34 and 35 in water and 33 in DMSO–water (1:3, v/v) medium showed entangled fibrillar networks with fibers of 20–50 nm in thickness. The helical nature of the gel fibers of hydrogel 32 was observed in TEM image. Incorporation of SWNTs in hydrogel 32 led to a change in the morphology of the 32-SWNT composite. The extent of the crisscrossed intertwined network increased in the aggregated structure of the 32-SWNT composite compared to the native gel 32. The gel-to-sol transition temperature (Tgel) was subsequently determined. For the hydrogels made of 32, 34 and 35 (at their CGC in water), the Tgel values were found to be 57, 61, and 65 °C, respectively, and 56 °C for the hydrogel made of 33 (at CGC in DMSO–water, 1:3 v/v).
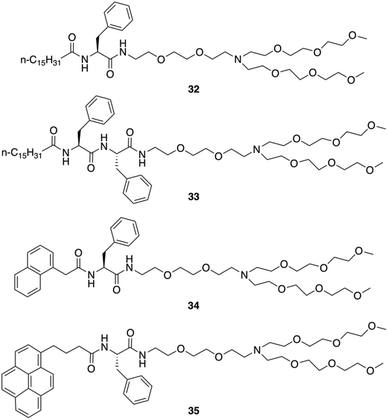 |
| | Fig. 26 Structures of amphiphilic gelators 32–35. Adapted with permission from ref. 91. Copyright 2016, John Wiley and Sons. | |
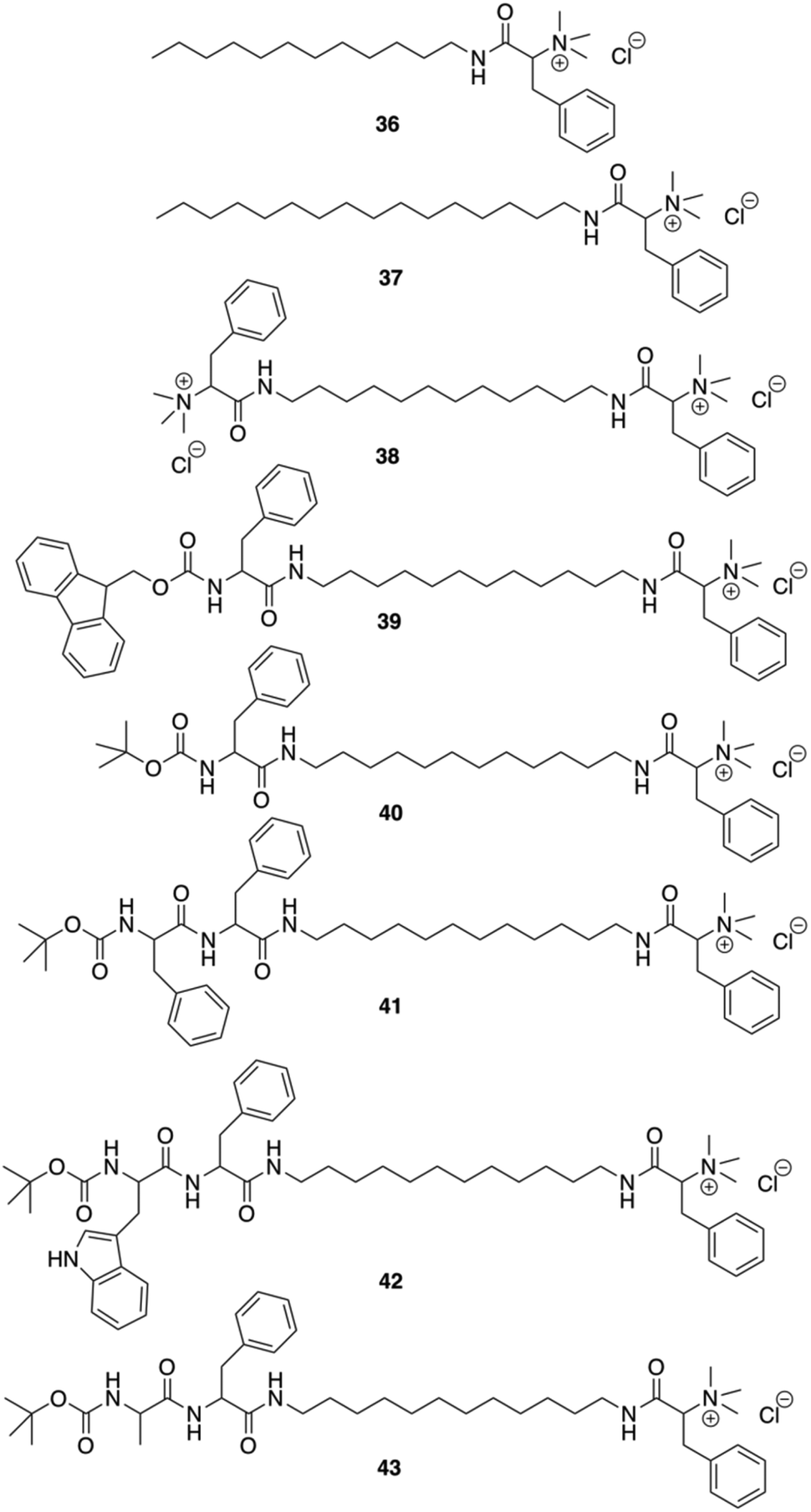
Das and co-workers were able to convert L-Phe based poor (C-16) or non-gelating (C-12 tail) amphiphiles to excellent gelators by simply incorporating N-terminal protected amino acid/dipeptide at the end of the alkyl tail (Fig. 27).92 It was shown that the amphiphilic gelator 40 consisting of a single L-Phe residue at the tail and an additional aromatic amino acid (L-Phe) in the dipeptide unit did not enhance the gelation efficiency, probably due to the loss in the HLB for amphiphile 41. The decrease of the gelation ability by increasing aromaticity was also observed for amphiphile 42 having a (tertiary butyloxycarbonyl) Boc-protected L-tryptophan-L-Phe dipeptide at the terminal end of the long chain. The extended aromaticity of indole moiety in tryptophan even converted a gelator (41) to a non-gelator (42). The poorer gelation ability might be due to the variation in the hydropathy indices of the amino acids. Interestingly, the alteration in the supramolecular arrangement of the self-assembled soft materials was used for in situ synthesis of different shapes of gold nanoparticles (GNPs). All gelators were capable of synthesizing GNP in situ at room temperature without any external reducing agents. Formation of GNP was evident from the corresponding surface plasmon resonance (SPR) peak in the UV-Vis spectra. The supramolecular nanoscale environment formed due to different modes of self-assembly by the tail modified amphiphilic gelators led to the synthesis of distinctly different shaped GNPs. Additionally, the biocompatibility of the tail-modified gelators (40 and 41) was tested against mammalian cells and results in marked improvement in biocompatibility towards mammalian cells.
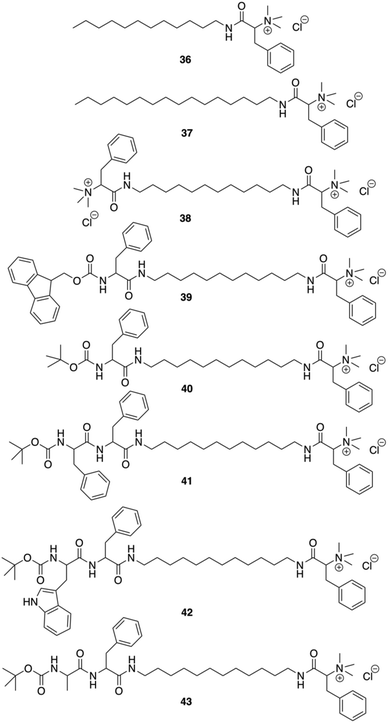 |
| | Fig. 27 Structures of amphiphilic ammonium-containing gelators 36–43. Adapted with permission from ref. 92. Copyright 2011, Royal Society of Chemistry. | |
Gels made from Phe-amide azo derivatives
Light constitutes a common external stimulus that can be absorbed by photochromic moieties and used to alter the self-assembly process of individual molecules or change the properties of supramolecular assemblies.93 In this respect, azobenzene is one of the most used photochromic groups due to its facile incorporation into organic molecules and its well-known controllable trans–cis photoisomerization. Usually, is the trans-isomer of the gelator the responsible for the gel formation. In many cases, the self-assembly pattern can subsequently change upon light irradiation due to the trans-to-cis isomerization, which is normally accompanied by a gel-to-sol transition. Nevertheless, it should be considered that the simple incorporation of switchable azo units into LMW gelators does not warrant a rapid, if any, macroscopic response of the gel material upon light irradiation because photo-induced isomerization is known to be very sensitive to the gel structure, especially if the azobenzene moieties are tightly packed.
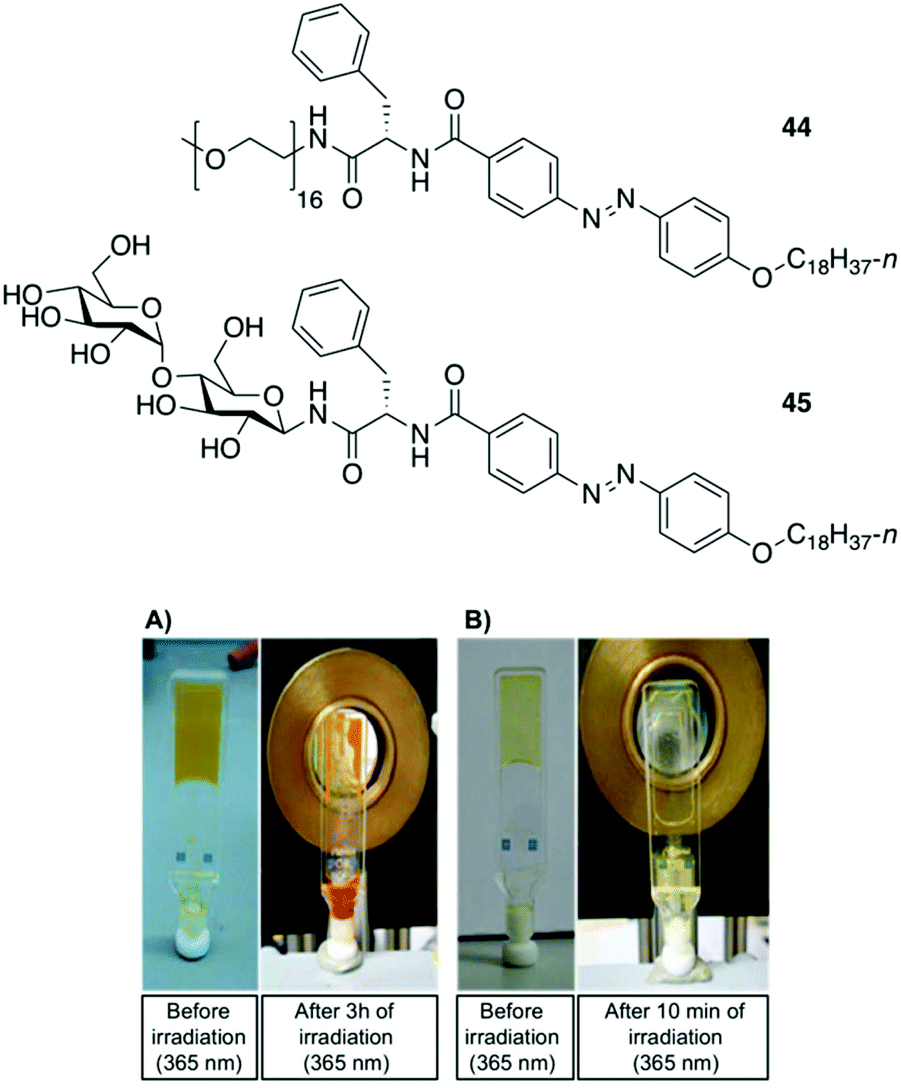
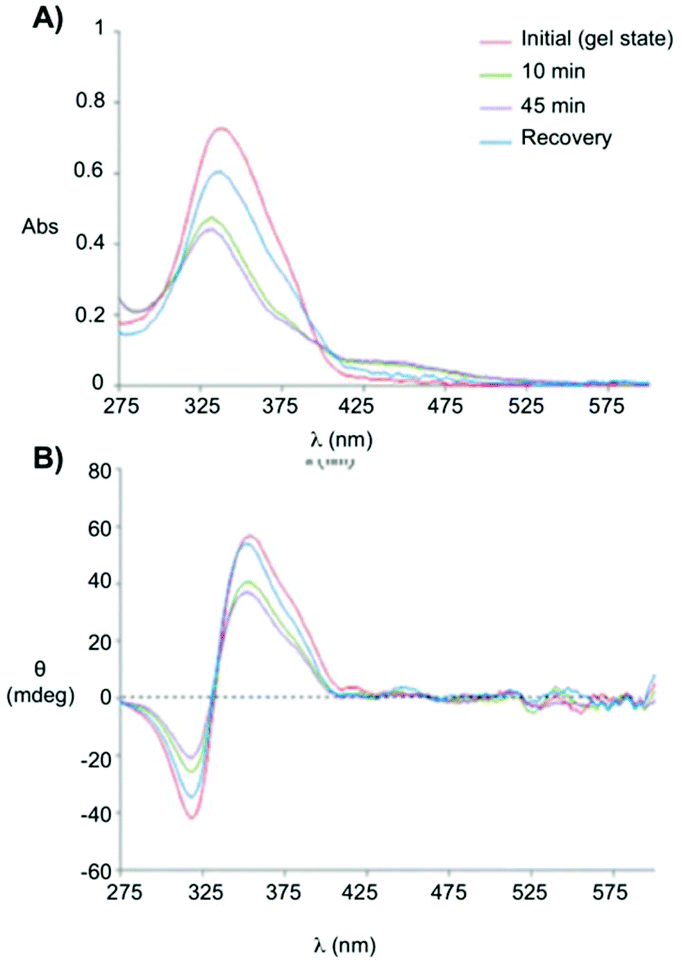
Clemente and co-workers used this knowledge and synthesized photoresponsive amphiphilic gelators 44–45 based on an azobenzene hydrophobic block and either poly(ethylene glycol) or maltose as polar heads, which were connected by L-Phe.94 The gelation ability of the final amphiphiles 44 and 45, as well as the intermediate molecules, were tested in different solvents, obtaining several gels even in DMSO. This results revealed that the gelation ability of these gelators can be moved toward more apolar solvents by using a sugar as polar head (45), while the incorporation of PEG units (44) facilitated the gelation of more polar solvents. The morphology of the corresponding xerogels displayed ribbons-like structures as established by scanning electronic microscopy. Gel-to-sol transition could be achieved by UV irradiation via trans-to-cis isomerization (Fig. 28), which was further confirmed by electronic circular dichroism (ECD) (Fig. 29). The cis-to-trans isomerization accompanied by the restoration of the gel state was regained in dark at room temperature. With the increase in irradiation time, the ellipticity values started to decrease due to the decrease in the chiral aggregation imposed by the cis-azobenzene moieties.
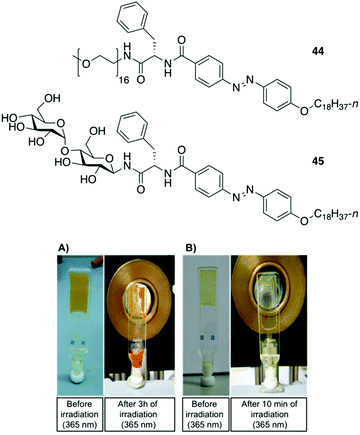 |
| | Fig. 28 Top: Structures of gelators 44 and 45. Bottom: (A) Gel made of 44 (5 wt% in DMSO) before irradiation and after 3 h of irradiation at 365 nm. (B) Gel made of 45 (0.5 wt% in 1-dodecanol) before irradiation and after 10 min of irradiation at 365 nm. Adapted with permission from ref. 94. Copyright 2015, Royal Society of Chemistry. | |
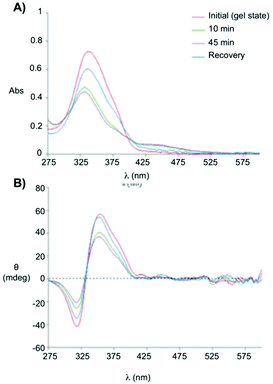 |
| | Fig. 29 (A) Absorption spectra and (B) ECD spectra of the gel made of 45 (0.5 wt% in 1 dodecanol) measured at room temperature, without irradiation, after 10 min, after 45 min of irradiation and recovery after 45 h of irradiation and keeping the sample in dark for 24 h at room temperature. Adapted with permission from ref. 94. Copyright 2015, Royal Society of Chemistry. | |
Gels made of Phe-amide ester derivatives
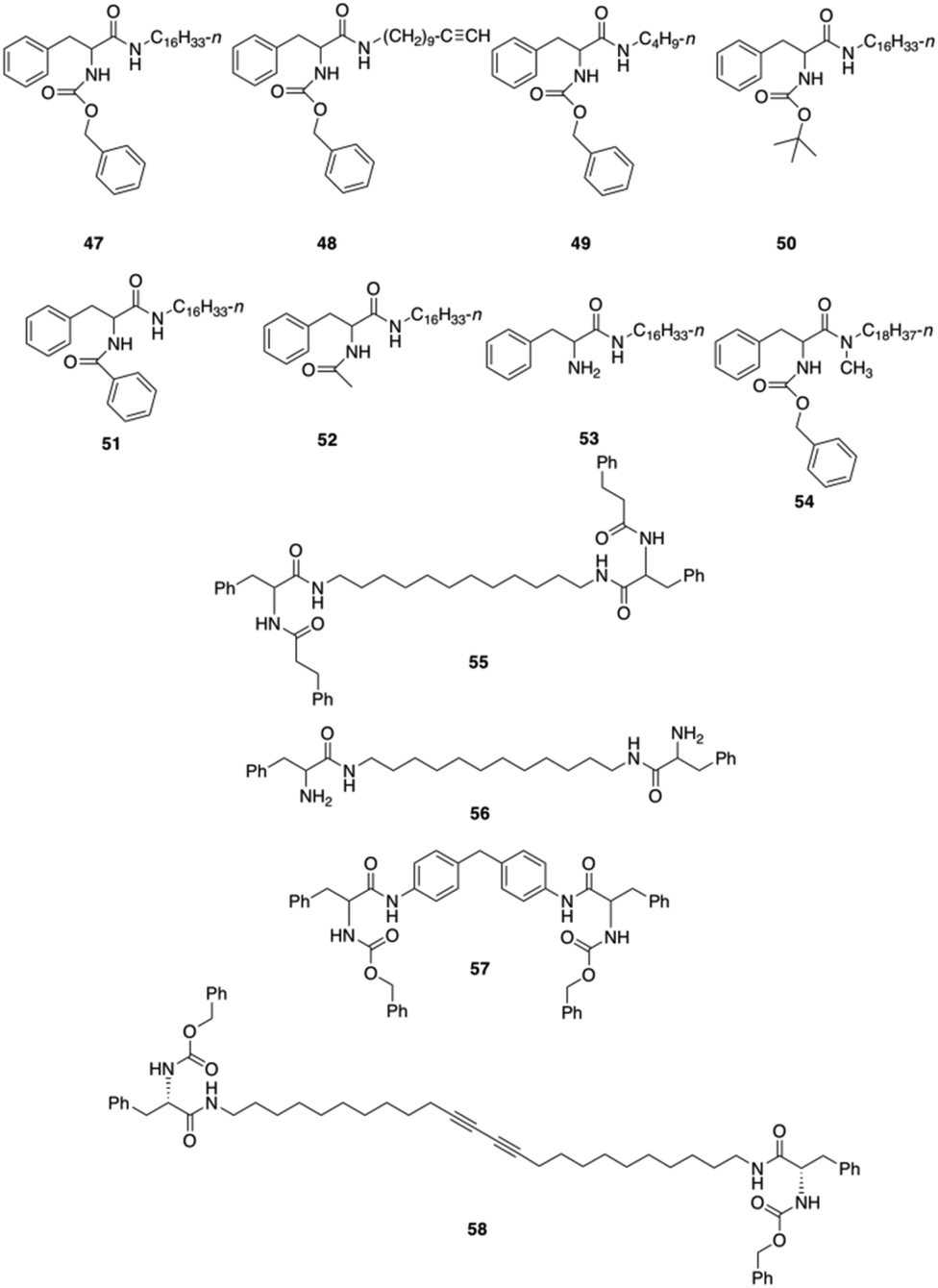
Bhattacharya and co-workers reported the synthesis of twelve L-Phe based mono- and bipolar gelators (47–58), and studied their structure–property relationship with respect to their gelation abilities in different solvents (Fig. 30).95 Interestingly, the authors found that dried gels from 47 were quite stable to mechanical disturbances and the corresponding gels did not transform into viscoelastic fluids or crystallize on prolonged storage.
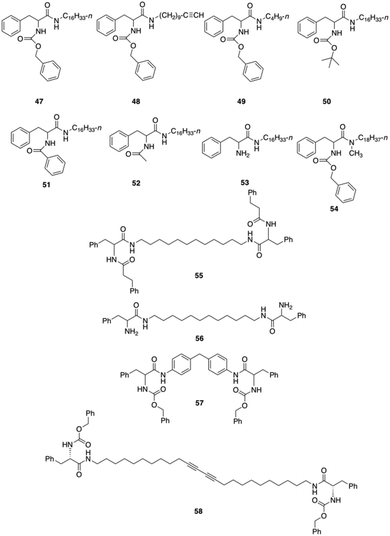 |
| | Fig. 30 Phe-based different mono and bipolar gelators 47–58. Adapted with permission from ref. 95. Copyright 1999, American Chemical Society. | |
Optical (Fig. 31) and electron microscopy (Fig. 32) were applied to gain further insights into the gel-forming systems. Compounds 47–49, when dissolved in polar, aprotic solvents, self-assembled forming macroscopic tapes. However in polar protic solvent, no gelation was obtained presumably due to the competition between solvent molecules and substrate molecules toward hydrogen bonding interactions. A detailed study on the significance of each of the functional units, such as, the effect of chain length, role of urethane, phenyl rings and amide linkages, etc. was investigated. The results showed that the mechanical strength of the gel is directly proportional to the chain lengths of the alkyl moiety. Gelator 47 bearing a long n-hexadecyl chain formed a stable gel toward mechanical disturbances, indicating that a better van der Waals stabilization is achieved with compounds having longer chains. To investigate the role of (benzyloxy) carbonyl group, it was replaced by a (tert-butyloxy)carbonyl group such as in 50, but no gelation could be observed despite the presence of a urethane linkage. In this situation, the hydrogen-bonding environment at the urethane is disturbed after the incorporation of a tert-butyl group, thus precluding the self-assembly. In addition, the interaromatic π–π stacking interactions via its flat phenyl rings from the benzyl group are no longer available which is essential for gel formation. To understand whether there is any role of primary amide group, gelator 54 was prepared and the primary amide type linkage on the C-terminal side of L-Phe was replaced by a secondary amide. In this case, no gelation could be observed in any of the above-mentioned solvents. Therefore, it was reasoned that primary amide group mediated intermolecular hydrogen bonding plays a crucial role in gel formation, which was further investigated by concentration dependent IR spectroscopy with compound 47. The shift of amide-I and amide-II bands with increasing concentration indicated the intermolecular hydrogen bonding. To understand the role of chirality, a racemic Phe derivative such as (±)-1 was synthesized. The racemic derivative did not produce any gel in the above solvents. May be attributed to the formation of planar aggregates hindering proper gelation. Thus, by using an enantiomerically pure amino acid core the intermolecular hydrogen bonding and aromatic stacking interactions reinforce the formation of nonplanar aggregates, which facilitate gel formation. Moreover, when a rigid spacer was used like in compound 57 no gelation was observed.
 |
| | Fig. 31 Optical micrographs of two specimens of gel formed from 47 in hexane–EtOAc (70:30 v/v): (A) view at 100 times magnification and (B) view of another sample at 300 times magnification. Adapted with permission from ref. 95. Copyright 1999, American Chemical Society. | |
 |
| | Fig. 32 Transmission electron microscopic images of the gel sample of 47 formed in hexane–EtOAc (70:30 v/v). Adapted with permission from ref. 95. Copyright 1999, American Chemical Society. | |
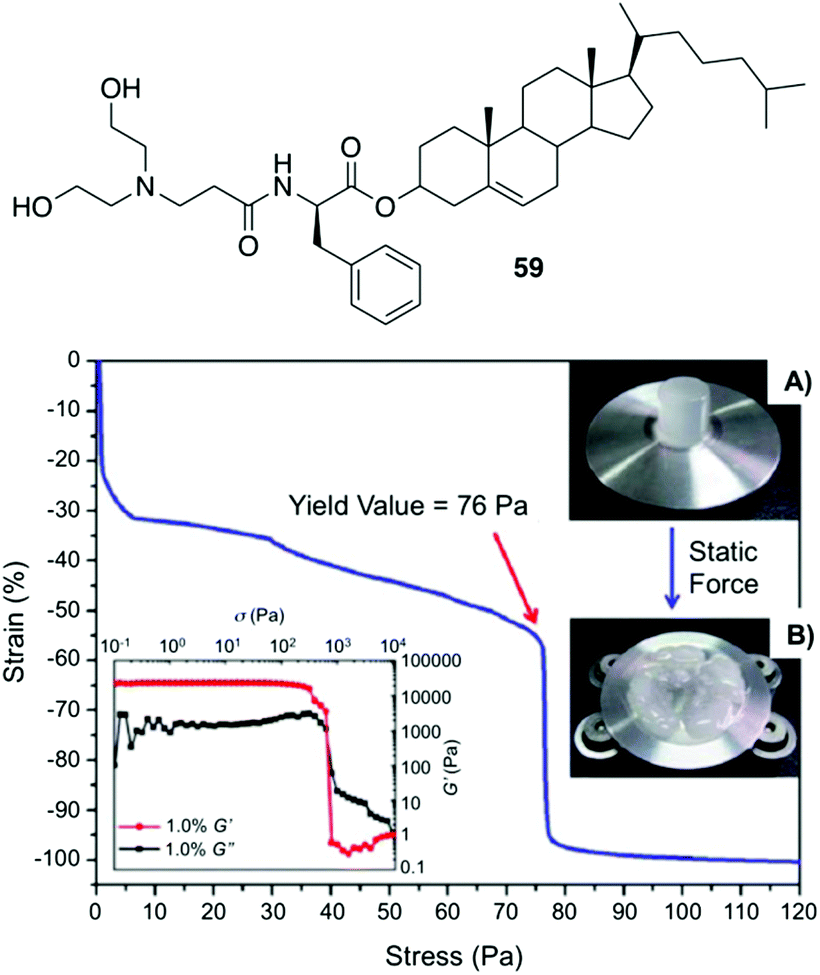
Fang and co-workers reported a number of thermo-reversible ionogels based on cholesteryl derivatives with Phe in the backbone, which produces stable gels in both neutral and acid medium.96 In this study, it was found that 76 Pa pressure (Fig. 33) is required for the breakage of the gel prepared with 1-butyl-3-methylimidazolium tetrafluoroborate (IL2) as solvent and the cholesteryl-based gelator 59. These ionogels can be easily converted to hydrogels by replacing the solvent with water. Interestingly, the cholesteryl derivatives were found to be more effective as gelators when D-Phe is used instead of L-Phe. Hence, it is clear that the configuration of the amino acid plays an important role in the gelation. Additionally, application of magnetic nanoparticles into these gels generates magnetic gel. Thus, when then a magnetic field was applied, the gel state could be converted to sol state (Fig. 34) due to magnetic field oriented movement of the particles inside the gel. However, the gel-state could be easily recovered by simple sonication followed by a heating–cooling cycle.
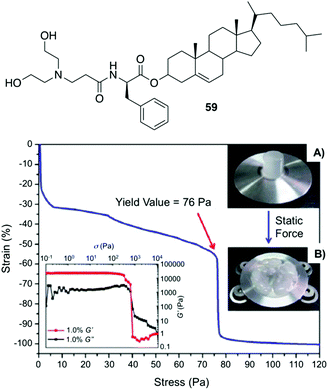 |
| | Fig. 33 Top: Structure of gelator 59. Bottom: Static compression strain vs. stress curve of a cylinder ionogel made of 59/IL2 at 10 mg mL−1 and its rheological behaviors (left inset), the cylinder ionogel before (A) and after (B) compression (right inset). Adapted with permission from ref. 96. Copyright 2012, Royal Society of Chemistry. | |
 |
| | Fig. 34 Preparation and phase behavior of the gel made of 59/IL2 (0.5%, w/v) under different conditions: (A) gelator 59; (B) IL2; (C) ionogel; (D) hydrogel; (E) gel under HCl atmosphere; (F) magnetic gel containing Fe3O4; and (G) magnetic gel under external magnetic field. Adapted with permission from ref. 96. Copyright 2012, Royal Society of Chemistry. | |
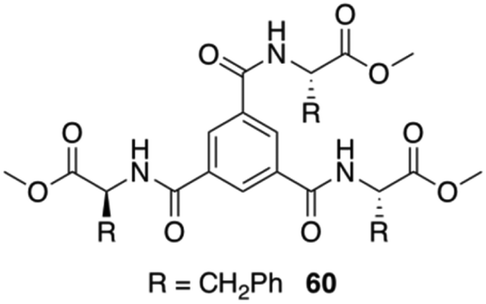
Maruyama reported the synthesis of supramolecular gelators comprising 1,3,5-benzenetricarboxylic acids and different amino acid methyl esters (i.e., glycine, L-alanine, L-valine, L-leucine, L-methionine, and L-Phe).97 The gelation behavior was examined by using eleven different types of ionic liquids. Benzenetricarboxamides self-assemble into helical structures, which is unfavored for the intermolecular π–π interactions. This study demonstrated that the amino acid residues play a crucial role in directing the molecular self-assembly of the benzenetricarboxamides. Self-assembly of these gelators in ionic liquids form entangled fibers as revealed by FESEM and confocal laser scanning microscopy. Interestingly, the observed gelation in different ionic liquids did not affect the intrinsic conductivity of the ionic liquids, hence preserving the ionic liquid characteristic even after the gelation. In particular, a 1,3,5-benzenetricarboxylic acid-based gelator 60 bearing L-Phe residues (Fig. 35) was expected to show good gelation property due to intermolecular π–π interactions between the phenyl rings of the L-Phe residues. However, this compound, compared to other gelators without L-Phe residues, displayed a limited gelation phenomenon remaining soluble in most of the ionic liquids. This poor gelation was attributed to the steric hindrance, exerted by the L-Phe residues. The helical assembly configuration along with steric effect precluded the efficiency of intermolecular interactions. Comparative studies with analogous compounds and FT-IR measurements confirmed that hydrogen bonding play a key role in the self-assembly of the gelator molecules in ionic liquids. In addition, the resulting ionogels possess reversible thermal transition properties and viscoelastic properties.
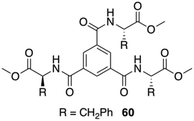 |
| | Fig. 35 Structure of compound 60. Adapted with permission from ref. 97. Copyright 2014, Royal Society of Chemistry. | |
Gels made of Phe-amide carbamates
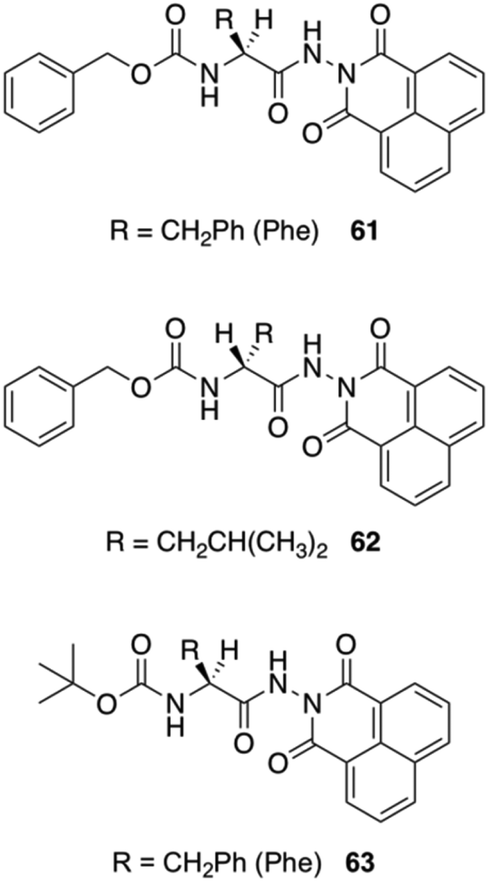
Jamart-Grégoire and co-workers carried out a series of studies in order to rationalize the steps during the gelation of an L-Phe derivative 61 in toluene.98 By using FT-IR and 1H NMR spectroscopy they found that the first step in the gelation process deals with the formation of hydrogen-bonded “head-to-tail” stacking-up of gelator molecules. The intensities of the IR peaks corresponding to free N–H bands were lowered with increasing concentration of the gelator, and the peak position corresponding to the carbonyl group was also shifted with the formation of hydrogen-bonded network. With increasing the temperature, the hydrogen-bonding network was getting disrupted in the gel state and the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequencies were comparable to their free ones. In NMR spectroscopy, the observation of downfield shift for N–H protons in the gel state supports the generation of a hydrogen-bonded network. The use of temperature dependent NMR spectroscopy showed significant sharpening of the peaks compared to the gel state. The authors demonstrated that the second step in the gelation process involves the intercolumnar π–π stacking interactions between hydrogen bonded columnar assemblies. The group studied the organogelation behavior of not only 61 but also other analogous derivatives 62–63 in different solvents (Fig. 36).99 The small structural variations between these molecules was enough to bring a drastic change in the gelation properties. The most efficient gelation was achieved in tetrachloroethylene using gelator 61. SEM images of the organogels revealed entangled 3D fibrillar networks. To check whether there is any role of chirality, they prepared the D-enantiomer 62, which exhibits the same behavior as the corresponding L-gelator. The circular-dichroism experiments showed an inversion of chirality from one enantiomer to the other. They also point out that δh appears to have primary importance in controlling whether hydrogen-bond networks of gelators is possible and further, that, a narrow δh domain is favored for gelation.
O stretching frequencies were comparable to their free ones. In NMR spectroscopy, the observation of downfield shift for N–H protons in the gel state supports the generation of a hydrogen-bonded network. The use of temperature dependent NMR spectroscopy showed significant sharpening of the peaks compared to the gel state. The authors demonstrated that the second step in the gelation process involves the intercolumnar π–π stacking interactions between hydrogen bonded columnar assemblies. The group studied the organogelation behavior of not only 61 but also other analogous derivatives 62–63 in different solvents (Fig. 36).99 The small structural variations between these molecules was enough to bring a drastic change in the gelation properties. The most efficient gelation was achieved in tetrachloroethylene using gelator 61. SEM images of the organogels revealed entangled 3D fibrillar networks. To check whether there is any role of chirality, they prepared the D-enantiomer 62, which exhibits the same behavior as the corresponding L-gelator. The circular-dichroism experiments showed an inversion of chirality from one enantiomer to the other. They also point out that δh appears to have primary importance in controlling whether hydrogen-bond networks of gelators is possible and further, that, a narrow δh domain is favored for gelation.
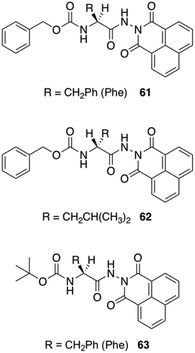 |
| | Fig. 36 Structure of carbamate-based gelators 61–63 studied by Jamart-Grégoire and co-workers. Adapted with permission from ref. 99. Copyright 2011, John Wiley and Sons. | |
Gels made of Phe-carbamate conjugates
Another common strategy to fabricate new gelators involves the attachment of Fmoc (i.e., fluorenylmethyloxycarbonyl) to the N-terminal side of an oligopeptide. The Fmoc group is easily available and widely used for N-terminal protection during peptide synthesis. Interestingly, Fmoc amino acids are also known to show anti-inflammatory properties, making them promising candidates for biomedical applications.100 The gelation ability of Fmoc protected amino acids have been investigated since the first Fmoc-dipeptide hydrogelator reported in 1995.101 In 2010, Nilsson and co-workers reported the dynamic transition of a self-assembled hydrogel amyloid-like fibrils into crystalline microtubes using Fmoc-p-nitro-Phe (Fmoc-4-NO2-Phe) 64 (Fig. 37).102 Although the self-assembly into amyloid-like fibrils was also observed for other Fmoc-Phe derivates, the conversion of Fmoc-4-NO2-Phe to crystalline microtubes over time was investigated in this work for the first time. CD spectroscopy was utilized to characterize the electronic structure of Fmoc-4-NO2-Phe fibrillar assemblies. Negatively stained TEM images revealed the fibrillar morphology of the hydrogel and the transition to crystals over time (Fig. 38). The crystals were investigated using SEM (Fig. 39). The conversion of the hydrogel made of 64 into a crystalline material was much faster than the gradual transformation of hydrogels made of other Fmoc-Phe derivatives into precipitates over a period of weeks to months. The crystal structure data indicated a critical role of the benzyl nitro group during fibril-to-crystal transformation. It was suggested that the polarity of the NO2 group might influence the crystallization process due to the polarization of the fundamental fibril units. In addition, Fmoc-2-NO2-Phe and Fmoc-3-NO2-Phe were investigated for comparative purposes. It was found that these two Phe derivatives also self-assembled into fibrils but much slower than Fmoc-4-NO2-Phe, which self-assembled into a hydrogel network within minutes while the fibril formation of the other isomers required 24 h. Due to the slow rate of assembly, neither a self-supporting hydrogel formation, nor a fibril to crystal transition was observed, even after weeks. This indicated that the exact position of the nitro group on the benzyl side chain plays a crucial role on the self-assembly of Fmoc-4-NO2-Phe. The structural similarities between the hydrogel fibrils and crystalline microtubes of Fmoc-4-NO2-Phe was investigated by X-ray powder diffraction (PXRD) analysis, using unassembled Fmoc-4-NO2-Phe as a control. The results showed that the molecular arrangements in each of these states were similar. When a sterically demanding PEG group was attached to the C-terminal side of Fmoc-4-NO2-Phe, it inhibited the gel-to-crystal transition by precluding the direct fibril–fibril fusion and further fibril lateral growth. Thus, the hydrogelation of Fmoc-4-NO2-Phe is a kinetically favored process, whereas the crystalline microtubes formation step leads to more thermodynamically stable state.
 |
| | Fig. 37 Images of Fmoc-4-NO2-Phe transition from optically transparent hydrogel to crystal precipitate over time: (A) 20 s, (B) 12 h, (C) 24 h, and (D) 48 h. Adapted with permission from ref. 100. Copyright 2010, American Chemical Society. | |
 |
| | Fig. 38 TEM images of the evolution Fmoc-4-NO2-Phe hydrogel fibrils to crystals over time: (A) 2 min (immediately after gel formation), (B) 10 min (arrows indicate higher order bundling of fibrils), (C) 60 min, and (D) 12–24 h. Reproduced with permission from ref. 100. Copyright 2010, American Chemical Society. | |
 |
| | Fig. 39 (A) SEM image of crystals of Fmoc-4-NO2-Phe formed from initial hydrogel states. (B) SEM image of Fmoc-4-NO2-Phe crystals illustrating the morphology of the crystalline microtube for mature crystals. Adapted with permission from ref. 100. Copyright 2010, American Chemical Society. | |
The same group also reported the efficient and rapid self-assembly of Fmoc-protected pentafluoro-Phe (Fmoc-F5-Phe) 65 into entangled fibrillar structures affording rigid supramolecular gels.103 The gelator was designed in order to know which effect, hydrophobic or electronic, was more important for hydrogelation. Therefore, Fmoc-Phe and Fmoc-Tyr were compared with 65 in this study. The influence of the N-terminal fluorenyl moiety in gelation was investigated when Fmoc was replaced by groups such as carboxybenzyl (Cbz) and tert-butoxycarbonyl (Boc). Due to the lack of π–π interaction in the case of Boc and weaker π–π interaction in the case of Cbz, no gelation was obtained. On the other hand, it was found that 65 has superior hydrogelation properties compared to Fmoc-Tyr, presumably due to electronic, steric, or hydrophobic differences via π–π interactions involving the perfluorinated phenyl ring. Circular dichroism (CD) spectroscopy was applied to evaluate the comparative structure of Fmoc-F5-Phe and Fmoc-Tyr hydrogels.
The similar absorbances in Fmoc-F5-Phe-based hydrogels compared to Fmoc-Tyr are consistent with the first proposed packing structure: Fmoc–Fmoc and F5-phenyl–F5-phenyl association in the context of fibril structures. Further investigations of the hydrogel using TEM, SEM and AFM techniques also indicated that gelator 65 packs into two energetically favorable geometries. The first one involves intermolecular π-stacking interactions between Fmoc fluorene groups and F5-phenyl side chains (Fig. 40). The hydrogen-bonding interactions were formed by the carbamate carbonyl and NH functionality, which further strengthened self-assembled structures. The CD and XRD data (Fig. 40) support a fibril structure involving Fmoc–Fmoc and F5-phenyl–F5-phenyl interactions along with the hydrogen-bonding interactions, which stabilize the assembled architecture.
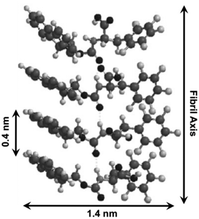 |
| | Fig. 40 Proposed packing model for Fmoc-F5-Phe self-assembly. Adapted with permission from ref. 103. Copyright 2010, The Royal Society of Chemistry. | |
In 2015, Thakur and co-workers found different factors that are crucial on the hydrogelation of fluorenylmethyloxycarbonyl Phe (FmocF) molecule.104 The roles of Fmoc, Fmoc and Phe covalent linkage, flexibility of Phe side chain, pH, and buffer ions were studied during the self-assembly that led to gelation. Phosphate buffer solutions of different pH values were used as solvent and the results demonstrated that the gel formation was very dependent on the pH of the solvent. It was observed that facile gel formation occurred above pH 7 whereas no gelation was achieved below pH 7. By only applying both heat and sonication, simultaneously, the gel formation was successful. However, when heat or sonication was used alone, it did not result in gel formation. In FT-IR studies the band at 1720 cm−1 was present in water and Tris buffer whereas in HEPES and phosphate buffer no band was visible, which suggests the involvement of carbamate carbonyl in hydrogen bonding in the self-assembly process. To further analyse the role of molecular contributors within FmocF in the gel formation they prepared few model compounds. In case of Fmoc-phenylglycine, no gel was obtained, which confirmed the important role of the extra methylene group. Further, the role of Fmoc was studied by using tert-butyloxycarbonyl-L-Phe under similar conditions. In this case, no gelation occurred, which implies the crucial role of the aromatic rings to form self-assembled higher order structures. Their results show that the FmocF hydrogelation is not dependent on one factor, but multiple factors plays important role behind the hydrogel formation. The overall experimental observations suggest four necessary components: (a) Fmoc as a hydrophobic gelator, (b) phenylalanine hydrophobic side chain and its flexibility, (c) pH and (d) buffer supporting ionic interactions. Moreover, dye release kinetics of the hydrogel proved its porous nature, which allows the free diffusion of small molecules across the gel matrix without specifically binding to the gel network.
In the same year, Chen and co-workers reported the use of Fmoc-Phe as hydrogelator to form supramolecular hydrogels based on intermolecular π–π stacking interactions and hydrogen bonding.105 Interestingly, it was possible to co-assemble the antimicrobial building block of Fmoc-Leu with Fmoc-Phe 66 which led to the formation of a supramolecular hydrogel with antimicrobial activity. The antimicrobial gel turned out to be not only biocompatible with normal mammalian cells but also showed selective Gram-positive antimicrobial activity involving cell wall and membrane disruption. Comparative UV-vis spectroscopic studies revealed the existence of antiparallel intermolecular π–π stacking interactions of Fmoc groups in the gel phase. The important role of hydrogen bonding in gel formation was proved by a red shift of CO stretching band in the FT-IR spectrum. The antimicrobial activity of the co-assembled hydrogel was evaluated by applying a pathogenic bacteria, S. aureus, over the hydrogel surface and measuring the optical density (OD) to determine the microbial proliferation (Fig. 41). In addition, the morphological changes of bacteria were studied to gain insight into the antimicrobial mechanism of the co-assembled hydrogel.
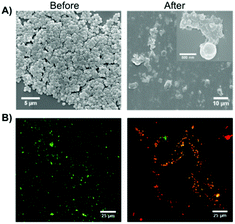 |
| | Fig. 41 (A) Representative SEM images and (B) overlapping fluorescence images for live/dead bacterial staining assay of S. aureus before and after contact with the co-assembled hydrogel for 2 h. Two fluorescent dyes were used in live/dead staining in which SYTO 9 with green color labeled both live and dead bacteria while propidium iodide with red color stained only dead bacteria. Adapted with permission from ref. 105. Copyright 2015, John Wiley and Sons. | |
Gels made of Phe-amide-amine derivatives
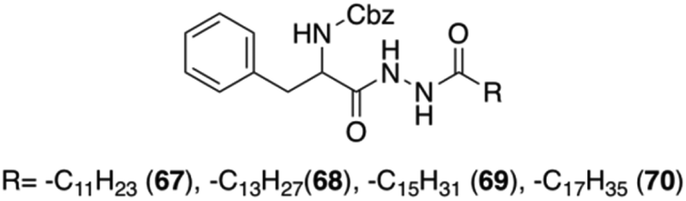
Sui and co-workers reported a new group of L-Phe based organogelators 67–70 having a dihydrazide moiety in the backbone (Fig. 42).106 These organogelators self-assembled in various organic solvents and formed thermally reversible physical supramolecular organogels at very low concentrations (<2 wt%).
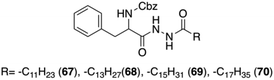 |
| | Fig. 42 Structures of Phe-based dihydrazide gelators 67–70. Adapted with permission from ref. 106. Copyright 2012, Springer Science+Business Media, Inc. | |
Scanning electron microscopy experiments were performed to study the self-assembly process leading to fibrillar structures in different solvents (Fig. 43). Moreover, FT-IR studies showed that intermolecular hydrogen bonding between N–H and CO of amide group and hydrophobic interactions between alkyl groups are the key factors for the gelation phenonmenon. Combining the results from XRD and FT-IR experiments, it was proposed that the gel aggregates are made up of a repeating bilayer unit within a tail-to-tail packing model.
 |
| | Fig. 43 SEM images of xerogel of 69 formed by (a) 1.5 wt% in chlorobenzene, (b) 2 wt% in n-hexanol, and (c) 1.5 wt% in benzene. Adapted with permission from ref. 106. Copyright 2012, Pleiades Publishing, Ltd. | |
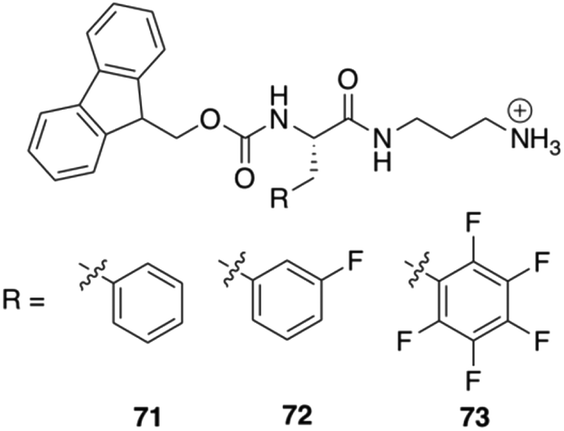
Nilsson and co-workers reported the synthesis of three C-terminal cationic Fmoc-Phe derivatives 71–73 (Fig. 44). The reason behind the synthesis of these derivatives was to identify Fmoc-Phe based molecules that can self-assemble into hydrogel without using any organic co-solvents or in situ pH modification. Addition of NaCl to the transparent aqueous solution of the cationic gelators produces hydrogel within 10–180 seconds. Rheological experiments were performed on these hydrogels in order to investigate the viscoelastic character of these materials. Hydrogels of the monofluorinated compound 72 had marginally higher storage and loss modulus values (G′ and G′′, respectively) than hydrogels of compounds 71 or 73 at the same concentrations. Fibrils or worm-like micelles of similar dimensions were obtained in TEM images of the various hydrogels. TEM images revealed that these cationic Fmoc-Phe derivatives assemble into distinctive sheet-based nanotube structures. Sheets were observed along with fibrils at lower concentrations (10 mM), but at higher concentrations (20 mM), those sheets folded into nanotubes. These nanotube structures are unique to C-terminal cationic Fmoc-Phe derivatives because the parent Fmoc-Phe carboxylic acids form only fibril/worm-like micelles structures. Interestingly, the nanotube formation by the cationic Fmoc-Phe derivatives is highly dependent on pH as the positive charge is very much necessary at the C-terminus for the formation of nanotube. Only fibrils are formed at basic pH where the positive charge is reduced and nanotube formation is suppressed (Fig. 45).
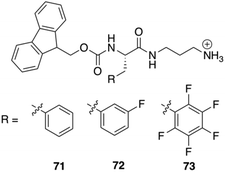 |
| | Fig. 44 Chemical structures of C-terminal cation-modified Fmoc-Phe derivatives 71–73. Reproduced with permission from ref. 107. Copyright 2017, American Chemical Society. | |
 |
| | Fig. 45 TEM images of hydrogels formed by compounds 71–73 at various concentrations obtained 15 min after hydrogelation: (A) 71 at 2.5 mM, (B) 71 at 5 mM, (C) 71 at 10 mM, (D) 72 at 2.5 mM, (E) 72 at 5 mM, (F) 72 at 10 mM, (G) 73 at 2.5 mM, and (H) 73 at 5 mM. Reproduced with permission from ref. 107. Copyright 2017, American Chemical Society. | |
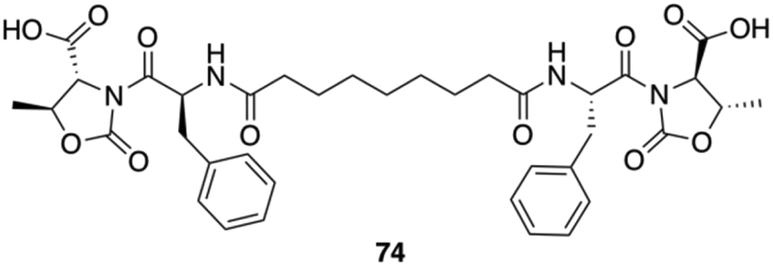
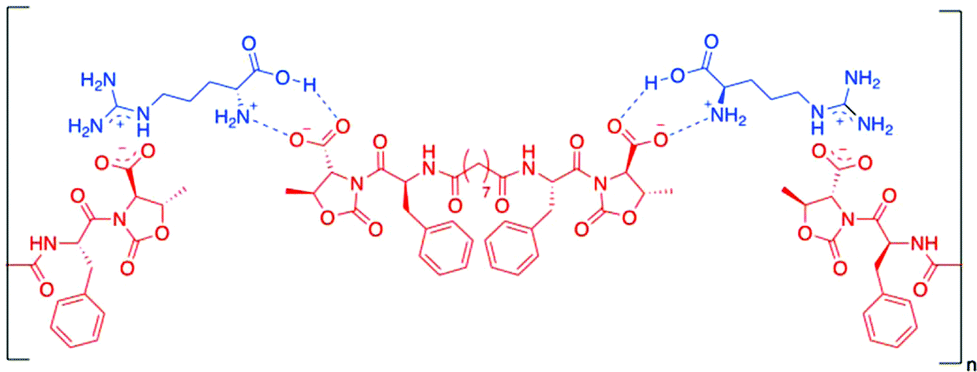
Gels made of Phe-diamide-acid derivatives
Tomasini and co-workers attempted to prepare amino acid mediated hydrogelation of a bolamphiphilic gelator 74 (Fig. 46). Although hydrogels formed in all cases under any conditions, strong and thermoreversible hydrogels are obtained in case of Arg. The hydrogels were prepared using the 74 both in 1% and in 2% w/w concentrations after addition of either 1 or 2 equivalents of each amino acid. In this particular case amino acids trigger the hydrogel formation via non-covalent cross link among the gelator molecules. As a result strong networks are formed with basic amino acids such as His and Arg. Fig. 47 shows a cross linking between the gelator molecules and Arg. The basicity of Arg allows a better dissolution of 74 where the electrostatic interactions between Arg and gelator molecule helps to form a supramolecular chain, thus mimicking a polymer. Apart from that the additional aromatic π–π stacking interactions construct a well-structured 3D network. Role of hydrogen bonding during gelation was confirmed by IR spectroscopy. The presence of strong N–H stretching bands below 3400 cm−1 suggests the formation of N–H⋯OC hydrogen bonds. Among different hydrogels the most promising one was obtained with 74 and Arg in a 1:1 ratio which is strong, elastic and thermoreversible. Moreover it has physiological pH and self-healing properties. So it is a good candidate for regenerative medicine applications.
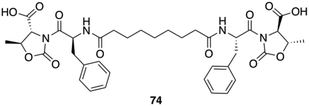 |
| | Fig. 46 Chemical structure of the gelator 74. Adapted with permission from ref. 108. Copyright 2016, Royal Society of Chemistry. | |
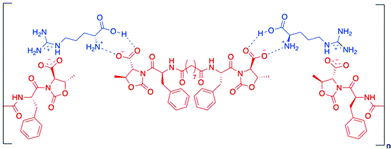 |
| | Fig. 47 Hypothetical interactions formed by molecule 74 and Arg. Adapted with permission from ref. 108. Copyright 2016, Royal Society of Chemistry. | |
In the same year, the same group prepared a pseudopeptide gelator Fmoc-L-Phe-D-Oxd-OH 75 (Fig. 48). They prepared four different hydrogels using that gelator either by changing the gelator concentration or adding graphene oxide (GO) to the water solution. The hydrogels were prepared by applying a new method where the gelation trigger was a pH slow variation induced by the hydrolysis of glucono-d-lactone (GdL). Rheological studies demonstrated that pure hydrogels are slightly stronger compared to GO-loaded hydrogels. This fact was also confirmed from the gel melting temperature. Hydrogels containing no GO show final melting points higher than the corresponding GO-loaded hydrogels. But the morphology of the hydrogel with and without the GO is almost same. The SEM images of the xerogels obtained after drying a sample of 75 hydrogels with or without GO do not show a dramatic difference between the two fiber networks. GO can interact with different pollutant molecules either through π–π interactions with the GO aromatic region or through hydrogen bond and electrostatic interactions due to its hydrophilic functional groups. So they considered cationic methylene blue (MB) and anionic eosin Y (EY) dyes and checked the hydrogels efficiency to trap these dyes. Out of these two dyes, MB is efficiently trapped (Fig. 49) by both the pure hydrogel and the GO-loaded hydrogel through π–π interactions. Apart from that, the gelator contains two carboxylic groups and GO is rich in epoxides, hydroxyl and carboxylic groups. All these groups efficiently interact with the cationic dye MB by electrostatic interactions, which causes efficient separation of MB by the hydrogel. In contrast, the removal of the anionic EY is achieved in less satisfactory yields, due to the unfavourable electrostatic interactions between the dye, the gelator and GO.
 |
| | Fig. 48 Chemical structure of the gelator 75. Adapted with permission from ref. 109. Copyright 2016, John Wiley and Sons. | |
 |
| | Fig. 49 MB solution in water flowing through a disposable hypodermic plastic syringes loaded with hydrogel of gelator 75: the recovered water is colourless. Adapted with permission from ref. 109. Copyright 2016, John Wiley and Sons. | |
One year later the same group used 74 and 75 to produce calcium-mediated hydrogel at physiological pH, which are strong and thixotropic hydrogels.110 In the presence of calcium chloride, the gelator solutions transforms into insoluble salts that get organized into fibers at a pH close to the physiological one. Addition of CaCl2 to the standing gelator solution forms the gel rapidly but after a few hours the gel collapses to form a precipitate. Interestingly, CaCl2 addition to stirred solutions of the gelators produces stable hydrogels. The morphological nature of the hydrogels was characterized using SEM and dense fibrous networks were obtained. Rheological measurements demonstrated that the hydrogels are thixotropic as they become liquid if a shear stress is applied and then they quickly recover the solid form on resting, with no variation of the Tgel. Being easily injectable, these hydrogels were biologically assessed by cell seeding and viability tests. They embedded Human gingival fibroblasts in 2% hydrogels and all of the hydrogels allowed the growth of encapsulated cells with a very good viability. Then, cytotoxicity and cytocompatibility studies show low toxicity of both the gelator solutions and the hydrogels. In addition, after 7 days when the 3D networks were completely formed, the number of living cells increased in all of the samples and the gelator toxicity is totally eliminated.
Conclusions
In this review, we have summarized the use of Phe and derivatives for the preparation of physical (or supramolecular) gels. Mainly, we have focused on the design of the Phe derivatives, elucidation of their self-assembly mechanisms, hierarchical structures and gelation abilities. We have elaborated on how the understanding at atomic level of this gelation illuminates their applications in chemical, material and biological sciences. The dynamic and reversible nature of physical gels as well as their responsiveness towards a variety of external stimuli make them interesting platforms for numerous applications including tissue engineering, environment remediation, nanodevices, and sensors. These applications have been also discussed in detail in the particular case of Phe derivatives. It seems clear that smart physical gels will continue to play an important role in the fabrication of functional nanomaterials. Keeping all these things in mind, it will be much easier to design and synthesize smart gelator molecules having multiple stimuli responsive behavior. In the future, the design of new gelator molecules and understanding of their stimuli-induced behavior can potentially unlock unprecedented applications in different areas such as drug delivery, tissue engineering, wound dressing, contact lenses, different chemo and biosensors, electronic applications and medicinal applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
T. Das acknowledges CSIR, India for research fellowship. D. Haldar thanks Alexander von Humboldt Foundation for financial support. D. D. Díaz thanks Deutsche Forschungsgemeinschaft (DFG) for the Heisenberg Professorship Award.
References
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- J. C. Tiller, Angew. Chem., Int. Ed., 2003, 42, 3072–3075 CrossRef CAS PubMed.
- A. Friggeri, B. L. Feringa and J. van Esch, J. Controlled Release, 2004, 97, 241–248 CrossRef CAS PubMed.
- Y. Huang, Y. Ding, T. Li and M. Yang, Anal. Methods, 2015, 7, 411–415 RSC.
- T. He, Z. Li, Z. Sun, S. Chen, R. Shen, L. Yi, L. Deng, M. Yang, H. Liua and Y. Zhang, RSC Adv., 2015, 5, 77296–77302 RSC.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. Tay, S. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS PubMed.
- F. Qu, Y. Zhang, A. Rasooly and M. Yang, Anal. Chem., 2014, 86, 973–976 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- S. Bera and D. Haldar, J. Mater. Chem. A, 2016, 4, 6933–6939 CAS.
- I. Irwansyah, Y.-Q. Li, W. Shi, D. Qi, W. R. Leow, M. B. Y. Tang, S. Li and X. Chen, Adv. Mater., 2015, 27, 648–654 CrossRef CAS PubMed.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS PubMed.
- B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5, 3452–3460 RSC.
- P. Chakraborty, B. Roy, P. Bairi and A. K. Nandi, J. Mater. Chem., 2012, 22, 20291–20298 RSC.
- S. J. Jhaveri, J. D. Mcmullen, R. Sijbesma, L.-S. Tan, W. Zipfel and C. K. Ober, Chem. Mater., 2009, 21, 2003–2006 CrossRef CAS PubMed.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS PubMed.
- P. K. Vemula and G. John, Chem. Commun., 2006, 2218–2220 RSC.
- D. K. Smith, Nat. Chem., 2010, 2, 162–163 CrossRef CAS PubMed.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- M. Mukai, H. Minamikawa, M. Aoyagi, M. Asakawa, T. Shimizu and M. Kogiso, J. Colloid Interface Sci., 2013, 395, 154–160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem.
Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- J. L. Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- S. R. Haines and R. G. Harrison, Chem. Commun., 2002, 2846–2847 RSC.
- H. Kobayashi, A. Friggeri, K. Koumoto, M. Amaike, S. Shinkai and D. N. Reinhoudt, Org. Lett., 2002, 4, 1423–1426 CrossRef CAS PubMed.
- F. M. Menger and A. V. Peresypkin, J. Am. Chem. Soc., 2003, 125, 5340–5345 CrossRef CAS PubMed.
- K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS PubMed.
- A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280–4288 CrossRef CAS PubMed.
- Z. Sun, Z. Li, Y. He, R. Shen, L. Deng, M. Yang, Y. Liang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 13379–13386 CrossRef CAS PubMed.
- M. Du, W. Song, Y. Cui, Y. Yang and J. Li, J. Mater. Chem., 2011, 21, 2228–2236 RSC.
- P. L. Zhu, X. H. Yan, Y. Su, Y. Yang and J. B. Li, Chem. – Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- X. H. Yan, Y. Cui, Q. He, K. W. Wang and J. B. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS PubMed.
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- T. M. Doran, D. M. Ryan and B. L. Nilsson, Polym. Chem., 2014, 5, 241–248 RSC.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- J.-B. Guilbaud, C. Rochas, A. F. Miller and A. Saiani, Biomacromolecules, 2013, 14, 1403–1411 CrossRef CAS PubMed.
- Y. Zimenkov, S. N. Dublin, R. Ni, R. S. Tu, V. Breedveld, R. P. Apkarian and V. P. Conticello, J. Am. Chem. Soc., 2006, 128, 6770–6771 CrossRef CAS PubMed.
- X.-R. Zhou, R. Ge and S.-Z. Luo, J. Pept. Sci., 2013, 19, 737–744 CrossRef CAS PubMed.
- J.-B. Guilbaud, E. Vey, S. Boothroyd, A. M. Smith, R. V. Ulijn, A. Saiani and A. F. Miller, Langmuir, 2010, 26, 11297–11303 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- L. Szkolar, J.-B. Guilbaud, A. F. Miller, J. E. Gough and A. Saiani, J. Pept. Sci., 2014, 20, 578–584 CrossRef CAS PubMed.
- A. Maslovskis, J.-B. Guilbaud, I. Grillo, N. Hodson, A. F. Miller and A. Saiani, Langmuir, 2014, 30, 10471–10480 CrossRef CAS PubMed.
- S. V. Stovbun, A. A. Skoblin and V. A. Tverdislov, Bull. Exp. Biol. Med., 2012, 152(6), 703–706 CrossRef CAS PubMed.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- H. B. Aiyappa, S. Saha, B. Garai, J. Thote, S. Kurungot and R. Banerjee, Cryst. Growth Des., 2014, 14, 3434–3437 CAS.
- J.-S. Shen, G.-J. Mao, Y.-H. Zhou, Y.-B. Jiang and H.-W. Zhang, Dalton Trans., 2010, 39, 7054–7058 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508–2511 CrossRef CAS.
- B. Weber, A. Serafin, J. Michie, C. Van Rensburg, J. C. Swarts and L. Bohm, Anticancer Res., 2004, 24, 763–770 CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- J. Shi, Y. Gao, Z. Yang and B. Xu, Beilstein J. Org. Chem., 2011, 7, 167–169 CrossRef CAS PubMed.
- R. Krieg, R. Wyrwa, U. Möllmann, H. Görls and B. Schönecker, Steroids, 1998, 63, 531–541 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- J. Sehnert, A. Hess and N. M. Nolte, J. Organomet. Chem., 2001, 637, 349–355 CrossRef.
- Z. Sun, Z. Li, Y. He, R. Shen, L. Deng, M. Yang, Y. Liang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 13379–13386 CrossRef CAS PubMed.
- J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508–2511 CrossRef CAS.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- K. Medini, B. W. Mansel, M. A. Williams, M. A. Brimble, D. E. Williams and J. A. Gerrard, Acta Biomater., 2016, 43, 30–37 CrossRef CAS PubMed.
- G. K. S. Prakash, F. Wang, M. Rahm, J. Shen, C. Ni, R. Haiges and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 11761–11764 CrossRef CAS PubMed.
- E. G. Cox, D. W. Cruickshank and J. A. S. Smith, Proc. Roy. Soc. (London), 1958, A247, 1–21 CrossRef.
- N. Boden, P. P. Davis, C. H. Stam and G. A. Wesselink, Mol. Phys., 1968, 25, 81–86 CrossRef.
- J. H. Williams, J. K. Cockcroft and A. N. Fitch, Angew. Chem., Int. Ed. Engl., 1992, 31, 1655–1657 CrossRef.
- J. M. Steed, T. A. Dixon and W. Klemperer, J. Chem. Phys., 1979, 70, 4940–4946 CrossRef CAS.
- E. Bartsch, H. Bertagnolli and P. Chieux, Ber. Bunsenges. Phys. Chem., 1986, 90, 34–46 CrossRef CAS.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. Raimondi, G. Calderoni, A. Famulari, L. Raimondi and F. Cozzi, J. Phys. Chem. A, 2003, 107, 772–774 CrossRef CAS.
- S.-M. Hsu, Y.-C. Lin, J.-W. Chang, Y.-H. Liu and H.-C. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921–1927 CrossRef CAS PubMed.
- S.-M. Hsu, F.-Y. Wu, T.-S. Lai, Y.-C. Lin and H.-C. Lin, RSC Adv., 2015, 5, 22943–22946 RSC.
- J.-L. Zhong, H. Pan, X.-Z. Luo, S.-G. Hong, N. Zhang and J.-B. Huang, Acta Phys.-Chim. Sin., 2014, 30, 1688–1696 CAS.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC.
- J. Šusteková, P. Drašar, D. Šaman and Z. Wimmer, Molecules, 2011, 16, 9357–9367 CrossRef PubMed.
- A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280–4288 CrossRef CAS PubMed.
- G.-F. Liu, D. Zhang and C.-L. Feng, Angew. Chem., Int. Ed., 2014, 53, 7789–7793 CrossRef CAS PubMed.
- W. Ji, G. F. Liu, M. X. Xu and C. L. Feng, Macromol. Chem. Phys., 2015, 216, 1945–1951 CrossRef CAS.
- G.-F. Liu, W. Ji and C.-L. Feng, Langmuir, 2015, 31, 7122–7128 CrossRef CAS PubMed.
- L. Frkanec, M. Jokic, J. Makarevic, K. Wolsperger and M. Zinic, J. Am. Chem. Soc., 2002, 124, 9716–9717 CrossRef CAS PubMed.
- X. Fu, N. Wang, S. Zhang, H. Wang and Y. Yang, J. Colloid Interface Sci., 2007, 315, 376–381 CrossRef CAS PubMed.
- S. Cao, X. Fu, N. Wang, H. Wang and Y. Yang, Int. J. Pharm., 2008, 357, 95–99 CrossRef CAS PubMed.
- X.-J. Fu, H. Zhang, S.-K. Zhou, S.-B. Liu, F.-Q. Guo, H. Wang and Y.-J. Yang, Helv. Chim. Acta, 2010, 93, 158–168 CrossRef CAS.
- U. K. Das, S. Banerjee and P. Dastidar, Chem. – Asian J., 2014, 9, 2475–2482 CrossRef CAS PubMed.
- P. Choudhury, D. Mandal, S. Brahmachari and P. K. Das, Chem. – Eur. J., 2016, 22, 5160–5172 CrossRef CAS PubMed.
- D. Das, S. Maiti, S. Brahmachari and P. K. Das, Soft Matter, 2011, 7, 7291–7303 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- M. J. Clemente, R. M. Tejedor, P. Romero, J. Fitremann and L. Oriol, New J. Chem., 2015, 39, 4009–4019 RSC.
- S. Bhattacharya and S. N. Ghanashyam Acharya, Chem. Mater., 1999, 11, 3121–3132 CrossRef CAS.
- J. Yan, J. Liu, P. Jing, C. Xu, J. Wu, D. Gao and Y. Fang, Soft Matter, 2012, 8, 11697–11703 RSC.
- Y. Ishioka, N. Minakuchi, M. Mizuhata and T. Maruyama, Soft Matter, 2014, 10, 965–971 RSC.
- F. Allix, P. Curcio, Q. N. Pham, G. Pickaert and B. Jamart-Grégoire, Langmuir, 2010, 26, 16818–16827 CrossRef CAS PubMed.
- P. Curcio, F. Allix, G. Pickaert and B. Jamart-Grégoire, Chem. – Eur. J., 2011, 17, 13603–13612 CrossRef CAS PubMed.
- R. M. Burch, M. Weitzberg, N. Blok, R. Muhlhauser, D. Martin, S. G. Farmer, J. M. Bator, J. R. Connor, C. Ko, W. Kuhn, B. A. McMillan, M. Raynor, B. G. Shearer, C. Tiffany and D. E. Wilkins, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 355–359 CrossRef CAS.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- W. Liyanage, W. W. Brennessel and B. L. Nilsson, Langmuir, 2010, 26, 16818–16827 CrossRef PubMed.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC.
- V. Singh, K. Snigdha, C. Singh, N. Sinha and A. K. Thakur, Soft Matter, 2015, 11, 5353–5191 RSC.
- I. Irwansyah, Y.-Q. Li, W. Shi, D. Qi, W. R. Leow, M. B. Y. Tang, S. Li and X. Chen, Adv. Mater., 2015, 27, 648–654 CrossRef CAS PubMed.
- C. Wang, W. Wei, Z. Li, X. Wang, H. Shen and Z. Sui, Russ. J. Phys. Chem. A, 2012, 86(1), 45–49 CrossRef CAS.
- A. Rajbhandary, D. M. Raymond and B. L. Nilsson, Langmuir, 2017, 33, 5803–5813 CrossRef CAS PubMed.
- N. Zanna, A. Merlettini and C. Tomasini, Org. Chem. Front., 2016, 3, 1699–1704 RSC.
- L. Milli, N. Zanna, A. Merlettini, M. D. Giosia, M. Calvaresi, M. L. Focarete and C. Tomasini, Chem. – Eur. J., 2016, 22, 12106–12112 CrossRef CAS PubMed.
- N. Zanna, S. Focaroli, A. Merlettini, L. Gentilucci, G. Teti, M. Falconi and C. Tomasini, ACS Omega, 2017, 2, 2374–2381 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
David
Díaz Díaz
a and
David
Díaz Díaz