
Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine – a new paradigm for tissue repair
I. M.
Bjørge†
 a,
S. Y.
Kim†
bc,
J. F.
Mano
a,
B.
Kalionis
d and
W.
Chrzanowski
*bc
a,
S. Y.
Kim†
bc,
J. F.
Mano
a,
B.
Kalionis
d and
W.
Chrzanowski
*bc
aDepartment of Chemistry, CICECO - Aveiro Institute of Materials, University of Aveiro, Aveiro, Portugal
bThe University of Sydney, The Australian Institute for Nanoscale Science and Technology, Sydney, NSW 2006, Australia. E-mail: wchrzanowski@sydney.edu.au
cThe University of Sydney, The Faculty of Pharmacy, Sydney, NSW 2006, Australia
dDepartment of Maternal-Fetal Medicine Pregnancy Research Centre, Royal Women's Hospital, Melbourne, Australia
First published on 14th November 2017
Abstract
Tissue regeneration by stem cells is driven by the paracrine activity of shedding vesicles and exosomes, which deliver specific cargoes to the recipient cells. Proteins, RNA, cytokines and subsequent gene expression, orchestrate the regeneration process by improving the microenvironment to promote cell survival, controlling inflammation, repairing injury and enhancing the healing process. The action of microRNA is widely accepted as an essential driver of the regenerative process through its impact on multiple downstream biological pathways, and its ability to regulate the host immune response. Here, we present an overview of the recent potential uses of exosomes for regenerative medicine and tissue engineering. We also highlight the differences in composition between shedding vesicles and exosomes that depend on the various types of stem cells from which they are derived. The conditions that affect the production of exosomes in different cell types are deliberated. This review also presents the current status of candidate exosomal microRNAs for potential therapeutic use in regenerative medicine, and in applications involving widely studied organs and tissues such as heart, lung, cartilage and bone.
 I. M. Bjørge | Isabel M. Bjørge is currently enrolled in the Doctoral Program of Materials Science and Engineering at the University of Aveiro, Portugal, under the supervision of Prof. João F. Mano. She has an Integrated Master's in Biomedical Engineering from the University of Minho, which included exchange periods at the Norwegian University of Science and Technology (Norway) and the University of Sydney (Australia). Isabel M. Bjørge is working with natural and synthetic-based polymers to create hierarchical bioencapsulation systems for tissue engineering purposes. |
 S. Y. Kim | Ms Sally Yunsun Kim received her Master of Philosophy (Pharmacy) at The University of Sydney and is a registered pharmacist. She is pursuing her Doctor of Philosophy degree and she has developed a technology to deliver stem cells to the airway via atomization with biomaterials. Her current research is focused on the targeted delivery of stem cells and extracellular vesicles to the lung to accelerate regeneration of tissue after injury. |
 J. F. Mano | João F. Mano is a Full Professor of Biotechnology at the Chemistry Department of University of Aveiro, Portugal, and director of the COMPASS Research Group, from CICECO – Aveiro Institute of Materials. His current research interests include the use of biomaterials and cells towards the progress of transdisciplinary concepts to be employed in regenerative and personalised medicine. In particular, he has been applying biomimetic and nano/micro-technology approaches to polymer-based biomaterials and surfaces in order to develop biomedical devices with improved structural and (multi-)functional properties, or in the engineering of microenvironments to control cell behaviour and organization, to be exploited clinically in advanced therapies or in drug screening. |
 B. Kalionis | Dr Bill Kalionis is a Senior Research Fellow at the Department of Maternal-Fetal Medicine Pregnancy Research Centre, Royal Women's Hospital, Parkville, Australia. He obtained his PhD in Biochemistry (University of Adelaide) and then carried out postdoctoral research on homeobox gene transcription factors in Drosophila melanogaster (Department of Biochemistry and Biophysics, UCSF, San Francisco, USA). After returning to Australia in 1990, his interest turned to a vital but poorly understood human organ; the placenta. Since 2005, his laboratory has focussed on the role of stem cells, and the extracellular vesicles they secrete, in normal and pathological placental development. |
 W. Chrzanowski | Dr Wojciech Chrzanowski is Health and Medicine Theme leader at the University of Sydney Nano Institute. He established Nanomedicine and Tissue Regeneration group and Nano-Bio-Characterisation facility within the Faculty of Pharmacy. He is a core member of Tissue Engineering and Regenerative Medicine nodes at the Charles Perkins Centre. He published excess of 140 publications predominantly in leading journals in the field. Dr Chrzanowski laboratory develops a frontier cell- and exosome-based approach targeted at lung tissue regeneration. He have assembled an unparalleled suite of microscopy systems dedicated to bio-nano-characterisation to gain fundamental understanding of cellular and biological processes. |
Cell-free strategies for tissue regeneration
Tissue engineering strategies often involve the use of stem cells due to their regenerative/reparative potential. These effects are still not fully understood but are generally attributed to stem cell trans-differentiation and cell fusion, paracrine effects, mitochondrial transfer, and finally, to the release of extracellular vesicles (EVs) by stem cells.1Recently, stem cell paracrine effects are considered to be primarily responsible for the regenerative potential and increasing interest has focussed on EVs and the bioactive molecules they release.2,3 Cell-free strategies using conditioned medium4–7 or EVs8 demonstrate that this may indeed be the case. Cell-free approaches may be advantageous when risk factors associated with stem cell use are considered. These can be intrinsic factors relating to cell origin, tumorigenic potential, differentiation and proliferation capacity, or extrinsic factors concerning cell handling, storage, transport conditions, among others.9 Furthermore, it is important to consider that stem cell yield decreases with donor age and that age also quantitatively impacts on performance.10 EVs, on the other hand, bypass a series of issues that arise with stem cell therapy. Moreover, EVs are easily stored and tested for optimal dosage and potency, all of which reduce the cost and time associated with stem cell expansion or collection from patients.2
Stem/progenitor cells transfer EVs containing functional regenerative signals to injured cells and organs.11 Examples of disease models used to demonstrate EV function in tissue regeneration include chronic wound,12 osteoarthritis,13 myocardial infarction,14 chronic kidney disease15 and lung diseases.16
Heterogenous compositions of proteins, lipids, and nucleic acids are selectively packaged by secreting stem cells in response to the microenvironment.11,17 The regenerative effects of EVs are, at least in part, attributed to the transfer of specific protein and microRNA (miRNA) cargos.18 There is increasing interest in identifying the subpopulations of EVs that have maximum therapeutic potential19,20 and studies show that miRNAs are enriched in the biologically active exosome fraction, rather than in the microvesicle/shedding vesicle fraction.18 Therefore, this review focuses on the role of exosomes and exosomal-miRNA in tissue regeneration and regenerative medicine.
Characterisation of exosomes and shedding vesicles
In 1967, shedding vesicles were described as platelet-dust by Peter Wolf.21 More than a decade later, Trams et al. found two populations of differently sized vesicles containing 5′-nucleotidase activity that were secreted by cells.22 These vesicles were derived from the plasma membrane present in cultures of normal and neoplastic cell lines. Pan et al.23 and Harding et al.24 studied the transferrin receptor during reticulocyte maturation and discovered that multivesicular endosomes (MVEs) contained bodies with a diameter of approximately 50 nm. Upon fusion of the MVEs with the plasma membrane, these vesicles were released from the cells and were later termed exosomes. These shedding vesicles (also called microvesicles or ectosomes) and exosomes are often collectively referred to as EVs.The secreted vesicles differ depending on the cell type, origin and state.25 EVs mediate a series of cellular functions such as the transport of materials and intercellular communication.26 EVs are described as highly specialised messenger molecules, which can deliver biological signals. Increasing evidence proposes that these messages are highly dependent on specific conditions. For example, an environmental stressor or drug treatment can alter EV production, size profile and composition. Furthermore, EVs can carry ‘undesired’ messages that contribute to the spreading of diseases.27 EVs have different origins; shedding vesicles (50 and 1000 nm) are formed at the cell surface via membrane budding, whereas exosomes (20–100 nm) originate from multivesicular bodies (MVBs) (250 to 1000 nm) (Fig. 1). MVBs are either degraded or fused with the plasma membrane, releasing intraluminal vesicles (ILVs). Upon fusion and release of the vesicles inside the MVB cargo, these vesicles are called exosomes.26,28
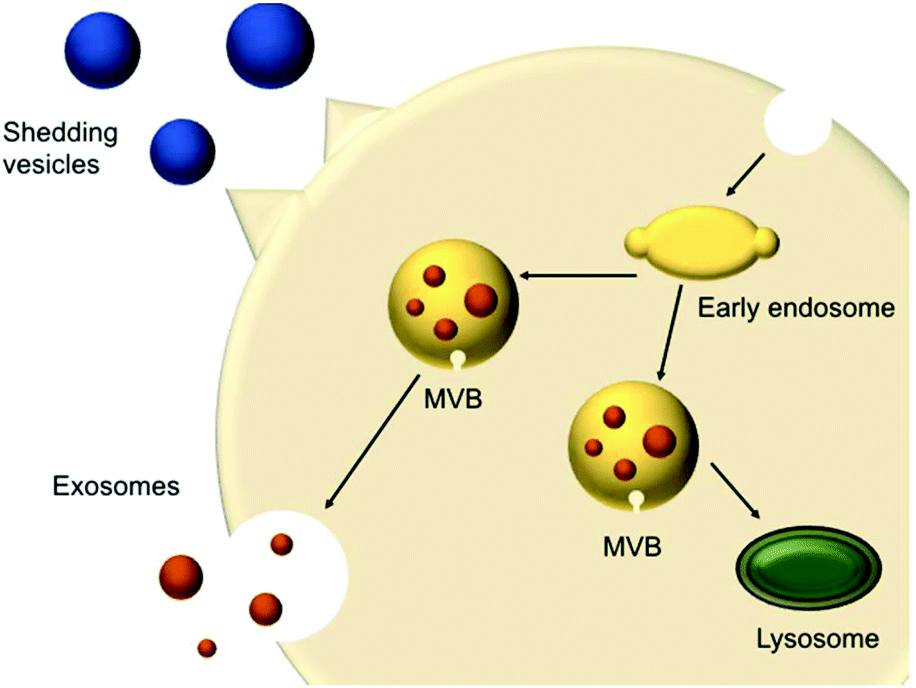 | ||
| Fig. 1 Schematic representation of extracellular vesicles production and release (adapted from Raposo and Stoorvogel).28 | ||
Shedding vesicles
Production of shedding vesicles (SVs) involves trafficking of biomolecules towards plasma membrane regions that are enriched in ceramide and lipid rafts (cholesterol-rich microdomains). Ceramide-enhanced membrane curvature results in protrusion and budding, followed by stalk fission and subsequent detachment.29,30The release of SVs may occur in resting cells, although this can be enhanced by several factors, including cell activation and exposure to proteins from activated complement cascades. Furthermore, cells subjected to irradiation, oxidative injury or under shear stress, show increased shedding.31 Membrane degradation and stimuli-driven increases in intracellular calcium are other impacting factors. Aside from calcium, phorbol ester activation of protein kinase C also enhances shedding in some cell types.32,33
Lipid raft-associated molecules tissue factor and flotillin and a high exposure of phosphatidylserine are typical markers of shedding vesicles.34 Depending on the cell type of origin, SVs may contain different plasma membrane proteins.35 Specific markers may therefore be required to identify SVs depending on the cell type of origin. The generic marker annexin V is used to identify SVs, whereas CD45 is commonly used for leukocyte-derived SVs. For platelet-derived SVs, CD42b/CD31- and CD62P are used, while endothelial-derived SVs are characterised by CD31+/CD42−, CD62E, CD31+/CD42− and CD144.36 Apart from the already mentioned cholesterol and ceramide, sphingomyelin is also present in SVs. Additionally, SVs contain integrins, selectins, CD40 ligand and metalloproteinases.37,38Table 1 presents typical markers for SVs derived from various human stem cell types. There are multiple databases that document molecular data of EVs, including shedding vesicles, such as Vesiclepedia39 and EVpedia.40 The miRandola 2017 database41 focuses on extracellular non-coding RNAs of EVs.
| SV type | Protein content | Known gene expression/RNA content | Ref. |
|---|---|---|---|
| Abbreviations: ASC, adipose MSCs; hWJMSC, human Wharton-Jelly MSCs; hBMSC, human bone marrow MSCs; hES, human embryonic MSCs; hiPSC-MSC, human induced pluripotent stem cell-derived MSCs; HLSC, human adult liver stem cell. | |||
| hASC-SV | CD105 and CD90 | miR29c and miR150 | 42 |
| hWJMSC-SV | CD9, CD44, CD63 and CD73 | miR-15a, -15b, and -16 | 43 |
| hBMSC-SV | CD44, CD29, α4- and α5 integrins, and CD73. | Cell differentiation genes (POLR2E, SENP2/SUMO1, RBL1, CXCR7 and LTA4H) | 44 and 45 |
| T cell internal antigen-1 (TIA), TIA-1-related (TIAR), AU-rich element binding protein (HuR), staufen1 and 2, and argonaute2 | mRNA involved in transcription (CLOCK, IRF6), cell proliferation (RBL1, SENP2) immune regulation (CRLF1, IL1RN), among others | ||
| miR-24, -103-1, -140, -143-5p, and -340 | |||
| miR-223, -451, and -564 | |||
| hES-SV | Oct-4 protein, Wnt-3 isoforms A and B | mRNA encoding Oct4, Nanog, Gata-4, Sox2, Klf4 and Lin28 | 46 and 47 |
| and miR-292, -294 and -295 | |||
| hiPSC-MSC-SV | CD9, CD24, CD63, CD81, integrins, glycoproteins, | Pluripotency-associated factors: OCT4, NANOG, and SOX2 | 48 and 49 |
| miRNAs from miR-302/367 cluster | |||
| HLSC-SV | α4 integrin, CD29, and CD44 | Cell proliferation genes: MATK, MRE11A, CHECK2, MYH11, VASP and CDK2 | 45 and 50 |
| Staufen2 | |||
| miR-451, -223, -24, -125b, -31, and -122 | |||
Various RNA types including mRNA51 and miRNA52 are contained in SVs and these nucleic acids modulate functions of target cells. A recent study showed microvesicles from adipose-derived stem cells promote angiogenesis via the delivery of microRNA-31 which, in turn inhibited anti-angiogenic gene HIF-1.53Table 2 contains some examples of SVs from different sources that have been used to improve tissue regeneration in animal models.
| SV source | Test platform | Outcome | Known mechanism | Ref. |
|---|---|---|---|---|
| hASCs | Ischemic murine flap models | Improved cell migration, survival, inflammation and angiogenesis resulting in improved wound healing | Altered expression of miR29c and miR150 and upregulated gene expression of SDF-1, CXCR4, CXCR7, CCL2, ANGPTL4 | 42 |
| hWJMSCs | Acute kidney injury (AKI) rats | Enhanced proliferation, and diminished renal cell apoptosis and inflammation. | Suppression of CX3CL1 expression | 43 |
| hBMSCs | Cisplatin- and glycerol-induced AKI mice | Increased proliferation and reduced apoptosis of tubular cells | Up-regulation of anti-apoptotic genes (Bcl-xL, Bcl2, and BIRC8) and down-regulation of genes involved in cell apoptosis (Casp1, Casp8, and LTA) | 44 and 62 |
| HLSCs | Rat model of 70% hepatectomy | Increased hepatocyte proliferation, associated with an accelerated morphological and functional recovery | mRNA transfer evidenced by human AGO2 expression | 50 |
Various RNA types including mRNA51 and microRNA52 are contained in SVs and these nucleic acids modulate functions of target cells. A recent study showed microvesicles from adipose-derived stem cells promote angiogenesis via the delivery of microRNA-31 which, in turn inhibited anti-angiogenic gene HIF-1.53Table 2 contains some examples of SVs from different sources that have been used to improve tissue regeneration in animal models.
Exosomes
Protein sorting in MVBs involves segregation of the ubiquitinated proteins into membrane domains (lipid rafts) by ESCRT-0. These lipid rafts are rich in cholesterol and sphingomyelin, and contain lysobisphosphatidic acid (LBPA) and phosphatidylinositol-3-phosphate.56–58 Segregation is followed by endosomal invagination, with subsequent cargo sorting, and abscission of the vesicles through further intervention of the ESCRT machinery to form ILVs within the multivesicular endosomes (MVEs).55 MVEs deliver misfolded proteins in the plasma membrane, activated growth-factor, hormone and cytokine receptors to lysosomes.
Alternatively, an ESCRT-independent mechanism leads to ILV formation.59 Alix (ALG-2-interacting protein X) binds directly with target molecules, where LBPA acts as a mediator, preventing degradation and inducing molecule sorting into ILVs.29,60 Furthermore, Alix supports endosomal membrane budding and abscission.61
Exosome secretion can occur in a constitutive or regulated manner, depending on the cell type of origin.63 In either case, MVBs move to the cell periphery, where they are tethered by tethering factors in conjunction with Rab GTPases.64,65 Fusion of the MVB membrane occurs through the vesicle-associated and acceptor membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs),66 and synaptotagmin family members.54 As with shedding vesicles, environmental factors such as oxygen level, disease, mechanical stress or media composition modulate exosome release and exosome composition. For some cell types, increased amounts of exosomes are produced and released under hypoxic and oxidative stress conditions.67Table 3 provides a few examples of conditions that impact on exosome production.
| Exosome source | Condition | Outcome | Ref. |
|---|---|---|---|
| Breast cancer cell lines: MCF7, SKBR3, and MDA-MB 231 | Modest (1%) and severe (0.1%) hypoxia | Increased secretion and elevated miR-210 levels | 68 |
| HeLa cells | Endoplasmic reticulum stress | Increased secretion, possibly mediated by IRE1 and/or PERK | 69 |
| Eosinophils | Asthma | Increased secretion | 70 |
| Dendritic cells | Activation of DCIR by anti-DCIR antibody or HIV-1 | Increased secretion | 71 |
| HEK 293 cells | Acidic pH | Higher isolation yield, increased exosomal protein, and RNA concentration | 72 |
| Melanoma cell lines | Preincubation with proton pump inhibitors (inhibition of extracellular acidification) | Reduced entry into target cells | 73 |
| Cardiac progenitor cells | Hypoxia (95% N2, 5% CO2) for 12 h | Upregulation of a range of miRNAs which contribute to cardiac repair | 74 |
| Cardiomyocytes | Hypoxia (near-zero O2 for 2 h or 1% O2 for various time points from 5 min to 24 h) | Two-fold increased secretion (at 1% O2 exposed for 2 h) and high content of pro-inflammatory cytokine (TNF-α) induced by HIF-1α, respectively. | 75 and 76 |
| Cardiomyocytes (H9C2) | Glucose starvation for 48 h | Enhanced secretion of exosomes, greater amount of protein, including protein species related to metabolic processes and signalling pathways oriented to promote energy acquisition, upregulation of miRNA related with cell proliferation, cell-cycle, MAPK signalling and protein transport. | 77 |
| Placental MSCs | Hypoxia (1, 3, 8% O2) | Increased secretion of exosomes by 3.3 and 6.7 folds at 1% or 3% O2 respectively; up to 4.6 times higher number of exosomal proteins identified for exosomes from 1% O2 compared to 3% or 8% O2. | 78 |
| Bone marrow MSCs | Serum starvation and 1% O2 for 40 h | Increased secretion of exosomes induced significantly higher tubule formation in human umbilical vein endothelial cells in vitro, indicating angiogenesis. | 79 |
| Exosome (Ex) type | Protein content | Known gene expression/RNA content | Ref. |
|---|---|---|---|
| Abbreviations: hUCMSC: human umbilical cord MSC, hPlaMSC: human placenta MSC, hEnSC: human endometrium MSCs, hMenSC: human menstrual MSCs, hES: human embryonic MSCs. | |||
| hUCMSC-Ex | CD9, CD63, and CD81 | miR-21, -23a, -125b, and -145 | 107–109 |
| hBMSC-Ex | CD9, CD63, CD81, Alix, Tsg101, flotillin-1, HSP70, β-actin, MHCII, and glyceraldehyde-3-phosphate dehydrogenase | Up-regulated: let-7a and miR-199b, -218, -148a, -135b, -203, -219, -299-5p, -302b | 12, 110 and 111 |
| Down-regulated: miR-221, -155, -885-5p, -181a, and -320c | |||
| tRNA CTC (Glu) | |||
| hiPSC-MSC-Ex | CD9, CD63 and CD81 | RNA content not directly characterized yet known to upregulate OPN, OCN, RUNX2 | 112 |
| hASC-Ex | CD63 and CD81 | miR-486-5p, -10a-5p, -10b-5p, -191-5p, -222-3p, -21-5p, and -22-3p | 111 |
| tRNA GCC (Gly) and tRNA CTC (Glu) | |||
| hPlaMSC-Ex | CD9 and CD63 | RNA content not directly characterized yet known to upregulate OCT4 and NANOG mRNA expression | 113 |
| hEnSC-Ex/hMenSC-Ex | CD63, TSG101, Hsp70, and Hsp90 | miR-21, -1275, -21-5p, -23-3p, -3940-5p, -4708-3p, -548ap-5p, and -642b-5 | 114 and 115 |
| hES-Ex | Fibronectin 1 (FN1), heat shock proteins, and fibrinogens | Let-7b and -7d miRNAs regulating HNF4A | 116–118 |
| miR-122, -1224-5p, -1228, -1234, -1237, -1238, -124, -150, -198, -296-5p, -572, -765, -940, -233, -451, -564 | |||
Cells transmit biological information to recipient injured cells through the biologically active lipids on EV membranes. The lipid contents of exosomes may also vary according to the cell type of origin. Exosomes are enveloped by a lipid bilayer that contains sphingomyelin, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, monosialotetrahex-osylganglioside (GM3) and phosphatidylinositol, which is similar in composition to the cell plasma membrane.86 Additionally, exosomes present cholesterol, ceramide, phospholipid phosphatidylserine and glyco-sphingolipids.80 Various signalling functions are contributed by bioactive lipids of stem cells including immunomodulation87 and anti-inflammatory activities88 and these have important roles in stem cell therapies. However, the particular lipids transported in EVs for specific regenerative medicine applications are yet to be identified.
A breakthrough in the history of exosome research was in 2006–7 when the research groups of Ratajczak et al. and Valadi et al. reported that exosomes contained mRNA and miRNA, which could be delivered to recipient cells and subsequently have a functional role.46,89 Exosomal miRNAs are important modulators of gene expression and cause physiological changes in recipient cells.90 Exosomes contain RNA in a size range less than 700 nt. Additionally, mRNA fragments, long non-coding RNA, piwi-interacting RNA, ribosomal RNA and fragments of tRNA-, vault- and Y-RNA are also present. Furthermore, mitochondrial DNA, single-stranded DNA, double-stranded DNA and oncogene amplification products are present in exosomes.80
Exosomal transfer of miRNAs is now generally accepted as a mechanism for intercellular communication.91,92 Recently, specific miRNAs carried by exosomes were identified that could be potentially used in therapeutic applications. Two critical miRNAs that regulate inflammation, miR-155 and miR-146a, enhanced and reduced inflammatory gene expression respectively when delivered by exogenous exosomes in endotoxin-injury models.93 Furthermore, exosomes are used as disease biomarkers, as is the case for exosomal miRNA biomarkers miR-375, miR-21 and miR-574 in prostate cancer.94
The miRNAs within exosomes are not randomly selected for packaging but are selectively packaged by the secreting cell, possibly via a regulated mechanism.45,95 More studies are required to understand how secreting cells selectively choose and package different cargos of RNA inside exosomes. Exosomal RNA profiles are reported to differ from those of the parent cells89 but Guduric-Fuchs et al. showed that inducing overexpression of miR-146a in parent cells resulted in increased levels of miR-146a in the extracellular vesicles released from those parent cells.96 Since nearly half of the genes in human cells are regulated by miRNA,97 altered exosomal miRNA levels could determine outcomes of disease as well as therapy.98
Extraction methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000g and the pellet is analysed by flow cytometry. A subsequent ultracentrifugation at 100000g may be used for purification, yet this can lead to the formation of unwanted aggregates.99
000g for 90 minutes. Since then, several variations have been reported for differential ultracentrifugation.102,103 However, the viscosity of the solution impacts on the yield, resulting in poor exosome recovery from biofluids. Changes in sedimentation distance, temperature, time, or speed may impact on the yield and should therefore be studied.104–106
000–20000g and the pellet is analysed by flow cytometry. A subsequent ultracentrifugation at 100000g may be used for purification, yet this can lead to the formation of unwanted aggregates.99
000g for 90 minutes. Since then, several variations have been reported for differential ultracentrifugation.102,103 However, the viscosity of the solution impacts on the yield, resulting in poor exosome recovery from biofluids. Changes in sedimentation distance, temperature, time, or speed may impact on the yield and should therefore be studied.104–106
ExoQuick™ (EQ) isolation kits comprise a family of reagents that rely on exosome precipitation. EQ solution is added to the exosome-containing sample and refrigerated overnight, after which the mixture is centrifuged at 1500g for 30 min for pellet retrieval.106 Other precipitation-based kits have also been developed such as Life Technologies™ Total Exosome Isolation Reagent, Exo-Spin™ and Invitrogen™ kits. In comparison with EQ, the average vesicle size from human serum samples was smaller and there was superior RNA recovery with the Life Technologies™ Total Exosome Isolation Reagent.119 Other precipitation-based methods (ExoSpin and Invitrogen kits) yielded higher BT474 cell-derived exosome content than density gradient method (PureExo®) and UC.120 Recently, a novel method has been proposed that exploits the presence of the negatively-charged lipid phosphatidylserine. Through titration with acetate, the surface charge of the cell-free supernatant is neutralized, resulting in exosome precipitation.121
Alvarez et al. analysed levels of mRNA, miRNA, and protein to determine which method was best for isolation of urinary exosomes. Whilst a modified EQ-based method led to a higher exosome yield and better quality mRNA and miRNA, UC-based methods resulted in higher protein purity.122 Similarly, for human serum samples, results showed similar exosomal miRNA profiles for the two methods, yet the miRNA yield was higher for EQ. Furthermore, miR-92a and miR-486-5p levels were statistically different for both methods.106
The ultrafiltration or density gradient centrifugation method was developed with monocyte-derived dendritic cells. It involves clarification of the supernatant, followed by ultrafiltration with a molecular weight cut-off at 500 kDa. The concentrate is diafiltered and ultracentrifuged with an underlying density cushion of 30% sucrose/deuterium oxide.83 When compared with UC for isolation of human tongue cancer cell line-derived exosomes, density gradient centrifugation yielded uniformly distributed sized particles, as well as enriched exosome markers.123
Yamada et al. studied combinations of UC followed by EQ or by density gradient centrifugation, in human milk exosome isolation. UC with EQ precipitation led to a rapid and increased exosome recovery, whereas UC with density gradient centrifugation led to samples with higher purity.124
Other exosome extraction methods include purification by high performance size exclusion liquid chromatography,125 size exclusion chromatography (SEC),104 and separation by sucrose density gradient due to their buoyant density of 1.23 to 1.16 g L−1.102 More recently, OptiPrepTM density gradient isolation method is progressively replacing the sucrose gradient method due to higher purity of the exosomes obtained.126
For rat and human blood plasma-derived exosomes, isolation by UC resulted in low yield and high sample contamination. In contrast, isolation by SEC led to purer samples, but a low vesicle yield.104
Quantification of exosomes derived from antigen-presenting cells is possible through immunoprecipitation technologies using antibody loaded magnetic cell beads. In this method, bead-exosome complexes are formed due to the immune-magnetic interaction between the beads and the exosomes containing human MHC class I molecules.127 Compared to the differential ultracentrifugation method, magnetic bead-based isolation of exosomes is designed to be fast, efficient and selective by the use of antibodies against the common exosomal markers – tetraspanins C9 or CD81.128 For human colon carcinoma cells, a study showed that EpCAM immunoaffinity capture-enriched exosomes expressed exosome markers and associated proteins more highly than the UC and OptiPrep methods.126
Overall, comparisons of SEC, magnetic beads, UC and EQ, for isolation of exosomes derived from ascites of clinical adenocarcinoma samples, showed the highest sample purity and RNA concentration was obtained by the EQ method, followed closely by SEC. Similarly, EQ showed the highest exosomal protein yield, whilst ultracentrifugation had the lowest.129
Besides regenerative medicine applications, exosomes are important biomarkers of disease as they are conveniently found in abundance in many biological fluids including blood, urine and saliva.25,130 However, extraction of exosomes from viscous fluids is challenging for several traditional isolation methods. Novel microfluidic-based isolation methods result in higher yields and purity, when compared with the aforementioned methods.130 Microfluidic strategies based upon acoustic,131 electrophoretic,132 and viscoelastic133 mechanisms emerge as highly effective approaches for isolation and fractionation of EV populations. These approaches reduce isolation time, reagent consumption, sample volume, and are more cost effective than conventional methods.134 Another significant advantage of these methods is their compatibility with biofluids.130
As described below, exosomal mRNA and miRNA are key modulators in tissue regeneration, affecting cellular behaviour and outcome through paracrine effects.135 Thus, more important than the yield of each extraction method, is mRNA and miRNA purity and the quality of extracted exosomes, which should drive the choice for the method of isolation. Thus, ExoQuick™ and microfluidic strategies may be the best exosome isolation options for tissue regeneration applications due to a greater mRNA and miRNA purity. One of the previously stated advantages of microfluidics is the smaller sample size. Whilst this may indeed be an advantage in the case of disease screening or exosome characterisation, it may act as an impediment for tissue engineering strategies, where a greater number of exosomes are required per study. Furthermore, when considering clinical trials this problem increases dramatically.
There is no consensus regarding the ideal method for exosome isolation and novel methods are likely to emerge that offer improved yield and/or purity. The outcome of exosome isolation is dependent on the cell source from which the exosomes are derived, and caution is needed in choosing a method since different extraction methods alter the exosomal protein and miRNA content. Thus, it is important to use a consistent isolation method when comparing exosome populations.126
Roles of EVs in regenerative medicine
An overview of the use of EVs and EV-miRNA in tissue engineering and regenerative medicine applications
Tissue engineering is a promising option for the regeneration of tissues and this technology can be combined with stem cell-based therapies since there is evidence that stem cells promote and regulate tissue regeneration. Current strategies use the cell and/or its by-products secreted into the culture medium, as the active biological agent(s).136 EVs are by-products that regulate intercellular communication, which is necessary for multicellular organisms to maintain their vital functions. These extracellular vesicles comprise the following sub-classes; apoptotic bodies, shedding vesicles and exosomes.25EVs are a cell-free alternative to current stem cell therapies with advantages of lower immunogenic response and preservation of biochemical activity upon storage.137–139 Furthermore, employing EVs bypasses important safety concerns associated with the engraftment of viable replicating cells, which have the potential for long term pathological transformation.140
MSC-derived exosomes and shedding vesicles have been investigated for a range of in vitro and in vivo applications involving lung, liver, kidney, and colon injury, as well as myocardial infarction, skin burns and defects, and cerebral artery occlusion.
Gatti et al.141 studied the effect of human bone marrow mesenchymal stem cell (hBMSC)-derived exosomes on acute and chronic kidney injury induced by ischemia–reperfusion injury in a rat model, and concluded that exosome administration led to a reduction of apoptosis and enhanced the proliferation of tubular epithelial cells. A rat model of acute kidney injury induced by cisplatin was used by Zhou et al.108 to demonstrate the beneficial effect of human umbilical cord MSC-derived exosomes (hUCMSC-Ex) in decreasing oxidative stress, inhibiting renal tubular apoptosis as well as reducing blood urea nitrogen and creatinine levels.
Li et al.107 used a carbon tetrachloride-induced murine liver fibrosis model and determined that hUCMSC-Ex reduced fibrous capsules formation at the surface, and decreased hepatic inflammation. Similarly, Yih142 observed that exosomes isolated from human embryonic stem cell-derived human mesenchymal stem cells, limited the extent of injury using the same murine drug-induced liver injury model. Moreover, in injury models induced by Acetaminophen and hydrogen peroxide treatment of immortalized murine transforming growth factor alpha transgenic hepatocyte cells, exosomes increased cell viability in the Acetaminophen model, and were cytoprotective in the hydrogen peroxide model.
In an induced stroke model, Xin et al.143 showed that rat bone marrow MSC-derived exosomes increased neurite remodelling, neurogenesis and angiogenesis in the ischemic boundary zone. Zhang et al. used a rat model for induced traumatic brain injury and verified that administration of exosomes increased angiogenesis and neurogenesis, and reduced neuroinflammation.144 Pascucci et al.145 studied the promotion of angiogenesis by horse adipose tissue MSCs-derived microvesicles (exosomes + shedding vesicles) through the scratch migration assay and rat aortic ring assay. Administration of microvesicles significantly increased cell migration after 24 hours, when compared to control medium (EndoGROTM basal medium, and 1:1 Dulbecco's modified Eagle medium (DMEM) and endothelial basal medium (EBM) for the scratch assay and ring assay, respectively).
Regarding the colon, Yang et al.146 showed protective effects of rat bone marrow mesenchymal stem cell-derived microvesicles in the 2,4,6-trinitrobenzene sulfonic acid-induced colitis model. Microvesicles attenuated inflammatory activity, oxidative perturbations and reduced apoptosis in a dose-dependent manner.
Exosomes also enhance MSC differentiation, as Takeda and Xu147 demonstrated the development of neuron-like morphology and the upregulation of neuronal markers in MSCs treated with neuronal cell line-derived exosomes. In a study reporting the potential of exosome therapy for invertebral disc degeneration, exosomes secreted by nucleus pulposus cells were found to promote differentiation of MSCs to a nucleus pulposus-like phenotype.148 The osteogenic potential of exosomes was demonstrated for the first time in vitro and in vivo when exosomes isolated from differentiated MSCs, induced lineage specific differentiation of naïve MSCs.149
The role of miRNA in tissue engineering and regenerative medicine is of interest because miRNAs influence a wide range of cell functions including differentiation and gene expression.150–153 Exosomal miRNA is thought to be the key factor in intracellular communication and regulation, with a preponderant role in the modulation of biological functions of acceptor cells.89,91,154 As proven by Valadi et al., exosomes can actively shuttle translatable RNAs between mast cells for the production of specific proteins.89 Since then, intensive investigations on the potential of miRNA in the repair and restoration of tissue function for regenerative therapies involving the heart,155 skin,156 lung,157 bone,158,159 cartilage,160 and neurons161 have been reported.
miRNAs present a half-life between 28 and 220 h, which is significantly greater than typical mRNAs (10 h).162 Delivering mRNA is problematic because they are unstable and are rapidly degraded by nucleases and therefore viral vectors and lipid nanoparticles have been tested as delivery vehicles. Cells and extracellular vesicles are also attractive delivery vehicles for miRNA delivery, improving circulation stability even in the presence of ubiquitous ribonucleases (RNases).151,163,164 However, studies have shown that RNase pre-treatment of shedding vesicles led to an inhibition of the biological effects.46,165 Additionally, exosomal miRNA has the advantage of being naturally stable enough to be a potential biomarker.166
Typical tissue contraction occurs during the proliferative phase of wound healing and is marked by the differentiation of fibroblasts into contractile α-smooth muscle actin (α-SMA)-expressing myofibroblasts, mediated by the TGF-β pathway. However, if myofibroblasts remain active, fibrotic diseases may arise.109,168 During the differentiation process, α-SMA is produced and cadherin-2 expression is switched to cadherin-11. Huang et al. showed the administration of keratinocyte-derived extracellular vesicles to fibroblasts resulted in opposite effects, by decreasing cadherin-2 expression, and simultaneously reducing α-SMA mRNA expression.168 Fang et al. also demonstrated that hUCMSC-Ex microRNAs (miR-21, -23a, -125b, and -145) reduced myofibroblast formation by impacting on the activity of the TGF-β/SMAD2 signalling pathway, which resulted in decreased α-SMA and collagen deposition.109 These findings suggest that extracellular vesicles could potentially promote skin tissue regeneration and minimise scar tissue formation.
For diabetic chronic wounds, the effect of human bone marrow-derived mesenchymal stem cells (hBMSCs) was beneficial, with increased endothelial angiogenesis as well as enhanced growth and migration of injured fibroblasts through the activation of several signalling pathways including Akt, Erk1/2, and STAT3.12 In a mouse model for type 2 diabetes, fibrocyte-derived exosomes led to accelerated wound closure and re-epithelialization, as well as increased collagen I and α-SMA expression.169 While this can be only hypothesized from these results, exosomes could also support fight against bacterial infection, which is perhaps regulated by their anti-inflammatory activity.
Similarly, Zhang et al.170 administered exosomes derived from human induced pluripotent stem cell-derived MSCs (hiPSC-MSC-Ex) in a rat full-thickness skin defect model and demonstrated enhanced wound healing, synthesis of collagen type I and III, as well as increased elastin secretion, and vascularization.
Exosomes derived from hUCMSC (hUCMSC-Ex) enhanced endothelial cell migration in cultured human umbilical vein endothelial cells (HUVEC), which occurred in a dose-dependent manner. Furthermore, in a skin-deep, second-degree burn model in rats, hUCMSC-Ex promoted cell proliferation and re-epithelialization, leading to a significant increase in the number of epidermal and dermal cells. Moreover, hUCMSC-Ex were more effective than human lung fibroblast-derived exosomes in the formation of healthy multi-layered skin tissue. This effect was attributed to the activation of Wnt/β-catenin signalling by hUCMSC-Ex, which is required for wound healing.171,172 A more recent study from the same group demonstrated that the administration of hUCMSC-Ex led to an initial increase in Wnt/β-catenin signalling for tissue repair, followed by an inhibition of the pathway at high cell density. The induction of YAP (YES-associated protein) phosphorylation is controlled by exosomal protein 14-3-3ζ at the Ser 127 site. These proteins are required for binding of YAP and p-LATS, which, under high cell density, form a complex. This consequently inhibits Wnt/β-catenin signalling, preventing dysplasia.173
Adipose mesenchymal stem cell-derived exosomes (ASCs-Ex) were used by Hu et al. to demonstrate that the internalisation of exosomes by fibroblasts led to increased cell migration and proliferation, as well as increased gene expression (i.e. collagen I, III, N-cadherin, and PCNA) and elastin protein production. Moreover, systemic administration of ASCs-Ex in a murine skin incision model resulted in their recruitment to soft tissue wound areas, which was associated with increased production of collagen I and III. This increase in collagen production occurs during the early healing stages, after which there may be an inhibition and reduction of scar tissue formation.174
A recent study analysed exosome levels in skin specimens and fibroblasts obtained from skin biopsies of patients with systemic sclerosis (SSc). Skin samples showed up-regulated mRNA levels of CD63 and CD9, typically used as exosome markers, when compared to normal skin samples. This was confirmed since protein levels of CD63, CD9, and CD81 were increased in cell lysates of SSc fibroblasts, and there was evidence of increased exosome secretion. Furthermore, SSc exosomes featured increased miR-142-3p levels, and decreased miR-150a and miR-196a levels. Media containing SSc exosomes contained increased mRNA levels of COL1A1 and COL1A2. However, serum exosome levels were decreased for both types of SSc skin specimens studied. In the same study, exosomes isolated from murine blood samples, and from human exosome-containing culture media, were administered to full-thickness wounds in mice. Collected serum samples were positive for CD63 and showed accelerated wound treatment.175
Transcription factor RUNX2, together with the bone morphogenetic protein 2 (BMP-2) RUNX2 and Wnt pathways, and osterix (OSX), are essential in osteogenesis.149,177 The miRNA cluster 23a∼27a∼24-2 negatively targets protein SATB2, a co-regulator of RUNX2. miRNA-27a, in particular, down regulates the RUNX2 activator Hoxa10. RUNX2 also increases the expression of miR-2861∼miR3960, which in turn target RUNX2 inhibitors (Hdac5 and Hoxa2) in BMP2-induced osteogenesis of ST2 stromal cells.150 Since, exosomal cargo is rich in different classes on RNAs and has been shown to impact on the above pathways, it is apparent that exosomes could aid bone tissue regeneration.
For example, miR-9 and miR-98 identified in exosomes are involved in endochondral ossification in a rat model, whilst cartilage development involves miR-140 and miR-455.178 Transcription factor SOX9 is necessary for chondrogenesis. miR-125 and -449a are negative regulators of MSC chondrogenic differentiation, whilst miR-23b positively regulates this process.150 Additionally, cartilage regeneration is characterised by fibrocartilage formation, which is associated with an increased amount of collagen type I, in place of hyaline cartilage.13
Tooi et al. studied the osteoblastic differentiation potential of normal, adult human dermal fibroblasts when cultured with human placenta mesenchymal stem cell-derived exosomes (PlaMSC-Ex). An increase in OCT4 and NANOG mRNA levels was observed for these stemness-related genes. In culture conditions that contained activator bone morphogenetic protein 2 (BMP-2) and PlaMSC-Ex, the mRNA expression of ALP and osteoblast-specific transcription factor OSX increased. Moreover, Alizarin red S-staining showed an increased mineral deposition after changing the culture medium to osteogenic induction medium.113
Studies show that exosomes derived from monocytes, platelet lysates and mineralizing pre-osteoblast MC3T3-E1 cells, induce osteogenic differentiation of hBMSCs.179–181 Addition of monocyte-derived exosomes increased expression of runt-related transcription factor 2 (RUNX2) and BMP-2, whilst platelet lysate-derived exosomes stimulated deposition of mineralised matrix, cell proliferation and migration.180,181 Moreover, pre-osteoblast MC3T3-E1 cell-derived exosomes expressed osteo-miRNAs (miR-1192, miR-680 and miR-302a) and altered the miRNA expression of the recipient cells to promote differentiation into osteoblasts. The β-catenin encoding gene, Ctnnb1, was up-regulated, together with miRNAs (miR-667-3p, miR-6769b-5p, miR-7044-5p, miR-7668-3p and miR-874-3p) that co-target Axin1, which negatively regulates the Wnt signalling pathway. This led to the up-regulation of osteogenic marker genes RUNX2 and alkaline phosphatase (ALP), as well as an increased matrix mineralization.179
Martins et al. induced osteogenic differentiation of hBMSCs and extracted exosome hBMSC-Ex to study osteogenic capability of these exosomes. He demonstrated that exosomes promoted osteoinduction, as determined by early ALP activation and overexpression of BMP-2.182
hUCMSC-derived secretion factors led to increased osteogenic differentiation of MSCs in vitro and enhanced rat ectopic bone formation as well as calvarial bone defect repair.183
A murine in vivo study involving subcutaneous implantation of hydrogels on the back of athymic nude mice showed the regenerative potential of hBMSC-Ex incorporated into type I collagen hydrogels, through the up-regulation of RUNX and OSX.149 In a rat critical-sized calvarial bone defect, hBMSC-Ex incorporated into a HyStem-HP hydrogel also led to the formation of new bone. hBMSC-Ex were characterised and they contained miR-196a, miR-27a and miR-206, which are critical for osteogenesis. miR-196a in particular induces the expression of ALP, OCN, OPN and RUNX2.184
hiPSC-MSC-Ex promoted proliferation and osteogenic differentiation in bone marrow MSCs from ovariectomized rats.112 Additionally, hiPSC-MSC-Ex led to enhanced proliferation, migration, and increased ALP expression of hBMSCs. This effect was partially attributed to phosphatidylinositol 3-kinase (PI3K)-Akt pathway, and the increased expression of positive effector genes (i.e. PDGFA, FGF1/2, FGFR1, COL1A1/2, and BCL2L1) as well as a decrease in the negative effector genes GSK3β and PTEN.185
In vivo studies involving hiPSC-MSC-Ex administration into critical-sized calvarial defects in ovariectomized rats, reported enhanced osteogenesis and angiogenesis.112 Increased new bone formation in another rat critical-sized calvarial bone defect was also induced by a combination of hiPSC-MSC-Ex and the osteoconductive biomaterial tricalcium phosphate (β-TCP).185
In a rat osteochondral defect model, exosomes derived from HuES9 human embryonic stem cell MSCs resulted in regeneration of hyaline cartilage and subchondral bone after 12 weeks.13 Furthermore, in mice with spontaneously developed polyarticular arthritis, oral administration of bovine milk-derived exosomes led to a decrease in ankle joint inflammation and cartilage depletion, and an overall delay in clinical development of arthritis.186
Numerous in vitro studies in the last decade were conducted to investigate the mechanism of the cardioprotective effects of stem cell-derived exosomes. A review by Chistiakov et al. highlighted that exosomal miRNAs have an important role in cardiac regeneration. Exosomes released by injured cardiomyocytes contain the cardiac-specific miRNAs, particularly miR-1, miR-133a and miR-208, and these triggered cardiac muscle cells and resident cardiac progenitor cells to initiate cardiac tissue repair.187 Regeneration of post-infarct myocardium is enhanced by exosomes derived from both cardiac and non-cardiac stem cells. Many studies report particular exosome-derived miRNAs responsible for the cardioprotective effects including MSC exosomal miR-21,188 cardiac progenitor cell exosomal miR-451,189 and induced pluripotent stem cell exosomal miR-21 and HIF-1α-regulated exosomal miR-210.190 Myocardial cells protected from oxidative stress-related apoptosis downregulate the PDCD4 gene through the action of exosomal miR-21 gene, which subsequently inhibits apoptosis. Taken together, these studies established exosomes as a novel therapeutic vehicle for ischemic cardiac disease.188,191
Further support for this notion came from studies demonstrating the cardioprotective effects of exosomes in various animal models. For example, exosomes from human umbilical cord MSCs improved cardiac regeneration and promoted angiogenesis in acute myocardial infarction models in rats.192,193 Extracellular vesicles derived from human bone marrow MSCs also promoted angiogenesis in a similar rat myocardial infarction model.194
Exosomes secreted by human CD34+ stem cells promoted angiogenic effects in the in vitro Matrigel tube-formation assay and these exosomes were subsequently shown in mouse models to induce angiogenesis in vivo.195 The angiogenic effects observed in this study were attributed to exosomes enriched in pro-angiogenic miRNAs, which are well-protected by the exosomal membrane and thus are more stable than molecules secreted directly into the extracellular matrix. In another study, exosomal miR-223, the most highly expressed miRNA in bone marrow-derived MSCs, was shown to downregulate Sema3A and Stat3 and reduce levels of pro-inflammatory cytokines in a sepsis-triggered cardiac injury model. The attenuation of cardiomyocyte death during sepsis and improved cardiac function were attributed to the action of miR-223, which downregulates numerous inflammation-related genes including Mef2C, PKnox1, CXCL2, CCL3, IL-6, NLRP3, TANK, DR6 and IRF4.196
MSCs modified to overexpress hepatocyte growth factor (HGF), an important cytokine for anti-apoptosis, angiogenesis and anti-inflammation, enhanced cell survival and paracrine effects of MSCs in a murine model of myocardial infarction. Conditioned medium from umbilical cord-derived MSCs that overexpress HGF increased expression of HGF, EGF, bFGF and VEGF compared to the non-modified MSCs. The effects of the conditioned medium were reduced cardiomyocyte apoptosis, enhanced angiogenesis and increased proliferation of cardiomyocytes.197
To date, few studies have investigated the therapeutic potential of small populations of cardiac progenitor cells that reside in the heart. Gray et al. noted that previous studies employed cardiac progenitor cell-derived exosomes generated under normoxic conditions, which does not mimic the microenvironment of post-infarct tissue. Cardiac progenitor cell-derived exosomes secreted under hypoxic conditions contained a different profile of miRNAs, which were attractive candidates for treating myocardial infarction. Seven miRNAs (miR-15b, -17, -20a, -103, -199a, -210 and -292) were upregulated in exosomes generated in cells exposed to hypoxia, which were not present in exosomes isolated from the normoxic cardiac progenitor cells. These hypoxia-induced miRNAs were associated with enhanced cardiac repair, since vesicle size, total RNA content and protein levels were similar between exosomes generated in hypoxic or normoxic conditions.74
In a similar study, extracellular vesicles isolated from cardiac progenitor cells cultured under hypoxic conditions were enriched in miR-210, miR-132 and miR-146a-3p. Functional in vitro assays revealed that miR-210 and miR-132 were involved with anti-apoptotic and proangiogenic activities. When these extracellular vesicles were injected into infarcted rat hearts, there was evidence of reduced cardiomyocyte apoptosis, enhanced angiogenesis and a significant in the left ventricular ejection fraction compared with controls.198
MSC exosomes also ameliorate chronic lung diseases including bronchopulmonary dysplasia, as recently reported.209 In a mouse model of hyperoxia-induced pulmonary hypertension and bronchopulmonary dysplasia, even a single dose of MSC exosomes restored lung architecture and led to significant long-term benefits in the lung function through immunomodulatory effects.209 The mRNA sequencing of the lung revealed that the proinflammatory M1 state of macrophages was suppressed while the anti-inflammatory M2-like state was augmented.
Numerous studies showed not only specific miRNAs contained in EVs are crucial for lung repair in vivo, particularly for influenza,210 hypoxia-induced pulmonary hypertension,211 and ventilator-induced lung injury,212 but also specific mRNAs are effective in endotoxin-induced acute lung injury, as detailed below.
Tan et al. analysed the regulation of miRNAs during early and late stages of repair after influenza infection in a mouse model.210 The miRNAs that play major roles in priming pulmonary tissues for repair and regeneration following influenza were identified, and particularly miR-290, miR-21, let-7 and miR-200 appeared to be highly involved in the regeneration process. Beyond regulating regeneration, miR-21 and let-7 have further benefits such as anti-inflammatory properties. These properties are of significant benefit since many lung conditions that require regeneration have concomitant, uncontrolled inflammation. miR-21 is also present in microvesicles of important stem cell types such as murine embryonic stem cells213 and human bone marrow mesenchymal stem cells.45
Lee et al. provided strong evidence that exosomes prevented the activation of hypoxic signalling, which underlies pulmonary inflammation and pulmonary hypertension. In a model of hypoxia-induced pulmonary hypertension, mice treated with exosomes derived from umbilical cord-derived MSCs showed suppression of hypoxia-induced pulmonary inflammation and vascular remodelling, in a dose-dependent manner. In that study, exosomes derived from mouse lung fibroblasts were used as a control and showed no protective effect against pulmonary hypertension, when compared with MSC-derived exosomes.211 The suppressive effect of the exosomes derived from umbilical cord-derived MSCs was attributed to their enriched levels of miR-16 and miR-21. miR-16 reduces SERT expression, which is a critical protein for the resolution of pulmonary oedema.214 Although let7b miRNA levels were similar in exosomes derived from fibroblasts and MSC-derived exosomes, the latter contained levels of let7b pre-miRNA that were ten times higher. The reduction of IL-6 pro-inflammatory cytokine by the MSC-derived exosomes led to de-activation of the STAT3 gene, which is a key mediator of hypoxic, pro-inflammatory signalling associated with pulmonary hypertension.211
In a mouse model of endotoxin-induced acute lung injury, reduced inflammation and prevention of pulmonary oedema formation in the injured alveoli followed intratracheal administration of human bone marrow MSC-derived microvesicles. A major finding of this study was that the microvesicles contained the mRNA for the keratinocyte growth factor (KGF). KGF was previously shown to restore alveolar fluid clearance in an ex vivo model of acute lung injury. Transfer of the mRNA from the microvesicles to the injured alveolar epithelium, and subsequent expression of KGF, was thought to be a key factor in the repair process.215
miRNA expression studies in acute lung injury report that overexpression of several miRNAs including miR-146 may be directly related to the host response that regulates macrophage function and inflammatory cytokine expression.216 An important functional role of miR-146 was suppression of TNF-α, IL-6, and IL-1β expression in alveolar macrophages through the inhibition of IRAK-1 and TRAF-6 in a mouse model of ventilator-induced lung injury.212 A recent study found miR-146a was critical for the immunomodulatory effects of exosomes derived from human umbilical cord MSCs in a mouse model of sepsis.217 Another study reported that miR-146a reduces inflammatory gene expression and inhibits endotoxin-induced inflammation in mice.93 miR-146a reduces microbial and mechanically induced inflammation in lung epithelia through the toll-like receptor signalling pathway.218 A recent review by Basu and Ludlow highlighted that the selection of RNA for recruitment into exosomes is highly precise and is regulated by proteins such as RNA binding complex ESCRT-II, which is present in the exosomal membrane.136 Interestingly, Song et al. showed that miR-146a could be selectively packaged into the exosomes of MSCs by pre-treating the cells with the pro-inflammatory cytokine IL-1β.217 Future studies that use exosomes to improve regeneration of the lung after injury should consider conditioning exosomes such that they are enriched in miR-146a.
After encapsulation of the anti-inflammatory drug curcumin into exosomes, curcumin had increased solubility and stability in vitro, and increased bioavailability in vivo.222,223 An additional benefit was that encapsulated curcumin protected mice from lipopolysaccharide-induced septic shock.221 Since, curcumin is effective in wound healing, its combination with exosomes, which have also been shown to promote functional skin regeneration, presents a unique opportunity to accelerate tissue regeneration beyond single delivery approaches. Another study showed that a doxorubicin-exosome delivery system resulted in inhibition of breast tumour growth in a nude mouse model.224
Exosomes also have the potential to deliver drugs across the blood–brain barrier (BBB). The encapsulation of the cancer drugs paclitaxel, doxorubicin and rhodamine 123 into exosomes was performed and the effectiveness of these exosomes for delivery across the BBB was demonstrated in a zebrafish (danio rerio) model.219 Another potential benefit of encapsulation was an increased cytotoxic effect of paclitaxel in cells, which was shown after loading paclitaxel into prostate cancer cell-derived extracellular vesicles and loading them into autologous prostate cancer cells.225 Finally, catalase-loaded exosomes showed neuroprotective effects in a mouse model, which demonstrated that exosomes are able to cross the BBB.226
Recently, exosomes conjugated with c(RGDyK) and loaded with curcumin were intravenously administered into an ischemic brain. Engineered exosomes targeted the lesion area and decreased both the inflammatory response and cellular apoptosis.227
The presence of mRNA and miRNA in exosomes suggests that these nucleic acids can be incorporated exogenously, transferred to recipient cells, and subsequently influence protein synthesis.220 Specific targeting can be performed by coupling the exosome with a biological recognition factor, which would interact with cell surface antigens or receptors.221
The first attempt at gene knockout using siRNA-loaded exosomes was by Alvarez-Erviti et al.228 This group used rabies virus glycoprotein (RVG)-targeted exosomes to deliver GAPDH siRNA to the brain, resulting in the knockout of the therapeutic target BACE1 in Alzheimer's disease. Cooper et al.229 loaded brain-targeting RVG-exosomes with siRNA to alpha-synuclein (α-Syn). Following exosome delivery, there were reduced α-Syn aggregates in the brains of mice, which is significant because α-Syn aggregates are associated with the neuropathological features of Parkinson's disease.
Mizrak et al.230 transfected HEK-293T cells with expression constructs that allowed the generation of secreted microvesicles containing protein–cytosine deaminase (CD) fused to uracil phosphoribosyltransferase (UPRT). The simultaneous injection of the prodrug 5-fluorocytosine, and microvesicles containing CD-UPRT-mRNA/protein, led to a regression in tumour size and decreased tumour growth. Transfecting the human embryonic kidney cell line 293 to produce GE11 peptide, which binds to epidermal growth factor receptor (EGFR) present in human tumours of epithelial origin, allowed Ohno et al.231 to isolate GE11-positive exosomes that target tumour cells. This group also transfected the cells with miRNA let-7a to obtain let-7a-containing exosomes, which targeted and inhibited tumour development in mice models.
Finally, Katakowski et al.232 isolated exosomes from MSCs transfected with miR-146b. This miR reduces glioma cell motility and invasion, and reduces the expression of EGFR. Following delivery of exosomes loaded with miR-146b, there was a decrease in the growth of 9L glioma cells in vitro, and a reduction of 9L tumour volume in a rat model of a primary brain tumour.
Summary
Despite significant investment and progress in regenerative medicine in the last decade, the search for effective approaches to regenerate functional tissues continues. Most synthetic and natural biomaterials including polymers, silk, keratin and ceramic that are used to support tissue regeneration unfortunately often fail to actively and dynamically aid formation of functional tissue structures. The introduction of stem cell therapy aimed to address this problem. Stem cells interact with body structures and respond to all biochemical and biomechanical cues leading to remodelling and regeneration of the tissues. To further enhance or guide cellular function, stem cell therapy is combined with the aforementioned biomaterials. However, in the recent years it becomes very clear that cells communicate and orchestrate tissue regeneration through the manufacture and deployment of paracrine biochemical signals in the form of EVs. Indeed, there is convincing evidence that stem cells without EVs do not yield any beneficial function. These discoveries gave rise to a new paradigm, where EVs are collected from stem cells (and other cells) and are used to actively regenerate tissues. The evidence suggests that EVs promote specific cellular responses through delivering biochemical cargos of RNAs, which are capable of reprogramming recipient cells. Currently, the focus is on non-coding RNAs, i.e. microRNAs, which promote the regeneration of skin, bone, heart, and lung tissue. With the growing library of the functional roles of exosomes carrying specific cargos of microRNAs, applications in regenerative medicine will continue to expand. However, the isolation and characterisation methods need to be standardised, but the field has yet to achieve consensus on these methods. When we achieve the level of matching the specific cargos to desired functional roles, EVs will eventually be a tool to communicate EVerything.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
WCh acknowledges the University of Sydney for the SOAR Fellowship. BK acknowledges the financial assistance of the Trevor Basil Kilvington Bequest.References
- X. Liang, Y. Ding, Y. Zhang, H.-F. Tse and Q. Lian, Cell Transplant., 2014, 23, 1045–1059 Search PubMed.
- F. Vizoso, N. Eiro, S. Cid, J. Schneider and R. Perez-Fernandez, Int. J. Mol. Sci., 2017, 18, 1852 CrossRef PubMed.
- M. Z. Ratajczak, M. Kucia, T. Jadczyk, N. J. Greco, W. Wojakowski, M. Tendera and J. Ratajczak, Leukemia, 2012, 26, 1166–1173 CrossRef CAS PubMed.
- M. Gnecchi, H. He, O. D. Liang, L. G. Melo, F. Morello, H. Mu, N. Noiseux, L. Zhang, R. E. Pratt, J. S. Ingwall and V. J. Dzau, Nat. Med., 2005, 11, 367–368 CrossRef CAS PubMed.
- L. Timmers, S. K. Lim, F. Arslan, J. S. Armstrong, I. E. Hoefer, P. A. Doevendans, J. J. Piek, R. M. El Oakley, A. Choo, C. N. Lee, G. Pasterkamp and D. P. V. de Kleijn, Stem Cell Res., 2008, 1, 129–137 CrossRef PubMed.
- X. Xia, P. W. Y. Chiu, P. K. Lam, W. C. Chin, E. K. W. Ng and J. Y. W. Lau, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864, 178–188 CrossRef CAS PubMed.
- S.-R. Park, J.-W. Kim, H.-S. Jun, J. Y. Roh, H.-Y. Lee and I.-S. Hong, Mol. Ther., 2017 DOI:10.1016/J.YMTHE.2017.09.023.
- Y. Nakamura, S. Miyaki, H. Ishitobi, S. Matsuyama, T. Nakasa, N. Kamei, T. Akimoto, Y. Higashi and M. Ochi, FEBS Lett., 2015, 589, 1257–1265 CrossRef CAS PubMed.
- C. A. Herberts, M. S. G. Kwa and H. P. H. Hermsen, J. Transl. Med., 2011, 9, 29 CrossRef PubMed.
- M. Fan, W. Chen, W. Liu, G.-Q. Du, S.-L. Jiang, W.-C. Tian, L. Sun, R.-K. Li and H. Tian, Rejuvenation Res., 2010, 13, 429–438 CrossRef PubMed.
- M. C. Deregibus, A. Iavello, C. Tetta and G. Camussi, in Adult Stem Cell Therapies: Alternatives to Plasticity, Humana Press, New York, NY, 2014, pp. 231–244 Search PubMed.
- A. Shabbir, A. Cox, L. Rodriguez-Menocal, M. Salgado and E. Van Badiavas, Stem Cells Dev., 2015, 24, 1635–1647 CrossRef CAS PubMed.
- S. Zhang, W. C. Chu, R. C. Lai, S. K. Lim, J. H. P. Hui and W. S. Toh, Osteoarthritis Cartilage, 2016, 24, 2135–2140 CrossRef CAS PubMed.
- M. Khan, E. Nickoloff, T. Abramova, J. Johnson, S. K. Verma, P. Krishnamurthy, A. R. Mackie, E. Vaughan, V. N. S. Garikipati, C. Benedict, V. Ramirez, E. Lambers, A. Ito, E. Gao, S. Misener, T. Luongo, J. Elrod, G. Qin, S. R. Houser, W. J. Koch and R. Kishore, Circ. Res., 2015, 117, 52–64 CrossRef CAS PubMed.
- A. van Koppen, J. A. Joles, B. W. M. van Balkom, S. K. Lim, D. de Kleijn, R. H. Giles and M. C. Verhaar, PLoS One, 2012, 7, e38746 CAS.
- F. F. Cruz and P. R. M. Rocco, Stem Cell Invest., 2017, 4, 78 CrossRef PubMed.
- P. J. Quesenberry, J. Aliotta, M. C. Deregibus and G. Camussi, Stem Cell Res. Ther., 2015, 6, 153 CrossRef PubMed.
- F. Collino, M. Pomatto, S. Bruno, R. S. Lindoso, M. Tapparo, W. Sicheng, P. Quesenberry and G. Camussi, Stem Cell Rev. Rep., 2017, 13, 226–243 CrossRef CAS PubMed.
- J. M. Aliotta, M. Pereira, S. Wen, M. S. Dooner, M. Del Tatto, E. Papa, L. R. Goldberg, G. L. Baird, C. E. Ventetuolo, P. J. Quesenberry and J. R. Klinger, Cardiovasc. Res., 2016, 110, 319–330 CrossRef CAS PubMed.
- R. Xu, D. W. Greening, A. Rai, H. Ji and R. J. Simpson, Methods, 2015, 87, 11–25 CrossRef CAS PubMed.
- P. Wolf, Br. J. Haematol., 1967, 13, 269–288 CrossRef CAS PubMed.
- E. G. Trams, C. J. Lauter, J. Norman Salem and U. Heine, Biochim. Biophys. Acta, Biomembr., 1981, 645, 63–70 CrossRef CAS.
- B. T. Pan, K. Teng, C. Wu, M. Adam and R. M. Johnstone, J. Cell Biol., 1985, 101, 942–948 CrossRef CAS PubMed.
- C. Harding, J. Heuser and P. Stahl, J. Cell Biol., 1983, 97, 329–339 CrossRef CAS PubMed.
- A. V. Vlassov, S. Magdaleno, R. Setterquist and R. Conrad, Biochim. Biophys. Acta, 2012, 1820, 940–948 CrossRef CAS PubMed.
- M. Alenquer and M. Amorim, Viruses, 2015, 7, 5066–5083 CrossRef CAS PubMed.
- S. Nomura, Int. J. Hematol., 2017, 105, 392–405 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- M. Nawaz, G. Camussi, H. Valadi, I. Nazarenko, K. Ekström, X. Wang, S. Principe, N. Shah, N. M. Ashraf, F. Fatima, L. Neder and T. Kislinger, Nat. Rev. Urol., 2014, 11, 688–701 CrossRef PubMed.
- E. Cocucci, G. Racchetti and J. Meldolesi, Trends Cell Biol., 2009, 19, 43–51 CrossRef CAS PubMed.
- J. Ratajczak, M. Wysoczynski, F. Hayek, A. Janowska-Wieczorek and M. Z. Ratajczak, Leukemia, 2006, 20, 1487–1495 CrossRef CAS PubMed.
- S. S. Sidhu, A. T. Mengistab, A. N. Tauscher, J. LaVail and C. Basbaum, Oncogene, 2004, 23, 956–963 CrossRef CAS PubMed.
- C. Théry, M. Ostrowski and E. Segura, Nat. Rev. Immunol., 2009, 9, 581–593 CrossRef PubMed.
- L. Biancone, S. Bruno, M. C. Deregibus, C. Tetta and G. Camussi, Nephrol., Dial., Transplant., 2012, 27, 3037–3042 CrossRef CAS PubMed.
- I. Del Conde, C. N. Shrimpton, P. Thiagarajan and J. A. López, Blood, 2005, 106, 1604–1611 CrossRef CAS PubMed.
- W. Jy, L. L. Horstman, J. J. Jimenez and Y. S. Ahn, J. Thromb. Haemostasis, 2004, 2, 1842–1843 CrossRef CAS PubMed.
- S. Mathivanan, H. Ji and R. J. Simpson, J. Proteomics, 2010, 73, 1907–1920 CrossRef CAS PubMed.
- H. F. G. Heijnen, A. E. Schiel, R. Fijnheer, H. J. Geuze and J. J. Sixma, Blood, 1999, 94, 3791–3799 CAS.
- H. Kalra, R. J. Simpson, H. Ji, E. Aikawa, P. Altevogt, P. Askenase, V. C. Bond, F. E. Borràs, X. Breakefield, V. Budnik, E. Buzas, G. Camussi, A. Clayton, E. Cocucci, J. M. Falcon-Perez, S. Gabrielsson, Y. S. Gho, D. Gupta, H. C. Harsha, A. Hendrix, A. F. Hill, J. M. Inal, G. Jenster, E.-M. Krämer-Albers, S. K. Lim, A. Llorente, J. Lötvall, A. Marcilla, L. Mincheva-Nilsson, I. Nazarenko, R. Nieuwland, E. N. M. Nolte-'t Hoen, A. Pandey, T. Patel, M. G. Piper, S. Pluchino, T. S. K. Prasad, L. Rajendran, G. Raposo, M. Record, G. E. Reid, F. Sánchez-Madrid, R. M. Schiffelers, P. Siljander, A. Stensballe, W. Stoorvogel, D. Taylor, C. Thery, H. Valadi, B. W. M. van Balkom, J. Vázquez, M. Vidal, M. H. M. Wauben, M. Yáñez-Mó, M. Zoeller and S. Mathivanan, PLoS Biol., 2012, 10, e1001450 CAS.
- D.-K. Kim, J. Lee, S. R. Kim, D.-S. Choi, Y. J. Yoon, J. H. Kim, G. Go, D. Nhung, K. Hong, S. C. Jang, S.-H. Kim, K.-S. Park, O. Y. Kim, H. T. Park, J. H. Seo, E. Aikawa, M. Baj-Krzyworzeka, B. W. M. van Balkom, M. Belting, L. Blanc, V. Bond, A. Bongiovanni, F. E. Borras, L. Buee, E. I. Buzas, L. Cheng, A. Clayton, E. Cocucci, C. S. Dela Cruz, D. M. Desiderio, D. Di Vizio, K. Ekstrom, J. M. Falcon-Perez, C. Gardiner, B. Giebel, D. W. Greening, J. C. Gross, D. Gupta, A. Hendrix, A. F. Hill, M. M. Hill, E. Nolte-'t Hoen, D. W. Hwang, J. Inal, M. V. Jagannadham, M. Jayachandran, Y.-K. Jee, M. Jorgensen, K. P. Kim, Y.-K. Kim, T. Kislinger, C. Lasser, D. S. Lee, H. Lee, J. van Leeuwen, T. Lener, M.-L. Liu, J. Lotvall, A. Marcilla, S. Mathivanan, A. Moller, J. Morhayim, F. Mullier, I. Nazarenko, R. Nieuwland, D. N. Nunes, K. Pang, J. Park, T. Patel, G. Pocsfalvi, H. del Portillo, U. Putz, M. I. Ramirez, M. L. Rodrigues, T.-Y. Roh, F. Royo, S. Sahoo, R. Schiffelers, S. Sharma, P. Siljander, R. J. Simpson, C. Soekmadji, P. Stahl, A. Stensballe, E. St pie, H. Tahara, A. Trummer, H. Valadi, L. J. Vella, S. N. Wai, K. Witwer, M. Yanez-Mo, H. Youn, R. Zeidler and Y. S. Gho, Bioinformatics, 2015, 31, 933–939 CrossRef CAS PubMed.
- F. Russo, S. Di Bella, F. Vannini, G. Berti, F. Scoyni, H. V. Cook, A. Santos, G. Nigita, V. Bonnici, A. Laganà, F. Geraci, A. Pulvirenti, R. Giugno, F. De Masi, K. Belling, L. J. Jensen, S. Brunak, M. Pellegrini and A. Ferro, Nucleic Acids Res., 2017 DOI:10.1093/nar/gkx854.
- N. T. Trinh, T. Yamashita, T. C. Tu, T. Kato, K. Ohneda, F. Sato and O. Ohneda, Biochem. Biophys. Res. Commun., 2016, 473, 1111–1118 CrossRef CAS PubMed.
- X. Zou, G. Zhang, Z. Cheng, D. Yin, T. Du, G. Ju, S. Miao, G. Liu, M. Lu and Y. Zhu, Stem Cell Res. Ther., 2014, 5, 40 CrossRef PubMed.
- S. Bruno, C. Grange, M. C. Deregibus, R. A. Calogero, S. Saviozzi, F. Collino, L. Morando, A. Busca, M. Falda, B. Bussolati, C. Tetta and G. Camussi, J. Am. Soc. Nephrol., 2009, 20, 1053–1067 CrossRef CAS PubMed.
- F. Collino, M. C. Deregibus, S. Bruno, L. Sterpone, G. Aghemo, L. Viltono, C. Tetta and G. Camussi, PLoS One, 2010, 5, e11803 Search PubMed.
- J. Ratajczak, K. Miekus, M. Kucia, J. Zhang, R. Reca, P. Dvorak and M. Z. Ratajczak, Leukemia, 2006, 20, 847–856 CrossRef CAS PubMed.
- D. Katsman, E. J. Stackpole, D. R. Domin, D. B. Farber and P. Khaw, PLoS One, 2012, 7, e50417 CAS.
- S. Bobis-Wozowicz, K. Kmiotek, M. Sekula, S. Kedracka-Krok, E. Kamycka, M. Adamiak, U. Jankowska, A. Madetko-Talowska, M. Sarna, M. Bik-Multanowski, J. Kolcz, D. Boruczkowski, Z. Madeja, B. Dawn and E. K. Zuba-Surma, Stem Cells, 2015, 33, 2748–2761 CrossRef CAS PubMed.
- G. Hu, Q. LiE, X. Niu, B. Hu, J. Liu, S. Zhou, S. Guo, H. Lang, C. Zhang, Y. Wang and Z. Deng, Stem Cell Res. Ther., 2015, 6, 10 CrossRef PubMed.
- M. B. Herrera, V. Fonsato, S. Gatti, M. C. Deregibus, A. Sordi, D. Cantarella, R. Calogero, B. Bussolati, C. Tetta and G. Camussi, J. Cell. Mol. Med., 2010, 14, 1605–1618 CrossRef CAS PubMed.
- M. Baj-Krzyworzeka, R. Szatanek, K. Weglarczyk, J. Baran, B. Urbanowicz, P. Brański, M. Z. Ratajczak and M. Zembala, Cancer Immunol. Immunother., 2006, 55, 808–818 CrossRef CAS PubMed.
- M. P. Hunter, N. Ismail, X. Zhang, B. D. Aguda, E. J. Lee, L. Yu, T. Xiao, J. Schafer, M.-L. T. Lee, T. D. Schmittgen, S. P. Nana-Sinkam, D. Jarjoura and C. B. Marsh, PLoS One, 2008, 3, e3694 Search PubMed.
- T. Kang, T. M. Jones, C. Naddell, M. Bacanamwo, J. W. Calvert, W. E. Thompson, V. C. Bond, Y. E. Chen and D. Liu, Stem Cells Transl. Med., 2016, 5, 440–450 CrossRef CAS PubMed.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 Search PubMed.
- C. Raiborg and H. Stenmark, Nature, 2009, 458, 445–452 CrossRef CAS PubMed.
- C. Raiborg, K. G. Bache, D. J. Gillooly, I. H. Madshus, E. Stang and H. Stenmark, Nat. Cell Biol., 2002, 4, 394–398 CrossRef CAS PubMed.
- M. Babst, Curr. Opin. Cell Biol., 2011, 23, 452–457 CrossRef CAS PubMed.
- K. Sobo, J. Chevallier, R. G. Parton, J. Gruenberg and F. G. van der Goot, PLoS One, 2007, 2, e391 Search PubMed.
- A. C. Theos, S. T. Truschel, D. Tenza, I. Hurbain, D. C. Harper, J. F. Berson, P. C. Thomas, G. Raposo and M. S. Marks, Dev. Cell, 2006, 10, 343–354 CrossRef CAS PubMed.
- S. Stuffers, C. Sem Wegner, H. Stenmark and A. Brech, Traffic, 2009, 10, 925–937 CrossRef CAS PubMed.
- J. H. Hurley and G. Odorizzi, Nat. Cell Biol., 2012, 14, 654–655 CrossRef CAS PubMed.
- S. Bruno, C. Grange, F. Collino, M. C. Deregibus, V. Cantaluppi, L. Biancone, C. Tetta and G. Camussi, PLoS One, 2012, 7, e33115 CAS.
- M. Colombo, G. Raposo and C. Théry, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- H. Cai, K. Reinisch and S. Ferro-Novick, Dev. Cell, 2007, 12, 671–682 CrossRef CAS PubMed.
- H. Cai, S. Yu, S. Menon, Y. Cai, D. Lazarova, C. Fu, K. Reinisch, J. C. Hay and S. Ferro-Novick, Nature, 2007, 445, 941–944 CrossRef CAS PubMed.
- C. Ungermann, K. Sato and W. Wickner, Nature, 1998, 396, 543–548 CrossRef CAS PubMed.
- S. Atienzar-Aroca, M. Flores-Bellver, G. Serrano-Heras, N. Martinez-Gil, J. M. Barcia, S. Aparicio, D. Perez-Cremades, J. M. Garcia-Verdugo, M. Diaz-Llopis, F. J. Romero and J. Sancho-Pelluz, J. Cell. Mol. Med., 2016, 20, 1457–1466 CrossRef CAS PubMed.
- H. W. King, M. Z. Michael and J. M. Gleadle, BMC Cancer, 2012, 12, 421 CrossRef CAS PubMed.
- S. Kanemoto, R. Nitani, T. Murakami, M. Kaneko, R. Asada, K. Matsuhisa, A. Saito and K. Imaizumi, Biochem. Biophys. Res. Commun., 2016, 480, 166–172 CrossRef CAS PubMed.
- C. Mazzeo, J. A. Cañas, M. P. Zafra, A. Rojas Marco, M. Fernández-Nieto, V. Sanz, M. Mittelbrunn, M. Izquierdo, F. Baixaulli, J. Sastre and V. del Pozo, J. Allergy Clin. Immunol., 2015, 135, 1603–1613 CrossRef CAS PubMed.
- C. M. Mfunyi, M. Vaillancourt, J. Vitry, T.-R. Nsimba Batomene, A. Posvandzic, A. A. Lambert and C. Gilbert, Virology, 2015, 484, 103–112 CrossRef CAS PubMed.
- J.-J. Ban, M. Lee, W. Im and M. Kim, Biochem. Biophys. Res. Commun., 2015, 461, 76–79 CrossRef CAS PubMed.
- I. Parolini, C. Federici, C. Raggi, L. Lugini, S. Palleschi, A. De Milito, C. Coscia, E. Iessi, M. Logozzi, A. Molinari, M. Colone, M. Tatti, M. Sargiacomo and S. Fais, J. Biol. Chem., 2009, 284, 34211–34222 CrossRef CAS PubMed.
- W. D. Gray, K. M. French, S. Ghosh-Choudhary, J. T. Maxwell, M. E. Brown, M. O. Platt, C. D. Searles and M. E. Davis, Circ. Res., 2015, 116, 255–263 CrossRef CAS PubMed.
- S. Gupta and A. A. Knowlton, Am. J. Physiol.: Heart Circ. Physiol., 2007, 292, H3052–H3056 CrossRef CAS PubMed.
- X. Yu, L. Deng, D. Wang, N. Li, X. Chen, X. Cheng, J. Yuan, X. Gao, M. Liao, M. Wang and Y. Liao, J. Mol. Cell. Cardiol., 2012, 53, 848–857 CrossRef CAS PubMed.
- N. A. Garcia, I. Ontoria-Oviedo, H. González-King, A. Diez-Juan, P. Sepúlveda and A. Rapraeger, PLoS One, 2015, 10, e0138849 Search PubMed.
- C. Salomon, J. Ryan, L. Sobrevia, M. Kobayashi, K. Ashman, M. Mitchell and G. E. Rice, PLoS One, 2013, 8, e68451 CAS.
- J. D. Anderson, H. J. Johansson, C. S. Graham, M. Vesterlund, M. T. Pham, C. S. Bramlett, E. N. Montgomery, M. S. Mellema, R. L. Bardini, Z. Contreras, M. Hoon, G. Bauer, K. D. Fink, B. Fury, K. J. Hendrix, F. Chedin, S. El-Andaloussi, B. Hwang, M. S. Mulligan, J. Lehtiö and J. A. Nolta, Stem Cells, 2016, 34, 601–613 CrossRef CAS PubMed.
- M. Yáñez-Mó, P. R.-M. Siljander, Z. Andreu, A. B. Zavec, F. E. Borràs, E. I. Buzas, K. Buzas, E. Casal, F. Cappello, J. Carvalho, E. Colás, A. C. Silva, S. Fais, J. M. Falcon-Perez, I. M. Ghobrial, B. Giebel, M. Gimona, M. Graner, I. Gursel, M. Gursel, N. H. H. Heegaard, A. Hendrix, P. Kierulf, K. Kokubun, M. Kosanovic, V. Kralj-Iglic, E.-M. Krämer-Albers, S. Laitinen, C. Lässer, T. Lener, E. Ligeti, A. Linē, G. Lipps, A. Llorente, J. Lötvall, M. Manček-Keber, A. Marcilla, M. Mittelbrunn, I. Nazarenko, E. Nolte-'t Hoen, T. A. Nyman, L. O'Driscoll, M. Olivan, C. Oliveira, É. Pállinger, H. A. del Portillo, J. Reventós, M. Rigau, E. Rohde, M. Sammar, F. Sánchez-Madrid, N. Santarém, K. Schallmoser, M. S. Ostenfeld, W. Stoorvogel, R. Stukelj, S. G. Van der Grein, M. H. Vasconcelos, M. H. M. Wauben and O. De Wever, J. Extracell. Vesicles, 2015, 4, 27066 CrossRef PubMed.
- C. Gutiérrez-Vázquez, C. Villarroya-Beltri, M. Mittelbrunn and F. Sánchez-Madrid, Immunol. Rev., 2013, 251, 125–142 CrossRef PubMed.
- S. S. Tan, Y. Yin, T. Lee, R. C. Lai, R. W. Y. Yeo, B. Zhang, A. Choo and S. K. Lim, J. Extracell. Vesicles, 2013, 2, 22614 CrossRef PubMed.
- H. G. Lamparski, A. Metha-Damani, J.-Y. Yao, S. Patel, D.-H. Hsu, C. Ruegg and J.-B. Le Pecq, J. Immunol. Methods, 2002, 270, 211–226 CrossRef CAS PubMed.
- S. Mathivanan, C. J. Fahner, G. E. Reid and R. J. Simpson, Nucleic Acids Res., 2012, 40, D1241–D1244 CrossRef CAS PubMed.
- P. A. Gonzales, G. Ma, T. Pisitkun, B. Ruttenburg and M. A. Knepper, Urinary Exosome Protein Database, https://hpcwebapps.cit.nih.gov/ESBL/Database/Exosome/, (accessed 4 November 2017) Search PubMed.
- D. Ha, N. Yang and V. Nadithe, Acta Pharm. Sin. B, 2016, 6, 287–296 CrossRef PubMed.
- L. Beer, M. Zimmermann, A. Mitterbauer, A. Ellinger, F. Gruber, M.-S. Narzt, M. Zellner, M. Gyöngyösi, S. Madlener, E. Simader, C. Gabriel, M. Mildner and H. J. Ankersmit, Sci. Rep., 2015, 5, 16662 CrossRef CAS PubMed.
- A. M. Campos, E. Maciel, A. S. P. Moreira, B. Sousa, T. Melo, P. Domingues, L. Curado, B. Antunes, M. R. M. Domingues and F. Santos, J. Cell. Physiol., 2016, 231, 1024–1032 CrossRef CAS PubMed.
- H. Valadi, K. Ekström, A. Bossios, M. Sjöstrand, J. J. Lee and J. O. Lötvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- C. Bang, S. Batkai, S. Dangwal, S. K. Gupta, A. Foinquinos, A. Holzmann, A. Just, J. Remke, K. Zimmer, A. Zeug, E. Ponimaskin, A. Schmiedl, X. Yin, M. Mayr, R. Halder, A. Fischer, S. Engelhardt, Y. Wei, A. Schober, J. Fiedler and T. Thum, J. Clin. Invest., 2014, 124, 2136–2146 CAS.
- M. Mittelbrunn, C. Gutiérrez-Vázquez, C. Villarroya-Beltri, S. González, F. Sánchez-Cabo, M. Á. González, A. Bernad and F. Sánchez-Madrid, Nat. Commun., 2011, 2, 282 CrossRef PubMed.
- X. Chen, H. Liang, J. Zhang, K. Zen and C.-Y. Zhang, Protein Cell, 2012, 3, 28–37 CrossRef CAS PubMed.
- M. Alexander, R. Hu, M. C. Runtsch, D. A. Kagele, T. L. Mosbruger, T. Tolmachova, M. C. Seabra, J. L. Round, D. M. Ward and R. M. O'Connell, Nat. Commun., 2015, 6, 7321 CrossRef CAS PubMed.
- M. Li, A. J. Rai, G. Joel DeCastro, E. Zeringer, T. Barta, S. Magdaleno, R. Setterquist and A. V. Vlassov, Methods, 2015, 87, 26–30 CrossRef CAS PubMed.
- B. J. Goldie, M. D. Dun, M. Lin, N. D. Smith, N. M. Verrills, C. V. Dayas and M. J. Cairns, Nucleic Acids Res., 2014, 42, 9195–9208 CrossRef CAS PubMed.
- J. Guduric-Fuchs, A. O'Connor, B. Camp, C. L. O'Neill, R. J. Medina and D. A. Simpson, BMC Genomics, 2012, 13, 357 CrossRef CAS PubMed.
- P. Munson and A. Shukla, Medicines, 2015, 2, 310–327 CrossRef CAS PubMed.
- G. Hu, K. M. Drescher and X.-M. Chen, Front. Genet., 2012, 3, 56 CAS.
- L. A. Hargett and N. N. Bauer, Pulm. Circ., 2013, 3, 329–340 CrossRef PubMed.
- R. M. Johnstone, M. Adam, J. R. Hammond, L. Orr and C. Turbide, J. Biol. Chem., 1987, 262, 9412–9420 CAS.
- D. D. Taylor and S. Shah, Methods, 2015, 87, 3–10 CrossRef CAS PubMed.
- C. Théry, A. Clayton, S. Amigorena and G. Raposo, Curr. Protoc. Cell Biol., 2006, 3.22.1–3.22.29 Search PubMed.
- C. Lässer, M. Eldh and J. Lötvall, J. Visualized Exp., 2012, e3037 Search PubMed.
- T. Baranyai, K. Herczeg, Z. Onódi, I. Voszka, K. Módos, N. Marton, G. Nagy, I. Mäger, M. J. Wood, S. El Andaloussi, Z. Pálinkás, V. Kumar, P. Nagy, Á. Kittel, E. I. Buzás, P. Ferdinandy and Z. Giricz, PLoS One, 2015, 10, e0145686 Search PubMed.
- F. Momen-Heravi, L. Balaj, S. Alian, P.-Y. Mantel, A. E. Halleck, A. J. Trachtenberg, C. E. Soria, S. Oquin, C. M. Bonebreak, E. Saracoglu, J. Skog and W. P. Kuo, Biol. Chem., 2013, 394, 1253–1262 CrossRef CAS PubMed.
- K. Rekker, M. Saare, A. M. Roost, A.-L. Kubo, N. Zarovni, A. Chiesi, A. Salumets and M. Peters, Clin. Biochem., 2014, 47, 135–138 CrossRef CAS PubMed.
- T. Li, Y. Yan, B. Wang, H. Qian, X. Zhang, L. Shen, M. Wang, Y. Zhou, W. Zhu, W. Li and W. Xu, Stem Cells Dev., 2013, 22, 845–854 CrossRef CAS PubMed.
- Y. Zhou, H. Xu, W. Xu, B. Wang, H. Wu, Y. Tao, B. Zhang, M. Wang, F. Mao, Y. Yan, S. Gao, H. Gu, W. Zhu and H. Qian, Stem Cell Res. Ther., 2013, 4, 34 CrossRef CAS PubMed.
- S. Fang, C. Xu, Y. Zhang, C. Xue, C. Yang, H. Bi, X. Qian, M. Wu, K. Ji, Y. Zhao, Y. Wang, H. Liu and X. Xing, Stem Cells Transl. Med., 2016, 5, 1425–1439 CrossRef PubMed.
- J.-F. Xu, G. Yang, X.-H. Pan, S.-J. Zhang, C. Zhao, B.-S. Qiu, H.-F. Gu, J.-F. Hong, L. Cao, Y. Chen, B. Xia, Q. Bi and Y.-P. Wang, PLoS One, 2014, 9, e114627 Search PubMed.
- S. R. Baglio, K. Rooijers, D. Koppers-Lalic, F. J. Verweij, M. Pérez Lanzón, N. Zini, B. Naaijkens, F. Perut, H. W. Niessen, N. Baldini and D. M. Pegtel, Stem Cell Res. Ther., 2015, 6, 127 CrossRef PubMed.
- X. Qi, J. Zhang, H. Yuan, Z. Xu, Q. Li, X. Niu, B. Hu, Y. Wang and X. Li, Int. J. Biol. Sci., 2016, 12, 836–849 CrossRef CAS PubMed.
- M. Tooi, M. Komaki, C. Morioka, I. Honda, K. Iwasaki, N. Yokoyama, H. Ayame, Y. Izumi and I. Morita, J. Cell. Biochem., 2016, 117, 1658–1670 CrossRef CAS PubMed.
- M. A. Lopez-Verrilli, A. Caviedes, A. Cabrera, S. Sandoval, U. Wyneken and M. Khoury, Neuroscience, 2016, 320, 129–139 CrossRef CAS PubMed.
- K. Wang, Z. Jiang, K. A. Webster, J. Chen, H. Hu, Y. Zhou, J. Zhao, L. Wang, Y. Wang, Z. Zhong, C. Ni, Q. Li, C. Xiang, L. Zhang, R. Wu, W. Zhu, H. Yu, X. Hu and J. Wang, Stem Cells Transl. Med., 2016, 5, 1–14 CrossRef PubMed.
- B. Zhang, Y. Yin, R. C. Lai, S. S. Tan, A. B. H. Choo and S. K. Lim, Stem Cells Dev., 2014, 23, 1233–1244 CrossRef CAS PubMed.
- W. Koh, C. Sheng, B. Tan, Q. Lee, V. Kuznetsov, L. Kiang and V. Tanavde, BMC Genomics, 2010, 11, S6 CrossRef PubMed.
- L. Guo, R. C. H. Zhao and Y. Wu, Exp. Hematol., 2011, 39, 608–616 CrossRef CAS PubMed.
- R. E. Crossland, J. Norden, L. A. Bibby, J. Davis and A. M. Dickinson, J. Immunol. Methods, 2016, 429, 39–49 CrossRef CAS PubMed.
- R. E. Lane, D. Korbie, W. Anderson, R. Vaidyanathan and M. Trau, Sci. Rep., 2015, 5, 7639 CrossRef PubMed.
- Z. Brownlee, K. D. Lynn, P. E. Thorpe and A. J. Schroit, J. Immunol. Methods, 2014, 407, 120–126 CrossRef CAS PubMed.
- M. L. Alvarez, M. Khosroheidari, R. K. Ravi and J. K. DiStefano, Int. Soc. Nephrol., 2012, 82, 1024–1032 CAS.
- Z. Zhang, C. Wang, T. Li, Z. Liu and L. Li, Oncol. Lett., 2014, 8, 1701–1706 CAS.
- T. Yamada, Y. Inoshima, T. Matsuda and N. Ishiguro, J. Vet. Med. Sci., 2012, 74, 1523–1525 CrossRef CAS PubMed.
- R. C. Lai, F. Arslan, M. M. Lee, N. S. K. Sze, A. Choo, T. S. Chen, M. Salto-Tellez, L. Timmers, C. N. Lee, R. M. El Oakley, G. Pasterkamp, D. P. V. de Kleijn and S. K. Lim, Stem Cell Res., 2010, 4, 214–222 CrossRef CAS PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ji, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56, 293–304 CrossRef CAS PubMed.
- A. Clayton, J. Court, H. Navabi, M. Adams, M. D. Mason, J. A. Hobot, G. R. Newman and B. Jasani, J. Immunol. Methods, 2001, 247, 163–174 CrossRef CAS PubMed.
- M. P. Oksvold, A. Neurauter and K. W. Pedersen, in RNA Interference, ed. M. Sioud, Humana Press, New York, NY, 2015, pp. 465–481 Search PubMed.
- D. D. Taylor, W. Zacharias and C. Gercel-Taylor, in Serum/Plasma Proteomics: Methods and Protocols, ed. R. Simpson and D. Greening, Humana Press, New York, 2011, vol. 728, pp. 235–246 Search PubMed.
- S. Gholizadeh, M. Shehata Draz, M. Zarghooni, A. Sanati-Nezhad, S. Ghavami, H. Shafiee and M. Akbari, Biosens. Bioelectron., 2017, 91, 588–605 CrossRef CAS PubMed.
- K. Lee, H. Shao, R. Weissleder and H. Lee, ACS Nano, 2015, 9, 2321–2327 CrossRef CAS PubMed.
- R. T. Davies, J. Kim, S. C. Jang, E.-J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- C. Liu, J. Guo, F. Tian, N. Yang, F. Yan, Y. Ding, J. Wei, G. Hu, G. Nie and J. Sun, ACS Nano, 2017, 11, 6968–6976 CrossRef CAS PubMed.
- P. Li, M. Kaslan, S. H. Lee, J. Yao and Z. Gao, Theranostics, 2017, 7, 789–804 CrossRef PubMed.
- O. G. De Jong, B. W. M. Van Balkom, R. M. Schiffelers, C. V. C. Bouten and M. C. Verhaar, Front. Immunol., 2014, 5, 608 Search PubMed.
- J. Basu and J. W. Ludlow, Expert Opin. Biol. Ther., 2016, 16, 489–506 CrossRef CAS PubMed.
- Y. Wu, W. Deng and D. J. Klinke II, Analyst, 2015, 140, 6631–6642 RSC.
- V. B. R. Konala, M. K. Mamidi, R. Bhonde, A. K. Das, R. Pochampally and R. Pal, Cytotherapy, 2016, 18, 13–24 CrossRef CAS PubMed.
- L. Sun, R. Xu, X. Sun, Y. Duan, Y. Han, Y. Zhao, H. Qian, W. Zhu and W. Xu, Cytotherapy, 2016, 18, 413–422 CrossRef CAS PubMed.
- R. C. Lai, T. S. Chen and S. K. Lim, Regener. Med., 2011, 6, 481–492 CrossRef PubMed.
- S. Gatti, S. Bruno, M. C. Deregibus, A. Sordi, V. Cantaluppi, C. Tetta and G. Camussi, Nephrol., Dial., Transplant., 2011, 26, 1474–1483 CrossRef CAS PubMed.
- T. C. Yih, in Exploring mesenchymal stem cell-derived exosomes and tocotrienol (T3) as therapeutic agents in drug-induced liver injury (DILI), National University of Singapore, 2014 Search PubMed.
- H. Xin, Y. Li, Y. Cui, J. J. Yang, Z. G. Zhang and M. Chopp, J. Cereb. Blood Flow Metab., 2013, 33, 1711–1715 CrossRef CAS PubMed.
- Y. Zhang, M. Chopp, Z. G. Zhang, M. Katakowski, H. Xin, C. Qu, M. Ali, A. Mahmood and Y. Xiong, Neurochem. Int., 2016 DOI:10.1016/j.neuint.2016.08.003.
- L. Pascucci, G. Alessandri, C. Dall'Aglio, F. Mercati, P. Coliolo, C. Bazzucchi, S. Dante, S. Petrini, G. Curina and P. Ceccarelli, Vet. J., 2014, 202, 361–366 CrossRef PubMed.
- J. Yang, X.-X. Liu, H. Fan, Q. Tang, Z.-X. Shou, D.-M. Zuo, Z. Zou, M. Xu, Q.-Y. Chen, Y. Peng, S.-J. Deng and Y.-J. Liu, PLoS One, 2015, 10, e0140551 Search PubMed.
- Y. S. Takeda and Q. Xu, PLoS One, 2015, 10, e0135111 Search PubMed.
- K. Lu, H. Li, K. Yang, J. Wu, X. Cai, Y. Zhou and C. Li, Stem Cell Res. Ther., 2017, 8, 108 CrossRef PubMed.
- R. Narayanan, C.-C. Huang and S. Ravindran, Stem Cells Int., 2016, 2016, 1–11 CrossRef PubMed.
- B. Peng, Y. Chen and K. W. Leong, Adv. Drug Delivery Rev., 2015, 88, 108–122 CrossRef CAS PubMed.
- C. K. Sen and S. Ghatak, Am. J. Pathol., 2015, 185, 2629–2640 CrossRef CAS PubMed.
- S. Yao, W. Zhao, Q. Ou, L. Liang, X. Lin and Y. Wang, Stem Cells Int., 2017, 2017, 1–13 CrossRef.
- I. Mencía Castaño, C. M. Curtin, G. P. Duffy and F. J. O'Brien, Sci. Rep., 2016, 6, 27941 CrossRef PubMed.
- A. Turchinovich, L. Weiz and B. Burwinkel, Trends Biochem. Sci., 2012, 37, 460–465 CrossRef CAS PubMed.
- F. H. Seeger, A. M. Zeiher and S. Dimmeler, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 1739–1746 CrossRef CAS PubMed.
- K. J. Miller, D. A. Brown, M. M. Ibrahim, T. D. Ramchal and H. Levinson, Adv. Drug Delivery Rev., 2015, 88, 16–36 CrossRef CAS PubMed.
- S. D. Alipoor, E. Mortaz, J. Garssen, M. Movassaghi, M. Mirsaeidi and I. M. Adcock, Mediators Inflammation, 2016, 2016, 5628404 Search PubMed.
- Y. Li, L. Fan, S. Liu, W. Liu, H. Zhang, T. Zhou, D. Wu, P. Yang, L. Shen, J. Chen and Y. Jin, Biomaterials, 2013, 34, 5048–5058 CrossRef CAS PubMed.
- Y.-H. Liao, Y.-H. Chang, L.-Y. Sung, K.-C. Li, C.-L. Yeh, T.-C. Yen, S.-M. Hwang, K.-J. Lin and Y.-C. Hu, Biomaterials, 2014, 35, 4901–4910 CrossRef CAS PubMed.
- S.-C. Tao, T. Yuan, Y.-L. Zhang, W.-J. Yin, S.-C. Guo and C.-Q. Zhang, Theranostics, 2017, 7, 180–195 CrossRef PubMed.
- X. S. Liu, M. Chopp, R. L. Zhang, T. Tao, X. L. Wang, H. Kassis, A. Hozeska-Solgot, L. Zhang, C. Chen and Z. G. Zhang, PLoS One, 2011, 6, e23461 CAS.
- Z. Zhang, Y.-W. Qin, G. Brewer and Q. Jing, Wiley Interdiscip. Rev.: RNA, 2012, 3, 593–600 CrossRef CAS PubMed.
- X. Chen, H. Liang, J. Zhang, K. Zen and C.-Y. Zhang, Trends Cell Biol., 2012, 22, 125–132 CrossRef CAS PubMed.
- N. Kosaka, H. Iguchi, Y. Yoshioka, F. Takeshita, Y. Matsuki and T. Ochiya, J. Biol. Chem., 2010, 285, 17442–17452 CrossRef CAS PubMed.
- M. C. Deregibus, V. Cantaluppi, R. Calogero, M. Lo Iacono, C. Tetta, L. Biancone, S. Bruno, B. Bussolati and G. Camussi, Blood, 2007, 110, 2440–2448 CrossRef CAS PubMed.
- L. Cheng, R. A. Sharples, B. J. Scicluna and A. F. Hill, J. Extracell. vesicles, 2014, 3, 23743 CrossRef PubMed.
- T. Velnar, T. Bailey and V. Smrkolj, J. Int. Med. Res., 2009, 37, 1528–1542 CrossRef CAS PubMed.
- P. Huang, J. Bi, G. R. Owen, W. Chen, A. Rokka, L. Koivisto, J. Heino, L. Häkkinen and H. Larjava, J. Invest. Dermatol., 2015, 135, 3051–3059 CrossRef CAS PubMed.
- A. Geiger, A. Walker and E. Nissen, Biochem. Biophys. Res. Commun., 2015, 467, 303–309 CrossRef CAS PubMed.
- J. Zhang, J. Guan, X. Niu, G. Hu, S. Guo, Q. Li, Z. Xie, C. Zhang and Y. Wang, J. Transl. Med., 2015, 13, 49 CrossRef PubMed.
- B. Zhang, X. Wu, X. Zhang, Y. Sun, Y. Yan, H. Shi, Y. Zhu, L. Wu, Z. Pan, W. Zhu, H. Qian and W. Xu, Stem Cells Transl. Med., 2015, 4, 513–522 CrossRef CAS PubMed.
- B. Zhang, M. Wang, A. Gong, X. Zhang, X. Wu, Y. Zhu, H. Shi, L. Wu, W. Zhu, H. Qian and W. Xu, Stem Cells, 2015, 33, 2158–2168 CrossRef CAS PubMed.
- B. Zhang, Y. Shi, A. Gong, Z. Pan, H. Shi, H. Yang, H. Fu, Y. Yan, X. Zhang, M. Wang, W. Zhu, H. Qian and W. Xu, Stem Cells, 2016, 34, 2485–2500 CrossRef CAS PubMed.
- L. Hu, J. Wang, X. Zhou, Z. Xiong, J. Zhao, R. Yu, F. Huang, H. Zhang and L. Chen, Sci. Rep., 2016, 6, 32993 CrossRef CAS PubMed.
- K. Nakamura, M. Jinnin, M. Harada, H. Kudo, W. Nakayama, K. Inoue, A. Ogata, I. Kajihara, S. Fukushima and H. Ihn, J. Dermatol. Sci., 2016, 84, 30–39 CrossRef CAS PubMed.
- Y. Qin, R. Sun, C. Wu, L. Wang and C. Zhang, Int. J. Mol. Sci., 2016, 17, 712 CrossRef PubMed.
- T. Gaur, C. J. Lengner, H. Hovhannisyan, R. A. Bhat, P. V. N. Bodine, B. S. Komm, A. Javed, A. J. van Wijnen, J. L. Stein, G. S. Stein and J. B. Lian, J. Biol. Chem., 2005, 280, 33132–33140 CrossRef CAS PubMed.
- J. Burke, R. Kolhe, M. Hunter, C. Isales, M. Hamrick and S. Fulzele, Stem Cells Int., 2016, 2016, 1–6 CrossRef PubMed.
- Y. Cui, J. Luan, H. Li, X. Zhou and J. Han, FEBS Lett., 2016, 590, 185–192 CrossRef CAS PubMed.
- K. Ekström, O. Omar, C. Granéli, X. Wang, F. Vazirisani and P. Thomsen, PLoS One, 2013, 8, e75227 Search PubMed.
- E. Torreggiani, F. Perut, L. Roncuzzi, N. Zini, S. Baglio and N. Baldini, Eur. Cells Mater., 2014, 28, 137–151 CrossRef CAS PubMed.
- M. Martins, D. Ribeiro, A. Martins, R. L. Reis and N. M. Neves, Stem Cell Rep., 2016, 6, 284–291 CrossRef CAS PubMed.
- K.-X. Wang, L.-L. Xu, Y.-F. Rui, S. Huang, S.-E. Lin, J.-H. Xiong, Y.-H. Li, W. Y.-W. Lee and G. Li, PLoS One, 2015, 10, e0120593 Search PubMed.
- Y. Qin, L. Wang, Z. Gao, G. Chen and C. Zhang, Sci. Rep., 2016, 6, 21961 CrossRef CAS PubMed.
- J. Zhang, X. Liu, H. Li, C. Chen, B. Hu, X. Niu, Q. Li, B. Zhao, Z. Xie and Y. Wang, Stem Cell Res. Ther., 2016, 7, 136 CrossRef PubMed.
- O. J. Arntz, B. C. Pieters, M. C. Oliveira, M. G. Broeren, M. B. Bennink, M. de Vries, P. L. van Lent, M. I. Koenders, W. B. van den Berg, P. M. van der Kraan and F. A. van de Loo, Mol. Nutr. Food Res., 2015, 59, 1701–1712 CAS.
- D. Chistiakov, A. Orekhov and Y. Bobryshev, Int. J. Mol. Sci., 2016, 17, 63 CrossRef PubMed.
- K. Luther, M. McGuinness, L. Haar, H. Xu, J. Chen, M. Medvedovic and W. K. Jones, FASEB J., 2016, 30, 719.1–719.1 Search PubMed.
- L. Chen, Y. Wang, Y. Pan, L. Zhang, C. Shen, G. Qin, M. Ashraf, N. Weintraub, G. Ma and Y. Tang, Biochem. Biophys. Res. Commun., 2013, 431, 566–571 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, Y. Li, L. Chen, X. Wang, W. Guo, X. Zhang, G. Qin, S. He, A. Zimmerman, Y. Liu, I. Kim, N. L. Weintraub and Y. Tang, Int. J. Cardiol., 2015, 192, 61–69 CrossRef PubMed.
- J. Xiao, Y. Pan, X. H. Li, X. Y. Yang, Y. L. Feng, H. H. Tan, L. Jiang, J. Feng and X. Y. Yu, Cell Death Dis., 2016, 7, e2277 CrossRef CAS PubMed.
- D. Nascimento, D. Mosqueira, L. Sousa, M. Teixeira, M. Filipe, T. Resende, A. Araújo, M. Valente, J. Almeida, J. Martins, J. Santos, R. Bárcia, P. Cruz, H. Cruz and P. Pinto-do-Ó, Stem Cell Res. Ther., 2014, 5, 5 CrossRef PubMed.
- J. Ma, Y. Zhao, L. Sun, X. Sun, X. Zhao, X. Sun, H. Qian, W. Xu and W. Zhu, Stem Cells Transl. Med., 2016, 6, 51–59 CrossRef PubMed.
- S. Bian, L. Zhang, L. Duan, X. Wang, Y. Min and H. Yu, J. Mol. Med., 2014, 92, 387–397 CrossRef CAS PubMed.
- S. Sahoo, E. Klychko, T. Thorne, S. Misener, K. M. Schultz, M. Millay, A. Ito, T. Liu, C. Kamide, H. Agrawal, H. Perlman, G. Qin, R. Kishore and D. W. Losordo, Circ. Res., 2011, 109, 724–728 CrossRef CAS PubMed.
- X. Wang, H. Gu, D. Qin, L. Yang, W. Huang, K. Essandoh, Y. Wang, C. C. Caldwell, T. Peng, B. Zingarelli and G.-C. Fan, Sci. Rep., 2015, 5, 13721 CrossRef PubMed.
- L. Zhao, X. Liu, Y. Zhang, X. Liang, Y. Ding, Y. Xu, Z. Fang and F. Zhang, Exp. Cell Res., 2016, 344, 30–39 CrossRef CAS PubMed.
- L. Barile, V. Lionetti, E. Cervio, M. Matteucci, M. Gherghiceanu, L. M. Popescu, T. Torre, F. Siclari, T. Moccetti and G. Vassalli, Cardiovasc. Res., 2014, 103, 530–541 CrossRef CAS PubMed.
- D. N. Kotton and E. E. Morrisey, Nat. Med., 2014, 20, 822–832 CrossRef CAS PubMed.
- L. Ionescu, R. N. Byrne, T. van Haaften, A. Vadivel, R. S. Alphonse, G. J. Rey-Parra, G. Weissmann, A. Hall, F. Eaton and B. Thebaud, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2012, 303, L967–L977 CrossRef CAS PubMed.
- S. Y. Kim, J. Burgess, Y. Wang, D. Weiss, K. Chan and W. Chrzanowski, J. Aerosol Med. Pulm. Drug Delivery, 2016, 29, 514–524 CrossRef CAS PubMed.
- N. Gupta, X. Su, B. Popov, J. W. Lee, V. Serikov and M. A. Matthay, J. Immunol., 2007, 179, 1855–1863 CrossRef CAS.
- J. Tibboel, R. Keijzer, I. Reiss, J. C. de Jongste and M. Post, COPD: J. Chronic Obstruct. Pulm. Dis., 2014, 11, 310–318 Search PubMed.
- M. A. Antunes, S. C. Abreu, F. F. Cruz, A. C. Teixeira, M. Lopes-Pacheco, E. Bandeira, P. C. Olsen, B. L. Diaz, C. M. Takyia, I. P. Freitas, N. N. Rocha, V. L. Capelozzi, D. G. Xisto, D. J. Weiss, M. M. Morales and P. R. Rocco, Respir. Res., 2014, 15, 118 CrossRef PubMed.
- A. Serrano-Mollar, M. Nacher, G. Gay-Jordi, D. Closa, A. Xaubet and O. Bulbena, Am. J. Respir. Crit. Care Med., 2007, 176, 1261–1268 CrossRef CAS PubMed.
- Y. E. Kim, W. S. Park, D. K. Sung, S. Y. Ahn, S. I. Sung, H. S. Yoo and Y. S. Chang, Pediatr. Res., 2016, 80, 415–424 CrossRef CAS PubMed.
- D. K. Sung, Y. S. Chang, S. Y. Ahn, S. I. Sung, H. S. Yoo, S. J. Choi, S. Y. Kim and W. S. Park, PLoS One, 2015, 10, e0135574 Search PubMed.
- A. Monsel, Y. Zhu, S. Gennai, Q. Hao, S. Hu, J.-J. Rouby, M. Rosenzwajg, M. A. Matthay and J. W. Lee, Am. J. Respir. Crit. Care Med., 2015, 192, 324–336 CrossRef CAS PubMed.
- G. R. Willis, A. Fernandez-Gonzalez, J. Anastas, S. H. Vitali, X. Liu, M. Ericsson, A. Kwong, S. A. Mitsialis and S. Kourembanas, Am. J. Respir. Crit. Care Med., 2017 DOI:rccm.201705-0925OC.
- K. Tan, H. Choi, X. Jiang, L. Yin, J. Seet, V. Patzel, B. P. Engelward and V. T. Chow, BMC Genomics, 2014, 15, 587 CrossRef PubMed.
- C. Lee, S. A. Mitsialis, M. Aslam, S. H. Vitali, E. Vergadi, G. Konstantinou, K. Sdrimas, A. Fernandez-Gonzalez and S. Kourembanas, Circulation, 2012, 126, 2601–2611 CrossRef CAS PubMed.
- K. Vaporidi, E. Vergadi, E. Kaniaris, M. Hatziapostolou, E. Lagoudaki, D. Georgopoulos, W. M. Zapol, K. D. Bloch and D. Iliopoulos, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2012, 303, L199–L207 CrossRef CAS PubMed.
- A. Yuan, E. L. Farber, A. L. Rapoport, D. Tejada, R. Deniskin, N. B. Akhmedov and D. B. Farber, PLoS One, 2009, 4, e4722 Search PubMed.
- R. Sessa and A. Hata, Pulm. Circ., 2013, 3, 315–328 CrossRef CAS PubMed.
- Y.-G. Zhu, X.-M. Feng, J. Abbott, X.-H. Fang, Q. Hao, A. Monsel, J.-M. Qu, M. A. Matthay and J. W. Lee, Stem Cells, 2014, 32, 116–125 CrossRef CAS PubMed.
- A. J. Goodwin, in Acute Lung Injury and Repair, ed. L. M. Schnapp and C. Feghali-Bostwick, Springer International Publishing, 2017, pp. 161–177 Search PubMed.
- Y. Song, H. Dou, X. Li, X. Zhao, Y. Li, D. Liu, J. Ji, F. Liu, L. Ding, Y. Ni and Y. Hou, Stem Cells, 2017, 35, 1208–1221 CrossRef CAS PubMed.
- Y. Huang, M. Crawford, N. Higuita-Castro, P. Nana-Sinkam and S. N. Ghadiali, FASEB J., 2012, 26, 3351–3364 CrossRef CAS PubMed.
- T. Yang, P. Martin, B. Fogarty, A. Brown, K. Schurman, R. Phipps, V. P. Yin, P. Lockman and S. Bai, Pharm. Res., 2015, 32, 2003–2014 CrossRef CAS PubMed.
- R. C. Lai, R. W. Y. Yeo, K. H. Tan and S. K. Lim, Biotechnol. Adv., 2013, 31, 543–551 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, S. Zhang, Z.-B. Deng, W. Grizzle, D. Miller and H.-G. Zhang, Adv. Drug Delivery Rev., 2013, 65, 342–347 CrossRef CAS PubMed.
- X. Zhuang, X. Xiang, W. Grizzle, D. Sun, S. Zhang, R. C. Axtell, S. Ju, J. Mu, L. Zhang, L. Steinman, D. Miller and H.-G. Zhang, Mol. Ther., 2011, 19, 1769–1779 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, X. Xiang, Y. Liu, S. Zhang, C. Liu, S. Barnes, W. Grizzle, D. Miller and H.-G. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- Y. Tian, S. Li, J. Song, T. Ji, M. Zhu, G. J. Anderson, J. Wei and G. Nie, Biomaterials, 2014, 35, 2383–2390 CrossRef CAS PubMed.
- H. Saari, E. Lázaro-Ibáñez, T. Viitala, E. Vuorimaa-Laukkanen, P. Siljander and M. Yliperttula, J. Controlled Release, 2015, 220B, 727–737 CrossRef PubMed.
- M. J. Haney, N. L. Klyachko, Y. Zhao, R. Gupta, E. G. Plotnikova, Z. He, T. Patel, A. Piroyan, M. Sokolsky, A. V. Kabanov and E. V. Batrakova, J. Controlled Release, 2015, 207, 18–30 CrossRef CAS PubMed.
- T. Tian, H.-X. Zhang, C.-P. He, S. Fan, Y.-L. Zhu, C. Qi, N.-P. Huang, Z.-D. Xiao, Z.-H. Lu, B. A. Tannous and J. Gao, Biomaterials, 2018, 150, 137–149 CrossRef CAS PubMed.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. A. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS PubMed.
- J. M. Cooper, P. B. O. Wiklander, J. Z. Nordin, R. Al-Shawi, M. J. Wood, M. Vithlani, A. H. V. Schapira, J. P. Simons, S. El-Andaloussi and L. Alvarez-Erviti, Mov. Disord., 2014, 29, 1476–1485 CrossRef CAS PubMed.
- A. Mizrak, M. F. Bolukbasi, G. B. Ozdener, G. J. Brenner, S. Madlener, E. P. Erkan, T. Ströbel, X. O. Breakefield and O. Saydam, Mol. Ther., 2013, 21, 101–108 CrossRef CAS PubMed.
- S. Ohno, M. Takanashi, K. Sudo, S. Ueda, A. Ishikawa, N. Matsuyama, K. Fujita, T. Mizutani, T. Ohgi, T. Ochiya, N. Gotoh and M. Kuroda, Mol. Ther., 2013, 21, 185–191 CrossRef CAS PubMed.
- M. Katakowski, B. Buller, X. Zheng, Y. Lu, T. Rogers, O. Osobamiro, W. Shu, F. Jiang and M. Chopp, Cancer Lett., 2013, 335, 201–204 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |