
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and fluorescent properties of boroisoquinolines, a new family of fluorophores†
Dénes Sóvária,
Attila Kormos b,
Orsolya Demeterb,
András Dancsóc,
György Miklós Keserűa,
Mátyás Milenc and
Péter Ábrányi-Balogh*a
b,
Orsolya Demeterb,
András Dancsóc,
György Miklós Keserűa,
Mátyás Milenc and
Péter Ábrányi-Balogh*a
aHungarian Academy of Sciences, Research Centre for Natural Sciences, Institute of Organic Chemistry, Medicinal Chemistry Research Group, 1519 Budapest, POB 286, Hungary. E-mail: abranyi-balogh.peter@ttk.mta.hu; Tel: +36 1 3826961
bHungarian Academy of Sciences, Research Centre for Natural Sciences, Institute of Organic Chemistry, Chemical Biology Research Group, 1519 Budapest, POB 286, Hungary
cEgis Pharmaceuticals Plc., Directorate of Drug Substance Development, 1475 Budapest, POB 100, Hungary
First published on 15th November 2018
Abstract
First representatives of a new family of isoquinolines, so called boroisoquinolines, were synthesized and characterized. The synthesis was based on the insertion of the difluoroboranyl group into the 1-methylidene-3,4-dihydroisoquinoline core. The optimization of the 2-difluoroboranyl-3,4-dihydroisoquinoline-1(2H)-ylidene core led to efficient fluorescence in a range of 400–600 nm with outstanding (>100 nm) Stokes shifts. The compounds might be suitable for reversible or irreversible labelling of proteins, particularly the cannabinoid receptor CB2.
Introduction
The isoquinoline core is represented in a large number of biologically active compounds containing alkaloids and also synthetic heterocycles (Fig. 1). The alkaloids can be found widely in various plants1–4 and marine invertebrates.5 A well-known pharmacologically significant compound is papaverine (1) that was isolated from opium poppies (Papaver somniferum) and was used as a smooth muscle relaxant.6 Other biologically active representatives are the members of the berberine (2) family that were isolated from various herbs.7,8 These plants (e.g. Coptis chinensis) have been used in traditional Chinese medicine for the treatment of infections, skin problems and diabetes.9–12 Among the latter compounds palmatine (3) should be mentioned as a widely used tetracyclic isoquinoline against, among others, hypertension and inflammation.13 From a marine sponge Jaspis sp. also, more complex isoquinoline natural products were isolated such as jasisoquinoline A and B (4a and 4b, respectively) that possess anticancer activity.14 Ripasudil (5, Glanatec®) activates the Rho-kinase, hence it is used in the treatment of glaucoma and increased eye-pressure.15,16 Furthermore, many isolated or synthesized isoquinoline derivatives exhibit a vast spectrum of biological activities including anti-HIV,17 antitubercular,18 antiplasmodial,19 anti-Alzheimer's20 and antifungal effects21 and some representatives are non-competitive AMPA [2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid] receptor antagonists.22 In addition, the commercially available drug solifenacin (Vesicare®, 6), a 1-aryl-tetrahydroisoquinoline derivative shows competitive cholinergic receptor antagonist effect used in the treatment of contraction of overactive bladder.23,24 Members of the 2′-(3,3-dimethyl-3,4-dihydroisoquinoline-1-ylidene)-1-ethane-1-one family (7) should be mentioned as non-toxic and specific nanomolar CB2 agonist, while other derivatives of this family can inhibit phosphodiesterase 7 (PDE7). On this basis, these compounds might be considered as preventive or treating agents for asthma, autoimmune diseases, rheumatoid arthritis, Crohn's disease or carcinomatous pain.25–28 Besides the pharmaceutical applications, isoquinoline derivatives were also utilized as chiral ligands for catalytic asymmetric reactions29 or as electrophosphorescent iridium complexes.30 Boranyls (8) and related compounds containing nitrogen–boron–oxygen structural element (NBO-complexes) are a recently developed family of fluorescent molecules (Fig. 2). The photophysical properties of several known NBO-complexes are summarized in Table 1. These compounds were designed to overcome the drawbacks of BODIPY's e.g. small Stokes-shift, difficult industrial synthesis.31 The synthesis of boranyls is usually starting from an imino compound containing a hydroxyl or an enolizable oxo group at the γ-position. The parent compounds are reacted with a trisubstituted borane, mostly BF3 or B(aryl)3 resulting in a disubstituted borane group inserted between the nitrogen and the oxygen atom. The photophysical characterization of these compounds showed acceptable Stokes-shifts with good ε and Φ values (Table 1, entry 1).32 It was observed that in the case of borobenzothiazoles33 (9) a push–pull system between a disubstituted amino and a nitrile group was required for an outstanding Stokes-shift (149–170 nm, entry 2), but the quantum yield of the compounds was weak. Minuti and co-workers designed boranyls for biological application by the ring opening of isoxazoles to 10 equipped with an acetylene tail in order to the click labeling of azidoproteins.34 Only one member of the boranyls derived from coumarins (11) showed weak fluorescence but on the contrary, the phosphorescence of those was remarkable (Table 1, entry 3).35 Boroquinolines (12, 13) were synthesized from salicylic aldehyde, aniline and phenylacetylene, showing a Stokes-shift of 80–200 nm and green fluorescence (Table 1, entries 4, 5).36,37 From naphthalene dialdehyde multiboranyls (14) were synthesized, but the larger molecule size caused lower fluorescence.38 Nevertheless, a nice red shift was obtained with a wide range of quantum yield (Table 1, entry 6). Related compounds, so called polyboranyls (15) were designed from m-phthalic aldehyde exhibiting large Stokes-shift with medium molar absorptivity and quantum yield (Table 1, entry 7).39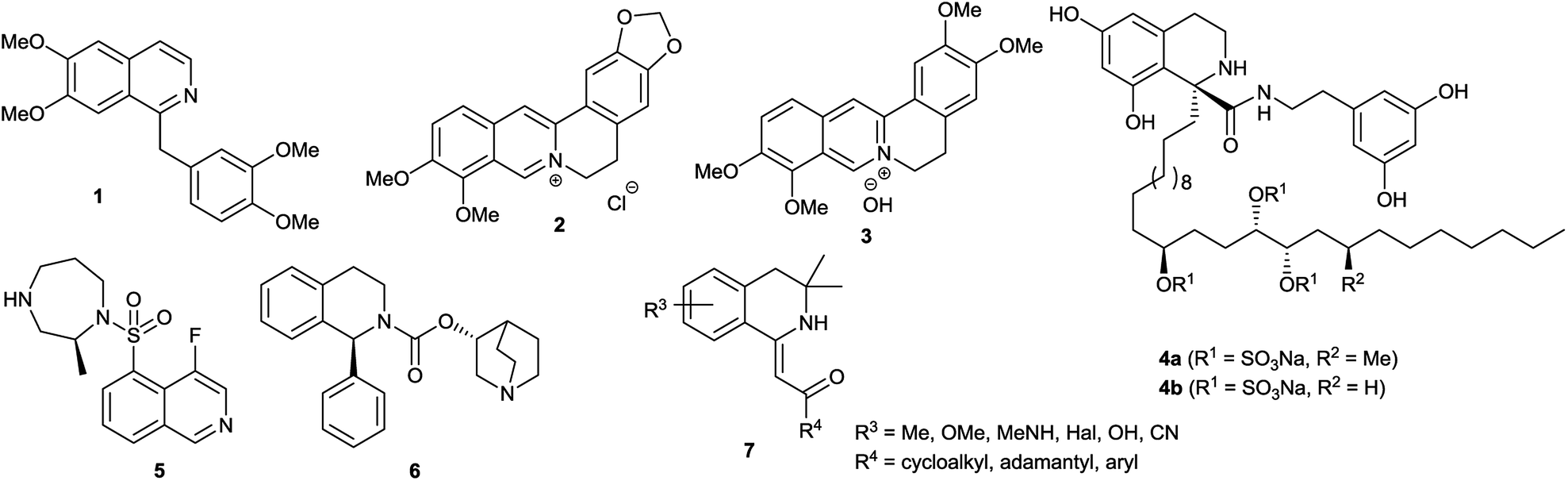 | ||
| Fig. 1 Structures of pharmaceutically active isoquinoline derivatives. | ||
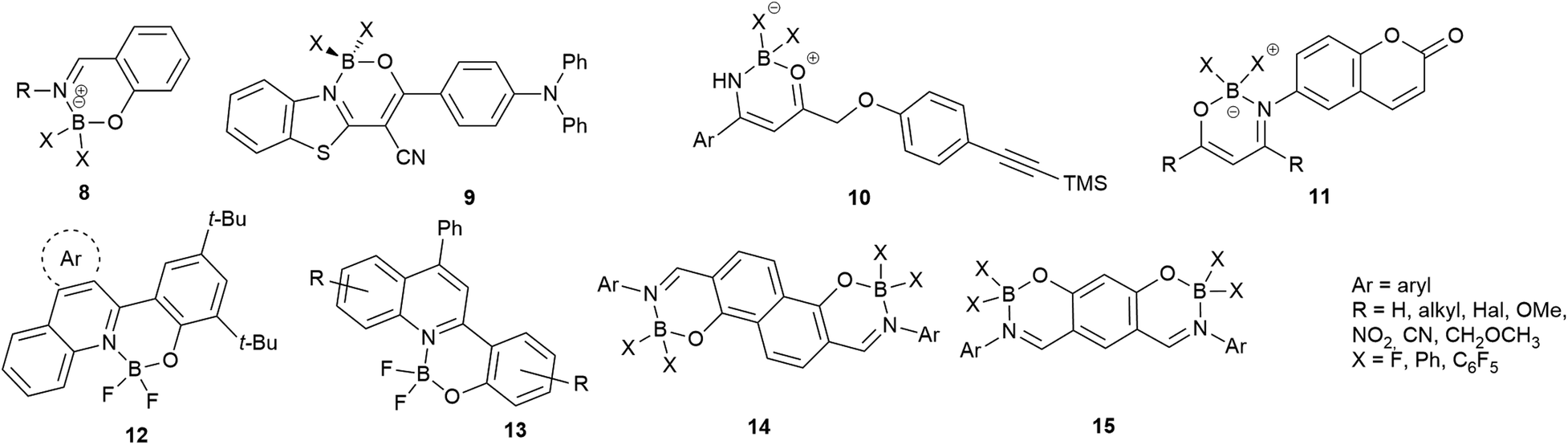 | ||
| Fig. 2 Boranyls (8) and related compounds containing nitrogen–boron–oxygen structural element (NBO-complexes, 9–15). | ||
| Entry | Solvent | λmaxabsz [nm] | λmaxem [nm] | Stokes-shift [nm] | ε [M−1 cm−1] | ΦF [−] | |
|---|---|---|---|---|---|---|---|
| a Only one member showed fluorescence. | |||||||
| 1 | 832 | CH2Cl2 | 344–387 | 380–432 | 14–68 | 13100–67200 | 0.22–0.73 |
| 2 | 933 | MeCN | 442–450 | 599–612 | 149–170 | — | 0.004–0.008 |
| 3 | 1135a | THF/dioxane | 331 | 420 | 89 | 33660 | — |
| 4 | 1236 | CHCl3 | 385–417 | 523–543 | 126–145 | 8317–11482 | 0.18–0.32 |
| 5 | 1337 | CHCl3 | 288–385 | 467–538 | 82–201 | 14300–38500 | 0–0.22 |
| 6 | 1438 | CH2Cl2 | 495–589 | 457–609 | 37–183 | 17300–148700 | 0–0.56 |
| 7 | 1539 | CH2Cl2 | 495–589 | 533–683 | 38–101 | 13100–44500 | 0–0.81 |
As a continuation of our efforts on the field of developing biologically interesting isoquinoline derivatives,40 the objective of our work was the synthesis of new fluorescent members of the 2′-(3,4-dihydroisoquinoline-1-ylidene)-1-ethan-1-one (7) family equipped with BR2 groups.
Results and discussion
The fluorescent molecules were envisaged to be obtained by the acylation of dihydroisoquinolines followed by the incorporation of a boranyl group. Initially, the starting 7-methoxy (16a), 6,7-dimethoxy (16b), 7-bromo (16c) and 7-fluoro-3,4-dihydroisoquinoline (16d) have been synthesized by slight modification of literature procedures.41,42 In most cases, the corresponding phenylacetonitriles were transformed to phenylethylamines using borane in THF,43 followed by the acylation with acetyl chloride. The ring closure was achieved with a modified Bischler–Napieralski reaction after Larsen using oxalyl chloride in the presence of FeCl3 and the subsequent reaction with sulphuric acid in MeOH.44 In the case of 7-pyrrolidinyl-1-methyl-3,4-dihydroisoquinoline (16e), the pyrrolidine group was inserted either with Buchwald–Hartwig reaction of 16c or with the aromatic nucleophilic substitution of 16d (Scheme 1). | ||
| Scheme 1 The synthesis of 1-methyl-7-pyrrolidine-1-yl-3,4-dihydroisoquinoline (16e). | ||
The Buchwald–Hartwig coupling was performed in the presence of Pd(OAc)2, JohnPhos and KOt-Bu in toluene leading to 16e in a yield of 40% (Scheme 1, Method A).45 In order to improve the outcome, several substitution reactions were tried using 16d (Method B, C and D).46,47 The best result was obtained reacting 16d with 6 equiv. of pyrrolidine in dioxane at 120 °C in a sealed tube for 3 days resulting in 16e in a yield of 70% (Method D). Notably, for the purification of 16e steam distillation and normal phase flash chromatography was required.
The synthesis of the 1-methylidene-1,2,3,4-tetrahydroisoquinolines (17) was accomplished by the slight modification of the method of Dannhardt et al. using BuLi or lithium-diisopropylamide (LDA) to deprotonate the methyl group of the 1-methyl-3,4-dihydroisoquinolines (16a–e) derivatives followed by the addition of the appropriate ester (Scheme 2).48 Heterocycles 17a–h were isolated in moderate yields (12–65%, Table 2, entries 1–8). In the case of 4-nitrobenzoic acid ethyl ester, the products (17i,j) could be isolated in traces with BuLi (3–5%), but using LDA the yield of 17j was raised to 54% (Table 2, entries 9, 10). Changing the methoxy group on the isoquinoline to pyrrolidine also lowered the yields (16–27%) possibly due to the interaction of the pyrrolidine nitrogen with the BuLi. The use of a larger amount of BuLi, or changing the base to sec-BuLi or BuLi–TMEDA did not give better results, but again, the application of LDA led to a significant raise in the isolated yields (20–72%, Table 2, entries 11–13).
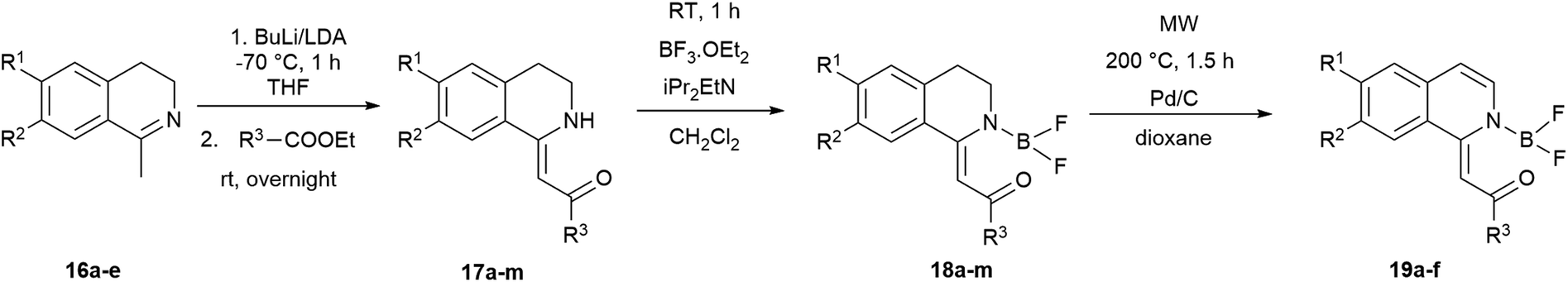 | ||
| Scheme 2 Synthesis of 1-methylidene-1,2,3,4-tetrahydroisoquinolines (17) and boroisoquinolines 18 and 19. | ||
| # | R1 | R2 | R3 | 17 a [%] | 18 a [%] | 19 a [%] | |
|---|---|---|---|---|---|---|---|
| a Isolated yields.b Yields with LDA, results with BuLi are in parentheses.c Pyrrolidine-1-yl. | |||||||
| 1 | a | MeO | H | Me | 46 | 77 | 73 |
| 2 | b | MeO | MeO | Me | 50 | 78 | 70 |
| 3 | c | MeO | H | Ph | 45 | 70 | 77 |
| 4 | d | MeO | MeO | Ph | 53 | 88 | 89 |
| 5 | e | MeO | H | 2-Thienyl | 58 | 50 | 52 |
| 6 | f | MeO | MeO | 2-Thienyl | 48 | 85 | 48 |
| 7 | g | MeO | H | C6F5 | 12 | 85 | — |
| 8 | h | MeO | MeO | C6F5 | 57 | 89 | — |
| 9 | i | MeO | H | 4-O2N–C6H4 | 5 | — | — |
| 10 | j | MeO | MeO | 4-O2N–C6H4 | 54 (3) b | 49 | — |
| 11 | k | Pyc | H | Me | 41 (26) b | 78 | — |
| 12 | l | Pyc | H | Ph | 72 (27) b | 97 | — |
| 13 | m | Pyc | H | C6F5 | 20 (16) b | 81 | — |
The difluoro-boroisoquinoline structure (18) was constructed by reacting compounds 17 with boron trifluoride diethyl etherate in the presence of 5 equiv. of diisopropylethylamine (DIPEA) in dichloromethane (Scheme 2).32 In order to synthesize fully aromatic derivatives with respect to its expected advantageous effect on the fluorescence, compounds 18a–f were dehydrogenated with Pd/C in dioxane at 200 °C for 90 min (Scheme 2). Boroisoquinolines 19a–f were isolated in good yields (48–89%, Table 2, entries 1–6). In the case of 18k–m, the pyrrolidine ring was aromatized during the reaction, a selective oxidation could not be developed. Dehydrogenating derivatives with C6F5 substituent produced only degradation products that could be detected by HPLC-MS with loss of various fluorine atoms. In the case of 18j, the nitro group was converted to amino function, and decomposition products were observed.
We aimed to synthesize further boranyl derivatives using triaryl boranes. A diphenyl-boroisoquinoline (20b) was prepared in a yield of 84% by refluxing 17b with triphenylborane in toluene at 90 °C (Scheme 3).31 In the case of B(C6F5)3 as the borylating agent, the formation of the desired bis(perfluorophenyl)-boroisoquinoline was observed depending on the starting compound either in a one-step reaction in toluene at 90 °C leading to 21l in a yield of 20%, or in a two-step procedure through the heating of tris(perfluorophenyl)-borosisoquinoline intermediate 22b at 200 °C resulting in 21b in a poor yield (3%) (Scheme 3).
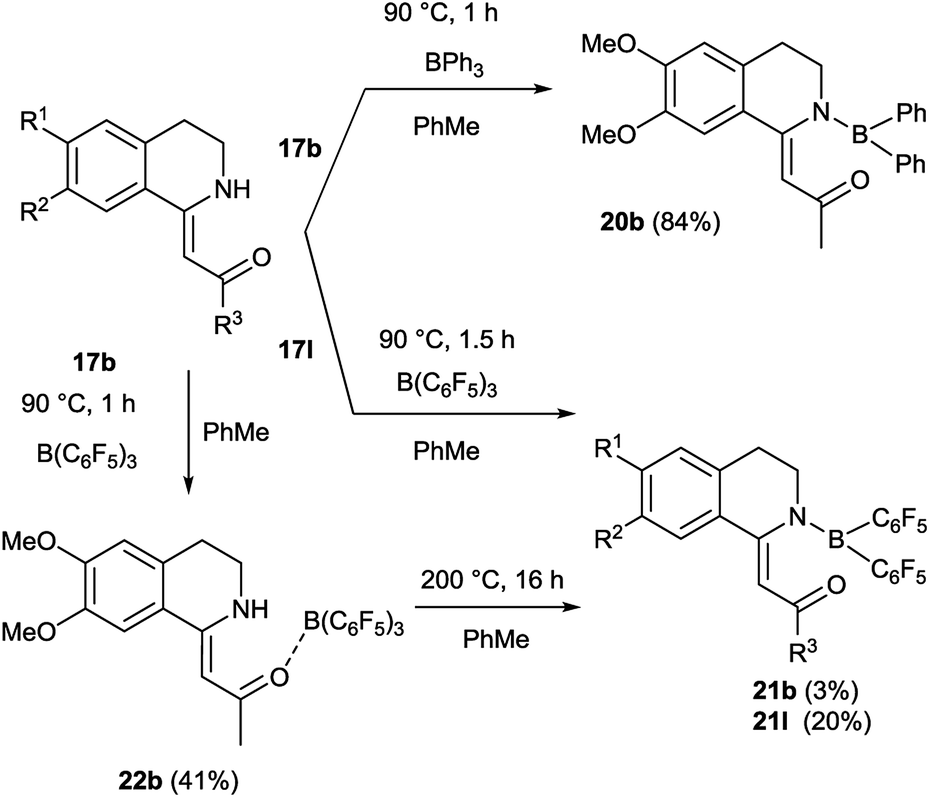 | ||
| Scheme 3 Synthesis of boroisoquinolines 20, 21, 22. | ||
In the cases of 18f and 19c the exact structure of the new heterocyclic family members was elucidated by single crystal X-ray diffraction measurements (Fig. 3). From the crystal structures it could be concluded, that until the nitrogen atom of 18f has pyramidal conformation, in 19c the nitrogen is planar. The bond lengths proved the boron–nitrogen bonding and the boron–oxygen coordination. The proposed electron-distribution and bonding in the NBO containing ring is shown on the figures next to the crystal structures. Representative bond lengths and bond angles are shown in Table 3. The detailed XRD data and ORTEP figures can be found in the ESI.†
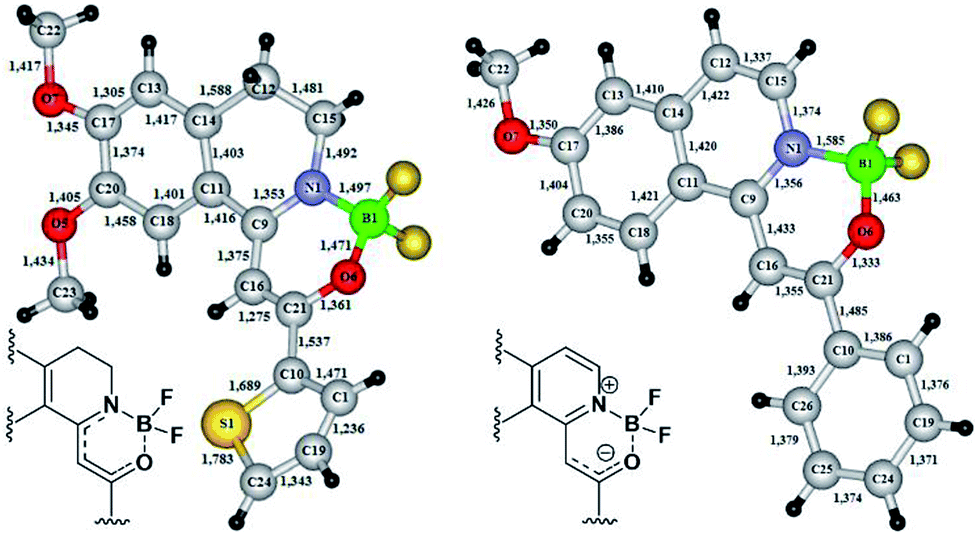 | ||
| Fig. 3 X-ray structures of boroisoquinolines 18f and 19c. | ||
| 18f | 19c | ||
|---|---|---|---|
| Bond length (Å) | Bond angle (°) | Bond length (Å) | Bond angle (°) |
| B1–N1 = 1.497 | C21–O6–B1 = 119.0 | C21–O6–B1 = 119.0 | |
| B1–O6 = 1.471 | C9–N1–C15 = 125.4 | C9–N1–C15 = 125.4 | |
| N1–C9 = 1.353 | C15–N1–B1 = 114.6 | B1–N1 = 1.585 | C15–N1–B1 = 114.6 |
| C9–C16 = 1.375 | N1–C9–C11 = 112.8 | B1–O6 = 1.463 | N1–C9–C11 = 112.8 |
| C16–C21 = 1.275 | O6–C21–C10 = 107.8 | N1–C9 = 1.365 | O6–C21–C10 = 107.8 |
| O6–C21 = 1.361 | C10–C21–C16 = 128.6 | N1–C15 = 1.374 | C10–C21–C16 = 128.6 |
| N1–C15 = 1.492 | F3–B1–F4 = 107.5 | C15–C12 = 1.337 | F3–B1–F4 = 107.5 |
| F3–B1–N1 = 108.6 | C9–C16 = 1.433 | F3–B1–N1 = 108.6 | |
| C9–N1–B1 = 119.5 | C16–C21 = 1.355 | C9–N1–B1 = 119.5 | |
| N1–C9–C16 = 120.6 | N1–C9–C16 = 120.6 | ||
| N1–C15–C12 = 110.8 | N1–C15–C12 = 110.8 | ||
| C9–C16–C21 = 121.1 | C9–C16–C21 = 121.1 | ||
| O6–C21–C16 = 123.5 | O6–C21–C16 = 123.5 | ||
| F3–B1–O6 = 105.8 | F3–B1–O6 = 105.8 | ||
| F4–B1–O6 = 109.8 | F4–B1–O6 = 109.8 | ||
| F4–B1–N1 = 114.4 | F4–B1–N1 = 114.4 | ||
| O6–B1–N1 = 110.3 | O6–B1–N1 = 110.3 | ||
The synthesis was followed by the extensive photophysical analysis of the new boroisoquinolines. The absorbance and fluorescence spectra were measured for compounds 18a–h,k–m, 19a–f, 20b, 21b,l and 22b (Table 4) in acetonitrile. It should be noted, that the nitro group of 18j was disadvantageous for the fluorescence, as the intensity was measured in the range of the noise.
| Entry | R1 a | R2 | R3 | BX | λmaxabs [nm] | λmaxexc [nm] | λmaxem [nm] | Δλ [nm] | |
|---|---|---|---|---|---|---|---|---|---|
| a Py = pyrrolidine-1-yl. | |||||||||
| 1 | 18a | MeO | H | Me | BF2 | 328 | 390 | 62 | |
| 2 | 18b | MeO | MeO | Me | BF2 | 321 | 339 | 431 | 110 |
| 3 | 18k | Py | H | Me | BF2 | 383 | 379 | 450 | 67 |
| 4 | 18c | MeO | H | Ph | BF2 | 357 | 411 | 54 | |
| 5 | 18d | MeO | MeO | Ph | BF2 | 361 | 380 | 483 | 122 |
| 6 | 18l | Py | H | Ph | BF2 | 408 | 406 | 537 | 129 |
| 7 | 21l | Py | H | Ph | B(C6F5)2 | 417 | 415 | 544 | 127 |
| 8 | 18e | MeO | H | 2-Thienyl | BF2 | 374 | 437 | 63 | |
| 9 | 18f | MeO | MeO | 2-Thienyl | BF2 | 383 | 448 | 65 | |
| 10 | 18g | MeO | H | C6F5 | BF2 | 334 | 343 | 435 | 101 |
| 11 | 18h | MeO | MeO | C6F5 | BF2 | 334 | 356 | 537 | 203 |
| 12 | 18m | Py | H | C6F5 | BF2 | 411 | 410 | 565 | 154 |
| 13 | 20b | MeO | MeO | Me | BPh2 | 337 | 387 | 453 | 116 |
| 14 | 21b | MeO | MeO | Me | B(C6F5)2 | 346 | 372 | 440 | 94 |
| 15 | 22b | MeO | MeO | Me | B(C6F5)3 | 357 | 368 | 465 | 108 |
| 16 | 19a | MeO | H | Me | BF2 | 355 | 410 | 55 | |
| 17 | 19b | MeO | MeO | Me | BF2 | 371 | 415 | 45 | |
| 18 | 19c | MeO | H | Ph | BF2 | 378 | 372 | 487 | 109 |
| 19 | 19d | MeO | MeO | Ph | BF2 | 396 | 458 | 62 | |
| 20 | 19e | MeO | H | 2-Thienyl | BF2 | 401 | 471 | 70 | |
| 21 | 19f | MeO | MeO | 2-Thienyl | BF2 | 410 | 480 | 70 | |
Regarding the different boranyls, although λmax-s of the di- or triarylboranyl derivatives (20b, 21b,l, 22b, Table 4, entries 7, 13–15) were uniformly higher than those of the difluoroboranyls, the Stokes-shift did not raise or even decreased. This observation has led to the conclusion that the fluoro derivatives should be developed further and this was supported by sterical factors too, namely it is presumed, that the extensive diarylboranyl group would be less favoured in a binding pocket of a protein than the smaller difluoroboranyl group.
On the isoquinoline ring electron donating substituents were chosen for the substitution envisioning that the electron donation will raise the fluorescence. This assumption was confirmed as the 6-methoxy derivatives showed the weakest fluorescence intensity and the lowest λmax-s, while the insertion of a second methoxy group or changing the methoxy to pyrrolidinyl initiated significant increase in intensity and also λmax-s (Table 4, entries 1–3, 4–6 and 10–12; representative spectra for 18c,d,l shown on Fig. 4) leading the fluorescence into the >500 nm range. In order to prove the electron donating effect of the pyrrolidine substituent, the fluorescence spectrum of 18l was measured in the presence of HCl (1 equiv.) (Fig. 4, grey spectrum) and a significant decrease of the fluorescence intensity was observed.
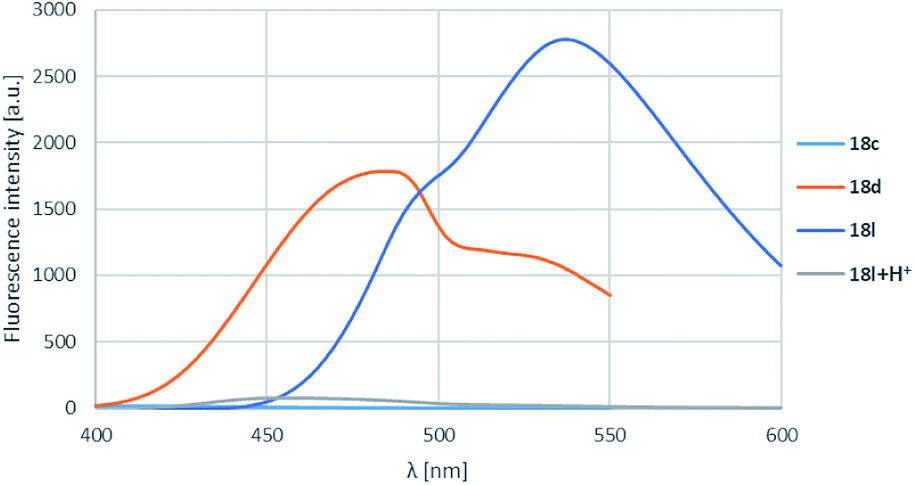 | ||
| Fig. 4 R1, R2 substituent effects for boroisoquinoline 18. | ||
The pyrrolidine substituent caused strikingly good fluorescence λmax-s depending on the substituent on the carbonyl group. It was found that the electron donating methyl group (18k) performed best, and as the electron withdrawal was increasing from the phenyl (18l) to the perfluorophenyl (18m) substituent, the fluorescence was shifted to the yellow range, but the intensity decreased significantly (Table 4, entries 3, 6, 12, Fig. 5).
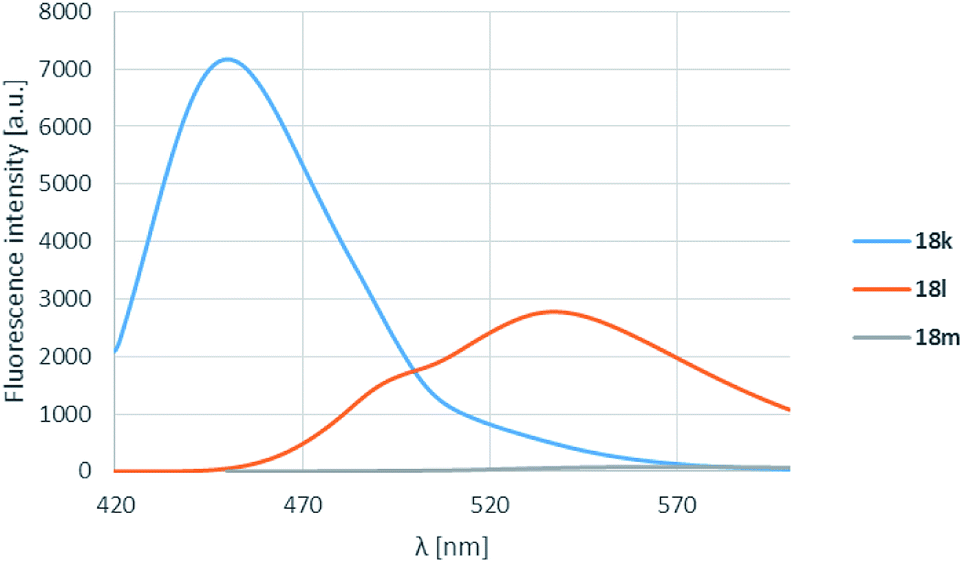 | ||
| Fig. 5 R3 substituent effect for boroisoquinolines 18. | ||
The effect of the dehydrogenation was also studied, and it was found that in the case of the 6-methoxy derivatives (18a,c,e and 19a,c,e) the extension of the aromatic system was advantageous for the λmax-s and the Stokes-shifts (Table 4, entries 1 and 16, 4 and 18, 8 and 20). It should be noted, that by 19c a remarkable Stokes-shift (115 nm) was measured (Table 4, entry 18). Meanwhile, in the case of the 6,7-dimethoxyboroisoquiolines, the oxidized species were better only for the thienyl derivative (18f and 19f, Table 4, entries 9 and 21).
The effect of various solvents for the absorbance, excitation and fluorescence was also investigated on boroisoquinoline 18l (Fig. 6). The solvent screen contained hexane, toluene, ethyl acetate, dichloromethane, dioxane, tetrahydrofuran, ethanol, acetonitrile and water. It could be concluded, that the absorbance and excitation spectra remained similar in the different solvents, the absorption maximum was not shifting significantly (Fig. 6a and b, respectively). It should be noted, that the largest intensity was reached in toluene, ethanol, ethyl acetate and dioxane, while in water (or PBS buffer) the absorbance was practically ceased because of aggregation of the molecules. Meanwhile the solvents seemed to influence the fluorescence more significantly (Fig. 6b).
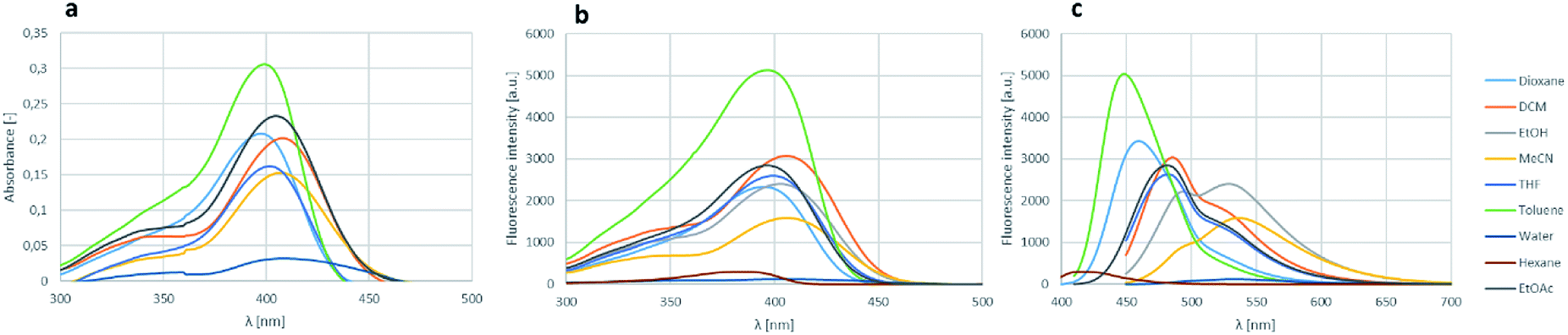 | ||
| Fig. 6 Solvent screen for the absorbance (a), excitation (b) and fluorescence (c) of 18l. | ||
The measured intensities were similar for the different solvents than those by the absorbance measurements, but in most solvents except MeCN and EtOH the fluorescence maximum decreased to the range under 500 nm narrowing the room between the excitation and the fluorescence. The loss of absorbance and fluorescence in aqueous medium might be an advantage targeting a protein in an aprotic environment e.g. in the cell membrane, if the fluorescence gets enhanced reaching the target, while stay hidden in the aqueous environment outside the membrane.
Furthermore, the photostability of compound 18l was investigated by measuring the fluorescence in acetonitrile after 0, 1, 2 and 4 h of irradiation with 360 nm light. As shown on Fig. 7, the fluorescence intensity was still around 60% after 4 h, that corresponds to outstanding photostability.
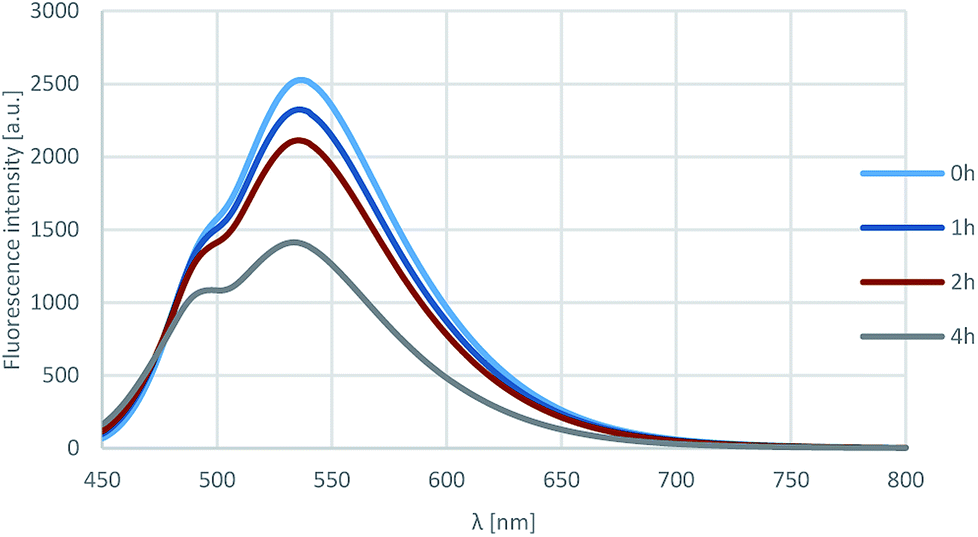 | ||
| Fig. 7 Results of the photostability test. | ||
Compounds showing outstanding Stokes-shift were studied forward measuring molar absorptivity (ε) and quantum yield (ΦF) (Table 5). It was found, that some of the boroisoquinolines, particularly 18b and 18l showed more than 0.3 (entries 1, 4) and a remarkable quantum yield of 0.84 belonged to 18k (entry 2). On the contrary, the fully aromatic 19c (entry 10) and the perfluorophenyl derivatives (18g,h,m; entries 6–8) has lower ΦF than 0.1, that might be explained with the dissipation of the electrons on the extended aromatic system, and the large, shielding substituents on the boron atom, respectively. Notably, the pyrrolidine substituent was advantageous for the ε and ΦF, but the larger substituents on either the carbonyl group or the boron atom decreased the above values, presumably because of the too extended electron system.
| Entry | λmaxabs [nm] | λmaxexc [nm] | λmaxem [nm] | Δλ [nm] | ε (λmaxabs) [×104 M−1 cm−1] | ΦF a [−] | |
|---|---|---|---|---|---|---|---|
| a Quantum yields relative to quinin sulfate (dyes 18b,g, 22b) or to Coumarin 153 (dyes 18k,d,l,g,h,m, 19c, 21l, 22b). | |||||||
| 1 | 18b | 321 | 339 | 431 | 110 | 2.20 | 0.32 |
| 2 | 18k | 383 | 379 | 450 | 67 | 5.47 | 0.84 |
| 3 | 18d | 361 | 380 | 483 | 122 | 3.06 | 0.18 |
| 4 | 18l | 408 | 406 | 537 | 129 | 3.58 | 0.35 |
| 5 | 21l | 417 | 415 | 544 | 127 | 3.71 | 0.17 |
| 6 | 18g | 334 | 343 | 435 | 101 | 2.75 | 0.0024 |
| 7 | 18h | 334 | 356 | 537 | 203 | 2.31 | 0.022 |
| 8 | 18m | 411 | 410 | 565 | 154 | 2.55 | 0.037 |
| 9 | 22b | 357 | 368 | 465 | 108 | 3.16 | 0.11 |
| 10 | 19c | 378 | 372 | 487 | 109 | 2.47 | 0.016 |
In order to find a possible biological application, compound 18h was reacted with N-acetylcysteine (23), as a thiol-surrogate in DMSO at 37 °C. The full conversion was reached after 72 h and the adduct was isolated in a yield of 75% (Scheme 4). It was proven, that the thiol group reacted with a fluorine atom on the aromatic ring. The photophysical properties of the conjugate were investigated, and although the intensity of the fluorescence was decreased, the shape of the excitation and fluorescence curve and the Stokes-shift remained similar (Fig. 8). Although the reaction conditions are not yet compatible with the conditions in human cells, this preliminary result projects that after careful optimization these compounds might be applicable for the covalent labelling of cysteines in proteins.
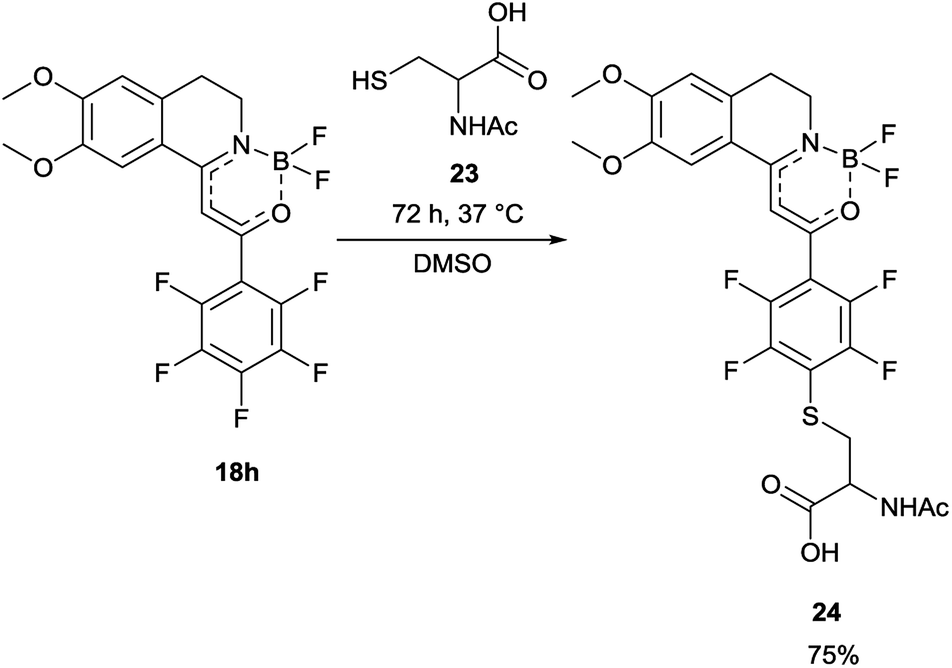 | ||
| Scheme 4 Reaction of 18h with N-acetylcysteine (23), as thiol-surrogate. | ||
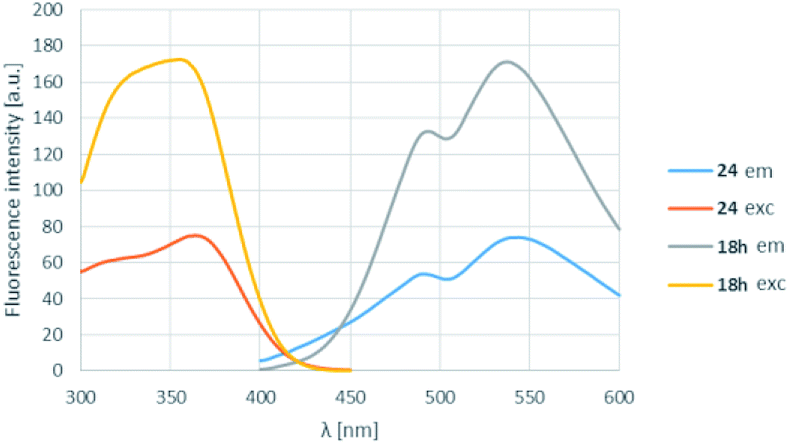 | ||
| Fig. 8 The change of the excitation and fluorescence spectra in the reaction of 18h with N-acetylcysteine (23). | ||
Conclusions
A new family of fluorophores, namely boroisoquinolines was developed, synthesized from 1-methyl-3,4-dihydroisoquinolines in a two or three step synthesis. The photophysical properties of the small library were extensively studied, and outstanding Stokes shifts with good molar absorptivity values were observed. Some of the dyes (18b,k,l) showed appreciable to excellent quantum yields with good photostability. The solvent screening results indicated low fluorescence in aqueous medium that might serve as an advantage using the compounds in aprotic conditions e.g. in cell membranes. We believe that upon further optimization these fluorescent dyes might be good choices for visualization of the labelling of the appropriate proteins (e.g. CB2).Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Á.-B. is grateful for the postdoctoral fellowship of the Hungarian Academy of Sciences and the Hungarian Science Foundation (OTKA PD124598).Notes and references
- K. W. Bentley, Chemistry and Biochemistry of Organic Natural Products, in The Isoquinoline Alkaloids, Hardwood Academic, Amsterdam, 1998, vol. 1 Search PubMed.
- K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC.
- D. L. Custódio and V. Florêncio Da Veiga Junior, RSC Adv., 2014, 4, 21864–21890 RSC.
- M. Iranshahy, R. J. Quinn and M. Iranshahi, RSC Adv., 2014, 4, 15900–15913 RSC.
- J. A. Joule, M. Alvarez and M. Salas, Heterocycles, 1991, 32, 759 CrossRef.
- M. S. Pathy and A. J. Reynolds, Postgrad. Med. J., 1980, 56, 488–490 CrossRef CAS PubMed.
- T. Schmeller, B. Latz-Brüning and M. Wink, Phytochemistry, 1997, 44, 257–266 CrossRef CAS PubMed.
- J. E. Park, T. D. Cuong, T. M. Hung, I. Lee, M. Na, J. C. Kim, S. Ryoo, J. H. Lee, J. S. Choi, M. H. Woo and B. S. Min, Bioorg. Med. Chem. Lett., 2011, 21, 6960–6963 CrossRef CAS PubMed.
- W. J. Kong, X. Y. Xing, X. H. Xiao, Y. L. Zhao, J. H. Wei, J. B. Wang, R. C. Yang and M. H. Yang, Appl. Microbiol. Biotechnol., 2012, 96, 503–510 CrossRef CAS PubMed.
- M. Wu, J. Wang and L. T. Liu, Chin. J. Integr. Med., 2010, 16, 188–192 CrossRef CAS PubMed.
- Q. Zhang, X. L. Piao, X. S. Piao, T. Lu, D. Wang and S. W. Kim, Food Chem. Toxicol., 2011, 49, 61–69 CrossRef CAS PubMed.
- S. Kang, Z. Li, Z. Yin, R. Jia, X. Song, L. Li, Z. Chen, L. Peng, J. Qu, Z. Hu, X. Lai, G. Wang, X. Liang, C. He and L. Yin, Nat. Prod. Res., 2015, 29, 2203–2206 CrossRef CAS PubMed.
- K. Bhadra and G. S. Kumar, Med. Res. Rev., 2011, 31, 821–862 CrossRef CAS PubMed.
- T. Yoshioka, M. Murai and H. Tasaki. European Patent EP2572717 (B1), 2016.
- H. Tanihara, T. Inoue, T. Yamamoto, Y. Kuwayama, H. Abe, A. Fukushima, H. Suganami and M. Araie, Acta Ophthalmol., 2016, 94, e26–e34 CrossRef CAS PubMed.
- H. Hidaka, US Pat. US20080064681 (A1), 2008.
- P. Cheng, N. Huang, Z.-Y. Jiang, Q. Zhang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Bioorg. Med. Chem. Lett., 2008, 18, 2475–2478 CrossRef CAS PubMed.
- P. Kumar, K. N. Dhawan, K. Kishor, K. P. Bhargava and R. K. Satsangi, J. Heterocycl. Chem., 1982, 19, 677–679 CrossRef CAS.
- S. Urverg-Ratsimamang, P. Rasoanaivo, H. Rafatro, B. Robijaona and A. Rakato-Ratsimamanga, Ann. Trop. Med. Parasitol., 1994, 88, 271–277 CrossRef.
- D. Kaufmann, A. Kaur Dogra, A. Tahrani, F. Herrmann and M. Wink, Molecules, 2016, 21, 1161 CrossRef PubMed.
- L. T. Liu, Y.-C. Lin, C.-L. J. Wang, M.-S. Lin, S.-C. Yen and H.-J. Chen, Bioorg. Med. Chem. Lett., 1996, 6, 1335–1338 CrossRef CAS.
- R. Gitto, M. L. Barreca, L. De Luca, G. De Sarro, G. Ferreri, S. Quartarone, E. Russo, A. Constanti and A. Chimirri, J. Med. Chem., 2003, 46, 197–200 CrossRef CAS PubMed.
- A. Ohtake, M. Ukai, T. Hatanaka, S. Kobayashi, K. Ikeda, S. Sato, K. Miyata and M. Sasamata, Eur. J. Pharmacol., 2004, 492, 243–250 CrossRef CAS PubMed.
- R. Naito, Y. Yonetoku, Y. Okamoto, A. Toyoshima, K. Ikeda and M. Takeuchi, J. Med. Chem., 2005, 48, 6597–6606 CrossRef CAS PubMed.
- M. Ogawa, Y. Takaoka and A. Ohhata. WO0210135 (A1), 2002.
- M. Ogawa, Y. Takaoka and A. Ohhata, US. Pat. US2004007643 (A1), 2004.
- A. Ohhata, Y. Takaoka, M. Ogawa, H. Nakai, S. Yamamoto and H. Ochiai, US Pat. US20050222138 (A1), 2005.
- K. Raitio, O. Salo, T. Nevalainen, A. Poso and T. Jarvinen, Curr. Med. Chem., 2005, 12, 1217–1237 CrossRef CAS PubMed.
- K. Murtagh, B. A. Sweetman and P. J. Guiry, Pure Appl. Chem., 2006, 78, 311–320 CAS.
- Z. Ma, J. Ding, B. Zhang, C. Mei, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2010, 20, 138–146 CrossRef CAS.
- D. Frath, S. Azizi, G. Ulrich, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 3414–3417 CrossRef CAS PubMed.
- J. Massue, D. Frath, G. Ulrich, P. Retailleau and R. Ziessel, Org. Lett., 2012, 14, 230–233 CrossRef CAS PubMed.
- L. Zhou, D. Xu, H. Gao, A. Han, Y. Yang, C. Zhang, X. Liu and F. Zhao, RSC Adv., 2016, 6, 69560–69568 RSC.
- L. F. Minuti, M. G. Memeo, S. Crespi and P. Quadrelli, Eur. J. Org. Chem., 2016, 821–829 CrossRef CAS.
- H. Doušová, P. Šimůnek, N. Almonasy and Z. Růžičková, J. Organomet. Chem., 2016, 802, 60–71 CrossRef.
- S. M. Elbert, P. Wagner, T. Kanagasundaram, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2017, 23, 935–945 CrossRef CAS PubMed.
- U. Balijapalli and S. K. Iyer, Eur. J. Org. Chem., 2015, 5089–5098 CrossRef CAS.
- M. Urban, K. Durka, P. Jankowski, J. Serwatowski and S. Luliński, J. Org. Chem., 2017, 82, 8234–8241 CrossRef CAS PubMed.
- D. Frath, K. Benelhadj, M. Munch, J. Massue and G. Ulrich, J. Org. Chem., 2016, 81, 9658–9668 CrossRef CAS PubMed.
- F. Haraszti, T. Igricz, G. Keglevich, M. Milen and P. Abranyi-Balogh, Curr. Org. Chem., 2018, 22, 912–922 CrossRef CAS.
- J. Václavík, J. Pecháček, B. Vilhanová, P. Šot, J. Januščák, V. Matoušek, J. Přech, S. Bártová, M. Kuzma and P. Kačer, Catal. Lett., 2013, 143, 555–562 CrossRef.
- R. Soni, F. K. Cheung, G. C. Clarkson, J. E. D. Martins, M. A. Graham and M. Wills, Org. Biomol. Chem., 2011, 9, 3290 RSC.
- M. Langlois, B. Bremont, S. Shen, A. Poncet, J. Andrieux, S. Sicsic, I. Serraz, M. Mathe-Allainmat, P. Renard and P. Delagrange, J. Med. Chem., 1995, 38, 2050–2060 CrossRef CAS PubMed.
- R. D. Larsen, R. A. Reamer, E. G. Corley, P. Davis, E. J. J. Grabowski, P. J. Reider and I. Shinkai, J. Org. Chem., 1991, 56, 6034–6038 CrossRef CAS.
- V. M. Manninen, W. A. E. Omar, J. P. Heiskanen, H. J. Lemmetyinen and O. E. O. Hormi, J. Mater. Chem., 2012, 22, 22971 RSC.
- L. Teuber and J. Larsen. WO 2004/087690 (A2), 2004.
- T. Di Franco, M. Stojanovic, S. C. Keller, R. Scopelliti and X. Hu, Helv. Chim. Acta, 2016, 99, 830–847 CrossRef CAS.
- G. Dannhardt, A. Bauer and U. Nowe, J. Prakt. Chem., 1998, 340, 256–263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopy and XRD data. For experimental procedures and compound characterization. CCDC 1854624 and 1854625. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08241c |
| This journal is © The Royal Society of Chemistry 2018 |