
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescence characteristics of rare-earth-doped barium hexafluorogermanate BaGeF6 nanowires: fast subnanosecond decay time and high sensitivity in H2O2 detection†
Gibin Georgea,
Machael D. Simpsona,
Bhoj R. Gautama,
Dong Fangb,
Jinfang Peng‡
c,
Jianguo Wenc,
Jason E. Davisd,
Daryush Ilaa and
Zhiping Luo *a
*a
aDepartment of Chemistry and Physics, Fayetteville State University, Fayetteville, NC 28301, USA. E-mail: zluo@uncfsu.edu
bCollege of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, P. R. China
cCentre for Nanoscale Materials, Argonne National Laboratory, Argonne, Illinois 60439, USA
dOak Ridge Institute for Science and Education, Oak Ridge Associated Universities, Oak Ridge, TN 37830, USA
First published on 26th November 2018
Abstract
Fluorides are promising host materials for optical applications. This paper reports the photoluminescent (PL) and cathodoluminescent (CL) characteristics of barium hexafluorogermanate BaGeF6 nanowires codoped with Ce3+, Tb3+ and Sm3+ rare earth ions, produced by a solvothermal route. The synthesized BaGeF6 nanowires exhibit uniform morphology and size distribution. X-ray diffraction divulges the one-dimensional growth of crystalline BaGeF6 structure, with the absence of any impurity phases. Visible luminescence is recorded from the nanowires in green and red regions, when the nanowires are codoped with Ce3+/Tb3+, and Ce3+/Tb3+/Sm3+, respectively, under a UV excitation source. The PL emission from the codoped BaGeF6 nanowires, when excited by a 254 nm source, originates from the efficient energy transfer bridges between Ce3+, Tb3+ and Sm3+ ions. The decay time of the visible luminescent emission from the nanowires is in the order of subnanoseconds, being one of the shortest decay time records from inorganic scintillators. The CL emission from the BaGeF6 nanowires in the tunable visible range reveals their potential use for the detection of high-energy radiation. The PL emissions are sensitive to H2O2 at low concentrations, enabling their high-sensitivity detection of H2O2 using BaGeF6 nanowires. A comparison with BaSiF6 nanowires is made in terms of decay time and its sensitivity towards H2O2.
Introduction
The luminescent property of a material may be significantly altered when it is prepared within a nanoscale domain, and further changes may be made by tailoring its morphology and doping. High luminescence has been reported for nanosized architectures with high aspect ratios, e.g., nanowires, nanofibers, nanorods, and nanowhiskers, etc., as they can guide the emission to the detector more efficaciously than bulk or other forms of architecture, when they are preferentially oriented.1–4 Fluorides are iconic materials for optical related applications due to their high quantum efficiency, high resistivity,5 a large optical-transmission domain and lower phonon energies, which in turn reduce the probability of non-radiative quenching from the excited states as compared to other halides or oxides.6 Although the photoluminescent (PL) emission from the pure fluoride matrices are in the near UV band, these materials are widely explored as fluorescent host matrices for rare-earth (RE) ions. RE3+ doping improves/tailors the luminescence of the host; however, the host also plays vital roles in tuning the luminescence emission bands of several RE3+ ions, since the crystal field of the host lattice and the local symmetry of the dopant in the crystal lattice determine the energy level splitting of the emissive ions.7 Besides the low power excitation, RE-doped materials exhibit sharp emissions, long lifetime, excellent photo-stability and large Stokes shift.8 These unique properties of RE3+ ions are connected to the electronic configuration in f-block elements, wherein, the 4f electrons in inner shells are shielded by the 5s and 5p subshells.9PL properties of the fluorides have gained intense research recently. RE3+-doped luminescent fluorides are envisaged for optical storage, biological labelling, non-invasive imaging, solid-state lasers, light-emitting diodes, theranostics/theragnostics, up-conversion lasers, solar cells, spectrum modifiers, photodynamic therapy remote controllers, optical waveguide amplifiers and temperature sensors.10 Moreover, fluorides have been widely used as wide-band gap insulating overlayers, gate dielectrics, insulators, and buffer layers in semiconductor-on-insulator structures in microelectronics and optoelectronic devices.11 Mn4+-activated complex fluorides, ABF6 phosphors (A = divalent element, B = tetravalent element), e.g., BaSiF6,12 BaGeF6,13 BaTiF6,14 BaSnF6,15 etc. and A2BF6 (A = monovalent element, B = tetravalent element) phosphors, e.g., K2TiF6,16 K2SiF6,17 K2GeF6,18 Na2SiF6,19 Na2GeF6,20 Na2TiF6,21 etc., are introduced as red emitting phosphors when excited by a blue light with better color rendering properties suitable for warm-white LED devices in the recent years, as replacements of conventional Ce3+:YAG phosphors.
Though Mn4+-doped complex hexafluorides are extensively studied as red phosphors, very little is known about the potentiality of these materials as the host matrices for RE ions and their structural and optical properties, although, Eu3+- and Sm3+-doped fluorides are also potential red emitting phosphors with a high color rendering index. But, the absorption bands of Eu3+ and Sm3+ ions are very narrow in blue or near-UV region, which is attributed to the parity-forbidden 4f–4f transitions. The absorptions bands of these red emitting RE3+ ions can be broadened by codoping with other RE3+ ions with broad absorption bands as sensitizers, for instance Ce3+, Tb3+, Dy3+, etc. Recently, we synthesized BaSiF6 nanowires codoped with RE3+ ions for the gamut emission and these nanowires exhibit efficient PL emission under UV excitation and the decay time was in the order of hundreds of picoseconds (subnanoseconds).22 Codoping of other complex fluorides with different luminescent RE3+ ions for the controlled emission has not been well addressed in literature so far. BaGeF6 is one such complex fluoride, whose PL characteristics are exclusively reported as a red-light emitting phosphor for warm white LED devices when doped with Mn4+ ions in the recent years.13,23,24 The PL emission properties of Mn4+ doped BaGeF6 red phosphors are analogous to the complex fluorides with the formula BaXF6 (X = Si, Ti, Sn, etc.).12–15 A warm white LED with a lower corrected color temperature (CCT = 4766 K) and higher color rendering index (Ra = 86.3) can be obtained by combining blue GaN chip with a mixture of BaGeF6:Mn4+ red phosphor and commercial YAG:Ce3+ yellow phosphor.24 Also, BaGeF6:Mn4+ red phosphor exhibit good thermal stability as the phosphor can retain 42.5% of the luminescence intensity at 448 K with respect to room temperature.24 PL and cathodoluminescent (CL) characteristics of nanosized RE3+-doped BaGeF6 have not been reported in the literature.
In this paper, we report the synthesis of RE-doped barium hexafluorogermanate BaGeF6 (BGF) nanowires through a solvothermal method. The PL emission bands of the nanowires are tailored by Ce3+, Tb3+ and Sm3+ doping and the RE3+ loading is optimized for efficient energy transfer between the ions, and the recorded lifetime of PL emissions are found in the order of subnanoseconds. The CL emission of these nanowires under high-energy irradiation is also studied for the potential application in high-energy radiation detection. In addition, the influences of several surrounding media on the PL characteristics of these nanowires have been studied and it is observed that the red PL emission is sensitive to H2O2, a reactive oxygen species, at low concentrations. H2O2 is used in a wide range of applications, such as disinfectant, antiseptic and oxidizer in food,25 pharmaceutical,26 biomedical,27 cosmetic,28 and chemical industries, involving biological and environmental implications.29,30 Besides, it is a propellant in rockets,31 and a precursor and a decomposition product of triacetone triperoxide, a progressively used explosive by terrorists.32 Therefore, application of these inorganic luminescent nanowires as an alternative to conventional analytical probes and organic luminescent materials for the detection of H2O2 is promising due to their simplicity and stability.
Experimental
Preparation of pure and doped BGF nanowires
The doped and undoped nanowires are prepared using a microemulsion containing cetyltrimethylammonium bromide (CTAB) as a surfactant and 1-pentanol as co-surfactant. Cyclohexane was used as the oil phase and all the precursors were dissolved/diluted in deionized water. The chemicals used were analytical grade as received without any purification. In the synthesis process, the first step was the preparation two identical solutions of 25 mL cyclohexane containing 1 mL of 1-pentanol (Sigma Aldrich) and 2 g of CTAB (VWR International, LLC.). The pure BGF nanowires were prepared by adding 1 mL of 1 M BaCl2·2H2O (VWR International, LLC.) dissolved in deionized water to one of the above solutions, and 1 mL of 1 M GeO2 (Sigma Aldrich) dissolved in 10% hydrofluoric acid (HF with concentration >40%, Sigma Aldrich) to the other solution. Both the precursor solutions were added drop wise at a rate of 2 mL h−1 under a vigorous stirring of 700 rpm, and the solutions became transparent after the addition. After 30 min of vigorous stirring, both the solutions were mixed together for another 10 min. The mixed solution was then transferred to 100 mL Teflon-lined autoclaves and heated at 120 °C for 12 h. After 12 h the solution was set to cool naturally to room temperature, washed with a mixture of 50% ethanol and 50% water for several times and dried at 80 °C for overnight. The doped nanowires were made using similar method, in which respective mol% of the metal chlorides RECl3 were added to BaCl2 solution with the subsequent replacement of BaCl2 and the aforesaid procedure was followed. The samples are notated by BGF:xCe–yTb–zSm, where x, y and z are the mol% of Ce3+, Tb3+ and Sm3+, respectively.Material characterization
The morphology of the BGF nanowires was identified using JEOL field-emission JXA-8530F Electron Probe Microanalyzer (EPMA), after coating the samples with ∼5 nm carbon film (Denton carbon coater). EPMA was equipped with an X-ray Energy-Dispersive Spectrometer (EDS) for chemical compositional analysis, and an xCLent IV Advanced B Hyperspectral CL Detector for the CL analysis. Samples for Transmission Electron Microscopy (TEM) were prepared in pure ethanol solutions, sonicated for 30 min to break the long nanowires and then dispersed on carbon-film supported grids. The grids were observed by using a FEI field-emission Tecnai F20ST instrument at 200 kV. The X-ray diffraction (XRD) patterns of the samples were obtained using Rigaku MiniFlex 600 X-ray diffractometer at a scanning rate of 0.075° min−1 in the range of 10–90°. The Fourier-Transform Infrared (FTIR) spectra of the samples were recorded by Shimadzu IRPrestige-21 in transmission mode by KBr pellet method in a wave number range of 4000–400 cm−1 for an average of 32 scans.The PL spectra of the samples were obtained in powder form, recorded using Shimadzu RF-5301PC spectroflurophotometer at room temperature. A time-correlated single photon counting (TCSPC) experiments were performed to measure the PL decay dynamics (Edinburgh instrument minitau). The samples were excited with 56 ps laser pulses at a wavelength of 405 nm and the emission was monitored at 594 nm. PL decay was detected using a single photon counting photomultiplier tube (PMT). The time resolution of the configuration was ∼100 ps. To test the H2O2 sensing characteristics, 5 mg of the nanowire phosphor was dispersed in 10 mL of deionized water and the concentration of H2O2 was subsequently adjusted in the solution and the PL emission spectra were recorded for different concentration of H2O2. To test the selectivity, the PL emissions from the nanowires in the presence of 100 μM CuSO4(Cu2+), 100 μM FeCl3(Fe3+), 100 μM glucose, HCl (2 pH) and NaOH (14 pH) were recorded.
Results and discussion
Morphology and structure
The SEM images of as synthesized pure BGF nanowires are shown in Fig. 1a and b. It is observed that the size and shape of the nanowires are consistently uniform. Similar to the BaSiF6 nanowires reported very recently,22 the synthesized BaGeF6 nanowires are long, typically in the range of 50–100 μm from low-magnification SEM images (Fig. 1a and S1†). The TEM images are shown in Fig. 1c and d. Without a surface coating in the TEM images, the average diameter is measured as 58 nm with a standard deviation of 12.3 nm, based on 30 measurements, giving the aspect ratio of several hundreds (Fig. S2†). A single nanowire, as marked in Fig. 1d, is magnified in Fig. 1e. A selected-area diffraction (SAD) pattern from the nanowire is inserted in Fig. 1e. At this orientation, the edge-on surface is (210), as the nanowire surface (110) is not at the edge-on orientation. However, the nanowire axis is identified as [001] direction. The EDS spectrum of pure BGF is shown in Fig. 1f that confirms the high purity of the nanowires (Table S1;† the element C is from the surface coating). A representative EDS spectrum of ternary doped nanowires is shown in Fig. S3.† TG analysis results of pure BGF nanowires is shown in Fig. S4.† The nanowires are stable up to 400 °C. However, the evaporation of adsorbed water and the crystallized water is observed until 300 °C, and then the weight is lost gradually until 600 °C by releasing a GeF4 gas to form BaF2 structure.33 It is observed that the phase transformation from rhombohedral BGF to cubic BaF2 is exothermic from the DTA curve. | ||
| Fig. 1 Pure BGF nanowires. (a and b) SEM images in low and high magnification; (c and d) TEM images; (e) enlarged image from (d) and SAD pattern showing crystallographic direction; and (f) EDS spectrum. | ||
Fig. 2 shows the XRD patterns of the as BGF nanowires and its RE-doped counterparts. The peaks in the XRD pattern of the nanowires is indexed to that of BGF (JCPDS data file no. 074-0924), which has a rhombohedral structure with a lattice parameter 4.948 Å. From the XRD data, pure crystalline nature of BGF nanowires can be observed, with well-defined sharp peaks with the highest intensity at (110) plane, implying the nanowires are grown along [001] direction with {110} surfaces exposed on the nanowires. The structure of BGF has a cesium chloride-like arrangement with Ba2+ cations and octahedrally arranged anions, GeF62−, having a rhombohedral angle of about 98.245°. Ba2+ ion is nearly equidistant from twelve fluorine ions making the coordination number as 12 and the Ge4+ ions are coordinated with six fluoride ions octahedrally.34 The crystal structure of BGF is described with the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. All the diffraction peaks of the RE3+-doped samples match with those of the pure BGF, which can be indexed to JCPDS 074-0924. Therefore, one can deduce that the crystalline structure of the doped BGF nanowires are pure, and the dopant ions occupied at the lattice sites of Ba2+ ions with octahedral symmetry.
m. All the diffraction peaks of the RE3+-doped samples match with those of the pure BGF, which can be indexed to JCPDS 074-0924. Therefore, one can deduce that the crystalline structure of the doped BGF nanowires are pure, and the dopant ions occupied at the lattice sites of Ba2+ ions with octahedral symmetry.
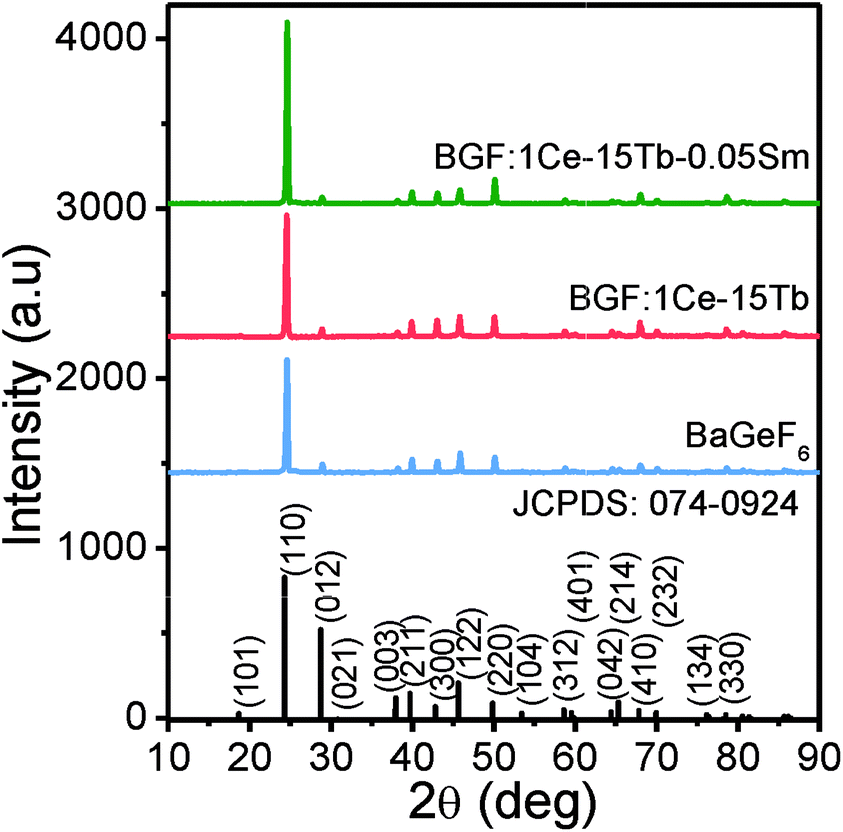 | ||
| Fig. 2 The XRD patterns of the pure and doped BGF nanowires. | ||
The nanowires are grown in one [001] direction and the overall growth process comprises of two subsequent processes, they are nucleation and growth. The CTAB/1-pentanol in oil phase forms microemulsions around the water droplets containing the precursors. When the two transparent precursor solutions are mixed together, long slender wormlike reverse micelle are formed by joining the water droplets. On heating, the water droplets collapse and adjacent drops containing the precursor solution join and thus nucleates are formed. By maintaining at a temperature above the ambient temperature, nucleates are grown as wires and the wire like morphology is retained under the restrain of CTAB/1-pentanol microemulsion. The overall chemical reactions involved in the formation of nanowires are:
| GeO2 + 6HF → H2GeF6 + 2H2O | (1) |
| H2GeF6 + BaCl2·2H2O → BaGeF6 + 2HCl + 2H2O | (2) |
The FTIR spectra of the as obtained samples are shown in Fig. 3. The FTIR spectra of all doped samples are analogous to pure BGF. There is no evidence of the peaks arising from the precursors used during the synthesis of the sample. The high intensity peak at 612 cm−1 is arising from the stretching of octahedral GeF62− ions.35 The peak at 3574 and 3582 cm−1 is attributed of –OH stretching from the adsorbed water and at 1,087, 1028 and 793 cm−1 can be attributed to several modes of vibrations in GeF62− ions.36 The significant shift of the peaks at 1087 and 1028 cm−1 of pure BGF samples to lower wavenumbers in doped BGF nanowires reveal the change in bond length in them, due to the difference in the electronegativity of the dopants and the Ba2+ ions. Moreover, the charge compensation between the trivalent RE3+ ions and the divalent Ba2+ ions is through defects such as interstitial fluoride ions and/or defect aggregations.37
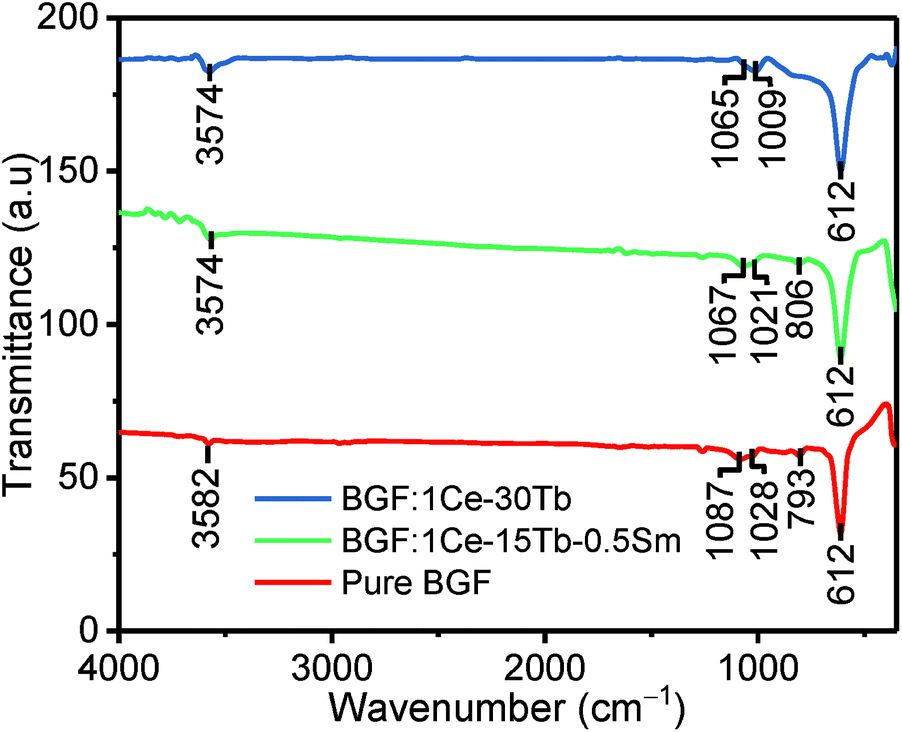 | ||
| Fig. 3 FTIR spectra of pure and doped BGF nanowires. | ||
Photoluminescence characteristics
The photoluminescence excitation (PLE) and PL spectra of unary, binary and ternary doped BGF nanowires are shown in Fig. 4. On comparing the PLE spectra of these RE3+-doped nanowires, one can observe that the excitation bands of ternary doped nanowires contain all excitation bands of individual RE3+ ions. The PLE spectrum of BGF:2Ce nanowires exhibits an absorption peak at 231 nm for the emission monitored at 336 nm, which is characteristic to the host absorption and the peaks at 254 and 280 nm are arising from 4f → 5d 2D multiplets transition of Ce3+ ions. The PL spectrum of BGF:2Ce nanowires displays a broad asymmetric emission band which constitutes two peaks in the UV region, when excited by a 254 nm source. In the BGF host matrix, Ce3+ ions replace Ba2+ ions with octahedral symmetry (D3d point symmetry38) and the crystal field splitting of 4f ground state in Ce3+ ion in the octahedral field is 2F5/2 and 2F7/2, respectively.39 One can expect two distinct emitting centers, as there are two ground state crystal field splitting and if the Ce3+ ions are close enough to form aggregates and the emission arises from different local structures and the respective emissions can be intense and broad.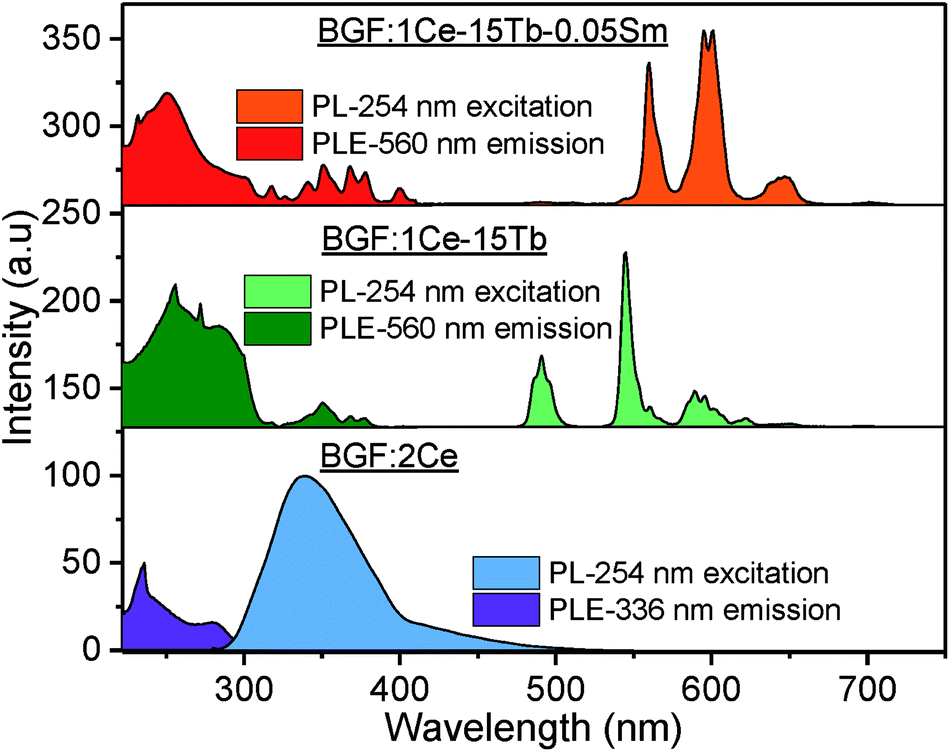 | ||
| Fig. 4 PLE (left) and PL (right) spectra of representative unary, binary and ternary doped BGF nanowires. | ||
On Ce3+ and Tb3+ codoping, PLE spectrum monitored at 544 nm emissions, exhibit several bands between 225–400 nm originated by 4f8 → 4f75d1 transition of Tb3+ ions.40 The broad excitation bands between 220–300 nm are due to 4f–5d transitions and the small peaks between 300–400 nm are due to 4f–4f transitions of Tb3+ ions. In the corresponding PL spectrum, the sharp peaks observed at 488, 544, 590 and 620 nm are originated from 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4, and 5D4 → 7F3 4f–4f transitions of Tb3+ ions, respectively.41 On comparing the PLE spectrum of BGF:1Ce–15Tb with that of BGF:1Ce–15Tb–0.05Sm nanowires, several additional peaks appeared in the latter, due to 6H5/2 → 4D3/2 (364 nm), 6H5/2 → 6P7/2 (378 nm), 6H5/2 → 4F7/2 (403 nm) and 6H5/2 → (6P, 4P)5/2 transitions of Sm3+ ions.42 As the excitation peaks are spanning in the UV region in the PLE spectrum, it can be concluded that both high and low energy UV light can be used as the excitation source of theses nanowires. Additionally, one can observe the overlapping of emission bands of Ce3+ with the excitation bands of Tb3+ and the emission bands of Tb3+ with the excitation bands of Sm3+, disclosing the apparent resonance type energy transfer between these ions.43 A detailed schematic of energy transfer bridge between Ce3+–Tb3+–Sm3+ ions is shown in Fig. S5.†
PL spectra of BGF:1Ce–yTb nanowires are shown in Fig. 5. The PL spectra consist of the weak emission peaks of Ce3+ ions in the UV region and the strong characteristic peaks of Tb3+ ions originated from 4f–4f transitions, when excited by 254 nm wavelength source. Even though, the weak 5d–4f UV emissions of Ce3+ ions are slightly changed with a subsequent change in the Tb3+ ion concentration, the significant sharp 4f–4f green emissions of Tb3+ ions are evident at every concentration of Tb3+ ions while the doping level of Ce3+ ions are kept constant as 1 mol%. Therefore, the corresponding spectra shown in Fig. 5 indicate the nonradiative energy transfer from Ce3+ to Tb3+ ions when doped in BGF matrix. The intensity of emission from Tb3+ ions are gradually increased with Tb3+ concentration and the most intense emission is observed from the nanowires doped with 15 mol% of Tb3+ ions, and thereafter the intensity is dropped. The emission lines from Ce3+ ions are completely vanished when Tb3+ doping level is increased to 30 mol%. The reduction in Tb3+ emission intensity at higher concentration above 15 mol% could be attributed to Tb3+–Tb3+ internal concentration quenching, during which the excitation energy is lost by the energy migration among the activator ions. The calculated energy transfer efficiency44 is more than ∼80% in all the Ce3+ and Tb3+ codoped nanowires, which could be accompanied to the low lattice phonon energy of the fluoride matrices. In addition, the emission from these binary doped nanowires does not alter significantly by changing the Ce3+ content (Fig. S6†).
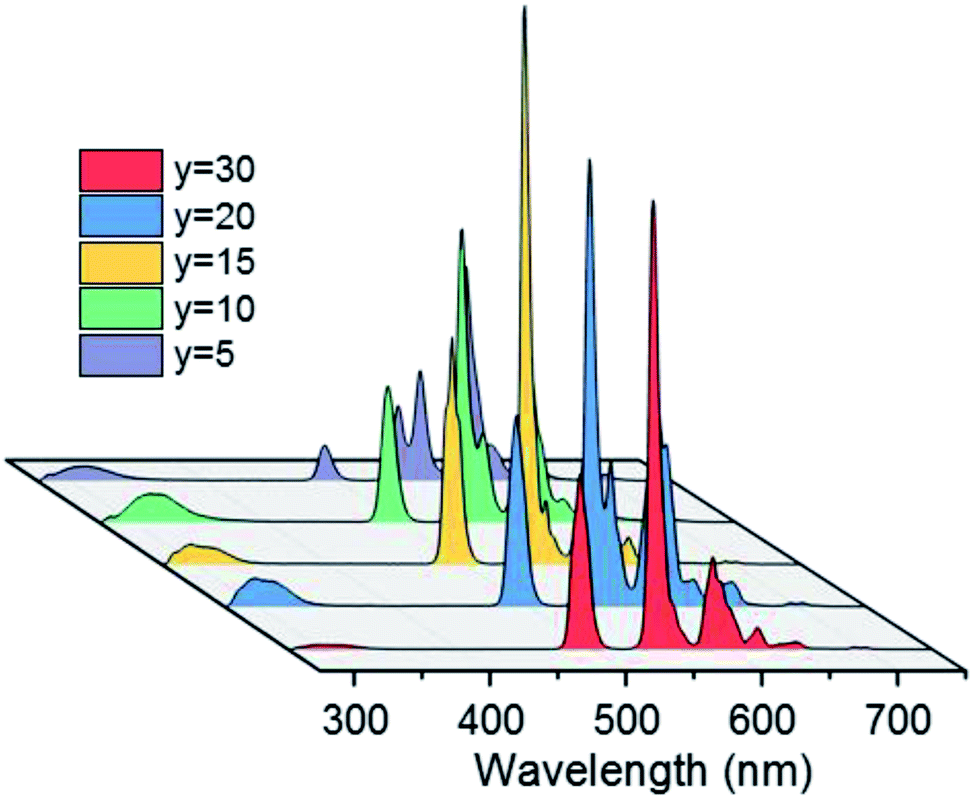 | ||
| Fig. 5 PL spectra of BGF:1Ce–yTb nanowires under 254 nm excitation. | ||
In the PL spectra of BGF:1Ce–15Tb–zSm nanowires as shown in Fig. 6, strong emissions in the orange-red band are observed. These emission bands are corresponding to 4G5/2 → 6HJ transitions of Sm3+ ions, under an excitation of 254 nm source. More precisely, the major emission bands peaking at 565, 604, and 653 nm are assigned to the 4G5/2 → 6H5/2, 4G5/2 → 6H7/2, and 4G5/2 → 6H9/2 electronic transitions of Sm3+ ions, respectively.45 The intensities of the emission from the Tb3+ ions are completely disappeared (when z = 0.5, 0.25, and 0.1) or insignificant (when z = 0.05, 0.02 and 0.01) in the presence of Sm3+ ions. The intensity of the orange-red emission is increased when the Sm3+ content is increased from 0.01 to 0.05 mol% and then decreased above 0.05 mol%, while the concentrations of Ce3+ and Tb3+ are kept constant. The weak emission line from Ce3+ ions at 335 nm is appeared in the spectra at all the concentrations. The characteristic emission lines of Sm3+ ions are very sensitive to its concentration. Increasing the concentration of Sm3+ ions above 0.5 mol% reduces the intensity of characteristic emission lines from Sm3+ ions to a large extend, as in Fig. S7,† due to the concentration quenching. Interestingly, a well-defined characteristic emission of Sm3+ ions did not appear, when the BGF nanowires are binary doped with Ce3+/Sm3+ or Tb3+/Sm3+, as in Fig. S8,† indicating the absence of a charge transfer bridge in those phosphors. The green emissions from Ce3+/Tb3+ and orange-red emissions from Ce3+/Tb3+/Sm3+ doped BGF nanowires can be clearly distinguished from the photographs of the nanowire phosphor dispersions in water under 254 nm UV excitation, as shown in Fig. S9.†
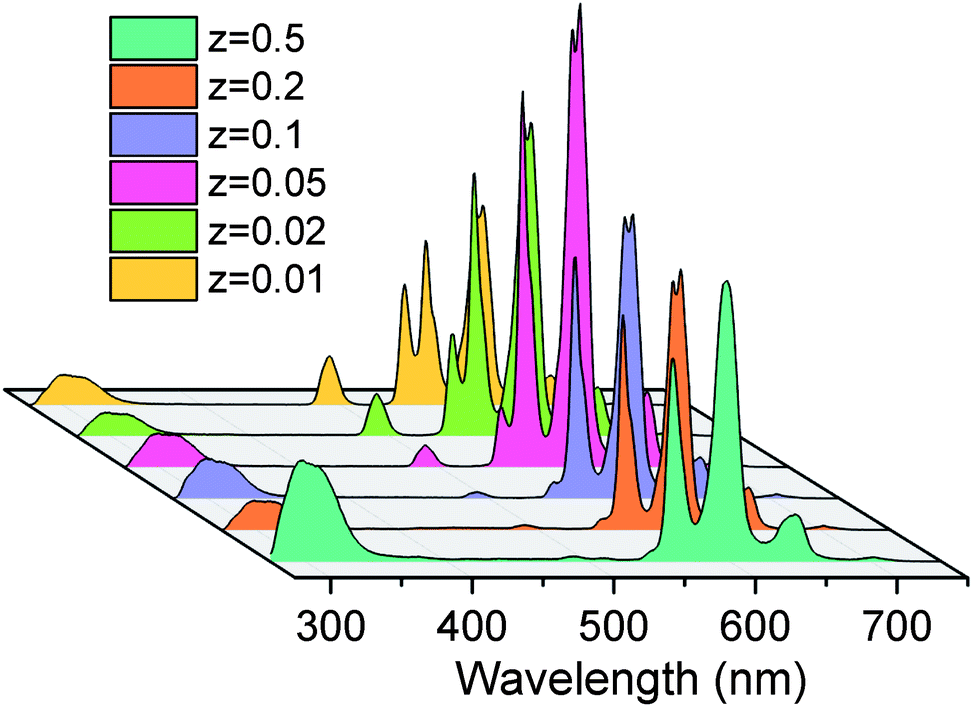 | ||
| Fig. 6 PL spectra of BGF:1Ce–15Tb–zSm nanowires under 254 nm excitation. | ||
The binary and ternary codoped BGF nanowires exhibit emission under 350 nm source, as shown in Fig. 7a and b, respectively. One can manifest the several excitation bands from the PLE spectra as shown in Fig. 4. The broad blue emission is accustomed to the 5D3 → 7Fj (j = 2, 3, 4) transitions of Tb3+ ions due to the relaxation of the ions excited by the direct source band and the orange-red emission bands can be ascribed to the 4G5/2 → 6HJ transition of Sm3+ ions. The 5D3 blue emission of Tb3+ is the highest when the concentration of Tb3+ is 5 mol% and it is decreased with the increase of Tb3+ content. In the case of ternary doped nanowires, as the Sm3+ content is 0.05 mol% or higher, the blue emission is the highest for a constant 1 mol% Ce3+ and 15 mol% Tb3+ doping, but it is reduced by increasing the Tb3+ content to 20 or 30 mol% as shown in Fig. 7b. This blue emission is not observed when excited by 254 nm source because the emission originate by the relaxation from 5D3 level is quenched by the cross-relaxation process, i.e., Tb3+(5D3) + Tb3+(7F6) → Tb3+(5D4) + Tb3+(7F0) accompanied by the subsequent enhancement of the emissions from the 5D4 level.46
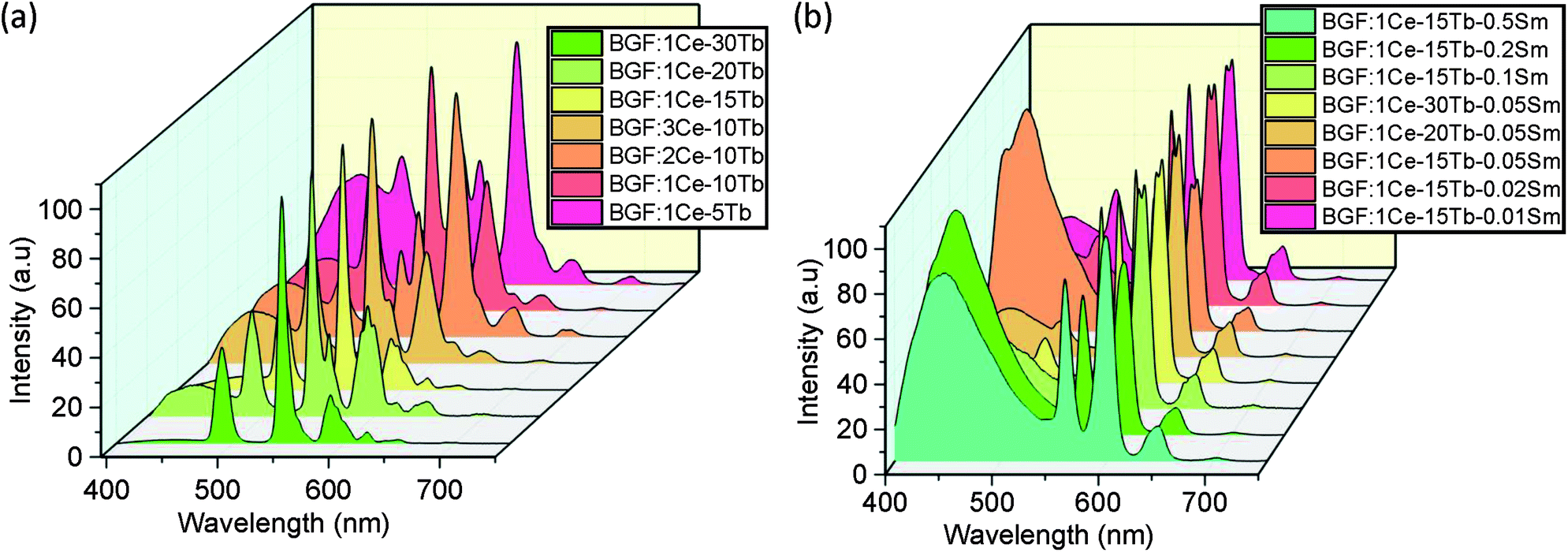 | ||
| Fig. 7 PL spectra of codoped BGF nanowires excited by 350 nm wavelength. | ||
The CIE diagrams and the corresponding coordinates of the nanowires when excited by 350 nm source are presented in Fig. S10† and Table 1, respectively. The emission from the nanowires can be tailored from blue, green and red region through white light by varying the level of doping. A slight change in the Sm3+ doping causes a significant change in the overall emission from red to blue. Therefore, a meticulous control on the dopant composition is required to produce blue, red, and white light emission from the nanowires.
| Sl no. | Sample composition | CIE (x) | CIE (y) |
|---|---|---|---|
| 1 | BGF:1Ce–5Tb | 0.321 | 0.309 |
| 2 | BGF:1Ce–10Tb | 0.313 | 0.382 |
| 3 | BGF:1Ce–15Tb | 0.303 | 0.478 |
| 4 | BGF:1Ce–20Tb | 0.343 | 0.413 |
| 5 | BGF:1Ce–30Tb | 0.318 | 0.538 |
| 6 | BGF:3Ce–10Tb | 0.309 | 0.378 |
| 7 | BGF:1Ce–15Tb–0.01Sm | 0.368 | 0.356 |
| 8 | BGF:1Ce–15Tb–0.02Sm | 0.377 | 0.354 |
| 9 | BGF:1Ce–15Tb–0.05Sm | 0.254 | 0.173 |
| 10 | BGF:1Ce–20Tb–0.05Sm | 0.405 | 0.345 |
| 11 | BGF:1Ce–30Tb–0.05Sm | 0.415 | 0.366 |
| 12 | BGF:1Ce–15Tb–0.1Sm | 0.353 | 0.304 |
| 13 | BGF:1Ce–15Tb–0.25Sm | 0.256 | 0.195 |
| 14 | BGF:1Ce–20Tb–0.05 Sm | 0.282 | 0.244 |
Photoluminescence decay dynamics
The time-resolved luminescent decay analysis of binary and ternary doped BGF nanowires are shown in Fig. 8. The lifetime for the intense emission at 544 nm from BGF:1Ce–yTb nanowires and 602 nm from BGF:1Ce–yTb–zSm is analyzed and presented in Table 2. The decay curves are fitted to three component exponential function as in eqn (3), as the adjusted R2 values are remarkably higher than the two-component fit.| I(t) = α1e(−t/τ1) + α2e(−t/τ2) + α3e(−t/τ3) | (3) |
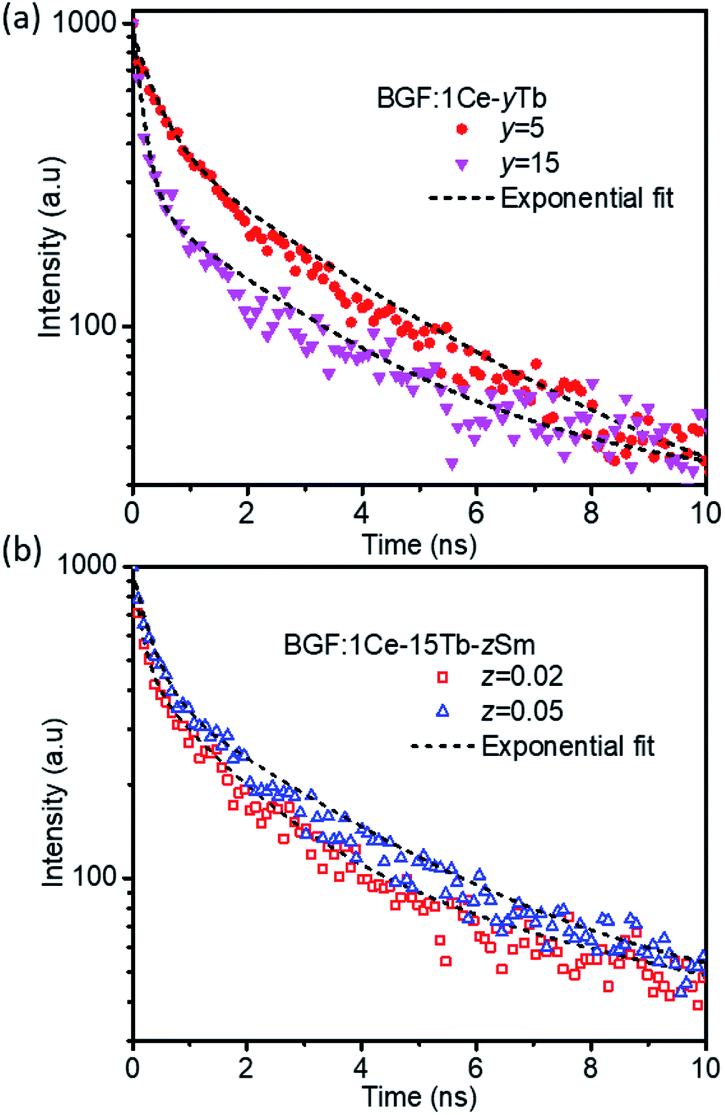 | ||
| Fig. 8 Lifetime curves of representative (a) BGF:1Ce–yTb and (b) BGF:1Ce–15Tb–zSm nanowires. | ||
| Sample | α1 | τ1 (ns) | α2 | τ2 (ns) | α3 | τ3 (ns) | Adj. R2 | Decay time from the plots (ns) |
|---|---|---|---|---|---|---|---|---|
| BGF:1Ce–5Tb | 513.15 | 0.381 | 402.52 | 2.89 | 25.12 | 38.92 | 0.99332 | 0.54 |
| BGF:1Ce–10Tb | 688.84 | 0.169 | 277.54 | 2.77 | 33.4 | 75.16 | 0.98545 | 0.32 |
| BGF:1Ce–15Tb | 708.61 | 0.137 | 251.14 | 2.21 | 32.32 | 74.14 | 0.97892 | 0.31 |
| BGF:1Ce–20Tb | 704.3 | 0.198 | 269.98 | 2.1 | 31.16 | 78.1 | 0.98289 | 0.37 |
| BGF:1Ce–30Tb | 730.53 | 0.157 | 232.87 | 2.476 | 32.84 | 111.36 | 0.97839 | 0.27 |
| BGF:3Ce–10Tb | 923.38 | 0.259 | 143.83 | 3.026 | 27.099 | 133.01 | 0.96734 | 0.32 |
| BGF: 1Ce–15Tb–0.01Sm | 601.81 | 0.204 | 347.33 | 2.65 | 31.57 | 126.05 | 0.98887 | 0.58 |
| BGF: 1Ce–15Tb–0.05Sm | 607.24 | 0.19 | 344.25 | 2.309 | 36.469 | 114.11 | 0.98658 | 0.62 |
| BGF:1Ce–20Tb–0.05Sm | 744.12 | 0.693 | 266.27 | 3.616 | 13.99 | 108.53 | 0.99648 | 1.57 |
| BGF:1Ce–30Tb–0.05Sm | 711.07 | 0.954 | 270.61 | 3.69 | 7.87 | 203.64 | 0.99718 | 2.63 |
| BGF: 1Ce–15Tb–0.1Sm | 655.21 | 0.516 | 343.89 | 3.183 | 25.915 | 131.13 | 0.99287 | 0.72 |
| BGF: 1Ce–15Tb–0.5Sm | 555.2 | 0.292 | 381.55 | 2.87 | 40.16 | 141.86 | 0.99018 | 0.75 |
Among the three components observed in the exponential fit, the first two components (τ1 and τ2) are remarkably faster than the third component. The fast decay time components τ1 and τ2 of the samples indicate two different recombination centers one on the distorted lattice sites close to the surface and the other from the shallow surface traps of the nanowires. While comparing the life time of BGF:xCe–yTb nanowires, as the Tb3+ content increases the decay lifetime is decreased monotonically, which strongly supports the energy transfer from the Ce3+ to Tb3+ ions. The non-radiative energy relaxation is significantly faster over cross-relaxation upon increasing the concentration of Tb3+ ions, as observed at several instances of sensitizer-activator involved luminescence.47,48 The decay time measured for the emissions from Sm3+ ions in BGF:xCe–yTb–zSm nanowires are longer than those measured from Tb3+ ions in BGF:xCe–yTb nanowires because of the longer travel of photons through several energy exchanges. The lifetime of Sm3+ emission is significantly affected on increasing the Tb3+ content, and it is increased from 0.58 ns to 2.67 ns while increasing the Tb3+ content from 15 mol% to 30 mol% for a constant Ce3+ and Sm3+ doping. Moreover, a subsequent increase in decay time is observed with increasing the Sm3+ content while Ce3+ and Tb3+ dopants kept constant.
The decay time observed in RE3+-doped BGF nanowires is several orders shorter than the decay time of Mn4+ doped BGF microrods, which was reported as ∼8.76 ms.13 The decay time in inorganic materials is dependent on the dopant occupancy in the lattice sites and crystal size effect.49 Mn4+ occupy a parity forbidden transition site of Ge4+ ions with inversion symmetry in BGF, whereas, the RE3+ dopants occupy Ba2+ site with center of symmetry. Luminescent ions occupy the lattice sites with inversion symmetry exhibit a longer decay time and the same occupy the center of symmetry sites has a shorter decay time, which is evident in the case of BaSiF6, where Mn4+ occupying Si4+ sites with a slow decay time in the order of micro- to milliseconds, whereas the RE3+ ions occupying Ba2+ has a fast decay time in the order of nanoseconds to subnanoseconds.22,50 The average decay time observed in the present study on binary and ternary doped BGF nanowires (0.3–0.75 ns) is shorter than the decay time reported for codoped BaSiF6 (0.8–1.0 ns) and BaF2 (1.5–2.5 ns) nanowires, in our previous study.22
The difference in the decay time of BaSiF6 and BaGeF6 nanophosphors is possibly due to the slightly different in their lattice distortions associated to the size difference of Si4+ (54 Å) and Ge4+ (67 Å) ions. One can appreciate the influence of the above difference in ionic radii on crystal lattice on comparing the lattice parameters of BaSiF6 (a = 4.851 Å, α = 98.206°) and BaGeF6 (a = 4.948 Å, α = 98.245°) unit cells, with a dissimilarity of 0.1668 on a scale of 0.75.51 Considering BGF:1Ce–yTb nanowires, the transitions 5D4 → 7F6 (488 nm) and 5D4 → 7F5 (at 544 nm) are electric-dipole allowed and magnetic-dipole allowed transitions, respectively.52 The ratio of electric dipole to magnetic dipole transition is a measure of local symmetry surrounding the Tb3+ ions as the electric dipole transitions are sensitive to the local symmetry of the dopant site,53 which is slightly higher in BaSiF6 nanowires. This higher ratio reveals higher symmetry of the dopant ions in the BaSiF6 lattice than those in the BaGeF6 lattice, and therefore a fast decay is expected, which is presumably similar to the reduction in decay time under high pressure due to the change in the local symmetry of the luminescent dopant ions.54
Cathodoluminescence characteristics
The CL spectra of the pure and codoped BGF nanowires are recorded under 10 kV and 100 nA electron beam, aimed to simulate the response of the nanowires towards high-energy radiation. The characteristic CL emission spectra are shown in Fig. 9a and b. The pure BGF nanowires emits a broad visible spectrum in the wide range of 300–750 nm when excited by an electron beam. Even though, there was no significant emission in PL studies, pure BGF samples exhibit CL emission. The CL emission in pure lattice is originated by the propagation of ionization initiated by the fast-primary electrons. Upon excitation, the primary electrons create secondary electrons and these secondary electrons leads to more ionization and continues to produce more secondary electrons. Ultimately the cloud of secondary electrons forms and excite the host lattice to generate visible emissions55 and hence, the luminescent mechanism and therefore the emission characteristics are different between PL and CL.46 In PL, the energy of photons used are only around 4–6 eV. However, for CL, the energy of fast electrons is 10 kV. Moreover, the CL emission recorded here are from a small area as compared with PL spectra and therefore the excitation energy on a single particle is much higher in CL. The ultraviolet and/or visible light usually excite the activator directly; the fast electrons as high-energy particles always excite the host lattice.55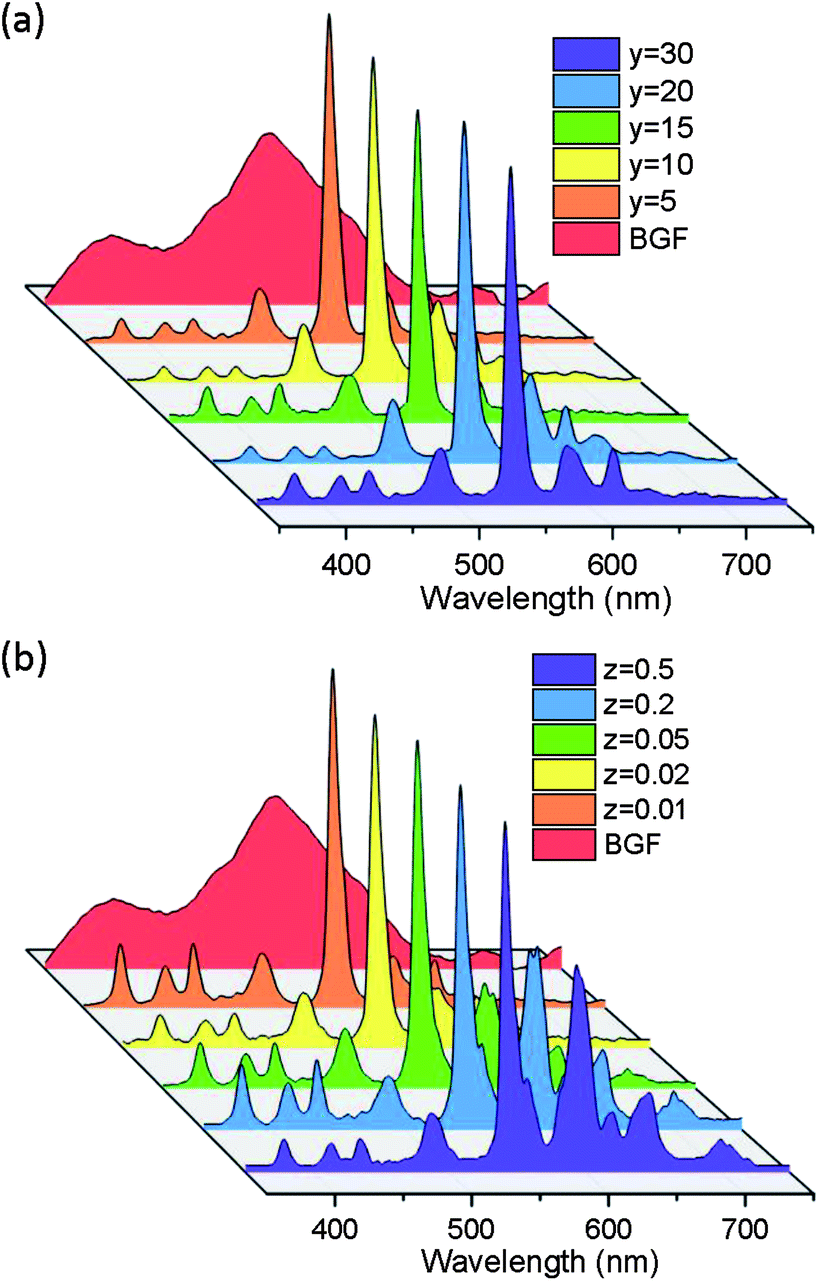 | ||
| Fig. 9 Cathodoluminescent emission of BGF:1Ce–yTb and (b) BGF:1Ce–15Tb–zSm nanowires. | ||
In the CL spectra of BGF:1Ce–yTb and BGF:1Ce–15Tb–zSm nanowires, the emission peaks originated from the host lattices are completely disappeared. The CL spectra of BGF:1Ce–yTb nanowires comprises the most intense emission lines at 544 nm, which is due to the Tb3+ ions as in the PL spectra. Since the emissions from the Ce3+ ions are completely disappeared in the CL spectra, the energy transfer from the host lattice to Ce3+ and then to Tb3+ ions are clearly recognized. All the possible transitions of Tb3+ are clearly visible in the CL spectra, which does not appear in the PL spectra. The several small peaks in the blue region <450 nm is due to 5D3 → 7Fj (j = 6, 5, 4, 3) transitions and the peaks in green region >450 are due to 5D4 → 7Fj (j = 6, 5, 4, 3) transitions of Tb3+ ions.55 The emission bands from Sm3+ ions are appeared in BGF:1Ce–15Tb–zSm nanowires, as shown in Fig. 9b. The strong emission lines characteristic to Tb3+ ions in all the CL spectra are due to the high doping level. Moreover, the emission intensities from Sm3+ ions in ternary doped BGF nanowires are remarkable though the level of doping of those ions is very minuscule and it is enhanced with the increase of Sm3+ content. The emission from Tb3+ and Sm3+ ions from these ternary doped nanowires are independent since the emission from Tb3+ ions are not compromised as observed in the PL spectra. Therefore, in the CL spectra of ternary doped nanowires, the simultaneous emission from both Tb3+ and Sm3+ ions are possibly due to the contemporary energy transfer to these ions from Ce3+ ions. The overall CL emission mechanism from the doped nanowires can be depicted as follows. The secondary electrons excite the host lattice and create many electron–hole pairs, leading to the formation of bound excitons. As no luminescence from bound exciton recombination is observed, these excitons, if formed, decay non-radiatively through a resonant or quasi-resonant transfer to the 4f shell of Sm3+ and/or Tb3+ ions and give their characteristic emission.56
Additionally, the CIE coordinates from the CL spectra of the nanowires are calculated and presented in Table 3. From the CIE diagram shown in Fig. S11,† majority of the nanowires exhibit an overall emission in green-orange region. It is obvious that the Ce3+–Tb3+ codoped nanowires expected to emit a green light and the Ce3+–Tb3+–Sm3+ codoped nanowires to emit orange light and the same can be observed in the chromaticity diagram.
| Sl no. | Sample composition | CIE (x) | CIE (y) |
|---|---|---|---|
| 1 | BGF:1Ce–10Tb | 0.3281 | 0.5205 |
| 2 | BGF:2Ce–10Tb | 0.3745 | 0.5164 |
| 3 | BGF:1Ce–15Tb | 0.3193 | 0.4910 |
| 4 | BGF:1Ce–20Tb | 0.3745 | 0.5162 |
| 5 | BGF:1Ce–30Tb | 0.3334 | 0.4970 |
| 6 | BGF: 1Ce–15Tb–0.01Sm | 0.3167 | 0.4724 |
| 7 | BGF: 1Ce–15Tb–0.02Sm | 0.3425 | 0.4947 |
| 8 | BGF: 1Ce–15Tb–0.05Sm | 0.3764 | 0.4806 |
| 9 | BGF:1Ce–20Tb–0.05Sm | 0.4244 | 0.4347 |
| 10 | BGF:1Ce–30Tb–0.05Sm | 0.3913 | 0.4778 |
| 11 | BGF:1Ce–15Tb–0.1Sm | 0.4058 | 0.4480 |
| 12 | BGF:1Ce–15Tb–0.5Sm | 0.4044 | 0.3711 |
Photoluminescence sensitivity towards H2O2 detection
The synthesized ternary RE3+ doped BGF nanowires are used as luminescent probe for the detection of H2O2, which is a model for the reactive oxygen species.57 The sensing performance of representative BGF:1Ce–15Tb–0.05Sm nanophosphors dispersed in water is tested by recording the fluorescent spectra in the presence of different concentrations of H2O2 (con. H2O2), as shown in Fig. 10a. The PL spectra for different con. H2O2 ranging from 25 μM to 3000 μM, clearly show a change in fluorescence nature of BGF:1Ce–15Tb–0.05Sm nanowires with respect to con. H2O2 even at the low concentrations. The calibration curve in the inset of Fig. 10a indicates that the relative fluorescence intensity (I/I0) has a logarithmic relationship I/I0 = 1.484 − 0.184 × ln(con. H2O2), where I and I0 are the fluorescence intensity in the presence and absence of H2O2, respectively. The corresponding calculated and residual values of the fit is presented in Table S2.† As shown in Fig. 10b, the quenching efficiency, defined as QE = (I0 − I)/I0 × 100%, obviously depicts that the increase in the H2O2 concentration x enhances the QE with the following relationship (the calculated and residual values are presented in Table S3†):| QE = −48.36 + 18.39 × ln(con. H2O2) | (4) |
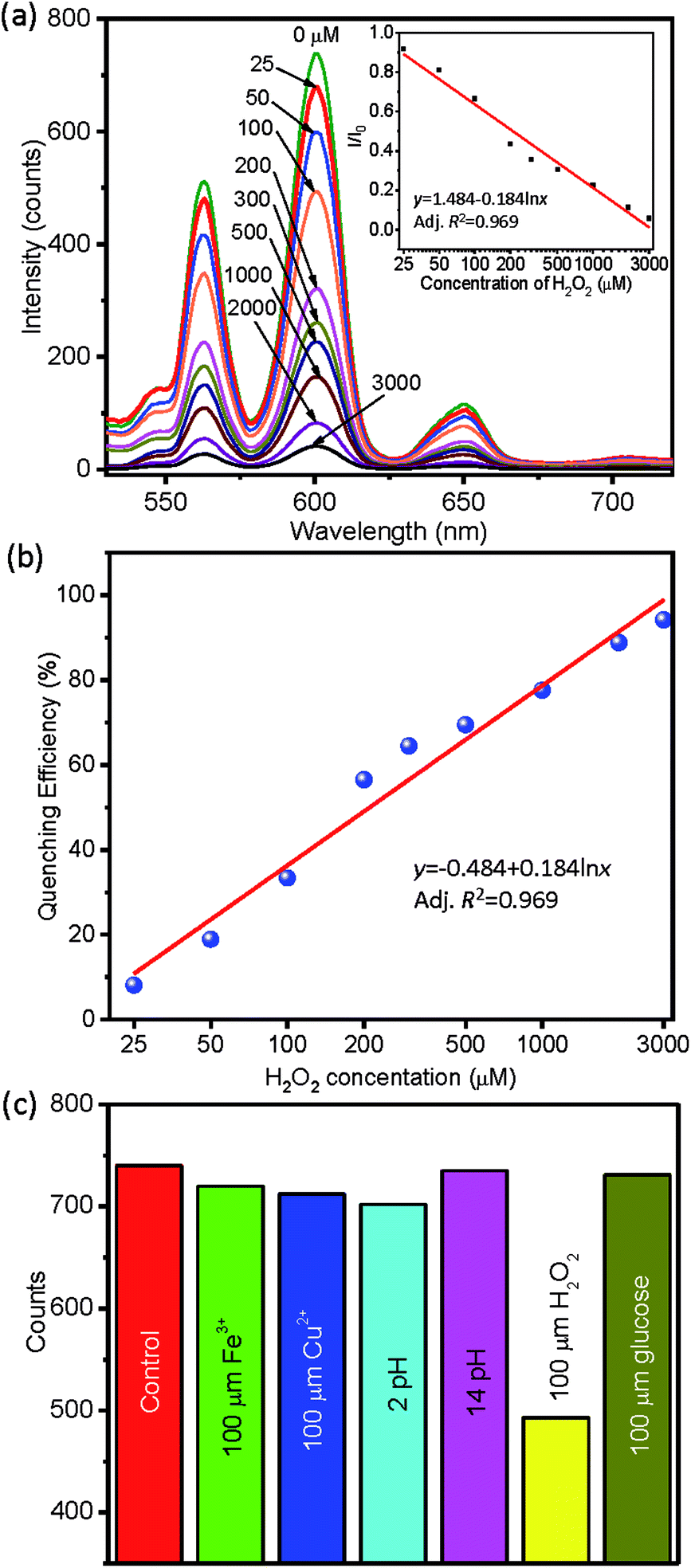 | ||
| Fig. 10 (a) PL response of nanowires towards H2O2 concentration, calibration curve is in the inset, (b) quenching efficiency, and (c) selectivity of BGF nanowires towards H2O2. | ||
The intensity of all transitions of Sm3+ are decreased simultaneously in the presence of H2O2, when excited by 254 nm source. The sensor is not only highly sensitive, it is also quantitative for x between 25–3000 ppm. Furthermore, the detection selectivity was tested with glucose, Fe3+, Cu2+, and pH, as shown in Fig. 10c. Their addition to BGF:1Ce–15Tb–0.05Sm dispersion leads to negligible modifications to the luminescence intensity (<5%), whereas the addition of H2O2 leads to a >40% decrease. This reveals the good selectivity of the sensor. The reduction in the intensity of the Sm3+ emission from the BGF:1Ce–15Tb–0.05Sm nanowires only in the presence of H2O2 can be attributed to the oxidation of Ce3+ to Ce4+ by eqn (5). Besides, Ln3+ ions in aqueous solutions exhibit a strong affinity towards negatively charged oxygen.58
| 2Ce3+ + H2O2 → 2Ce4+ + 2OH− | (5) |
With an increase of Ce4+ ions, the actual charge transfer bridge between Ce3+, Tb3+ and Sm3+ is disrupted and ultimately the intensity of emissions from Sm3+ ions are decreased.
The sensitivity of binary doped green emitting BGF:1Ce–30Tb and representative binary (Ce3+ and Tb3+) and ternary (Ce3+, Tb3+ and Eu3+) doped BaSiF6 nanowires towards H2O2 are further studied and presented in Fig. S12.† In both the cases, the ternary doped nanowires are more sensitive than their binary doped counterparts, which reveals that the low concentration of sensitizer ions (Sm3+ or Eu3+) are favorable for effective sensing of H2O2 using these nanowires. On comparing the sensitivity towards H2O2 between BGF and BaSiF6 nanowires, BGF nanowires are more sensitive towards H2O2. Moreover, the binary doped BGF nanowires display more sensitivity than the ternary doped BaSiF6 nanowires (Fig. S12d†). Hence, codoped BGF nanowires are promising for the detection of H2O2 at the low concentrations. In addition, the sensitivity and detection limit may be further optimized by controlling the level of dopants in the nanowires and the content of nanowires in the water dispersion.59
Conclusions
A pure phase BGF nanowires with high aspect ratios was fabricated through a solvothermal method. The nanowires exhibited visible PL emissions when codoped with varying Ce3+, Tb3+, and Sm3+ rare-earth ions, under 254 nm excitation. The XRD analysis revealed the crystalline nature of the nanowires with rhombohedral structure and the structure type is not affected by the rare-earth dopants. The PL emission monitored when excited by 254 nm source is related to the energy transfer between Ce3+–Tb3+–Sm3+ ions. The visible emission from the codoped BGF nanowires when excited by 350 nm source was also recorded. The decay time of codoped BGF nanowires was found to be very short in the order of subnanoseconds, which is one of the shortest decay time records observed so far. The strong visible CL emission from the BGF nanowires reveals their potential use for the field emission displays and the detection of high-energy radiation. The visible PL emissions from the ternary doped BGF nanowires are sensitive to H2O2 at low concentrations and thus they can be used for H2O2 detection with high sensitivity.Authors' contributions
G. George conducted the sample synthesis, and PL and EPMA/CL measurements, and prepared the manuscript; M. D. Simpson conducted the sample processing and preparation; B. R. Gautam conducted the decay dynamics; D. Fang attended the experimental design and data analysis; J. Peng, and J. Wen conducted the TEM work; J. E. Davis and D. Ila conducted the research on scintillation; and Z. Luo designed the experiment, analysed the experimental data and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the U.S. National Science Foundation (NSF) for research funding, award no. HRD 1436120, and ORAU-Directed Research and Development Grant. The instrumentation was supported by the U.S. Department of Defense grants W911NF-14-1-0060, W911NF-15-1-0566 and W911NF-09-1-0011, and NSF MRI Program DMR 1626376. Use of the Center for Nanoscale Materials, an Office of Science user facility, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- Y. S. Zhao, P. Zhan, J. Kim, C. Sun and J. Huang, ACS Nano, 2010, 4, 1630–1636 CrossRef CAS PubMed.
- X. Wang, C. J. Summers and Z. L. Wang, Nano Lett., 2004, 4, 423–426 CrossRef CAS PubMed.
- J. H. He, R. S. Yang, Y. L. Chueh, L. J. Chou, L. J. Chen and Z. L. Wang, Adv. Mater., 2006, 18, 650–654 CrossRef CAS.
- Z. Luo, J. G. Moch, S. S. Johnson and C. C. Chen, Curr. Nanosci., 2017, 13, 364–372 CAS.
- Y. Huang, H. You, G. Jia, Y. Song, Y. Zheng, M. Yang, K. Liu and N. Guo, J. Phys. Chem. C, 2010, 114, 18051–18058 CrossRef CAS.
- Y.-P. Du, X. Sun, Y.-W. Zhang, Z.-G. Yan, L.-D. Sun and C.-H. Yan, Cryst. Growth Des., 2009, 9, 2013–2019 CrossRef CAS.
- E. Nieboer, C. K. Jørgensen, R. D. Peacock and R. Reisfeld, Rare Earths, Springer-Verlag, Berlin Heidelberg, 1975 Search PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199 CrossRef CAS PubMed.
- S. Wang, J. Feng, S. Song and H. Zhang, CrystEngComm, 2013, 15, 7142–7151 RSC.
- C. Altavilla, Upconverting Nanomaterials: Perspectives, Synthesis, and Applications, CRC Press, Boca Raton, 2016 Search PubMed.
- N. Kumam, L. P. Singh, S. K. Srivastava and N. R. Singh, J. Lumin., 2018, 203, 59–66 CrossRef CAS.
- Q. Zhou, Y. Zhou, F. Lu, Y. Liu, Q. Wang, L. Luo and Z. Wang, Mater. Chem. Phys., 2016, 170, 32–37 CrossRef CAS.
- D. Sekiguchi and S. Adachi, Opt. Mater., 2015, 42, 417–422 CrossRef CAS.
- Y. Zhou, Q. Zhou, Y. Liu, Z. Wang, H. Yang and Q. Wang, Mater. Res. Bull., 2016, 73, 14–20 CrossRef CAS.
- R. Hoshino, T. Nakamura and S. Adachi, ECS J. Solid State Sci. Technol., 2016, 5, R37–R43 CrossRef CAS.
- Q. Zhou, H. Tan, Q. Zhang, N. Wang, Q. Wei, Z. Wang, M. Rong and Q. Wang, RSC Adv., 2017, 7, 32094–32099 RSC.
- H. F. Sijbom, R. Verstraete, J. J. Joos, D. Poelman and P. F. Smet, Opt. Mater. Express, 2017, 7, 3332–3365 CrossRef CAS.
- S. Adachi and T. Takahashi, J. Appl. Phys., 2009, 106, 013516 CrossRef.
- T.-C. Lang, T. Han, L.-L. Peng and M.-J. Tu, Mater. Chem. Front., 2017, 1, 928–932 RSC.
- Z. Wang, Y. Liu, Y. Zhou, Q. Zhou, H. Tan, Q. Zhang and J. Peng, RSC Adv., 2015, 5, 58136–58140 RSC.
- Y. K. Xu and S. Adachi, J. Electrochem. Soc., 2011, 158, J58–J65 CrossRef CAS.
- G. George, S. L. Jackson, Z. R. Mobley, B. R. Gautam, D. Fang, J. Peng, D. Luo, J. Wen, J. E. Davis, D. Ila and Z. Luo, J. Mater. Chem. C, 2018, 6, 7285–7294 RSC.
- Q. Zhou, Y. Zhou, Y. Liu, L. Luo, Z. Wang, J. Peng, J. Yan and M. Wu, J. Mater. Chem. C, 2015, 3, 3055–3059 RSC.
- F. Hong, H. Cheng, G. Liu, X. Dong, W. Yu and J. Wang, Inorg. Chem., 2018, 57, 9892–9901 CrossRef CAS PubMed.
- R. A. B. Silva, R. H. O. Montes, E. M. Richter and R. A. A. Munoz, Food Chem., 2012, 133, 200–204 CrossRef CAS.
- I. Kim, N. Yamashita and H. Tanaka, J. Hazard. Mater., 2009, 166, 1134–1140 CrossRef CAS PubMed.
- M. Schäferling, D. B. M. Grögel and S. Schreml, Microchim. Acta, 2011, 174, 1 CrossRef.
- J. Bogacki, P. Marcinowski, E. Zapałowska, J. Maksymiec and J. Naumczyk, Environ. Technol., 2017, 38, 2589–2600 CrossRef CAS PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- Y. Wang, J. Huang, C. Zhang, J. Wei and X. Zhou, Electroanalysis, 1998, 10, 776–778 CrossRef CAS.
- N. Luo, G. H. Miley, R. J. Gimlin, R. L. Burton, J. Rusek and F. Holcomb, J. Propul. Power, 2008, 24, 583–589 CrossRef CAS.
- M. E. Germain and M. J. Knapp, Inorg. Chem., 2008, 47, 9748–9750 CrossRef CAS PubMed.
- G. Brauer, Handbook of Preparative Inorganic Chemistry, Academic Press, London, 1963, vol. 1 Search PubMed.
- J. L. Hoard and W. B. Vincent, J. Am. Chem. Soc., 1940, 62, 3126–3129 CrossRef CAS.
- D. Gantar, A. Rahten and B. Volavšek, Thermochim. Acta, 1985, 96, 207–212 CrossRef CAS.
- T. E. Mallouk, B. Desbat and N. Bartlett, Inorg. Chem., 1984, 23, 3160–3166 CrossRef CAS.
- C. Lorbeer, F. Behrends, J. Cybinska, H. Eckert and A.-V. Mudring, J. Mater. Chem. C, 2014, 2, 9439–9450 RSC.
- E. van der Kolk, P. Dorenbos, C. W. E. van Eijk, A. P. Vink, C. Fouassier and F. Guillen, J. Lumin., 2002, 97, 212–223 CrossRef CAS.
- B. F. Aull and H. P. Jenssen, Phys. Rev. B, 1986, 34, 6640–6646 CrossRef CAS.
- C. Duan, Z. Zhang, S. Rösler, S. Rösler, A. Delsing, J. Zhao and H. T. Hintzen, Chem. Mater., 2011, 23, 1851–1861 CrossRef CAS.
- H. Yin, Y. Li, J. Bai, M. Ma and J. Liu, J Materiomics., 2017, 3, 144–149 CrossRef.
- M. G. Ha, J.-S. Jeong, K.-R. Han, Y. Kim, H.-S. Yang, E. D. Jeong and K. S. Hong, Ceram. Int., 2012, 38, 5521–5526 CrossRef CAS.
- Z. Xia and W. Wu, Dalton Trans., 2013, 42, 12989–12997 RSC.
- P. I. Paulose, G. Jose, V. Thomas, N. V. Unnikrishnan and M. K. R. Warrier, J. Phys. Chem. Solids, 2003, 64, 841–846 CrossRef CAS.
- G. Seeta Rama Raju and J. S. Yu, Spectrochim. Acta, Part A, 2014, 124, 383–388 CrossRef CAS PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin Heidelberg, 2012 Search PubMed.
- X. Zhang and M. Gong, Ind. Eng. Chem. Res., 2015, 54, 7632–7639 CrossRef CAS.
- W. Lü, H. Xu, J. Huo, B. Shao, Y. Feng, S. Zhao and H. You, Dalton Trans., 2017, 46, 9272–9279 RSC.
- L. Wang, R.-J. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951–2009 CrossRef CAS PubMed.
- D. Sekiguchi, J. Nara and S. Adachi, J. Appl. Phys., 2013, 113, 183516 CrossRef.
- K. Persson, Materials Data on BaSiF6 (SG:166) by Materials Project, 2014, DOI:10.17188/1270502.
- T. R. Faulkner and F. S. Richardson, Mol. Phys., 1978, 36, 193–914 CrossRef CAS.
- J. J. H. A. van Hest, G. A. Blab, H. C. Gerritsen, C. de Mello Donega and A. Meijerink, J. Phys. Chem. C, 2017, 121, 19373–19382 CrossRef CAS PubMed.
- A. Lazarowska, S. Mahlik, M. Grinberg, G. Li and R.-S. Liu, Sci. Rep., 2016, 6, 34010 CrossRef CAS PubMed.
- X. Liu and J. Lin, J. Mater. Chem., 2007, 18, 221–228 RSC.
- P. T. Diallo, P. Boutinaud, R. Mahiou and J. C. Cousseins, Phys. Status Solidi A, 1997, 160, 255–263 CrossRef CAS.
- E. J. Pereira, C. M. Smolko and K. A. Janes, Front. Pharmacol., 2016, 7, 457 Search PubMed.
- F. S. Richardson, Chem. Rev., 1982, 82, 541–552 CrossRef CAS.
- N. Duée, C. Ambard, F. Pereira, D. Portehault, B. Viana, K. Vallé, D. Autissier and C. Sanchez, Chem. Mater., 2015, 27, 5198–5205 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Low-magnification image of BaGeF6 nanowires, EDS results of BaGeF6 nanowires, mechanism of energy transfer, PL spectra of binary doped BaGeF6 nanowires and photographs of PL emission at different doping. See DOI: 10.1039/c8ra07806h |
| ‡ Present address: Southwest Jiaotong University, Chengdu 610031, P. R. China. |
| This journal is © The Royal Society of Chemistry 2018 |