
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-electron redox chemistry of p-nitro- and p-cyanobenzene diazohydroxides
James P. McEvoy *a,
Nhan V. Phamb,
Hong T. Leb,
Micah M. Fernandezb,
Ryan P. Farmerb and
Surendra N. Mahapatrob
*a,
Nhan V. Phamb,
Hong T. Leb,
Micah M. Fernandezb,
Ryan P. Farmerb and
Surendra N. Mahapatrob
aCentre for Biomedical Sciences, School of Biological Sciences, Royal Holloway University of London, Egham, Surrey, TW20 0EX, UK. E-mail: james.mcevoy@rhul.ac.uk
bDepartment of Chemistry, Regis University, Denver, Colorado 80221, USA
First published on 13th December 2018
Abstract
The electrochemistry of aryl diazonium salts is usually dominated by one-electron reduction, loss of dinitrogen, and the grafting of aryl radicals onto the electrode. In contrast, p-nitro- and p-cyanobenzene diazonium salts dissolved in mixed aqueous-acetonitrile solvents showed diffusion-limited, quasi-reversible, two-electron cyclic voltammetry. Voltammetric pH-dependence and spectrophotometric kinetic studies suggest that the basicity and low polarity of the aqueous solvent mixture has revealed the biologically-significant interconversion of diazohydroxide and diazene.
Introduction
Diazonium ion chemistry has intrigued generations of synthetic, physical and industrial chemists since the discovery of the Sandmeyer reaction in 1884.1,2 In the biological sphere, the redox chemistry of the diazonium ion in the aqueous phase is the basis of its nitrosating action upon DNA3 and is closely connected with nitrosamine's carcinogenicity.4 Electron transfer is also important in diazonium's reaction with hemoglobin.5 Contemporary investigations into the redox chemistry of arene diazonium species have been led by Pinson and co-workers, whose studies have demonstrated the covalent modification of electrode surfaces by the capture of aryl radicals arising from the one-electron reduction of the diazonium ion and loss of dinitrogen.6 Surfaces that have been covalently modified by “grafting” in this manner include glassy carbon (GC) and platinum.6,7In contrast to this behaviour, we report here the diffusional, quasi-reversible, two-electron redox chemistry of p-nitrobenzene diazonium (p-NBD) and p-cyanobenzene diazonium (p-CBD) salts in mixed aqueous-acetonitrile (ACN) solutions. These results, along with spectrophotometric measurements, suggest the previously unobserved and biologically relevant interconversion of diazohydroxide and diazene species.
Results and discussion
In either aqueous phosphate buffer or in pure ACN, the cyclic voltammetry (CV) of all the diazonium derivatives tested was dominated by one large, broad and irreversible reductive wave (Fig. 1, dashed lines), reminiscent of the polarographic results previously obtained in sulfolane.8 These signals disappeared after the first cycle because of fouling caused by the electro-grafting of aryl radicals onto the electrode, which passivated the GC surface.6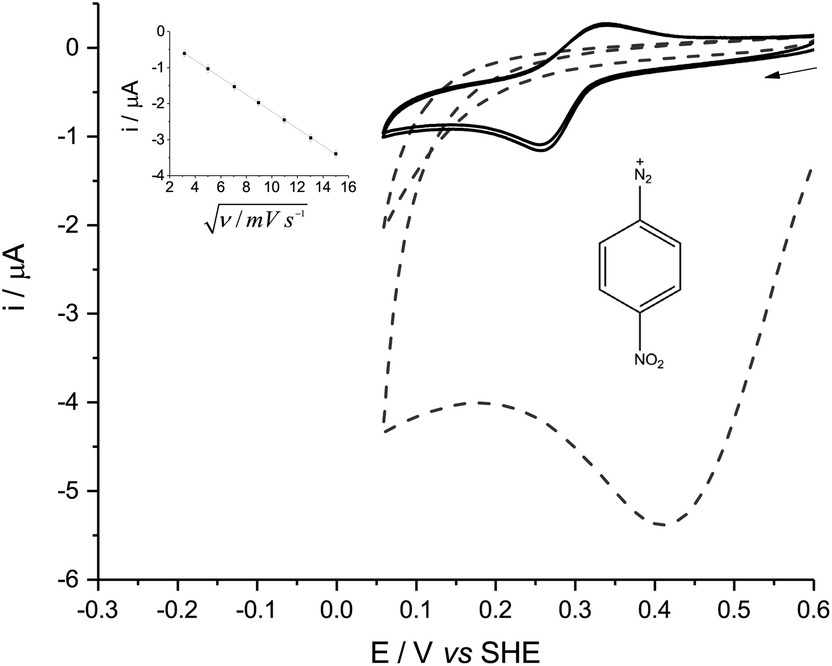 | ||
| Fig. 1 Solid lines: CV of 1 mM p-NBD in 50% (v/v) ACN-aqueous phosphate buffer (0.05 M, pHobs 8.0) at a GC working electrode. Dashed lines: CV of 1 mM p-NBD in pure ACN. The scan rate was 50 mV s−1 in each case, and the first two scans are shown. An arrow indicates the scan direction. Inset: the reductive peak current of p-NBD in 50% (v/v) ACN-phosphate buffer solution plotted against the square root of the scan rate ν, with a straight line of best fit. | ||
p-NBD and p-CBD gave very different, quasi-reversible voltammograms in mildly alkaline aqueous buffers which contained between 30% and 70% ACN by volume (Fig. 1, solid lines). The voltammetric peaks were best defined in a 50% (v/v) ACN-aqueous mixture, giving midpoint potentials around 0.28 V (p-NBD) and 0.19 V (p-CBD) when the aqueous component was buffered at pHobs = 8.0 (Fig. 2). Fig. 2 shows that the quasi-reversible p-CBD signal was superimposed on an irreversible reductive wave centered around 0.5 V, whereas p-NBD only showed the quasi-reversible response.
 | ||
| Fig. 2 CV of 1 mM p-NBD (solid lines) and p-CBD (dashed lines) in 50% (v/v) ACN-aqueous phosphate buffer (0.05 M, pHobs 8.0) at a GC working electrode. The scan rate was 50 mV s−1 in each case and the first two scans are shown. An arrow indicates the scan direction. Inset: variation of p-NBD oxidative peak potential with pHobs in 50% (v/v) ACN phosphate buffer solution at a scan rate of 50 mV s−1, with a straight line of best fit. Data markers show the average of three readings after electrode polishing. | ||
The quasi-reversible signal persisted over many scans with little evidence of fouling. This was particularly the case with p-NBD, whose reductive peak current fell by less than 3% between the first and second scans (Fig. 2, solid lines). None of the other diazonium derivatives showed any reversible electrochemistry in the potential window from −0.5 V to +1.0 V. The CV peak current of p-NBD was directly proportional to the square root of the scan rate from 10 to 225 mV s−1 (Fig. 1, inset), showing that the signal was due to a species diffusing to the electrode surface, not adsorbed to the surface.9
CV of p-NBD was carried out across the pHobs range over which this redox couple was visible. The oxidative peak potential showed a change of −27.6 mV per pHobs unit (Fig. 2, inset), which is consistent with a 2e−/H+ coupled process.9 The peak separation at 50 mV s−1 was ca. 40 mV at 8.2 < pHobs < 9.0. Ferrocenecarboxylic acid (FCA), a one-electron standard, gave a CV peak separation of over 70 mV under the same conditions, although precedented reactivity between p-NBD and FCA prevented their simultaneous study.5 Taken together, these data are consistent with the diazonium compound yielding a quasi-reversible, two-electron signal.9
In order to identify the electroactive species responsible for the quasi-reversible CVs, we studied the reaction kinetics of the p-NBD and p-CBD salts in 50% (v/v) ACN-aqueous solution (Fig. 3 and 4).
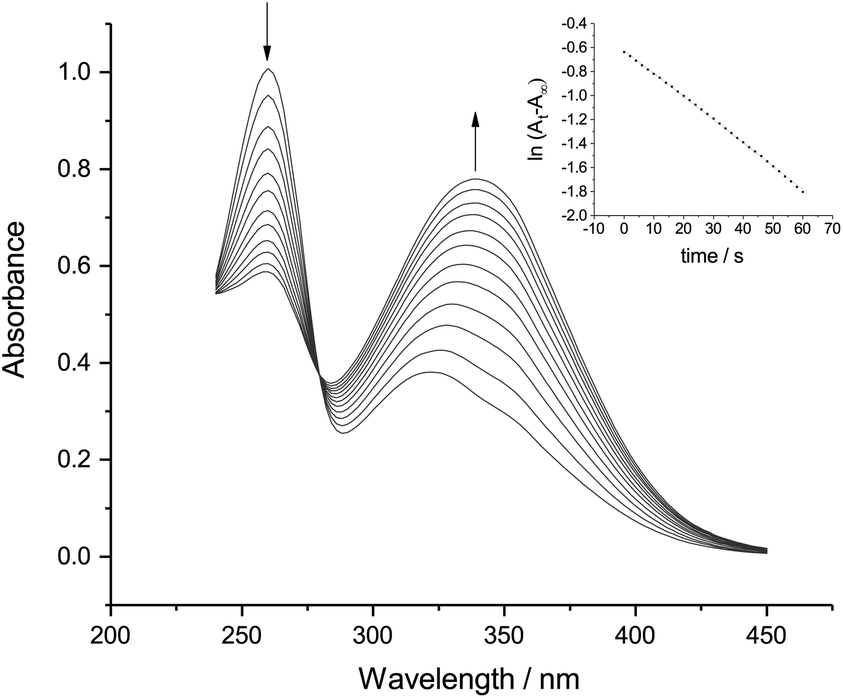 | ||
| Fig. 3 UV-Vis spectra of p-NBD tetrafluoroborate (70 μM) in 50% (v/v) ACN-aqueous phosphate buffer (0.05 M, pHobs 7.8). Scans were repeated every five seconds at 22.5 ± 0.5 °C. Inset: first-order rate plot of the disappearance of the p-NBD peak at 260 nm. | ||
 | ||
| Fig. 4 UV-Vis spectra of p-CBD tetrafluoroborate (70 μM) in 50% (v/v) ACN-aqueous phosphate buffer (0.05 M, pHobs 7.8). Scans were repeated every two minutes at 22.5 ± 0.5 °C. Inset: first-order rate plot of the disappearance of the p-CBD peak at 268 nm. | ||
Fig. 3 and 4 both show a shrinking diazonium ion peak, at 260 nm and 268 nm respectively, with a λmax identical to that seen in purely aqueous solution.10 Accompanying this was a growing peak at higher wavelengths, with isosbestic points at 280 nm for p-NBD and 276 nm for p-CBD. The major product of p-NBD reaction showed a λmax of 350 nm after a few minutes.
These results resemble those obtained in purely aqueous solution11,12 and correspond to the combination of the diazonium and hydroxide ions.
The first-order rate constant measured for p-NBD in 50% ACN buffered at pHobs 7.8 (Fig. 3) was kobs = 1.94 × 10−2 s−1, which matches the rate of reaction in water at pH 8.7.12 The pH-indicator acidity function of 50% ACN–water solutions has been found to exceed pHobs by about 0.9 pH units,13 and it is this increased effective pH, rather than the barely increased nucleophilicity of the hydroxide ion in 50% ACN14 or the small salt effect,15 which explains the accelerated rate of p-NBD reaction in 50% ACN relative to water. The rate constant measured for p-CBD in 50% ACN at pHobs 7.8 (Fig. 4) was kobs = 2.14 × 10−3 s−1, ten times smaller than the p-NBD value. This was a bigger difference than is seen in water,16 presumably because of differential solvation effects.
Reaction of p-NBD with hydroxide in purely aqueous alkaline solution yields first the syn and then the anti-diazotate,11,12 whose extinction coefficient at λmax = 330 nm has been reported as 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 430 M −1 cm−1.15 Reaction of p-NBD in 50% ACN–NaOH (aq., 0.05 M) gave the same bright yellow product, with an extinction coefficient of 1.3 × 104 M−1 cm−1 at its λmax of 346 nm (data not shown). Given the lower hydrogen-bonding acidity of 50% ACN relative to pure water,17 this modestly negative solvatochromism suggests that water preferentially stabilizes the diazotate ion's polar ground state by donating hydrogen bonds.18 Importantly, these products of strongly alkaline and purely aqueous reaction did not give a reversible voltammetric signal.
430 M −1 cm−1.15 Reaction of p-NBD in 50% ACN–NaOH (aq., 0.05 M) gave the same bright yellow product, with an extinction coefficient of 1.3 × 104 M−1 cm−1 at its λmax of 346 nm (data not shown). Given the lower hydrogen-bonding acidity of 50% ACN relative to pure water,17 this modestly negative solvatochromism suggests that water preferentially stabilizes the diazotate ion's polar ground state by donating hydrogen bonds.18 Importantly, these products of strongly alkaline and purely aqueous reaction did not give a reversible voltammetric signal.
The pale yellow, reversibly electroactive product of p-NBD reaction with hydroxide in 50% ACN at pHobs 7.8 (Fig. 3) showed an apparent extinction coefficient of 11850 M−1 cm−1 at its final λmax of 350 nm. This extinction coefficient is lower than that of the anti-diazotate and corresponds more closely to that of the diazohydroxide11 whose formation has been recently inferred by electrochemical means.19 Aryl diazonium compounds with highly electron-withdrawing substituents on the benzene ring, such as the nitro and cyano groups, demonstrate a faster rate of aqueous hydroxide combination to produce a more acidic diazohydroxide.16,20 This product is indeed almost absent under the conditions of purely aqueous reaction because of rapid proton loss21 but is expected to be stabilized with respect to the anionic diazotate in the less polar 50% ACN solvent.22
The reactions we propose to explain our observed results are shown in Scheme 1, along with other known reactions. The left-hand reaction in Scheme 1 shows the rapid conversion of the diazonium ion A to the syn-diazohydroxide B, before the beginning of the cyclic voltammetric experiment. The reactions in the lower part of the scheme show the quasi-reversible electrochemical cycle, comprising the two-electron reduction of B to the diazene G, and the two-electron oxidation of G back to B. The base-catalyzed elimination of hydroxide from methyldiazohydroxide to form diazomethane23 suggests that the leaving group in the reductive half-reaction B → G may be OH− itself.
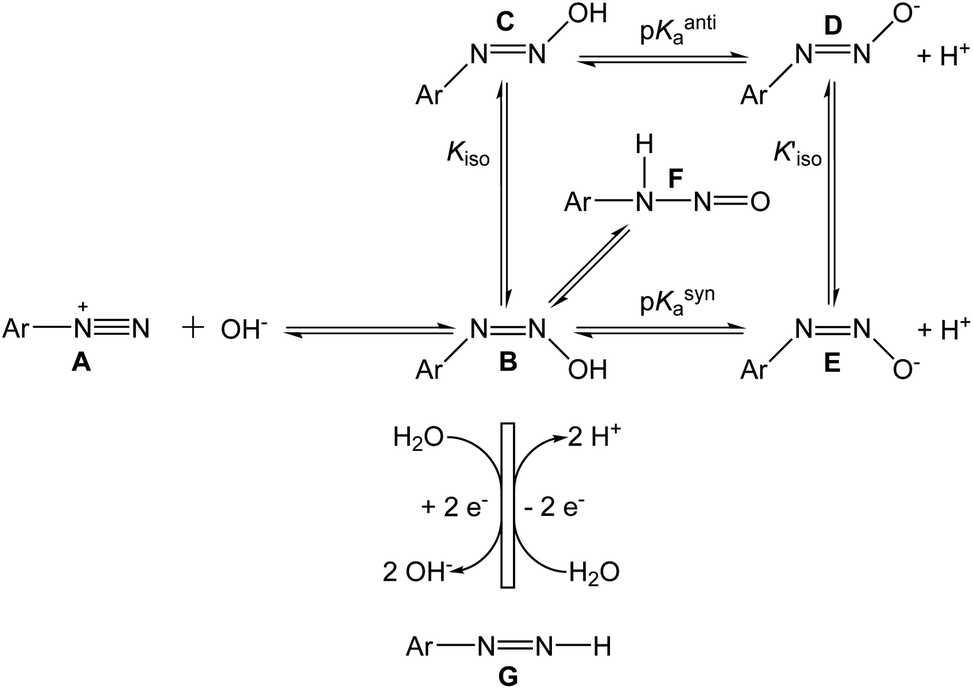 | ||
| Scheme 1 Proposed reactions for p-CBD and p-NBD (A). | ||
Phenyldiazenes are typically unstable in oxygen24 but the p-nitrophenyl derivative persists25 and the two-electron reduction of p-NBD by direct hydride transfer has been reported,26 making it a plausible partner in the electrochemical reactions. The addition of small quantities of sodium borohydride attenuated the electrochemical signals (data not shown), a result compatible with this interpretation.
No reversible voltammetry of p-NBD hydrolysed in 50% ACN (Fig. 1 and 2) was observed above pHobs 9.0, at which value the pH-indicator acidity function is around 9.9.13 Our identification of the diazohydroxide as the oxidized species in these experiments therefore places the pKa of p-NBD hydroxide in 50% ACN at approximately 8.9, corresponding to an increase of about 2.5 pH units from the consensus pKa value of 6.4 in pure water.27 This increase is at the upper end of pKa increases observed in carboxylic acids in 50% ACN–water mixtures.28–30
Kiso for p-NBD diazotate has been reported to be 600, meaning that the anti product is strongly favored over the syn.20 A value for the equilibrium constant Kiso has not been reported31 but p-NBD diazohydroxide syn–anti isomerization is about ten times slower than the isomerization of the diazotate.21 We conclude therefore that the major, electrochemically active species in the present work is the syn-diazohydroxide B in equilibrium with the nitrosamine F. The λmax of anti-diazohydroxide is around 315 nm in water, but the UV-Vis spectrum of B has not been reported.11 The pH-dependent, irreversible, one-electron reduction of a diazohydroxide has been observed polarographically in purely aqueous solution.32
The two-electron redox process we describe is undoubtedly accompanied by one-electron, homolytic dediazonation reactions leading to electrode fouling. This was visible over the timescale of our CV experiments in the case of p-CBD (Fig. 2, dashed lines) and, by poising the working electrode for five minutes at 0.52 V in a solution of p-NBD in 50% ACN to graft nitrobenzene onto the GC surface,33 we observed the reduction of the adsorbed nitro group at 0.56 V when we scanned in fresh buffer (data not shown). Aryldiazonium salts also undergo spontaneous dediazonation in aqueous buffers through the decomposition of diazoanhydrides, which are formed from aryldiazotates and aryldiazonium ions.1 The one-electron reduction of the diazohydroxide or the diazotate, however, appears unlikely, since the aryldiazotate (azoxy) radical has no known stability in aqueous solution. The oxidation of nitrosamines occurs at potentials far higher than those used in our experiments.34
Our discovery of the reversible, two-electron redox chemistry of p-NBD and p-CBD derivatives is biologically significant. A diazene intermediate has been implicated in both phenylhydrazine-induced hemolytic anemia35 and in the metabolic oxidation of isoniazid, a tuberculosis drug, from a hydrazine to a diazohydroxide.36 Aryldiazenes have been generated in situ by the spontaneous hydrolysis and decarboxylation of aryldiazene carboxylate esters37 and by the reduction of aryldiazonium tetrafluoroborates by hydride donors,26 but their instability has limited diazene characterization to UV-Vis spectroscopy and NMR of disproportionation products.26,38 Mixed ACN-aqueous solvents allow previously unavailable access to p-NBD and p-CBD diazenes by accelerating the combination of diazonium and hydroxide ions and stabilizing the neutral diazohydroxide product.
Experimental
Preparation of diazonium salts
p-NBD tetrafluoroborate was obtained from Sigma Aldrich. Seven other p-substituted arenediazonium tetrafluoroborate salts (substituents = H, F, Cl, Br, CH3, CN and OCH3) were synthesized from the corresponding aniline derivatives using either tert-butylnitrite/BF3·Et2O39 or nitrosonium tetrafluoroborate (NO+BF4−).40 The diazonium derivatives were further recrystallized by dissolving a sample (0.5–1.0 g) in a minimum volume of dry acetone and adding a large volume of hexane for crystallization. This procedure was repeated three to four times until the crystalline material was nearly colorless or, in the case of p-NBD, pale yellow. The recrystallized diazonium tetrafluoroborate salts were air-dried and stored desiccated at 4 °C. The purity of the salts was checked by melting point, 1H-NMR and UV-Vis spectra in dry ACN.Preparation of solutions for cyclic voltammetry
For experiments in pure ACN, 0.1 M tetrabutyl ammonium perchlorate was used as a supporting electrolyte. For experiments in aqueous or mixed aqueous-ACN solvents, stock diazonium salt solutions (0.1 M) were freshly prepared in dry, spectroscopic grade ACN and diluted to 1.0 mM in phosphate or HEPES buffer. The pH of ACN-aqueous buffer solutions was measured with a Mettler Toledo pH meter and electrode calibrated with standard, aqueous solutions and reported as the uncorrected meter readings, pHobs. After the solution had developed a pale yellow (p-NBD, rapidly) or orange colour (p-CBD, after approx. 20 minutes) the mixture was transferred from the bench to the working compartment of the electrochemical cell.Cyclic voltammetry
CV was performed at room temperature on an argon-purged solution using an alumina-polished GC working electrode (BASi), a Ag/AgCl reference electrode (Mettler Toledo), and a platinum wire auxiliary electrode. Electrochemical data were collected and analyzed with a Metrohm Autolab PGSTAT128N potentiostat and Nova 2.1 software. All potentials, E, have been reported vs. the standard hydrogen electrode (SHE) by the addition of 0.197 V to the measured E vs. Ag/AgCl.41 CV plots of current, i, against E vs. SHE were made with Origin 2017 (OriginLab). Ferrocenecarboxylic acid (FCA) was obtained from Sigma Aldrich.UV-Vis spectral titrations
Reactions were initiated with the injection of 15–25 μL of 10 mM stock diazonium salt solution in ACN into 3.0 mL of nitrogen-purged 50% (v/v) ACN-aqueous buffer. The loss of the diazonium ion was monitored at 260 nm using a Beckman DU-7 rapid scanning spectrophotometer equipped with a thermostatted cell holder held at 22.5 ± 0.5 °C. Spectral and kinetic plots were made with Origin 2017; those of ln(At − A∞) were linear through four half-lives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants (to SNM) from the Department of Energy (Rocky Flats), Pittsburgh Memorial National College Grant program, the Western Alliance to Expand Student Opportunities (WAESO) and the Regis College Faculty Development Committee. SNM thanks Dr Thomas Bowie and Dr Christopher Roberts for their encouragement and continuing support, and Prof. Fraser Armstrong for hosting his sabbaticals in Oxford. We are indebted to this article's reviewers for their useful suggestions and to Dr William T. Miller, SJ for promoting undergraduate research and for his inspiration.Notes and references
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
- S. Rayat, M. Qian and R. Glaser, Chem. Res. Toxicol., 2005, 18, 1211–1218 Search PubMed.
- C. N. Zink, N. Soissons and J. C. Fishbein, Chem. Res. Toxicol., 2010, 23, 1223–1233 Search PubMed.
- M. P. Doyle, S. N. Mahapatro, J. K. Guy, M. R. Hester, C. M. Van Zyl and K. L. Boundy, Inorg. Chem., 1987, 26, 3387–3392 CrossRef CAS.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- J. Ghilane, M. Delamar, M. Guilloux-Viry, C. Lagrost, C. Mangeney and P. Hapiot, Langmuir, 2005, 21, 6422–6429 CrossRef CAS PubMed.
- R. Elofson and F. Gadallah, J. Org. Chem., 1969, 34, 854–857 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- E. S. Lewis and H. Suhr, Chem. Ber., 1958, 91, 2350–2358 CrossRef CAS.
- E. Lewis and H. Suhr, J. Am. Chem. Soc., 1958, 80, 1367–1371 CrossRef CAS.
- C. Ritchie and D. Wright, J. Am. Chem. Soc., 1971, 93, 2425–2428 CrossRef CAS.
- F. Jordan, J. Phys. Chem., 1973, 77, 2681–2683 CrossRef CAS.
- S. Minegishi and H. Mayr, J. Am. Chem. Soc., 2003, 125, 286–295 CrossRef CAS PubMed.
- R. J. W. Le Fèvre, R. Roper and I. H. Reece, J. Chem. Soc., 1959, 4104–4109 RSC.
- C. Ritchie and D. Wright, J. Am. Chem. Soc., 1971, 93, 6574–6577 CrossRef CAS.
- J. Barbosa and V. Sanz-Nebot, J. Chem. Soc., Faraday Trans., 1994, 90, 3287 RSC.
- H. Lu and S. C. Rutan, Anal. Chem., 1996, 68, 1381–1386 CrossRef CAS.
- A. Sienkiewicz, M. Szymula, J. Narkiewicz-Michalek and C. Bravo-Díaz, J. Phys. Org. Chem., 2014, 27, 284–289 CrossRef CAS.
- J. S. Littler, Trans. Faraday Soc., 1963, 59, 2296–2304 RSC.
- V. Štěrba, in The Chemistry Of Diazonium And Diazo Groups, ed. S. Patai, Wiley, New York, 1978, vol. 1, pp. 71–93 Search PubMed.
- E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef CAS.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn, 2012 Search PubMed.
- P. C. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2367–2376 CrossRef CAS.
- E. M. Kosower, P. C. Huang and T. Tsuji, J. Am. Chem. Soc., 1969, 91, 2325–2329 CrossRef CAS.
- C. E. McKenna and T. G. Traylor, J. Am. Chem. Soc., 1971, 93, 2313–2314 CrossRef.
- E. S. Lewis and M. P. Hanson, J. Am. Chem. Soc., 1967, 89, 6268–6272 CrossRef CAS.
- J. Barbosa, D. Barrón, R. Bergés, V. Sanz-Nebot and I. Toro, J. Chem. Soc., Faraday Trans., 1997, 93, 1915–1920 RSC.
- J. Barbosa, J. L. Beltrán and V. Sanz-Nebot, Anal. Chim. Acta, 1994, 288, 271–278 CrossRef CAS.
- J. Barbosa, R. Bergés, I. Toro and V. Sanz-Nebot, Talanta, 1997, 44, 1271–1283 CrossRef CAS PubMed.
- P. D. Goodman, T. J. Kemp and P. Pinot de Moira, J. Chem. Soc., Perkin Trans. 2, 1981, 1221–1229 RSC.
- E. R. Atkinson, H. H. Warren, P. I. Abell and R. E. Wing, J. Am. Chem. Soc., 1950, 72, 915–918 CrossRef CAS.
- M. Delamar, G. Désarmot, O. Fagebaume, R. Hitmi, J. Pinson and J. M. Savéant, Carbon, 1997, 35, 801–807 CrossRef CAS.
- L. Coffacci, L. Codognoto, L. F. Fleuri, G. P. P. Lima and V. A. Pedrosa, Food Anal. Methods, 2013, 6, 1122–1127 CrossRef.
- H. A. Itano, K. Hirota and K. Hosokawa, Nature, 1975, 256, 665–667 CrossRef CAS.
- I. G. Metushi, T. Nakagawa and J. Uetrecht, Chem. Res. Toxicol., 2012, 25, 2567–2576 Search PubMed.
- P. C. Huang and E. M. Kosower, J. Am. Chem. Soc., 1968, 90, 2362–2367 CrossRef CAS.
- W. T. Evanochko and P. B. Shevlin, J. Am. Chem. Soc., 1978, 100, 6428–6432 CrossRef CAS.
- M. P. Doyle and W. J. Bryker, J. Org. Chem., 1979, 44, 1572–1574 CrossRef CAS.
- J. L. Bahr, J. Yang, D. V. Kosynkin, M. J. Bronikowski, R. E. Smalley and J. M. Tour, J. Am. Chem. Soc., 2001, 123, 6536–6542 CrossRef CAS PubMed.
- D. Ives and G. Janz, Reference Electrodes: Theory and Practice, Academic Press, New York, 1961 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |