
Co3O4 nanoneedle arrays as a multifunctional “super-reservoir” electrode for long cycle life Li–S batteries†
Zhi
Chang‡
a,
Hui
Dou‡
*a,
Bing
Ding
a,
Jie
Wang
a,
Ya
Wang
a,
Xiaodong
Hao
a and
Douglas R.
MacFarlane
*b
aJiangsu Key Laboratory of Materials and Technology for Energy Conversion, College of Material Science and Engineering, Nanjing University of Aeronautics and Astronautics, Nanjing, 210016, P. R. China. E-mail: dh_msc@nuaa.edu.cn
bAustralian Centre for Electromaterials Science, Monash University, Clayton, Victoria 3800, Australia. E-mail: Douglas.MacFarlane@monash.edu
First published on 10th November 2016
Abstract
Lithium–sulfur (Li–S) batteries are highly attractive as energy storage devices due to their low cost and high energy density. The undesired capacity degradation caused by the polysulfide shuttle, however, has hindered their commercialization. Herein, a Co3O4 nanoneedle array on carbon cloth (CC@Co3O4) nanocomposite has been prepared and demonstrated for the first time as a multifunctional “super-reservoir” electrode to prolong the cycle life of Li–S batteries. Owing to the polar surface of the Co3O4 nanoneedle array, soluble lithium polysulfides (Li2Sn, 4 < n < 8) can be effectively absorbed and then transformed to insoluble Li2S2/Li2S which evenly covers the surface of the Co3O4 nanoneedle during the discharge process. Further, during the charge process, the Co3O4 nanoneedle can catalyze the electrochemical transformation of Li2S2/Li2S into soluble polysulfides. A high initial capacity of 1231 mA h g−1 at 0.5C and a slow capacity decay of 0.049%/cycle at 2.0C over 500 cycles were achieved; excellent rate performance was also obtained.
1. Introduction
Lithium–sulfur (Li–S) batteries are emerging as one of the most promising energy storage devices due to their high specific capacity (1675 mA h g−1) and theoretical energy density (2600 W h kg−1).1–3 However, sulfur cathodes still suffer from several inherent challenges that hinder their commercialization.4–6 For example, Li–S batteries are known to suffer from low utilization of active materials and rapid capacity decay during the cycling process. The notable “shuttle effects” caused by the dissolution of lithium polysulfides in the organic electrolyte combined with the large volumetric expansion during cycling and the uncontrolled deposition of Li2S2/Li2S are believed to be the main reasons for the low coulombic efficiency and rapid capacity fade.7–9Tremendous efforts have been devoted to circumventing the problem caused by soluble polysulphides. The most prevalent approach is to encapsulate sulfur in the pores of different host materials, including mesoporous carbon,10–12 microporous carbon spheres,13–15 carbon nanotubes (CNTs),16–20 graphene oxides,21–26 hollow porous carbon spheres27,28 and MXene.29 This strategy improves the kinetics/reversibility and alleviates the capacity fading; however, it still fails to adequately resolve the short cycle life problem.30 The weak interaction between non-polar carbon materials and Li2Sn (4 < n < 8) leads to loss of the active material from the cathode and finally reduces the cycle life of the battery.31,32 Another promising approach is to bind the polysulfides with functional groups (such as oxygen-containing and/or nitrogen-containing groups) on the surface of the host materials.21,33,34 Nevertheless, the electronic conductivity of these functionalised materials can be much lower than that of the corresponding carbon host, which is deleterious to battery performance. Moreover, the interaction between the functional groups and the polysulfides is not strong enough to completely entrap soluble polysulphides. The pioneering work by Cui and Nazar et al. on oxides for confining sulfur suggested that the efficiently reversible conversion of Li2S2/Li2S into soluble polysulfides also has great significance for the electrochemical performance of Li–S batteries.31,32,35 Therefore, multifunctional host materials that can promote the even deposition and decomposition of Li2S2/Li2S are highly desired to develop long-life Li–S batteries.
Co3O4 is a promising transition metal oxide electrocatalyst in a number of reactions. For example, it was reported that Co3O4 nanocrystals on graphene displayed high catalytic activity for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER).36 Co3O4 nanocrystals with different shapes and crystal planes also showed catalytic properties for methane catalytic combustion.37 Co3O4 used as a catalytic electrode for Li–S batteries has not been reported so far.
Herein, an electrode consisting of bottlebrush-like Co3O4 nanoneedle arrays on flexible carbon cloth fibers (CC@Co3O4) has been prepared and demonstrated for the first time as a multifunctional “super-reservoir” to prolong the cycle life of Li–S batteries. The strong affinity of Co3O4 for the soluble lithium polysulfides (Li2Sn) can effectively accelerate the polysulfide conversion during the discharge process. The widely distributed active sites of the nanoneedle array afford the continuous deposition of Li2S2/Li2S during the discharge process. The Li2S2/Li2S can be readily transformed to the polysulfide species during the charge process due to the possible electrocatalytic activity of the Co3O4 nanoneedles. We show that, as a multifunctional “super reservoir” for Li–S batteries, this special design of CC@Co3O4 combining the polar surface, massive active sites and good conductivity exhibits high initial capacity, slow capacity decay and excellent rate performance.
2. Experimental
2.1 Material synthesis
2.2 Material characterization
The morphologies were characterized by scanning electron microscopy (SEM, JEOL JSM-6380LV FE-SEM) and transmission electron microscopy (TEM, FEI, Tecnai-20). X-ray diffraction (XRD) measurements were carried out with a Bruker-AXS D8 DISCOVER diffractometer using Cu Kα radiation. The X-ray photoelectron spectroscopy (XPS) analysis was performed on a Perkin-Elmer PHI 550 spectrometer with Al Kα (1486.6 eV) as the X-ray source. The relative content of polysulfides was recorded using an ultraviolet-visible spectrophotometer (Cary 60 UV-Vis). Raman spectra were recorded on a HORIBA Scientific LabRAMHR Raman spectrometer system with a 532.4 nm laser. The N2 adsorption–desorption isotherm was obtained by using a Micromeritics BK122T-B analyzer. The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method. The pore size distribution was obtained from the Barrett–Joyner–Halenda (BJH) desorption branch of the isotherm.2.3 Electrochemical measurements
The CC@Co3O4 nanoneedle arrays were directly used as the cathode, with Li foil as the anode in CR2016 coin cells, assembled in an argon-filled glovebox, in which both the moisture and oxygen contents were controlled to be less than 1 ppm. The CC@Co3O4 was cut into a piece of 0.5 × 0.5 cm2 and was dried in a vacuum oven at 60 °C overnight prior to use. 20 μL of Li2S8 electrolyte was introduced into the coin cells, corresponding to about 4.1 mg cm−2 of sulfur mass loading, which is comparable with that in the previous work (Table S1†). The coin cells were galvanostatically charged/discharged at different current densities between 1.7 and 3.0 V (vs. Li/Li+) by using a CT2001A cell test instrument (LAND Electronic Co.). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted with a CHI 600A electrochemical workstation at a scanning rate of 0.02 mV s−1.3. Results and discussion
The CC@Co3O4 nanoneedle arrays were prepared with a facile hydrothermal method, in combination with a thermal treatment, as shown in Fig. 1a. Owing to its excellent mechanical strength and high electrical conductivity, flexible carbon cloth was selected as a substrate. SEM and TEM were used to identify the morphology of the CC@Co3O4. As shown in Fig. 1b and c, the SEM images show that bottlebrush-like Co3O4 nanoneedles uniformly grow on the carbon fibers. The Co3O4 nanoneedle displays a length of about 3 μm. The structure of the Co-precursor nanoneedle arrays on carbon cloth (CC@Co-precursor) was perfectly retained after heat treatment (Fig. S1†). The bottlebrush-like structure of the Co3O4 nanoneedle array enlarges the specific area, exposes more active sites and allows fast diffusion of the electrolyte to the active surface of the electrode.40 The TEM image elucidates that the Co3O4 nanoneedle consists of nanoparticles of about 30 nm (Fig. 1d). The CC@Co3O4 exhibits a BET surface area of 75.6 m2 g−1 and a pore volume of 0.26 cm3 g−1 (Fig. S2†). The crystalline structure of the Co3O4 nanoneedle was characterized by XRD. Fig. 1e shows the XRD pattern of the CC@Co3O4, which contains eight diffraction peaks attributed to the (111), (220), (311), (222), (400), (422), (511) and (440) planes of the cubic Co3O4 phase (JCPDS card no. 42-1467). The peaks at 26.21° and 43.61° are typical graphite (002) and (100) reflection ascribed to the carbon cloth.38 The mechanism of the CC@Co3O4 as a multifunctional “super-reservoir” to prolong the cycle life of Li–S batteries is schematically demonstrated in Fig. 1f. In the discharge process, the active material Li2S8 in the form of catholyte can be absorbed on the surface of the Co3O4 nanoneedle arrays due to the strong affinity of Co3O4 for the soluble polysulfides. Then Li2S8 was effectively reduced to insoluble Li2S2/Li2S and evenly distributed because of widely exposed active sites of the Co3O4 nanoneedle. The diameter of the Co3O4 nanoneedle increases due to the continuous accumulation of Li2S2/Li2S. During the subsequent charge process, the deposited Li2S2/Li2S could be readily transformed to the polysulfide species, which makes the diameter of the Co3O4 nanoneedle decrease and morphology of the Co3O4 nanoneedle recovers. Meanwhile, the resulting soluble polysulfides could be again absorbed on the surface of the Co3O4 nanoneedle, eventually realizing a highly reversible redox process and effectively prolonging the cycle life.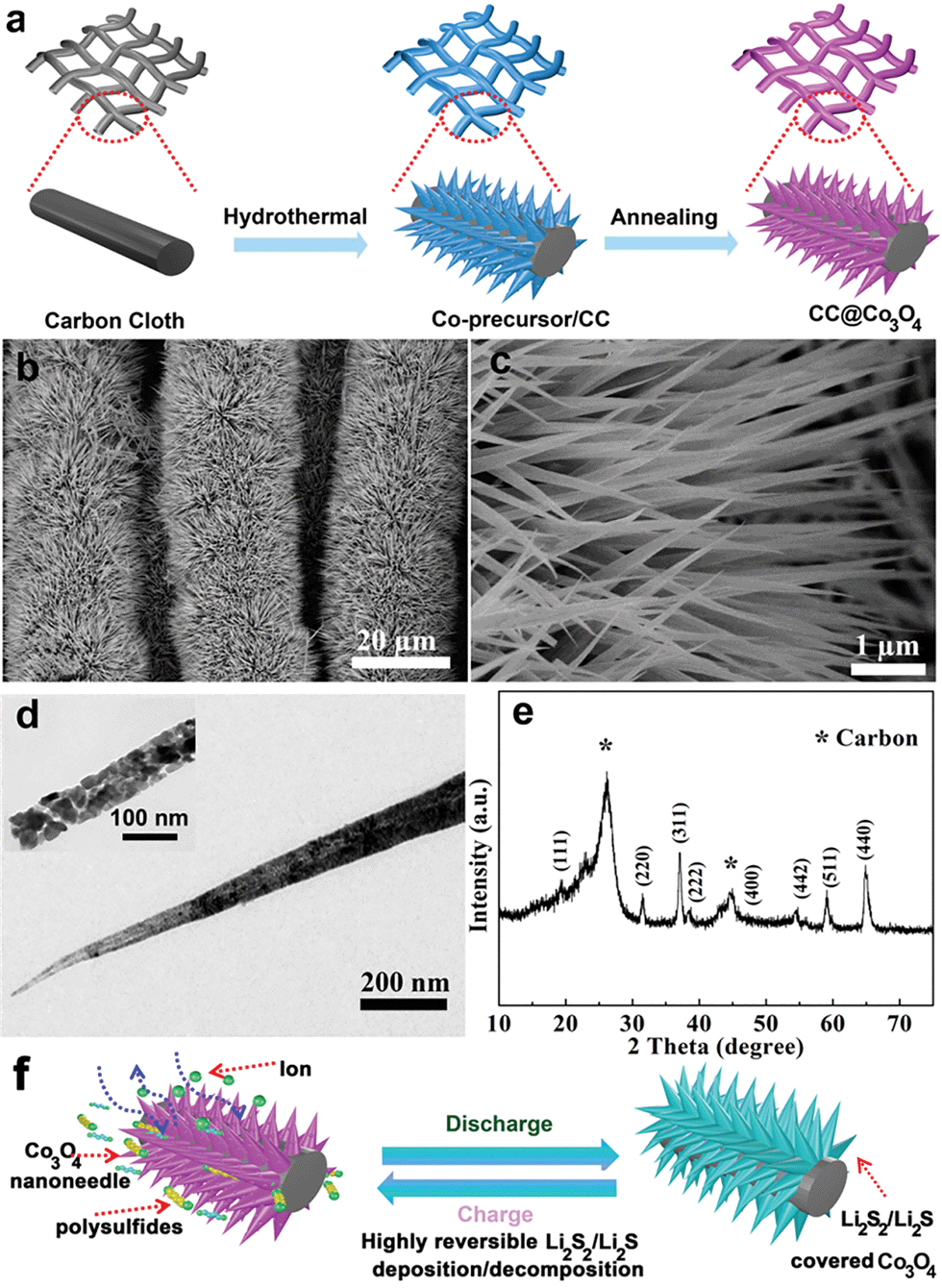 | ||
| Fig. 1 (a) Schematic illustration of the preparation route of the CC@Co3O4. Physical characterization of the CC@Co3O4: (b and c) SEM images. (d) TEM images (inset: under higher magnification at 100 nm) and (e) XRD pattern. (f) The electrochemical process of the CC@Co3O4 during the cycling process. | ||
The free-standing CC@Co3O4 “super reservoir” was directly used as an electrode in a coin cell with 0.2 M Li2S8 catholyte as the electrolyte. As shown in Fig. 2a, the galvanostatic charge/discharge profiles of the first cycle for the CC@Co3O4 at current rates of 0.2, 0.5 and 1C (1C = 1675 mA h g−1) show well defined plateaus. The low polarization of the CC@Co3O4 electrode with increasing current rate and cycling number (Fig. S3†) indicates favourable kinetics and good electrolyte accessibility.41,42Fig. 2b, c and e display the cycling performance at different rates. For comparison, the CC and CC@Co3O4-1 in which the carbon cloth is not fully covered with Co3O4 nanoneedles were also used as the reservoirs and evaluated under the same conditions. Despite the relatively high discharge capacities of 900 mA h g−1 in the first two cycles at 0.5C, the capacities of the CC reservoir rapidly decay to less than 400 mA h g−1 after 160 cycles (Fig. S4†). The severe capacity fading is attributed to the continued shuttle effect of polysulfides and the random and irreversible deposition of the insoluble lithium sulfides (Li2S/Li2S2).7 The CC@Co3O4-1 reservoir shows enhanced performance due to the effect of the Co3O4 (Fig. 2b). By sharp contrast, the CC@Co3O4 “super reservoir” shows an initial capacity of 1231 mA h g−1 at 0.5C and sustains about 987 mA h g−1 even after 200 cycles with a capacity fade of only 0.14% per cycle. At a higher current density of 1C, the CC@Co3O4 “super reservoir” delivers an exceptional reversible capacity of nearly 700 mA h g−1 after 280 cycles with a very small capacity degradation rate of 0.06% (Fig. 2c). Even at a current density as high as 2C (Fig. 2e), the CC@Co3O4 “super reservoir” still delivers an initial discharge capacity of 621 mA h g−1, and stabilizes at 476 mA h g−1 after 500 cycles. Rate performances of the CC@Co3O4 and CC@Co3O4-1 reservoirs are summarized in Fig. 2d. The CC@Co3O4 delivers a reversible discharge capacity of 1120, 1010, 830, and 610 mA h g−1 at 0.2, 0.5, 1 and 2C, respectively. When the current density was switched abruptly from 2 to 0.2C, the original capacity is largely recovered. The rate performance of the CC@Co3O4 is much better than that of the CC@Co3O4-1. Furthermore, the electrochemical performance of the CC@Co3O4 achieved in this work is competitive compared to other excellent studies reported previously (Table S1†).31,43,44 The excellent electrochemical performance can be ascribed to the advantages of the CC@Co3O4. The conductive bottlebrush-like structure of CC@Co3O4 is robust and flexible, which could accommodate the precipitation/decomposition of the active material during cycling as a reservoir. More importantly, the Co3O4 nanoneedle shows strong affinity to the polysulfides. As shown in the results of the adsorption experiment (Fig. S5†), the color of the Li2S8 solution treated with CC@Co3O4 fades more quickly than the one treated with CC@Co3O4-1, while the color of the Li2S8 treated with CC is nearly unchanged, which indicates that CC@Co3O4 is a super polysulfide reservoir. The strong affinity of Co3O4 nanoneedles to the polysulfides can effectively improve the reversibility of the redox process and promote the precipitation/decomposition of Li2S2/Li2S; it can thus further enhance the stability and prolong the cycling life.
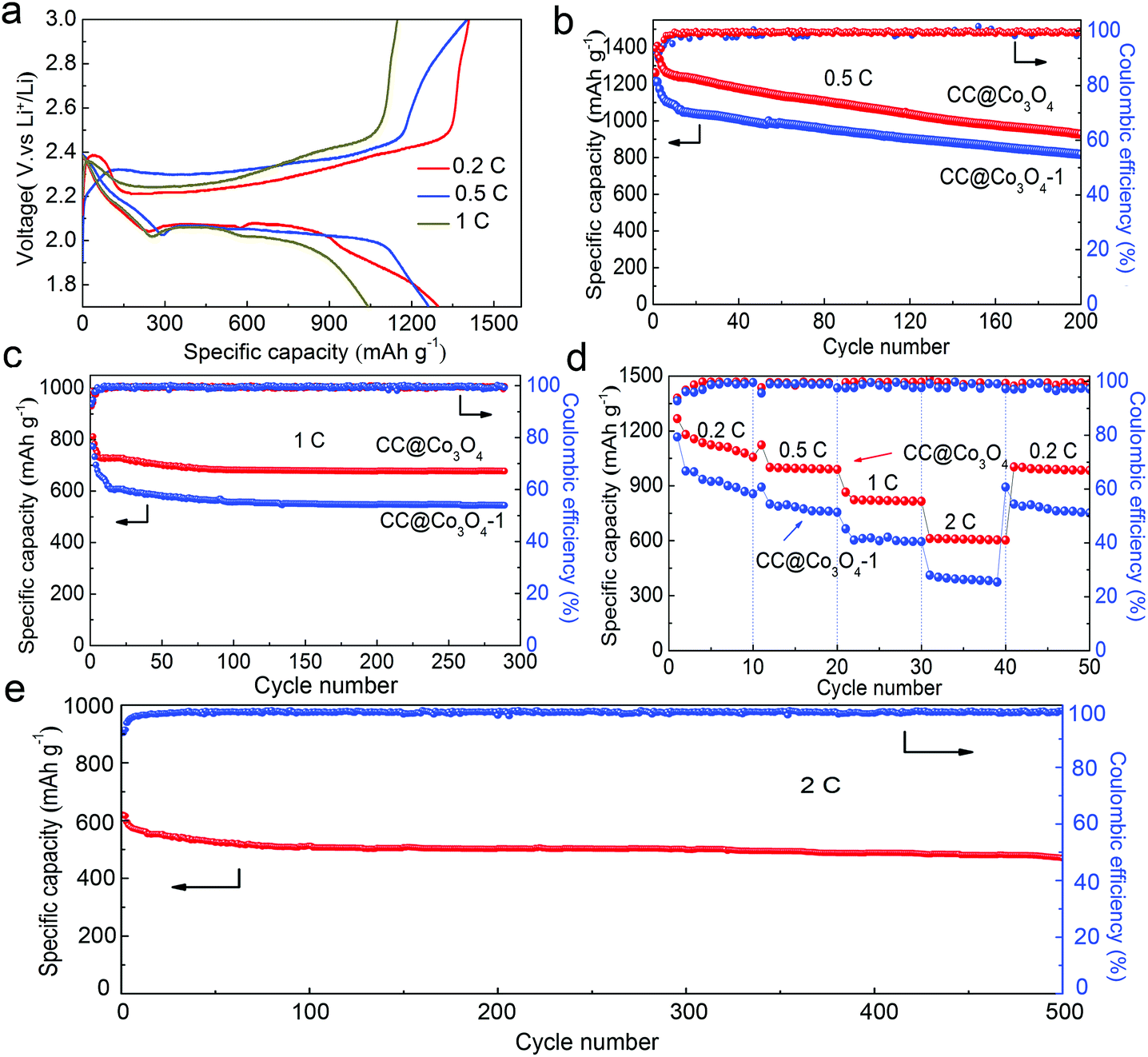 | ||
| Fig. 2 Electrochemical performance of Li–S batteries using CC@Co3O4 and CC@Co3O4-1 as reservoirs. (a) Galvanostatic charge/discharge profiles of the first cycle for CC@Co3O4 at current rates of 0.2, 0.5, and 1C. (b, c, and e) Cycling performance and coulombic efficiency at 0.5, 1 and 2C. (d) Rate performance of CC@Co3O4 and CC@Co3O4-1. | ||
From the viewpoint of electrochemical transitions, the charge/discharge profile of the CC@Co3O4 “super reservoir” can be described in terms of the following voltage states: fresh cathode, the partly discharged state of intermediate polysulfides Li2Sn (n < 8) at 2.0 V, fully discharged state of Li2S2/Li2S at 1.7 V, partly charged state of intermediate polysulfides Li2Sn (n < 8) at 2.4 V and fully charged state of Li2S8 (even S8) at 2.8 V (Fig. 3a). The electrochemical transition of the active material during the first cycle at 0.5C was characterized by ex situ Raman and UV-Vis absorption spectra. It is clear that all CC@Co3O4 electrodes in different voltages show obvious Raman peaks from Co3O4 (Fig. 3b). Compared with the fresh CC@Co3O4 electrode, the electrode in the partly discharged state of 2.0 V displays four new Raman peaks ascribed to Li2S8 (456 cm−1), Li2S6 (510 cm−1 and 398 cm−1) and Li2S4 (203 cm−1) respectively.42 In the fully discharged state of 1.7 V, the Raman spectrum shows strong signals from Li2S2 (450 cm−1) and Li2S (381 cm−1). In addition, no signals from other sulfur species are discovered, which validates the fact that the polysulfides are completely transformed to the solid Li2S2 and Li2S.45 In the partly charged state of 2.4 V, the Raman peaks from Li2S2 and Li2S become weaker than those at 1.7 V, and peaks from polysulfides appear again due to the decomposition of the solid Li2S2 and Li2S. In the fully charged state of 2.8 V, only peaks from polysulfides can be found and no peaks from Li2S2 and Li2S exist, implying thorough decomposition of Li2S2/Li2S. It is notable that the intensity of the Co3O4 peaks undergoes a periodic change during the cycling process. With the proceeding of the discharge/charge process, the intensity of the Co3O4 peaks gradually becomes weak/strong. This could be ascribed to the improved reversible deposition/decomposition of the Li2S2/Li2S on the Co3O4 nanoneedle. In the discharged state, the Co3O4 nanoneedle is evenly covered with the Li2S2/Li2S induced by the strong affinity of Co3O4 for the soluble polysulfides; thus the intensity of Co3O4 peaks becomes weak. In the charged state, the solid Li2S2/Li2S decomposes, and the Co3O4 nanoneedle exposes and hence the intensity of Co3O4 peaks becomes strong. The content of polysulfides in the extracts of CC@Co3O4 at different voltages was characterized by UV-Vis absorption spectra. The concentration of the polysulfides decreases/increases gradually during the discharge/charge process (Fig. S6†). It is the lowest in the fully discharged voltage, and nearly equals to that of the fresh electrode in the fully charged voltage. Digital photos of the CC@Co3O4 extracts show the same trend (Fig. S7†). The colour of the extracts fades/deepens gradually. The extract is nearly colourless in the fully discharged state. The Raman and UV-Vis results indicate a fact of highly reversible electrochemical transition of the active material.
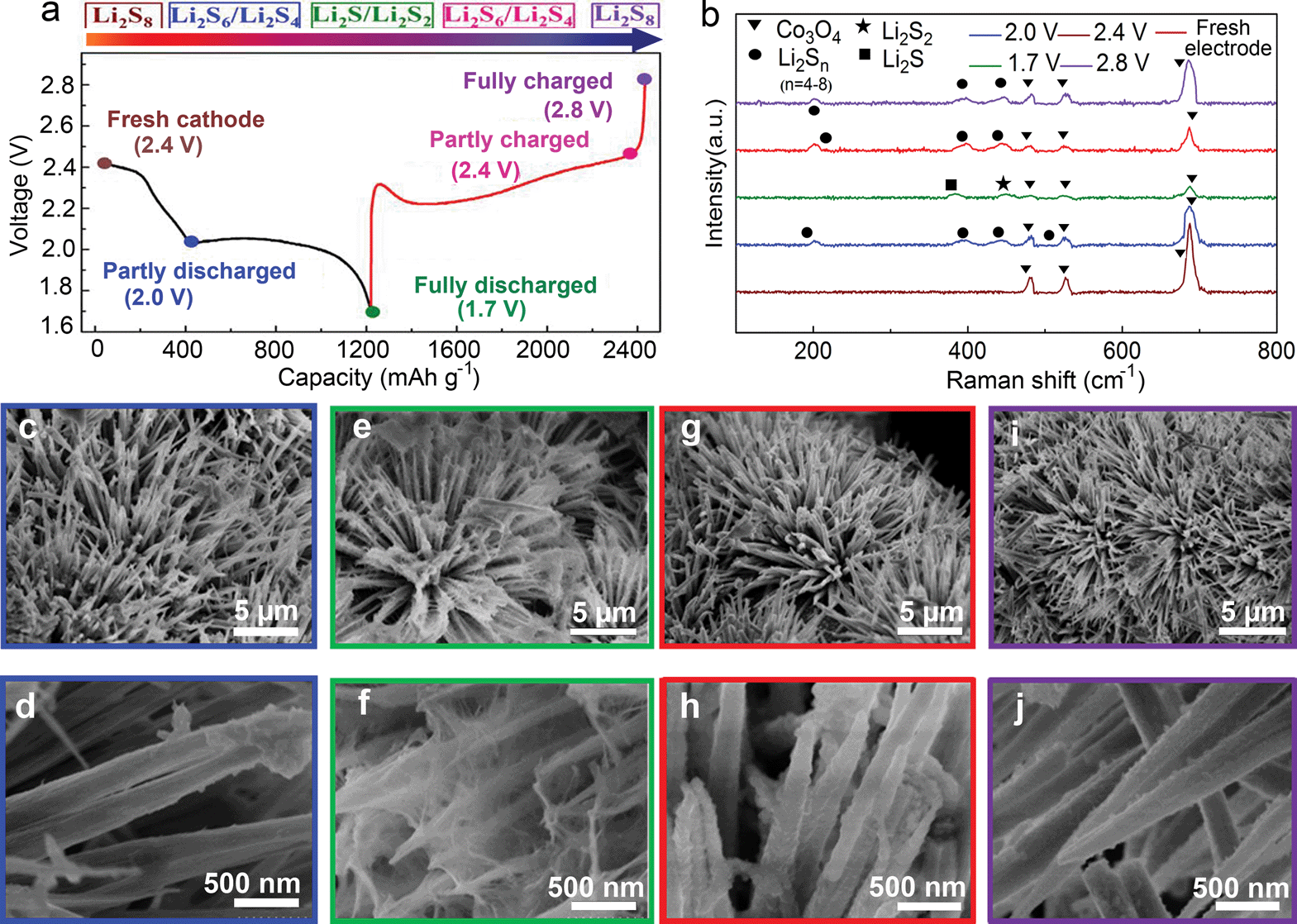 | ||
| Fig. 3 (a) The first galvanostatic charge/discharge profile of the CC@Co3O4 at 0.5C (brown, blue, green, red, and purple dots represent electrodes in fresh, partly discharged, fully discharged, partly charged and fully charged states, respectively). (b) Raman spectra of the washed CC@Co3O4 in different discharge/charge voltages. (c–j) SEM images of the CC@Co3O4 in different charge/discharge voltages ((c, d) 2.0 V; (e, f) 1.7 V; (g, h) 2.4 V; (i, j) 2.8 V). | ||
Morphologies of the CC@Co3O4 “super reservoir” in different charge/discharge voltages are shown in Fig. 3c–j. Compared with the fresh CC@Co3O4 “super reservoir” (Fig. 1c and d), it can be observed that a few tiny particles of Li2S2/Li2S deposit on the CC@Co3O4 nanoneedle at 2.0 V (Fig. 3c and d). It is notable that Co3O4 nanoneedles could afford the uniform deposition of Li2S2/Li2S due to the excellent affinity to polysulfides and high surface area, which is similar to the uniform deposition of Li2S2/Li2S on the conductive tin-doped indium oxide and Ti4O7 as reported in previous work.31,32 As observed in Fig. 3e and f, deposited Li2S2/Li2S forms a spider web-like structure in the fully discharged state of 1.7 V. The three-dimensional (3D) deposition of Li2S2/Li2S could improve the electrochemical performance.46 Meanwhile, the original bottlebrush-like morphology of CC@Co3O4 can be clearly identified. In the partly charged state of 2.4 V, it is interesting that the 3D spider web-like Li2S2/Li2S disappears (Fig. 3g and h). In the fully charged state at 2.8 V, the Co3O4 nanoneedles recover the original morphology with a nearly smooth surface as shown in Fig. 3i and j. This indicates effective decomposition of Li2S2/Li2S, which can be ascribed to the possible catalytic effect of Co3O4.
To further understand the role of the Co3O4 nanoneedle array, the morphologies of CC and CC@Co3O4-1 in different charged/discharged states at 0.5C were checked (Fig. S8a†). In the case of CC@Co3O4-1, in the fully discharged state of the first cycle the solid Li2S2/Li2S mostly accumulates on the Co3O4 nanoneedle array, and not on the void space of the CC fibres. This indicates that Co3O4 is favourable for the preferential and selective deposition of the sulfur species (Fig. S8b and c†), which can be ascribed to the strong affinity of polar Co3O4 to the polysulfides and widely distributed active sites of the Co3O4 nanoneedle array.31,32,47 In the case of CC, the Li2S2/Li2S randomly deposits and accumulates on the electrode and is present even in the fully charged state as shown in Fig. S8d;† this again emphasizes the crucial role of the Co3O4 nanoneedle in accelerating the electrochemical process.
XPS was used to confirm the chemical interaction between the sulfur species and Co3O4. The S, C, O and Co signals are present in the XPS survey of the CC@Co3O4 in the fully discharged state in Fig. S9a.† Different sulfur bindings in the fully discharged state are observed in the S 2p XPS spectrum (Fig. S9b†). The peak at 170.1 eV is assigned to sulfate species arising from sulfur oxidation in air.34 The peak at 169.4 eV corresponds to Co–S bonding, indicating the presence of chemical interaction between solid Li2S2/Li2S and Co3O4.48,49 Peaks at 168.7 and 167.3 eV correspond to S–O and S–S bonding. Peaks at 163.8 and 165.2 eV are assigned to S 2p1/2 and S 2p3/2, respectively.50 In the Co 2p3/2 spectra, as shown in Fig. 4a, the peak at 783.0 eV in both the charged and discharged states can be ascribed to the shakeup satellite. Co 2p3/2 in the discharged state exhibits two major peaks at 781.7 and 780.1 eV corresponding to Co2+ and Co3+, in good accordance with the reported data for Co3O4.51,52 However, in the fully charged state, the Co 2p3/2 peaks shift to a lower binding energy of 780.9 and 779.8 eV, respectively, which could be ascribed to the electron transfer from polysulfides to the Co atoms, indicating the strong chemical interaction between Co3O4 and the polysulfides.53 The chemical interaction between Co3O4 and polysulfides could effectively avoid the diffusion of the resulting polysulfides into the electrolyte, therefore increasing the utility of the active material.
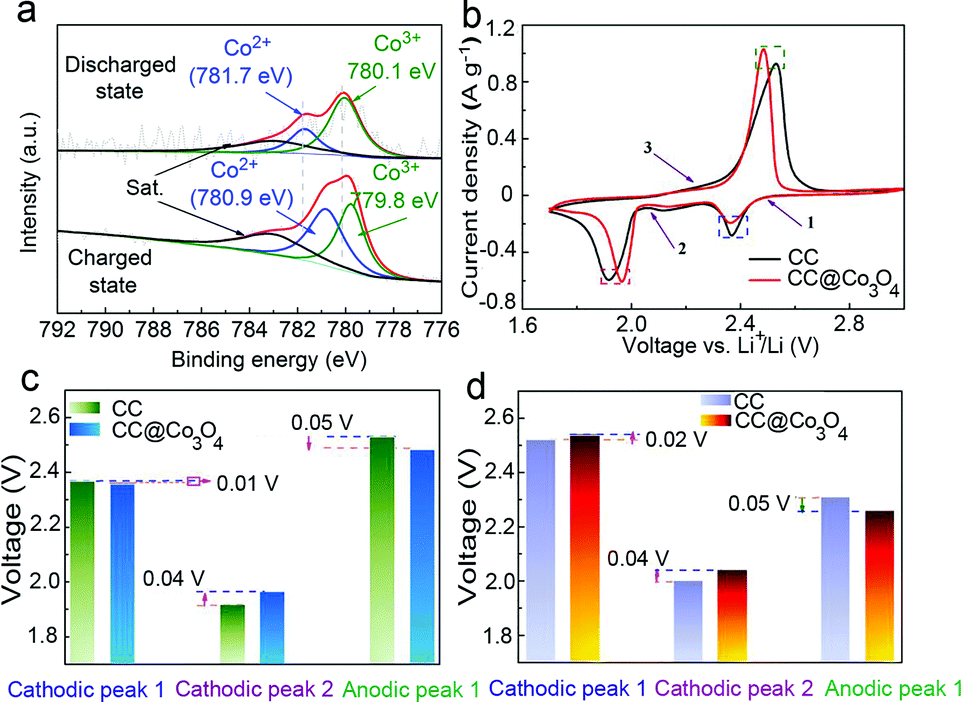 | ||
| Fig. 4 (a) The Co 2p3/2 XPS spectra of the CC@Co3O4 in the fully charged and discharged states of the first cycle at 0.5C. (b) CV curves of CC and CC@Co3O4. (c) Peak voltages of CC and CC@Co3O4; (d) onset voltages of CC and CC@Co3O4. | ||
CV curves were recorded at a scan rate of 0.02 mV s−1 (Fig. 4b) in order to investigate the electrocatalytic activity of the Co3O4. The CV curve of the CC@Co3O4 “super reservoir” displays two cathodic peaks at 2.36 (peak 1) and 1.96 V (peak 2) and one anodic peak at 2.48 V (peak 3), consistent with the charge/discharge profiles. Compared with the CC, the CC@Co3O4 “super reservoir” indicates sharper redox peaks, suggesting better redox reversibility of polysulfides. In addition, the cathodic peak 2 for the CC@Co3O4 has a positive shift of 0.04 V (Table S2†), and the anodic peak 3 negatively shifts 0.05 V compared with those for the CC reservoir, indicating greatly suppressed electrochemical polarization of the CC@Co3O4 “super reservoir” (Fig. 4c). Moreover, in the case of the CC@Co3O4, the increased onset voltage of 0.02 and 0.04 V for the cathodic peaks 1 and 2 and the decreased onset voltage of 0.05 V for the anodic peak 3 (Fig. 4d and Table S3†) further verify the better kinetics of polysulfides. These accelerated kinetics of polysulfides could be ascribed to not only the strong adsorption of Co3O4 nanoneedle arrays to polysulfides, but also the catalytic effect of Co3O4 to promote the decomposition of Li2S2/Li2S. Recently, Dong et al. also reported improved performance of sulfur cathodes due to the catalytic effect of Co.54 The dual functions of Co3O4 nanoneedles to accelerate the redox process highly improve the stability and prolong the cycling life of Li–S batteries.
To get a more comprehensive understanding of the effects of CC@Co3O4 nanoneedle arrays on the stability of Li–S batteries, the electrodes after long-term cycling were further characterized. The EIS spectra of the CC and CC@Co3O4 “super reservoir” cycled for 300 cycles at 0.5C were recorded. Obviously, the CC electrode exhibits an extra-flattened semicircle, which could be due to the severe irreversible deposition of insoluble products on the surface of the CC electrode (Fig. S10 and Table S4†). By contrast, the CC@Co3O4 “super reservoir” shows lower charge-transfer resistance and Warburg diffusion resistance, indicating the more rapid electronic/ionic transport thanks to the efficiently reversible deposition/decomposition of the Li2S2/Li2S. The Rct is 336 Ω for the CC reservoir, while 116 Ω for the CC@Co3O4 “super reservoir”. The obviously decreased arc radius of impedance of the CC@Co3O4 “super reservoir” shows that the energy barrier for the electrode reactions is smaller than that of the CC reservoir, that is, the impedance value of the Faraday current is decreased and the reaction speed is increased. In order to evaluate the catalytic effect of Co3O4, Rct values at different temperatures for CC and CC@Co3O4 as reservoirs were measured (Fig. S11†) and then the Arrhenius plots (ln(1/Rct) ∼ 1/T) (Fig. S12†) were drawn based on the Rct of CC (Table S5†) and CC@Co3O4 (Table S6†). The reaction activation energies for CC and CC@Co3O4 are respectively 226.5 and 87.5 kJ mol−1 according to the Arrhenius plots (Fig. S12†). This indicates that the reaction activation energy for Li–S batteries is greatly reduced due to the catalytic effect of Co3O4. This is mainly due to the possible catalytic activity of Co3O4, which can decrease the energy barrier of the electrode reaction and accelerate the reaction of the electrode. Charge transfer at the CC@Co3O4 interface is much faster than that at the CC interface, indicating considerable enhancement of the redox kinetics of polysulfides, which could be ascribed to the possible catalytic effect of the CC@Co3O4 electrode.41,55
SEM images of the CC@Co3O4 “super-reservoir” in fully discharged and charged states of the 100th, 200th, 300th and 500th cycles at 0.5C are shown in Fig. S13.† For all the CC@Co3O4 electrodes after cycling for different cycles, Li2S2/Li2S uniformly deposits on the Co3O4 nanoneedle and retains a similar spider web-like structure to that of the first cycle in the fully discharged state. The original bottlebrush-like morphology of the CC@Co3O4 can be clearly identified. While in fully charged states, the spider web-like Li2S2/Li2S disappears, and the Co3O4 nanoneedles just display a little rough surface but without obvious accumulation of Li2S2/Li2S bulks even after 500 cycles. TEM was further employed to monitor the morphology of the CC@Co3O4 “super-reservoir”. As shown in Fig. 5a and b, there is a layer of Li2S2/Li2S uniformly distributed on the Co3O4 nanoneedle in the fully discharged state of the 100th cycle, and it nearly disappears in the fully charged state. With prolonged cycles, the morphology does not change significantly. Even at the 500th cycle, the layer of Li2S2/Li2S is uniform in the fully discharged state, and mostly disappears in the charged state. These SEM and TEM results further indicate effective decomposition of insoluble Li2S2/Li2S ascribed to the catalytic effect of Co3O4 in the long cycling process. The conductive bottlebrush-like structure of the CC@Co3O4 is robust and flexible, which can not only accommodate the solubilization/precipitation of the active material, but also effectively improve the reversibility of the precipitation/decomposition process during cycling, and therefore enhance the utilization of the active materials and maintain the long cycling stability of the Li–S batteries.
 | ||
| Fig. 5 TEM images of the CC@Co3O4 at 0.5C (a, c, e, and g) in fully discharged states of the 100th, 200th, 300th and 500th cycles. (b, d, f, and h) in fully charged states of the 100th, 200th, 300th and 500th cycles. | ||
4. Conclusions
In conclusion, the CC@Co3O4 nanoneedle array with a bottlebrush-like structure was employed as an efficient multifunctional “super-reservoir” to prolong the cycle life of Li–S batteries. The special 3D structure array provides uniform active sites for the continuous deposition of insoluble Li2S2/Li2S during the discharge process. The inherent polar surface of Co3O4 nanoneedles has a high affinity to polysulfides, which is beneficial to the kinetics of the polysulfide conversion and improves the stability of the batteries. Moreover, the decomposition of insoluble Li2S2/Li2S can be accelerated due to the possible electrocatalytic activity of the Co3O4 and therefore further prolong the cycling life of Li–S batteries. In addition, the carbon cloth as a backbone employed here can act as a conductive core to provide efficient transport of electrons for the Co3O4 nanoneedle array. The characterization for the CC@Co3O4 in different charge/discharge voltages convincingly indicates highly reversible precipitation/decomposition of the active material, validating the dual functions of Co3O4 nanoneedles to accelerate the redox process. This special “super-reservoir” electrode is robust and flexible, leading to greatly improved performance of Li–S batteries. A high initial capacity of 1231 mA h g−1 at 0.5C and a slow capacity decay of 0.049% per cycle at 2.0C for 500 cycles were achieved. Excellent rate performance was also obtained. This work demonstrates that the concept of employing a catalytic host material will improve the electrochemical performance of high-energy Li–S batteries.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program) (No. 2014CB239701), National Natural Science Foundation of China (No. 51372116, 51672128), Natural Science Foundation of Jiangsu Province (BK20151468, BK2011030) and Fundamental Research Funds for the Central Universities of NUAA (NJ20160104). Z. Chang is grateful to the Foundation of Graduate Innovation Centre in NUAA (kfjj20150612).Notes and references
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- P. G. Bruce, L. J. Hardwick and K. M. Abraham, MRS Bull., 2011, 36, 506–512 CrossRef CAS.
- J. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325–332 CrossRef PubMed.
- Y.-S. Su, Y. Fu, T. Cochell and A. Manthiram, Nat. Commun., 2013, 4, 2985–2993 Search PubMed.
- H. D. Shin, M. Agostini, I. Belharouak, J. Hassoun and Y.-K. Sun, Carbon, 2016, 96, 125–130 CrossRef CAS.
- G. He, X. L. Ji and L. Nazar, Energy Environ. Sci., 2011, 4, 2878–2883 CAS.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- S. R. Chen, Y. P. Zhai, G. L. Xu, Y. X. Jiang, D. Y. Zhao, J. T. Li, L. Huang and S. G. Sun, Electrochim. Acta, 2011, 56, 9549–9555 CrossRef CAS.
- D. S. Jung, T. H. Hwang, J. H. Lee, H. Y. Koo, R. A. Shakoor, R. Kahraman, Y. N. Jo, M. S. Park and J. W. Choi, Nano Lett., 2014, 14, 4418–4425 CrossRef CAS PubMed.
- Z. Chang, H. Dou, B. Ding, J. Wang, Y. Wang, G. Y. Xu and C. Li, New J. Chem., 2016, 40, 7680–7686 RSC.
- Z. Li, Y. Jiang, L. Yuan, Z. Yi, C. Wu, Y. Liu, P. Strasser and Y. Huang, ACS Nano, 2014, 8, 9295–9303 CrossRef CAS PubMed.
- X. B. Cheng, J. Q. Huang, Q. Zhang, H. J. Peng, M. Q. Zhao and F. Wei, Nano Energy, 2014, 4, 65–72 CrossRef CAS.
- J. Guo, Y. Xu and C. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 CAS.
- H. B. Wu, S. Wei, L. Zhang, R. Xu, H. H. Hng and X. W. Lou, Chem.–Eur. J., 2013, 19, 10804–10808 CrossRef CAS PubMed.
- G. Y. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- H. L. Wang, Y. Yang, Y. Y. Liang, J. T. Robinson, Y. G. Li, A. Jackson, Y. Cui and H. J. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- M. Q. Zhao, Q. Zhang, J. Q. Huang, G. L. Tian, J. Q. Nie, H. J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410–3418 Search PubMed.
- Y. X. Wang, L. Huang, L. C. Sun, S. Y. Xie, G. L. Xu, S. R. Chen, Y. F. Xu, J. T. Li, S. L. Chou, S. X. Dou and S. G. Sun, J. Mater. Chem., 2012, 2, 4744–4750 RSC.
- L. C. Yin, J. L. Wang, F. J. Lin, J. Yang and Y. Nuli, Energy Environ. Sci., 2012, 5, 6966–6972 CAS.
- F. F. Zhang, X. B. Zhang, Y. H. Dong and L. M. Wang, J. Mater. Chem., 2012, 22, 11452–11454 RSC.
- C. Zhang, H. B. Wu, C. Yuan, Z. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 124, 9730–9733 CrossRef.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- J. Jiang, J. Zhu, W. Ai, X. Wang, Y. Wang, C. Zou, W. Huang and T. Yu, Nat. Commun., 2015, 6, 8622–8631 CrossRef CAS PubMed.
- H. Yao, G. Zheng, P. C. Hsu, D. Kong, J. J. Cha, W. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943–3952 CAS.
- Q. Pang, D. Kundu, M. Cuisinier and L. Nazar, Nat. Commun., 2014, 5, 4759–4767 CrossRef CAS PubMed.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou and Z. Liu, Nano Lett., 2015, 15, 5137–5142 CrossRef CAS PubMed.
- S. Yuan, J. L. Bao, L. Wang, Y. Xia, D. G. Truhlar and Y. Wang, Adv. Energy Mater., 2015, 6, 1733–1741 Search PubMed.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682–5688 CrossRef PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS PubMed.
- L. Shen, Q. Che, H. Li and X. Zhang, Adv. Funct. Mater., 2014, 24, 2630–2637 CrossRef CAS.
- F. Zhang, C. Yuan, X. Lu, L. Zhang, Q. Che and X. Zhang, J. Power Sources, 2012, 203, 250–256 CrossRef CAS.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588–598 CrossRef CAS.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 6, 519–527 CrossRef PubMed.
- H. Yao, K. Yan, W. Li, G. Zheng, D. Kong, Z. W. Seh, V. K. Narasimhan, Z. Liang and Y. Cui, Energy Environ. Sci., 2014, 7, 3381–3390 CAS.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760–7771 CrossRef CAS PubMed.
- G. Babu, K. Ababtain, K. Y. S. Ng and L. M. R. Arava, Sci. Rep., 2015, 5, 8763–8770 CrossRef CAS PubMed.
- J. J. Chen, R. M. Yuan, J. M. Feng, Q. Zhang, J. X. Huang, G. Fu, M. S. Zheng, B. Ren and Q. F. Dong, Chem. Mater., 2015, 27, 2048–2055 CrossRef CAS.
- L. C. Gerber, P. D. Frischmann, F. Y. Fan, S. E. Doris, X. Qu, A. Scheuermann, K. A. Persson, Y. M. Chiang and B. A. Helms, Nano Lett., 2015, 16, 549–555 CrossRef PubMed.
- Z. Wei Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331–1337 CrossRef PubMed.
- Z. Li, C. Li, X. Ge, J. Ma, Z. Zhang, Q. Li, C. Wang and L. Yin, Nano Energy, 2016, 23, 15–26 CrossRef CAS.
- C. Ouyang, X. Wang and S. Wang, Chem. Commun., 2015, 51, 14160–14163 RSC.
- G. Xu, J. Yuan, X. Tao, B. Ding, H. Dou, X. Yan, Y. Xiao and X. Zhang, Nano Res., 2015, 8, 3066–3074 CrossRef CAS.
- R. Nie, J. Shi, W. Du, W. Ning, Z. Hou and F. S. Xiao, J. Mater. Chem. A, 2013, 1, 9037–9045 CAS.
- J. Zhu, K. Kailasam, A. Fischer and A. Thomas, ACS Catal., 2011, 1, 342–347 CrossRef CAS.
- Q. Pang, D. Kundu and L. F. Nazar, Mater. Horiz., 2016, 3, 130–137 RSC.
- Y. J. Li, J. M. Fan, M. S. Zheng and Q. F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 CAS.
- H. Al Salem, G. Babu, C. V. Rao and L. M. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta07202j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |