
Polyaniline coated Fe3O4 hollow nanospheres as anode materials for lithium ion batteries†
Xiaoliang
Wang‡
a,
Yanguo
Liu‡
b,
Hongyan
Han
c,
Yanyan
Zhao
d,
Wuming
Ma
d and
Hongyu
Sun
 *de
*de
aCollege of Science, Hebei University of Science and Technology, Shijiazhuang 050018, PR China
bSchool of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao 066004, PR China
cHangzhou Branch of Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Hangzhou 310018, PR China
dNational Center for Electron Microscopy in Beijing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, PR China
eDepartment of Micro- and Nanotechnology, Technical University of Denmark, Kongens Lyngby 2800, Denmark. E-mail: hsun@nanotech.dtu.dk
First published on 12th April 2017
Abstract
Polyaniline (PANI) coated Fe3O4 hollow nanospheres (h-Fe3O4@PANI) have been successfully synthesized and investigated as anode materials for lithium ion batteries (LIBs). The structure and composition analyses have been performed by employing X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS). The results show a good combination between h-Fe3O4 and PANI with a conductive state. When evaluated as anode materials for LIBs, the h-Fe3O4@PANI nanocomposites exhibit excellent LIB performance with enhanced reversible capacity, good cycling performance and rate capability as compared to h-Fe3O4 and solid Fe3O4 (s-Fe3O4) nanospheres. The improved electrochemical performance of the nanocomposite is considered due to the hollow nature of the products and the coated PANI layers.
1. Introduction
Owing to their high energy density, long cycling life, and environmental friendliness, rechargeable lithium ion batteries (LIBs) have achieved great commercial success in portable electronic devices and stationary energy storage systems.1,2 However, the specific energy of current LIBs is still insufficient for many applications due to the low capacity of electrode materials (the capacity of commercial graphite anode is 372 mA h g−1).3,4 In recent years, transition metal oxides have been widely studied as anodes for their high theoretical capacities.5,6 Among these materials, iron oxide, especially Fe3O4, is considered as one of the most promising anodes due to its high specific capacity (∼926 mA h g−1), nontoxicity, natural abundance, low cost, and high electronic conductivity.7–10 Iron oxide anodes also show safety benefits compared with graphite because of their higher potential, which reduces lithium dendrite deposition on the anodes during cycling.11 When matched with suitable materials with a higher voltage platform than commercial LiCoO2 cathodes (∼3.6 V), iron oxides show potential application in electric vehicle or hybrid electric vehicle fields.12 However, the practical using of Fe3O4 material mainly suffers from drastic volume change during charging/discharging cycles, which results in pulverization, subsequent electrical connectivity loss, and thus rapid capacity fading.Many approaches have been proposed to overcome the limitations, such as construction of nanostructured materials with a suitable morphology, anchoring or embedding active materials in a conducting medium (carbon nanotube, reduced graphene oxide, or conducting polymers). The nanostructure facilitates electron and ion transportation, provides more active sites for the storage of lithium ions than its bulk counterpart, and accommodates the local volume change upon charging/discharging cycles.1,13 The incorporation of conducting materials not only lowers the charge-transfer resistance during electrochemical reactions, but also prevents cracking and the breakdown of active materials caused by the volume variation.14–18 Among different conducting materials, polyaniline (PANI) is attractive because of its high conductivity, good environmental stability, and commercial viability with low production cost. Anodes based on metal oxides and PANI usually possess excellent lithium storage properties. For example, an urchin-like Fe2O3 structure coated with PANI shows a large reversible capacity, high rate capability, and excellent cycling stability after 100 cycles.19 Fe2O3 nanorods capped with PANI possess a high capacity of 778 mA h g−1 at a current of 1.0 Ag−1 after 100 cycles.20 Although a wide range of methods including macromolecule-induced self-assembly,21in situ surface polymerization,22,23 plasma polymerization,24 and electropolymerization25 have been employed to synthesize PANI modified Fe3O4 nanostructures, which show excellent electromagnetic wave absorption and pseudocapacitive properties, to the best of our knowledge, there is no report on lithium storage properties of Fe3O4/PANI anodes.
In this work, we synthesized PANI layer coated Fe3O4 hollow nanospheres (h-Fe3O4@PANI) via a facile one-pot hydrothermal and subsequent in situ polymerization method. The as-prepared nanocomposite presents two structural superiorities, in which the hollow inner can reduce the internal stress during the electrochemical process, provide void spaces for active materials from the electrode during volume expansion and the PANI layer can effectively enhance the conductivity. In this way the h-Fe3O4@PANI nanocomposite exhibits excellent cycling performance, reversible capacity, and rate capability as compared with h-Fe3O4 and solid Fe3O4 (s-Fe3O4) nanospheres, making it promising for application in LIBs.
2. Experimental
2.1 Synthesis of h-Fe3O4 nanospheres and h-Fe3O4@PANI composites
All the chemicals were used as received without further purification. The h-Fe3O4 nanospheres were synthesized by a facile one-pot hydrothermal method. In a typical synthesis, iron(III) chloride hexahydrate (FeCl3·6H2O, 0.75 g), sodium citrate (Na3C6H5O7, 1.8 g), and urea (CO(NH2)2, 0.45 g) were dissolved in 50 mL Milli-Q water. Sodium polyacrylate ((C3H3NaO2)n, 0.45 g) was added into the solution under continuous stirring for 1.5 h and sealed in a 100 mL Teflon-lined stainless-steel autoclave at 200 °C for 5 h. The product was then collected by centrifugation and dried in a vacuum oven at 60 °C for 12 h. h-Fe3O4@PANI composites were synthesized by using in situ polymerization of the aniline monomer in the presence of h-Fe3O4 nanospheres. Typically, h-Fe3O4 nanospheres (0.8 g) were dispersed in ethanol (500 mL) by sonication. Subsequently, aniline (C6H5NH2, 0.8 mL) was injected into the above suspension. Then HCl aqueous solution (25.0 mL, 1.21 M) containing ammonium peroxydisulfate ((NH4)2S2O8, APS) (2.48 g) was added into the ethanol suspension. For full polymerization of aniline, the mixture was mechanically stirred at 0 °C for 24 h. After washing with distilled water and ethanol several times, the product was dried under vacuum at 50 °C for 8 h.2.2 Materials characterization
A powder X-ray diffractometer (XRD) with Cu Kα irradiation (λ = 1.5418 Å, Bruker Model D8) was used to detect the crystallographic information for the samples. The morphology and microstructure of the products were examined using field-emission scanning electron microscopy (FESEM; Hitachi-S5500, 5 kV; Zeiss Merlin, 5 kV) and transmission electron microscopy (TEM; JEOL, JEM-2100, 200 kV; FEI, Tecnai G2 20, 200 kV). X-ray photoelectron spectroscopy (XPS, Escalab 250, Al Kα) was employed to probe the surface chemical states. Thermal gravimetric analysis (TGA, Netzsch-STA 449C) was measured from room temperature to 800 °C at a heating rate of 10 °C min−1 under an air flow. Fourier transform infrared spectroscopy (FTIR) spectra were recorded on a PE (Spotlight 3000) spectrometer in the 400–4000 cm−1 region to investigate the functional groups of the samples. The Brunauer–Emmett–Teller (BET) surface area of the products was analyzed by using nitrogen adsorption–desorption isotherms at 77 K in a Micromeritics ASAP 2010 system. The adsorption isotherm was used to determine the pore size distribution via the Barret–Joyner–Halender (BJH) method. The average pore size was determined based on the nitrogen adsorption volume at arelative pressure of 0.994.2.3 Electrochemical measurements
The working electrodes with a typical thickness of 30–40 μm were prepared by mixing the active materials, conductive carbon black and carboxymethyl cellulose in a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the N-methyl-2-pyrrolidone solvent, with the following addition of a suitable amount of deionized water. The obtained slurry was then coated on a Cu foil (current collector) and dried under vacuum at 80 °C overnight. The obtained electrodes were punched and dried again at 120 °C for 6 h. The loading mass of the active materials on the Cu foil was about 2.5–3 mg cm−2 for each electrode. Testing cells (CR 2032 coin-type half cells) were assembled inside an Ar-filled glove box by using a lithium metal foil as the counter electrode and the reference electrode and microporous polypropylene as the separator. The electrolyte used was 1 M lithium hexafluorophosphate (LiPF6) dissolved in a mixture of ethylene carbonate, propylene carbonate, and diethyl carbonate (3:1:1 in volume ratio). Cyclic voltammogram (CV) curves were recorded between 0.05 and 3 V (vs. Li+/Li) at a scan rate of 0.5 mV s−1. Galvanostatic charge and discharge of the cells were performed at a current density of 100 mA g−1 between a voltage window of 0.05 and 3 V (vs. Li+/Li). For the rate performance test, the discharge current gradually increased from 50 mA g−1 to 100, 500, 1000, 2000, 4000, and 5000 mA g−1, and then decreased to 50 mA g−1. Electrochemical impedance spectroscopy was conducted at open circuit potential (OCP) by applying an alternating current voltage of 5 mV over a frequency range of 100 kHz to 0.01 Hz. All the electrochemical tests were performed symmetrically at room temperature.
:1:1 in the N-methyl-2-pyrrolidone solvent, with the following addition of a suitable amount of deionized water. The obtained slurry was then coated on a Cu foil (current collector) and dried under vacuum at 80 °C overnight. The obtained electrodes were punched and dried again at 120 °C for 6 h. The loading mass of the active materials on the Cu foil was about 2.5–3 mg cm−2 for each electrode. Testing cells (CR 2032 coin-type half cells) were assembled inside an Ar-filled glove box by using a lithium metal foil as the counter electrode and the reference electrode and microporous polypropylene as the separator. The electrolyte used was 1 M lithium hexafluorophosphate (LiPF6) dissolved in a mixture of ethylene carbonate, propylene carbonate, and diethyl carbonate (3:1:1 in volume ratio). Cyclic voltammogram (CV) curves were recorded between 0.05 and 3 V (vs. Li+/Li) at a scan rate of 0.5 mV s−1. Galvanostatic charge and discharge of the cells were performed at a current density of 100 mA g−1 between a voltage window of 0.05 and 3 V (vs. Li+/Li). For the rate performance test, the discharge current gradually increased from 50 mA g−1 to 100, 500, 1000, 2000, 4000, and 5000 mA g−1, and then decreased to 50 mA g−1. Electrochemical impedance spectroscopy was conducted at open circuit potential (OCP) by applying an alternating current voltage of 5 mV over a frequency range of 100 kHz to 0.01 Hz. All the electrochemical tests were performed symmetrically at room temperature.
3. Results and discussion
The XRD patterns of the as-prepared h-Fe3O4@PANI and h-Fe3O4 are shown in Fig. 1. All diffraction peaks of h-Fe3O4 and the h-Fe3O4@PANI composite are in good agreement with the standard pattern of magnetite Fe3O4 with a cubic inverse spinel structure (JCPDS card No. 19-0629). No other peaks from possible impurities are observed, indicating the high purity of the products. It is worth noting that no distinct diffraction peaks corresponding to PANI are detected which may due to its amorphous nature. The FTIR spectra (Fig. 2) show a similar trend for the h-Fe3O4@PANI composite and PANI. For PANI, the characteristic bands located at 1572 and 1477 cm−1 are ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the quinoid and benzene rings, demonstrating the presence of a conducting state of PANI (emeraldine salt).26 The bands located at 1300, 1123, and 832 cm−1 correspond to the C–N stretching vibrations of the secondary aromatic amine, CN stretching vibrations (–NquinoidN–), and out-of-plane bending vibrations of the C–H band in the aromatic ring, respectively.27 In addition, the bands between 800 and 500 cm−1 are attributed to the C–H vibration of benzene rings.28 Remarkably, the FTIR spectrum of the h-Fe3O4@PANI composite shows combined characteristics of both PANI and h-Fe3O4.29 The intense vibration band for PANI (∼1123 cm−1) related to the CN stretching in aromatic amines decreases. The possible reason is that the benzoid ring structure in the initial PANI polymer is transferred into semiquinoid or quinoid ones upon the compositing with h-Fe3O4 nanospheres.30
C stretching vibration of the quinoid and benzene rings, demonstrating the presence of a conducting state of PANI (emeraldine salt).26 The bands located at 1300, 1123, and 832 cm−1 correspond to the C–N stretching vibrations of the secondary aromatic amine, CN stretching vibrations (–NquinoidN–), and out-of-plane bending vibrations of the C–H band in the aromatic ring, respectively.27 In addition, the bands between 800 and 500 cm−1 are attributed to the C–H vibration of benzene rings.28 Remarkably, the FTIR spectrum of the h-Fe3O4@PANI composite shows combined characteristics of both PANI and h-Fe3O4.29 The intense vibration band for PANI (∼1123 cm−1) related to the CN stretching in aromatic amines decreases. The possible reason is that the benzoid ring structure in the initial PANI polymer is transferred into semiquinoid or quinoid ones upon the compositing with h-Fe3O4 nanospheres.30
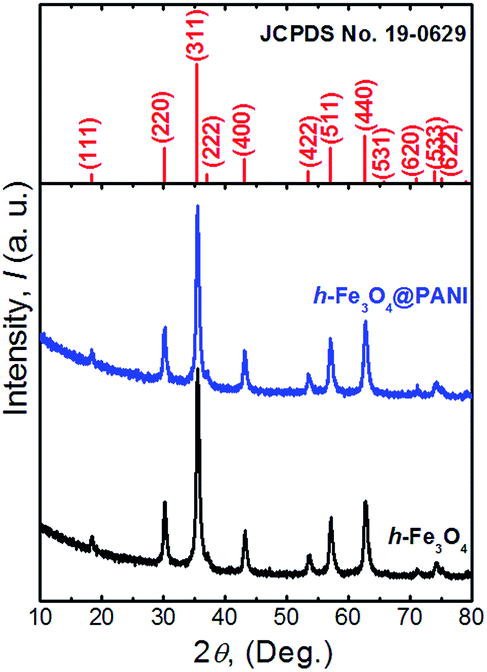 | ||
| Fig. 1 XRD patterns of the as-prepared h-Fe3O4, h-Fe3O4@PANI and the standard pattern of the Fe3O4 phase. | ||
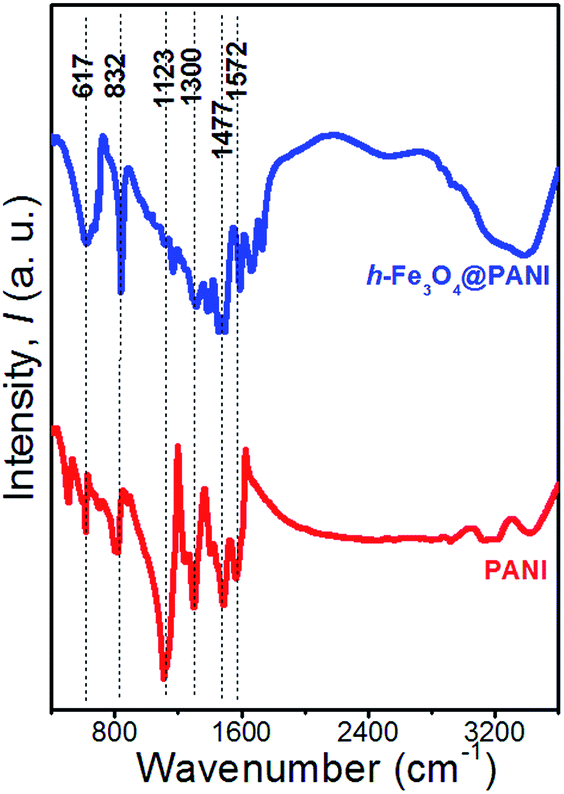 | ||
| Fig. 2 FTIR spectra of PANI and h-Fe3O4@PANI samples. | ||
The thermal behavior of the as-prepared h-Fe3O4@PANI composite and pure PANI is investigated by TGA and derivative thermogravimetric (DTG) curves (Fig. S1†). The small weight loss before 100 °C is attributed to the evaporation of moisture, gaseous content and trace ethanol in the sample. As the temperature rises, the rapid mass loss of about 40.5 wt% taking place in the range of 100–400 °C is ascribed to the combustion of PANI and oxidation of Fe3O4 to Fe2O3 (4Fe3O4 + O2 = 6Fe2O3) in the air atmosphere.31–33 According to the change in weight and the reaction equation, the estimated content of Fe3O4 and PANI in the composite is about 48.8 wt% and 51.2 wt%, respectively.
The low and high magnification FESEM images (Fig. 3a and b) of the as-prepared h-Fe3O4@PANI composite confirm the formation of a large number of nanospheres with a rough surface. From the typical low magnification TEM images (Fig. 3c–f), it can be found that the nanospheres show a hollow nature with an average diameter in the range of 200–300 nm. The core–shell structure is unambiguously resolved from the magnified TEM images (Fig. 3g and h). The PANI with a lower contrast shell is coated on to the h-Fe3O4 nanospheres. It is further showed that the nanospheres are assembled by primary particles with a size of 10–20 nm and there exists a porous gap between each particle. The selected area electron diffraction (SAED) pattern (inset in Fig. 3c) shows the polycrystalline nature of the h-Fe3O4@PANI.
 | ||
| Fig. 3 (a and b) FESEM images, (c–h) TEM images, (i and j) HRTEM images of the h-Fe3O4@PANI sample. The inset in (c) shows the SAED pattern of the h-Fe3O4@PANI sample. The white and blue arrows in (b) and (g) show the existence of PANI coatings. | ||
Fig. 3i and j show HRTEM images of an individual nanosphere. The lattice spacings of d ∼ 2.55 Å, 2.93 Å and 4.87 Å correspond to the (311), (220) and (111) planes of Fe3O4. The PANI layer is amorphous because no lattice fringe is observed. This is in agreement with the XRD results. The high conductive PANI coated layer can be a key factor to enhance the electrochemical performance not only because the conductivity of the composites is improved but they can also play a role as a buffering material due to its flexibility.18,19 Typical FESEM and TEM images of the h-Fe3O4 sample are shown in Fig. S2.† The EDS spectra (Fig. S3 and S4†) and the elemental mapping images (Fig. S5†) of the two samples further confirm the distribution of Fe, O, N and agree well with the TEM results.
The specific surface areas and the porous nature of the samples are determined by measuring nitrogen adsorption–desorption isotherms at 77 K (Fig. 4a and b). Both samples exhibit type II adsorption–desorption isotherms according to IUPAC classification, indicating that the samples are composed of nanoparticles.34 It is also shown that only a large adsorption is observed at relative pressures close to 1. Therefore the adsorption mainly occurs in the interparticle voids with a mesopore range. The corresponding BET specific surface areas are ∼109.4 and 124.8 m2 g−1 for h-Fe3O4@PANI and h-Fe3O4, respectively. The adsorption isotherm was used to determine pore size distribution based on the BJH method (Fig. 4b). The pore size of the h-Fe3O4@PANI and h-Fe3O4 samples distribute in 5–30 nm, which reveals the existence of mesopores. The mesopores mainly originate from the aggregation of primary nanoparticles within h-Fe3O4@PANI and h-Fe3O4 nanospheres as shown by the TEM results. The surface chemical states of the h-Fe3O4@PANI composite and h-Fe3O4 are determined by XPS (Fig. 5a–d, S6–S8†). The survey spectrum of the h-Fe3O4@PANI composite (Fig. 5a) shows the presence of Fe, O, N, and C peaks, which is in agreement with previous EDS results.
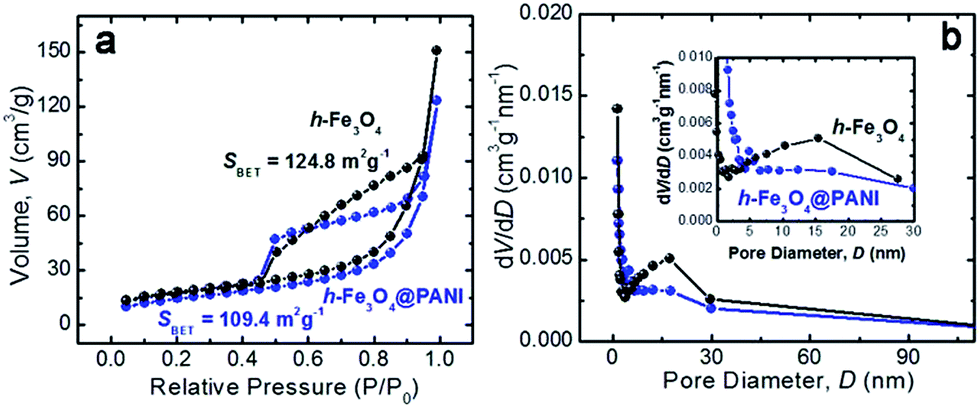 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms and ((b) & inset) the corresponding pore size distribution curves of h-Fe3O4 (black) and h-Fe3O4@PANI (blue) samples. | ||
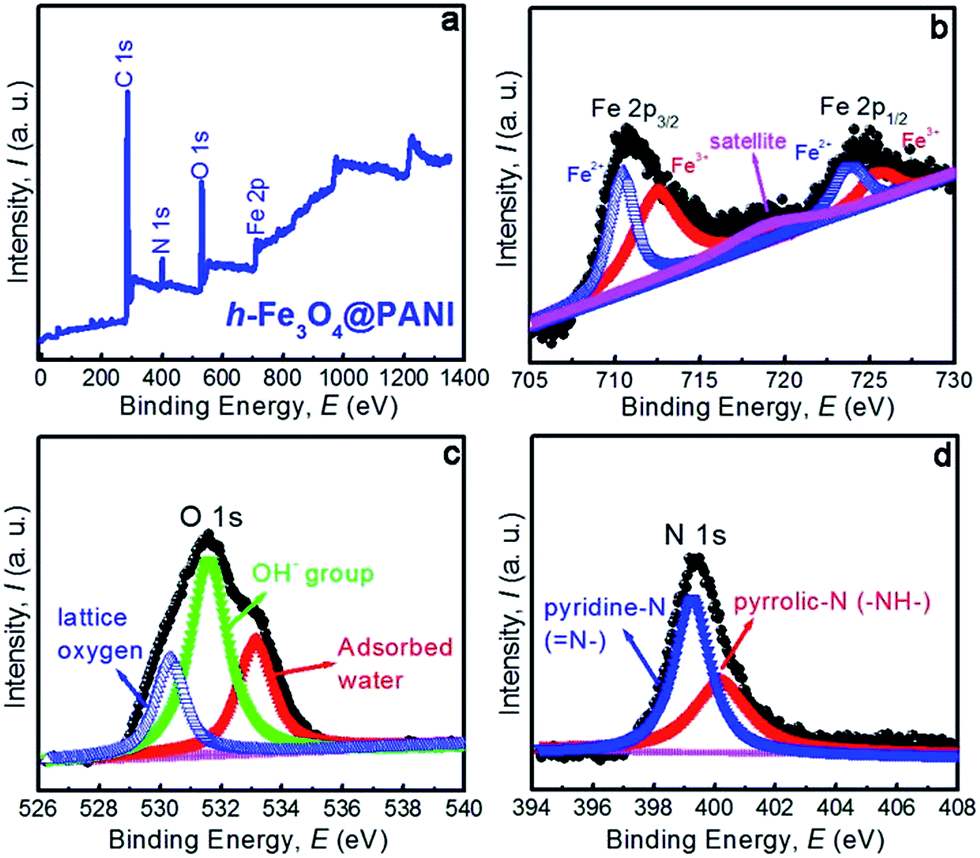 | ||
| Fig. 5 (a) XPS survey spectra of the h-Fe3O4@PANI sample, (b–d) high-resolution XPS spectra of the Fe 2p, O 1s, and N 1s regions, respectively. | ||
The de-convoluted scan of Fe 2p (Fig. 5b) shows two distinct binding energy peaks located at 711.64 and 724.72 eV corresponding to the electronic states of Fe 2p3/2 and Fe 2p1/2 respectively, which can be fitted with two spin–orbit doublets and a shakeup satellite. The doubles are characteristic of the peaks of Fe2+ and Fe3+. The results are consistent with the reported values of Fe3O4.35–37 The Fe3+/Fe2+ ratio calculated from the peak area is 2.04, which is close to 2:1 in Fe3O4. Moreover, the satellite peak situated at about 719.4 eV is a characteristic peak of Fe3+ in γ-Fe2O3, suggesting that the surface of h-Fe3O4 is partly oxidized.35,36 The O 1s peak (Fig. 5c) is deconvoluted into three components with peaks at 530.38, 531.66 and 533.12 eV, which can be assigned to lattice oxygen, oxygen vacancies or defects related OH-group, and molecular water in the sample or adsorbed on the surface. The present results are in good agreement with those of the previous report.38 The de-convolution of the N 1s peak (Fig. 5d) shows two distinct curves, related to different nitrogen forms. The peak at 399.2 eV corresponds to pyridine-N (N–),39 and the peak at 400.1 eV may be assigned to pyrrolic-N (–NH–).40 The results are also consistent with the data as demonstrated in the literature.41,42
The electrochemical properties of the assembled cell are investigated by CV between 0.05 and 3 V at a scan rate of 0.5 mV s−1. The CV curves of the first three cycles for the h-Fe3O4@PANI cell are shown in Fig. S9.† In the first cycle, there is a large reduction peak at 0.47 V, which can be ascribed to the irreversible conversion from Fe3O4 to Fe/Li2O upon Li uptake.43,44 Moreover, the anodic peak at 1.72 V is attributed to the oxidation of the metallic iron into Fe3O4.43,44 In the subsequent cycles, both the cathodic and anodic peaks are shifted towards a higher potential due to the irreversible change of Fe3O4 from the spinel to the rock salt type structure during the initial insertion of lithium ions.45 In addition, the current of the cathodic and anodic peaks decreased obviously, which is due to the formation of a solid electrolyte interphase (SEI) film.45 In the CV graph of the h-Fe3O4 cell (Fig. S10†), a pair of cathodic and anodic peaks appears at 0.63 and 1.68 V in the first scan. In the following second and third cycles, the cathodic peak shifts to 0.75 V, while the anodic peak position does not show any obvious change. The galvanostatic charge and discharge measurements of the assembled cells are performed at a rate of 100 mA g−1 in the voltage range of 0.05–3 V (versus Li/Li+) at room temperature. Fig. 6a shows the charge–discharge voltage profiles of the h-Fe3O4@PANI cell for the first three cycles. The first discharge profile exhibits three plateaus (∼1.44 V, 0.91 V and 0.77 V) and a following slope. The three plateaus represent a three-step lithium ion insertion process. The slope is attributed to the formation of an SEI film and lithium ion insertion into the polymer material. The characters of multiple discharge plateaus have also been observed in the Fe3O4/graphene sheet composite and spinel transition metal oxide anodes.37,46 The plateau around 0.77 V confirms the existence of a conversion process for the lithiation of the h-Fe3O4@PANI electrode. In the charging state, the reactions occur in the reverse direction and Li+ is released at a voltage of ∼1.5 V. The initial discharge and charge capacities are 1800 and 1350 mA h g−1 for h-Fe3O4@PANI electrodes respectively. The values are found to be much higher than those of h-Fe3O4 nanospheres and the results recently reported by other groups, such as 1300–990 mA h g−1 for hierarchical Fe3O4@PPy nanocages,47 972–880 mA h g−1 for hollow Fe3O4 microspheres48–50 and 1550–1080 mA h g−1 for other nanostructured iron oxide anodes.51,52 The irreversible capacity loss arising during the first cycle is due to the incomplete conversion reaction and the formation of the SEI layer. The charge–discharge voltage profiles of the h-Fe3O4 cell for the first three cycles are also shown in Fig. S11.† In the first discharge curve, there is one clear potential plateau at ∼0.69 V due to the conversion from Fe3O4 to Fe, followed by a sloping curve down to the cutoff voltage, which is in agreement with that of the reported results for Fe3O4 anodes.53,54Fig. 6b shows the comparison of cycling performance of the h-Fe3O4@PANI, h-Fe3O4, and s-Fe3O4 cells at a current density of 100 mA g−1 in a voltage range of 0.05–3 V (versus Li+/Li) up to 50 cycles. It can be seen that the h-Fe3O4@PANI cell exhibits much better cycling performance. It is also found that the h-Fe3O4@PANI cell possesses the highest lithium storage capacity, and the reversible capacity reaches ∼1090 mA h g−1 after 50 cycles, which decreases to ∼956 mA h g−1 for the h-Fe3O4 cell and 335 mA h g−1 for the s-Fe3O4 cell. It is noticed that the reversible capacity of the h-Fe3O4@PANI and h-Fe3O4 cells is higher than the theoretical value of Fe3O4. We first evaluated the contribution of PANI to the capacity of h-Fe3O4@PANI and tested the capacity of pristine PANI at the same current density. The result shows that the capacity is only 38 mA h g−1 for PANI, which is obviously lower than the theoretical capacity of Fe3O4 and comparable with the values reported elsewhere.20,55–58 Therefore the effect of PANI addition on the overall specific capacity can be considered as negligible, and the additional reversible capacity beyond the theoretical capacity originates from the Fe3O4 electrode. Actually, this phenomenon is generally observed in other conversion based electrodes.59 There are several different mechanisms that have been proposed to reveal the possible origin of the extra capacity. For example, Laruelle et al. showed that the polymeric gel-like film formed at the grain boundaries between nanoparticles at a low potential range enables extra lithium storage on its surface through a capacitive way.60 Maier et al. proposed that the interfaces between the dispersively distributed metal nanoparticles and lithium binary compound matrix allow for the storage of lithium ions on the lithium compound side, and electrons are localized on the metallic side.61,62 Hu and co-workers demonstrated that the contribution to the extra capacity comes from the reversible reaction of LiOH and Li to form Li2O and LiH.63 Apart from those, the reactions of the Li2O phase64 and the further evolution of the SEI film65 are also found to be related with the additional capacities. In our case, the extra capacity was maintained with the cell cycling, which is in agreement with Laruelle's work. Considering the similar reaction processes of Fe3O4 and CoO60 electrodes, we think that the formation of a polymeric gel-like film is responsible for the extra capacity. Moreover, the contributions from other reversible side reactions cannot be neglected.63,66 It should be noted that a full understanding of the extra capacity relies on the experimental ability to in situ identify nanoscale structures present at the electrode/salt interfaces under real working conditions.
 | ||
| Fig. 6 Electrochemical performance of the h-Fe3O4@PANI sample: (a) galvanostatic charge/discharge curves of h-Fe3O4@PANI for the first three cycles between 0.05 V and 3.00 V (vs. Li+/Li) at a current density of 100 mA g−1, (b) cycling performance and (c) rate capability of the h-Fe3O4@PANI (blue lines), h-Fe3O4 (black lines) and s-Fe3O4 (red lines) electrodes at different current densities between 50 and 5000 mA g−1. | ||
Compared to h-Fe3O4 and s-Fe3O4 cells, the improved capacity and cycle life of the h-Fe3O4@PANI cell may be considered due to the large specific surface area and porous nature of nanospheres which is more convenient and accessible for electrolyte diffusion and intercalation of lithium ions into the active phases. The presence of the PANI layers also reduces the exposed surface of active material to the electrolyte and results in a more conductive and stable SEI film, which is favorable to the reversible capacity improvement.1 The subsequent coulombic efficiency quickly increases to 98%, 96% and 90% for h-Fe3O4@PANI, h-Fe3O4 and s-Fe3O4 electrodes, respectively. The electrochemical performance of the electrodes at different current densities between 50 and 5000 mA g−1 is shown in Fig. 6c. The charge/discharge rates are programmable modified from 50 mA g−1 to 100, 500, 1000, 2000, 4000, 5000 and then back to 50 mA g−1 for 10 cycles. The reversible capacity of h-Fe3O4@PANI cell varies from 1220.7 mA h g−1 to 714.6 mA h g−1 at current densities of 50 mA g−1 and 1000 mA g−1, respectively, while the reversible capacity of the h-Fe3O4 cell drops from 1139.4 mA h g−1 to 670.4 mA h g−1, which are lower than but still close to those of the h-Fe3O4@PANI cell. However, at higher current densities of 2000, 4000, and 5000 mA g−1, the h-Fe3O4@PANI cell exhibits remarkably higher capability than that of the h-Fe3O4 cell. The reversible capacities of the h-Fe3O4@PANI cell are 642 and 549.5 mA h g−1 at the current densities of 2000 and 4000 mA g−1. Even at a high rate of 5000 mA g−1, the h-Fe3O4@PANI cell still shows a capacity as high as 439 mA h g−1. In contrast, the capacity retention of the h-Fe3O4 cell at high current densities (2000 to 5000 mA g−1) is inferior. For example, the capacity is 408, 169.2, and 32 mA h g−1 at the current densities of 2000, 4000, and 5000 mA g−1, respectively. When the rate returns to the initial 50 mA g−1 after 80 cycles, the h-Fe3O4@PANI and h-Fe3O4 cells can go back to their original capacities to some extent as illustrated in Fig. 6c. For the s-Fe3O4 cell, the rate performance is inferior to the h-Fe3O4@PANI and h-Fe3O4 cells in the whole current density range. The results show the positive influence of PANI addition and the importance of hollowed nanostructures inrate performance enhancement, especially at higher current densities. Specifically, the PANI layer offers a rapid conductive network connecting the h-Fe3O4 spheres, while the porous structure can reduce the effective diffusion distance for lithium ions and electrons, thus leading to good rate capability. The reversible capacity and rate capability of the present h-Fe3O4@PANI composite anodes is found to be much higher than the values recently reported by other groups as shown in Table S1.†
EIS is carried out at OCP to determine the lithium ion transfer behavior in the cells after the 1st and 50th cycles. As shown in Fig. 7, all the Nyquist plots of the h-Fe3O4@PANI, h-Fe3O4 and s-Fe3O4 cells exhibit a single semicircle in the high to medium frequency region and an inclined line in the low frequency region, which are attributed to the charge-transfer process and the lithium ion diffusion process inside the electrode material.15–17,51,52 The semicircle diameter of the h-Fe3O4@PANI cell is found to be much smaller than those of the h-Fe3O4 and s-Fe3O4 cells, indicating that the addition of PANI enhances the charge transfer process, which is beneficial for improving rate capability. This is in good agreement with the existing reports that PANI can greatly reduce electron transfer resistance in the composite.58 This will lead to high utilization of the cell even under high rate discharge conditions.67 After the 50th cycle, the charge-transfer resistances increase, which are attributed to the formation of SEI films,68 inducing the lower conductivity of the electrodes. The morphology and structure of the h-Fe3O4@PANI electrodes after the rate capability testing (50 cycles) were also characterized by TEM observations (Fig. S12†). The sample still maintains the initial morphology and structure after the cycling test, demonstrating the good stability of the nanocomposite structures during charge/discharge cycling.
 | ||
| Fig. 7 Nyquist plots of the h-Fe3O4@PANI (blue), h-Fe3O4 (black) and s-Fe3O4 (red) electrodes after the first (solid) and the 50th (hollow) cycles at the current density of 100 mA g−1 with an amplitude of 5 mV over the frequency range of 100 kHz and 0.01 Hz. | ||
The good lithium storage properties of h-Fe3O4@PANI nanocomposite can be attributed to the following reasons. (1) The hollow nature of the Fe3O4 nanospheres provides extra active sites for the storage of lithium ions, which is beneficial for enhancing the specific capacity. (2) The addition of PANI can accommodate the local volume change upon charge/discharge cycling and is able to alleviate the problem of pulverization and aggregation of the electrode material, hence improving the cycling performance. (3) The PANI has a good electrical conductivity and serves as the conductive channels between h-Fe3O4, which decrease the inner resistance of LIBs. Moreover, the porous structure can reduce the effective diffusion distance for lithium ions and electrons, also resulting in good rate capabilities.
4. Conclusions
In summary, h-Fe3O4@PANI nanocomposites have been successfully synthesized. When used as the anode materials for LIBs, the as-prepared h-Fe3O4@PANI nanocomposites delivered high capacity, good cycling stability and high rate capability as compared to h-Fe3O4 and s-Fe3O4 nanospheres. The good electrochemical performance can be attributed to the hollow nature of the products, the addition of PANI, and the unique assembled architecture. We believe that the lithium storage properties of the h-Fe3O4@PANI nanocomposites can be further optimized by tailoring the inner space and PANI thickness. The excellent electrochemical performance combined with the low-cost Fe element endows h-Fe3O4@PANI nanocomposites with high potential in next generation LIBs.Acknowledgements
The authors would like to appreciate the financial support from the Chinese National Natural Science Foundation (51401114 and 51571054). This work made use of the resources of the National Center for Electron Microscopy in Beijing.Notes and references
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E63 CrossRef CAS PubMed.
- J. F. V. Bulow, H. L. Zhang and D. E. Morse, Adv. Energy Mater., 2012, 2, 309–315 CrossRef.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- T. H. Kim, J. S. Park, S. K. Chang, S. Choi, J. H. Ryu and H. K. Song, Adv. Energy Mater., 2012, 2, 860–872 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- S. Mitra, P. Poizot, A. Finke and J. M. Tarascon, Adv. Funct. Mater., 2006, 16, 2281–2287 CrossRef CAS.
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495–6502 CAS.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- S.-H. Yu, S. H. Lee, D. J. Lee, Y.-E. Sung and T. Hyeon, Small, 2016, 12, 2146–2172 CrossRef CAS PubMed.
- N. Mahmood, T. Y. Tang and Y. L. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- F. Bonaccorso, L. Colombo, G. H. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- S. Han, D. Q. Wu, S. Li, F. Zhang and X. L. Feng, Small, 2013, 9, 1173–1187 CrossRef CAS PubMed.
- Y. G. Liu, Z. Y. Cheng, H. Y. Sun, H. Arandiyan, J. P. Li and M. Ahmad, J. Power Sources, 2015, 273, 878–884 CrossRef CAS.
- H. Y. Sun, Y. G. Liu, Y. L. Yu, M. Ahmad, D. Nan and J. Zhu, Electrochim. Acta, 2014, 118, 1–9 CrossRef CAS.
- H. Y. Sun, M. Ahmad, J. Luo, Y. Y. Shi, W. C. Shen and J. Zhu, Mater. Res. Bull., 2014, 49, 319–324 CrossRef CAS.
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, S. J. Chang, Y. K. Han, Y. B. Lee, H. U. Lee, S. Kwon, G. Lee, C. S. Lee and Y. S. Huh, Adv. Mater., 2013, 25, 6250–6255 CrossRef CAS PubMed.
- S. B. Wang, L. W. Hu, Y. J. Hu and S. Q. Jiao, Mater. Chem. Phys., 2014, 146, 289–294 CrossRef CAS.
- J. Huang, Q. Li, D. Li, Y. Wang, L. Dong, H. Xie, J. Wang and C. X. Xiong, Langmuir, 2013, 29, 10223–10228 CrossRef CAS PubMed.
- S. H. Xuan, Y. J. Wang, K. C. Leung and K. Y. Shu, J. Phys. Chem. C, 2008, 112, 18804–18809 CAS.
- Y. P. Sun, F. Xiao, X. g. Liu, C. Feng and C. G. Jin, RSC Adv., 2013, 3, 22554–22559 RSC.
- Y. R. Wang, H. G. Wei, J. M. Wang, J. R. Liu, J. Guo, X. Zhang, B. L. Weeks, T. D. Shen, S. Y. Wei and Z. H. Guo, J. Mater. Chem. A, 2015, 3, 20778–20790 CAS.
- L. K. Joy, V. Sooraj, U. S. Sajeev, S. S. Nair, T. N. Narayanan, N. Sethulakshmi, P. M. Ajayan and M. R. Anantharaman, Appl. Phys. Lett., 2014, 104, 121603 CrossRef.
- C. Vallés, P. Jiménez, E. Muñoz, A. M. Benito and W. K. Maser, J. Phys. Chem. C, 2011, 15, 10468–10474 Search PubMed.
- G. G. Kumar, C. J. Kirubaharan, S. Udhayakumar, C. Karthikeyan and K. S. Nahm, Ind. Eng. Chem. Res., 2014, 53, 16883–16893 CrossRef.
- G. H. Xu, D. D. Xu, J. N. Zhang, K. X. Wang, Z. M. Chen, J. F. Chen and Q. Xu, J. Colloid Interface Sci., 2013, 411, 204–212 CrossRef CAS PubMed.
- A. Mukhopadhyay, N. Joshi, K. Chattopadhyay and G. De, ACS Appl. Mater. Interfaces, 2012, 4, 142–149 CAS.
- A. Kellenberger, E. Dmitrieva and L. Dunsch, J. Phys. Chem. B, 2012, 116, 4377–4385 CrossRef CAS PubMed.
- A. Jafari, S. FarjamiShayesteh, M. Salouti and K. Boustani, J. Magn. Magn. Mater., 2015, 379, 305–312 CrossRef CAS.
- S. Nasrazadani and A. Raman, Corros. Sci., 1993, 34, 1355–1365 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, Wiley-VCH Verlag GmbH & Co, Weinheim, Germany, 2003, p. 402 Search PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of porous solids and powders: surface area, pore size and density, Springer, Netherlands, 2004, p. 43 Search PubMed.
- Q. Gao, A. W. Zhao, Z. B. Gan, W. Y. Tao, D. Li, M. F. Zhang, H. Y. Guo, D. P. Wang, H. H. Sun, R. R. Mao and E. H. Liu, CrystEngComm, 2012, 14, 4834–4842 RSC.
- X. D. Zhu, K.-X. Wang, D.-J. Yan, S.-R. Le, R.-J. Ma, K.-N. Sun and Y.-T. Liu, Chem. Commun., 2015, 51, 11888–11891 RSC.
- X. F. Meng, Y. L. Xu, X. F. Sun, J. Wang, L. L. Xiong, X. F. Du and S. C. Mao, J. Mater. Chem. A, 2015, 3, 12938–12946 CAS.
- L. Li, Y. Li, S. Y. Gao and N. Koshizaki, J. Mater. Chem., 2009, 19, 8366–8371 RSC.
- F. Jaouen, J. Herranz, M. Lefevre, J. P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CAS.
- R. L. Arechederra, K. Artyushkova, P. Atanassov and S. D. Minteer, ACS Appl. Mater. Interfaces, 2010, 2, 3295–3302 CAS.
- E. T. Kang, K. G. Neoh and K. L. Tan, Prog. Polym. Sci., 1998, 23, 277–324 CrossRef CAS.
- K. Wang, Y. Huang, D. Wang, Y. Zhao, M. Wang, X. Chen, X. Qina and S. Li, RSC Adv., 2015, 5, 107247–107253 RSC.
- H. Liu, G. X. Wang, J. Z. Wang and D. Wexler, Electrochem. Commun., 2008, 10, 1879–1882 CrossRef CAS.
- B. Jin, A. H. Liu, G. Y. Liu, Z. Z. Yang, X. B. Zhong, X. Z. Ma, M. Yang and H. Y. Wang, Electrochim. Acta, 2013, 90, 426–432 CrossRef CAS.
- L. S. Shen, H. W. Song, H. Cui, X. Wen, X. L. Wei and C. X. Wang, CrystEngComm, 2013, 15, 9849–9854 RSC.
- K. He, S. Zhang, J. Li, X. Q. Yu, Q. P. Meng, Y. Z. Zhu, E. Y. Hu, K. Sun, H. Yun, X. Q. Yang, Y. M. Zhu, H. Gan, Y. F. Mo, E. A. Stach, C. B. Murray and D. Su, Nat. Commun., 2016, 7, 11441 CrossRef CAS PubMed.
- J. Liu, X. J. Xu, R. Z. Hu, L. C. Yang and M. Zhu, Adv. Energy Mater., 2016, 6, 1600256 CrossRef.
- D. Wang, L. Jiang, X. Wei and C. Song, Mater. Lett., 2015, 138, 164–166 CrossRef CAS.
- T. H. Kim and H. K. Song, Electrochim. Acta, 2013, 105, 47–52 CrossRef CAS.
- H. S. Lim, B. Y. Jung, Y. K. Sun and K. D. Suh, Electrochim. Acta, 2012, 75, 123–130 CrossRef CAS.
- S. Chaudhari and M. Srinivasan, J. Mater. Chem., 2012, 22, 23049–23056 RSC.
- X. Jiang, X. L. Yang, Y. Zhu, Y. F. Yao, P. Zhao and C. Z. Li, J. Mater. Chem. A, 2015, 3, 2361–2369 CAS.
- S. Q. Wang, J. Y. Zhang and C. H. Chen, J. Power Sources, 2010, 195, 5379–5381 CrossRef CAS.
- Y. Han, Y. Wang, L. Li, Y. Wang, L. Jiao, H. Yuan and S. Liu, Electrochim. Acta, 2011, 56, 3175–3181 CrossRef CAS.
- M. Y. Feng, J. H. Tian, H. M. Xie, Y. L. Kang and Z. Q. Shan, J. Solid State Electrochem., 2015, 19, 1773–1782 CrossRef CAS.
- Q. Zhou, L. Liu, Z. F. Huang, L. G. Yi, X. Y. Wang and G. Z. Cao, J. Mater. Chem. A, 2016, 4, 5505–5516 CAS.
- G. Wang, J. Peng, L. L. Zhang, J. Zhang, B. Dai, M. Y. Zhu, L. L. Xia and F. Yu, J. Mater. Chem. A, 2015, 3, 3659–3666 CAS.
- R. Ma, M. Wang, D. T. Dam, Y. J. Yoon, Y. Chen and J. M. Lee, ChemElectroChem, 2015, 2, 503–507 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. Rosa Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dollé, L. Dupont and J.-M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621–625 CrossRef CAS.
- Y. F. Zhukovskii, P. Balaya, E. A. Kotomin and J. Maier, Phys. Rev. Lett., 2006, 96, 058302 CrossRef PubMed.
- Y.-Y. Hu, Z. Liu, K.-W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L.-S. Du, K. W. Chapman, P. J. Chupas, X.-Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
- S.-Y. Lee, K.-Y. Park, W.-S. Kim, S. Yoon, S.-H. Hong, K. Kang and M. Kimn, Nano Energy, 2016, 19, 234–245 CrossRef CAS.
- S. J. Rezvani, R. Gunnella, A. Witkowska, F. Mueller, M. Pasqualini, F. Nobili, S. Passerini and A. D. Cicco, ACS Appl. Mater. Interfaces, 2017, 9, 4570–4576 CAS.
- A. Ponrouch, P.-L. Taberna, P. Simon and M. R. Palacín, Electrochim. Acta, 2012, 61, 13–18 CrossRef CAS.
- Z. H. Yang and H. Q. Wu, Solid State Ionics, 2001, 143, 173–180 CrossRef CAS.
- M. Pfanzelt, P. Kubiak, S. Jacke, L. Dimesso, W. Jaegermann and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2012, 159, A809 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: TGA and DTG plots, additional SEM and TEM images, XPS spectra, electrochemical results, and Table. See DOI: 10.1039/c7se00139h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |