
Highly active, durable and pH-universal hybrid oxide nanocrystals for efficient oxygen evolution†
Yahia H.
Ahmad‡
a,
Kamel A.
Eid‡
a,
Siham Y.
AlQaradawi
 a and
Nageh K.
Allam
*b
a and
Nageh K.
Allam
*b
aDepartment of Chemistry and Earth Sciences, College of Arts and Sciences, Qatar University, Doha 2713, Qatar
bEnergy Materials Lab (EML), School of Sciences and Engineering, The American University in Cairo, New Cairo 11835, Egypt. E-mail: nageh.allam@aucegypt.edu
First published on 10th April 2017
Abstract
The development of an active, durable, and low-cost catalyst remains hitherto a grand challenge in energy conversion technologies. Herein, hybrid Ptx–NiMnO3 nanocrystals (NCs) (x = 0.1, 0.5, and 1.0 mM) were synthesized by a modified citrate-gel approach and utilized as efficient catalysts towards the oxygen evolution reaction (OER). The results showed that the size of Ptx–NiMnO3 NCs decreases with increasing the initial concentration of Pt. Meanwhile, the OER activity and durability of the as-synthesized Pt–NiMnO3 under different pH conditions are found to be Pt-content-dependent. Particularly, Pt1–NiMnO3 showed superior OER efficiency relative to its counterparts Pt0.5–NiMnO3, Pt0.1–NiMnO3, NiMnO3, and commercial Pt/C at different pH values, ascribed to the size, morphology, and composition effects as confirmed via XRD, XPS and electrochemical characterization.
Introduction
The anodic oxygen evolution reaction (OER) is one of the most promising sustainable and environmentally benign energy conversion technologies.1 Hitherto, the sluggish OER kinetics, complex electron transfer, and high polarization potential preclude its practical application in commercialized fuel cell devices.1 Until now, Pt-based materials are still the most popular catalysts for anodic and cathodic reactions; meanwhile Ru- and Ir-based catalysts are known for their superior activity towards the OER at an earlier overpotential (η).2–7 However, the scarcity, high-cost, and instability of these noble metals are the main factors that preclude their practical large scale deployment especially in automobile fuel cells.2,4 Various efforts are dedicated to solve this issue, which culminated in controlling the morphology and composition of Pt-, Ir-, and Ru-based catalysts in addition to noble-metal free catalysts.1–4,8–19 For instance, RuCo, IrO2, PtCu, and PtRe synthesized by different chemical approaches manifested outstanding catalytic activities towards the OER.8–13,20,21 Recently, Pt@Rudendritic and Pt@Rucuboctahedra synthesized using CO as a reducing agent were more active and durable than PtRu, Pt, and Ru NCs towards the OER, which can be ascribed to the composition and morphology effects.22 Alternatively, various earth abundant transition metals and their oxides emerged as propitious candidates for the OER, owing to their variable oxidation states, high activity, and low-cost.8,23–26 Among them, Ni and Mn along with their oxides are favorable alternative catalysts to precious noble metals for the OER, ascribed to their great intrinsic redox properties and high activity.27–31 In particular, Ni- and Mn-based materials provide abundant catalytic sites and promote charge transfer along with retarding the adsorption of the reaction intermediates in the OER.28 Various developed Ni- and Mn-based catalysts including but not limited to NiFe2O4, NiCo2O4, Mn3O4, and MnCo2O4, manifested a significant increment in the OER performance.8,32–40 For example, NiCo double hydroxide nanosheets prepared by a hydrothermal approach revealed an η of 0.367 V at 10 mA cm−2 with great stability in 1 M KOH owing to their high surface area and active sites.41Conversely, merging Ni with Mn in the form of hybrid binary NiMnO3 oxides can result in a higher OER activity compared to their monometallic oxide counterparts owing to their high electrical-conductivity, redox activities, and magnetic merits.29,32,42–47 Additionally, modifying the electronic structure of Ni with Mn plausibly has an electronic effect on the Mn–O, resulting in a decrease in the binding energies of the intermediates over the NiMnO3 surface. Accordingly, NiMn-based oxides with different structures such as nanoflakes, hollow nanorods, and hollow urchin-like structures are prepared by various chemical approaches such as solvo-thermal, self-assembly, and template-based methods.32,42–48 Recently, NiMnO3/NiMn2O4 was fabricated via a two-step annealing method, with the assistance of pollen to tailor the surface oxidation states of Ni/Mn, which showed high activity for the OER in KOH solution.49 Particularly, 0.61NiMnO3/NiMn2O4 exhibited a superior OER activity with an η of 0.38 V at 10 mA cm−2 that was interestingly substantially lower than that of IrO2 (0.420 V).49 Despite the great progress in this area of research, the catalytic activity and durability of the as-developed NiMnO3 NCs are not up to the level to be implemented in fuel cells owing to transformations of Ni2+/Ni3+ and Mn3+/Mn4+.49 This, however, can be circumvented by integration of Pt ions into the spinel of NiMnO3 that can rigorously improve the OER performance owing to its unique catalytic properties and the modified electronic structure of Ni/Mn, which could prevent their possible transformation.12,13,29,46,47,49
Therefore, developing an active, durable, and inexpensive catalyst for the OER vulnerable to different pH values is still a grand challenge and not highlighted enough in the literature.50,51 There are few previously reported active and durable catalysts towards the OER such as porous graphite carbon@oxidized carbon cloth.3,50–52 To this end, we report, herein, the synthesis of Pt–NiMnO3 NCs with a well-defined shape and composition via a modified citrate-gel method. The catalytic performance of the as-obtained Pt–NiMnO3 is investigated towards the OER under various pH conditions.
Experimental
Chemicals and materials
K2PtCl4, Ni(NO3)2·6H2O, Mn(NO3)2·4H2O, and citric acid monohydrate (C6H8O7·H2O) are purchased from BDH chemicals (London, UK) and used without further purification.Synthesis of NiMnO3
NiMnO3 is prepared by a modified citrate-gel approach with slight modifications.47 Briefly, 10 mmol of citric acid was added drop-wise to a 50 mL aqueous solution containing 5 mmol of Ni(NO3)2·6H2O and 5 mmol of Mn(NO3)2·4H2O under vigorous stirring. Then, the pH of the solution was adjusted to 6 via the addition of NH4OH and kept under stirring at 80 °C for 4 h. The as-formed gel was dried at 100 °C for 24 h before annealing at 450 °C for 5 h.Preparation of Ptx–NiMnO3 NCs
Ptx–NiMnO3 NCs are obtained by slow drop-wise addition of an aqueous solution of 50 mL 10 mM citric acid (10 mmol) containing different amounts of K2PtCl4i.e. (0.005, 0.025 and 0.05 mmol) into an aqueous solution containing 5 mmol of both Ni(NO3)2·6H2O and Mn(NO3)2·4H2O under stirring at room temperature. The gel is formed at pH 6 under stirring at 60 °C for 4 h, and then the final product was obtained after drying at 100 °C for 24 h followed by annealing at 450 °C for 5 h. The final products were kept for further characterization.Materials characterization
The Ptx–NiMnO3 size, morphology, and composition were investigated using a field emission scanning electron microscope (FESEM, Philips XL-30, FEI Co., USA) equipped with an energy dispersive X-ray spectrometer (EDX). The powder wide angle X-ray diffraction patterns (XRD) were recorded on an X'Pert-Pro MPD diffractometer (PANalytical Co., Netherlands) with a Cu Kα X-ray source (λ = 1.540598 Å). X-ray photoelectron spectroscopy (XPS) measurements were performed using a Kratos Axis Ultra XPS with a monochromatic Al Kα radiation source (1486.6 eV) in a UHV environment (ca. 5 × 10−9 Torr).Electrochemical investigation
All the electrochemical measurements were carried out using a GAMRY electrochemical analyzer (GAMRY Reference-3000, GAMRY Co., USA). A conventional three-electrode cell including a saturated calomel electrode (SCE), a Pt wire, and a modified glassy carbon electrode (GCE, 5 mm in diameter) as reference, counter and working electrodes, respectively was used. The working electrodes were covered with 8 μL of 2 mg mL−1 of each catalyst followed by drop-casting 4 μL of Nafion (0.05%) and drying at room temperature before electrochemical measurements.The OER measurements were performed in different electrolytes including 1 M KOH, 0.5 M H2SO4, and 0.1 M phosphate buffer saline (PBS) (pH 7.4). Polarization curves were obtained using linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1 after cleaning and activation in a N2-saturated electrolyte solution for 100 cycles at a scan rate of 100 mV s−1. The electrochemical stability tests were performed via chronopotentiometric measurements at 10 mA cm−2 for five hours. This is in addition to the chronoamperometric tests performed over the course of 5 hours. Electrochemical impedance spectroscopy (EIS) measurements were carried out in each electrolyte solution at frequencies ranging from 0.1 Hz to 100 kHz with an AC voltage pulse of 10 mV amplitude. The current densities (j) were normalized in reference to the geometric surface area of the working electrode. All the measured potentials were converted to the reversible hydrogen electrode (RHE) by using the following equation
| E(RHE) = E(SCE) + 0.242 + (0.059 × pH) |
Results and discussion
The hybrid binary NiMnO3 and Ptx–NiMnO3 oxides were synthesized by a citrate-gel method via mixing Ni and Mn precursors with the corresponding amount of Pt precursor in the presence of citric acid as a complexing agent at pH = 6 followed by drying before annealing at 450 °C for 5 h. Fig. 1a displays the FESEM images of NiMnO3 oxides. Note that a high yield of NiMnO3 in an octahedral shape is obtained with an average size of 950 nm (Fig. 1b). Fig. 1c shows the FESEM image of Pt1–NiMnO3 synthesized via the citrate-gel method in the presence of 1 mmol percentage of Pt. The as-prepared Pt1–NiMnO3 has a well-defined octahedral shape with high yield. Interestingly, the average diameter of Pt1–NiMnO3 NCs is 43 nm which is only 4.5% that of NiMnO3 NCs, Fig. 1d. This implies a significant decrease in the sizes of NCs upon the incorporation of Pt into the spinel structure of NiMnO3. The concentration of Pt salt was varied between 0.1 and 0.5 mmol in order to decipher the formation mechanism of PtNiMnO3. The FESEM images showed the formation of PtNiMnO3 with a high yield in an octahedral structure with sizes in the range of 270 nm, Fig. 1e and f. Likewise, octahedral Pt–NiMnO3 NCs with 390 nm size are formed upon using 0.1 mmol percentage of Pt precursor, Fig. 1g and h. These results substantiate the decrease in the sizes of Pt–NiMnO3 NCs upon increasing the amount of incorporated Pt in the spinel oxide. This is plausibly ascribed to the potential for limited growth originating from the catalytic effect of Pt along with insertion of Pt into the NiMnO3 lattice.49,53 The EDX analysis was conducted to confirm the composition of the as-formed NiMnO3 and Pt–NiMnO3 NCs, Fig. 2a. The EDX analysis revealed an atomic ratio of Ni/Mn/O of 29.11/25.16/45.74, promoting the formation of NiMnO3, Fig. 2a. Meanwhile, the atomic ratios of Pt/Ni/Mn/O are about 1.10/22.25/20.09/56.56, 0.87/27.30/27.06/44.78, and 0.26/25.19/21.89/52.56 upon using 0.1, 0.5, and 1 mol percentages of Pt precursors, respectively implying the formation of Ptx–NiMnO3, Fig. 2a. These results clearly warranted that the composition of the obtained NCs can be easily tailored by varying the concentration of the Pt-precursor. Elemental mapping analysis was further performed to get more insights into the composition of the as-obtained Pt1–NiMnO3 NCs, Fig. 2b. It is clear that Pt, Ni, Mn, and O are well distributed throughout the entire formed NCs, implying the formation of Pt1–NiMnO3. The atomic ratio of Pt/Ni/Mn/O is approximately 1.15/22.20/20.13/56.52 which is in agreement with the EDX analysis. | ||
| Fig. 1 (a, c, e and g) SEM images and (b, d, f and h) particle size distribution of NiMnO3, Pt1–NiMnO3, Pt0.5–NiMnO3, and Pt0.1–NiMnO3 respectively. | ||
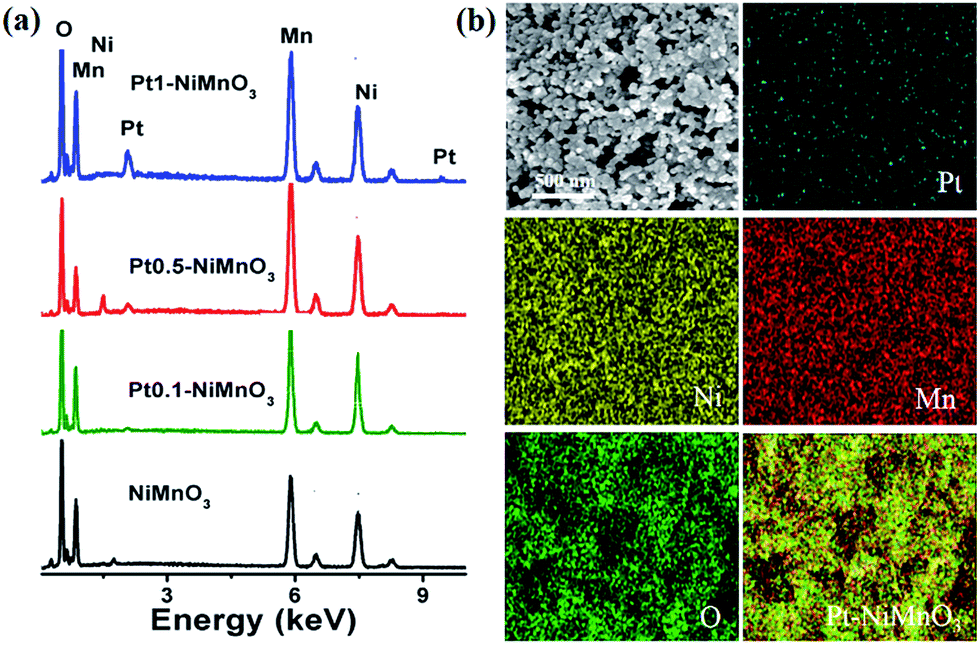 | ||
| Fig. 2 (a) EDX analysis of the as-synthesized NiMnO3, Pt1–NiMnO3, Pt0.5–NiMnO3, and Pt0.1–NiMnO3. (b) Elemental mapping analysis of Pt0.1–NiMnO3. | ||
Based on the obtained results, the fabrication mechanism of Pt–NiMnO3 NCs is possibly ascribed to the dissolution/recrystallization and subsequent Ostwald ripening growth (Scheme 1).49,54 In particular, Pt is feasibly nucleated promptly at the initial stage of the reaction owing to its positive reduction potential, resulting in facilitating nucleation of other metal precursors to form nanocrystallites (Scheme 1). These were consequently recrystallized into rhombohedral structures in order to minimize their surface energies, followed by subsequent Ostwald ripening growth to form PtNiMnO3 octahedral NCs. Hence, Pt-nuclei trigger fast recrystallization of NiMnO3 due to their spontaneous strong interaction, which hampers the NC aggregation that explains the decrease in the particle size. In contrast, the absence of Pt salt led to a sluggish diffusion–recrystallization with a high tendency to agglomerate during the growth step and yielded larger particle sizes (Scheme 1).
 | ||
| Scheme 1 The proposed formation mechanism of NiMnO3 and Pt–NiMnO3. | ||
Fig. 3 shows the XRD analysis of the as-fabricated NiMnO3 and Pt1–NiMnO3 NCs. The planes of {101}, {112}, {110}, {120}, {220}, {231}, {130}, and {211} endorsed the formation of NiMnO3 in agreement with ICDD: 98-003-1853. After addition of Pt salt, only the {111} Pt facet appeared at 2θ of 39°, implying the successful formation of Pt–NiMnO3 which is consistent with the EDX and mapping analysis. Interestingly, the NiMnO3 planes are slightly shifted positively owing to the expansion of its lattice structure due to the insertion of the Pt atom.53,55,56 Moreover, the absence of individual peaks for Ni and Mn oxides can be attributed to the full merging of Pt into the lattice structure of NiMnO3 without undesired phases, indicating the high purity of the obtained PtNiMnO3.
 | ||
| Fig. 3 XRD analysis of the as-synthesized NiMnO3 and Pt1–NiMnO3. | ||
The surface composition and metal valence state of the as-synthesized NiMnO3 and Pt–NiMnO3 NCs are investigated using XPS analysis, Fig. 4. Obviously, Pt–NiMnO3 NCs show the inherent peaks of Pt4f, Ni2p, Mn2p, and O1s. Meanwhile, NiMnO3 NCs are marked with the presence of Ni2p, Mn2p, and O1s. In particular, the determined binding energies of Pt4f7/2 and Pt4f5/2 are about (70.66, 70.87, and 70.98 eV) and (74, 74.23, and 74.4 eV) for Pt0.1–NiMnO3, Pt0.5–NiMnO3, and Pt1–NiMnO3, respectively (Fig. 4). Intriguingly, the binding energies of Pt4f are blue-shifted with increasing the amount of embedded Pt in the lattice of NiMnO3.53,56 Likewise, the binding energies of Ni2p, Mn2p, and O1s in the obtained Pt–NiMnO3 NCs are also shifted to higher values relative to NiMnO3. This implies the merging of Pt into the lattice structure of NiMnO3, hence promoting the fabrication of Pt–NiMnO3.53,56 Based on these results, Pt–NiMnO3 NCs are expected to depict a higher catalytic performance than NiMnO3 along with retarding the adsorption of the reaction intermediates, due to the blue shift in the binding energy of Pt4f.48,50
 | ||
| Fig. 4 XPS spectra of the as-synthesized NiMnO3, Pt1–NiMnO3, Pt0.5–NiMnO3, and Pt0.1–NiMnO3: (a) Pt 4f, (b) Ni 2p, (c) Mn 2p, and (d) O 1s. | ||
Inspired by the well-defined morphology and composition of the as-obtained Pt1–NiMnO3 NCs, their electrochemical activity and durability are benchmarked towards the OER relative to Pt0.5–NiMnO3, Pt0.1–NiMnO3, and NiMnO3 NCs under different pH values. Fig. 5a depicts the linear sweep voltammetry (LSV) curves of the as-obtained catalysts measured in 1.0 M KOH at a scan rate of 5 mV s−1. Notably, Pt1–NiMnO3 NCs exhibited a maximum current density at the earliest potential relative to its counterparts. Particularly, Pt1–NiMnO3 exhibited a current density of 35.5 mA cm−2 at 1.94 V, which is 1.1 times that of commercial Pt/C (33.0 mA cm−2) at 2.05 V, 1.57 times that of Pt0.5–NiMnO3 (22.5 mA cm−2) at 2.54 V, 1.71 times that of Pt0.1–NiMnO3 (20.67 mA cm−2) at 2.55 V, and 2.35 times that of NiMnO3 (15.08 mA cm−2) at 2.56 V. Additionally, the onset potential of Pt1–NiMnO3 (1.59 V) is lower than that of Pt/C (1.7 V), Pt0.5–NiMnO3 (2.14 V), Pt0.1–NiMnO3 (2.23 V), and NiMnO3 (2.31 V), Fig. 5a. Meanwhile, the estimated overpotential (ηOER) at 10 mA cm−2 for Pt1–NiMnO3 (0.44 V) is significantly inferior to that of Pt/C (0.61 V), Pt0.5–NiMnO3 (1.13 V), Pt0.1–NiMnO3 (1.18 V), and NiMnO3 (1.21 V) by 0.17, 0.69, 0.74, and 0.76 V, respectively (Fig. 5b). The Tafel plot for each catalyst was investigated to envisage the OER kinetics, Fig. 5c. The evaluated Tafel slope of Pt1–NiMnO3 (0.061 mV dec−1) is substantially lower than that of Pt0.5–NiMnO3 (0.11 mV dec−1), Pt0.1–NiMnO3 (0.32 mV dec−1), NiMnO3 (0.38 mV dec−1), and Pt/C (0.22 V mV dec−1). The Tafel slopes of the as-synthesized nanocatalysts were measured at a current density of 1 mA cm−2 for further verification of the stability of Pt1–NiMnO3. The Tafel slope of Pt1–NiMnO3 is also found to be lower than those of the other counterparts and that of the Pt/C catalyst (Fig. S1a†).
 | ||
| Fig. 5 (a) LSV curves of the as-obtained catalysts benchmarked in 1 M KOH at a scan rate of 5 mV s−1, (b) overpotential measured at a current density of 10 mA cm−2, (c) Tafel plots, (d), Nyquist impedance plots, and (e) chronopotentiometric curves with a constant current density of 10 mA cm−2 in 1.0 M KOH. | ||
This implies that the OER occurs more easily on the surface of Pt1–NiMnO3 compared to other counterparts, reflecting the momentous effect of incorporated Pt on improving the OER activity. It is noteworthy that the OER activity of Pt1–NiMnO3 (0.44 V) at 10 mA cm−2 is found to be superior to that previously reported for NiMn2O4 (0.610 V), Ni6MnO8 (0.480 V), NiCo2O4 nanoneedles (0.565 V), NiMnO3 (0.67 V), NiMn2O4 (0.560), 0.42NiMnO3/NiMn2O4 (0.51 V), IrO2 (0.450 V), and IrO2 (0.480 V) measured under similar conditions.46,49,57–59
The durability of the catalyst is an indispensable factor in the OER.1 Intriguingly, the potential required to obtain a current density of 10 mA cm−2 on Pt1–NiMnO3 increased by only 0.019 V after a 5 h durability test, which is significantly lower than that of Pt0.5–NiMnO3 (0.044 V), Pt0.1–NiMnO3 (0.05 V), and NiMnO3 (0.12 V) at 1.83 V, Fig. 5e. This implies that Pt1–NiMnO3 is 6.3, 2.63, and 2.3 times more stable than Pt0.5–NiMnO3, Pt0.1–NiMnO3, and NiMnO3 correspondingly. The chronoamperometric tests (i–t) for 5 h at a potential of 1.6 V also revealed the superior stability of Pt1–NiMnO3 compared to Pt/C and NiMnO3 (Fig. S2a†). Moreover, the XRD results revealed that Pt1–NiMnO3 maintained its crystalline structure after accelerated durability tests without any leaching of Pt. Note that NiMnO3 showed two additional peaks at 2θ of 26.2° and 37° attributed to the oxidation of Ni2+ to Ni3+ (Fig. S3a†).49 This is owing to the significant effect between Pt and NiMnO3, which makes the reaction intermediate species bind less over the surface of Pt1–NiMnO3. Fig. 5d shows the Nyquist impedance plots of the as-synthesized catalysts measured in 1.0 M KOH. Obviously, Pt1–NiMnO3 NCs display a minimum resistance compared to Pt/C, Pt0.5–NiMnO3, Pt0.1–NiMnO3, and NiMnO3 NCs, implying their high conductivity and fast OER kinetics. This significant decline in the resistance possibly arises from the composition effect owing to incorporation of Pt into a highly stable lattice of NiMnO3. Fig. S4† shows the amount of O2 evolved during OER measurements over 300 min. The measured amounts of O2 evolved during measurements using Pt–NiMnO3 and NiMnO3 almost corresponded to a quantitative faradaic yield over the course of the reaction time (Fig. S4†) in agreement with previously published reports.3,60
To get more insights into the catalytic performance of Pt1–NiMnO3 NCs, their activity and durability were measured under various pH values, which is a grand challenge in the OER.1–3,50Fig. 6a demonstrates the LSV of Pt1–NiMnO3, relative to NiMnO3 tested in PBS (∼pH 7.4). Noticeably, Pt1–NiMnO3 possesses a current density of 20.57 mA cm−2 at 2.42 V which is 1.6-fold that of Pt/C (13 mA cm−2) at 2.27 V, and 3.45-fold that of NiMnO3 (5.95 mA cm−2) at 2.57 V, Fig. 6a. Moreover, Pt1–NiMnO3 exhausted an ηOER of 0.86 V in order to provide a current density of 10 mA cm−2 which is lower than that of Pt/C (0.94 V), while NiMnO3 needed a potential of 2.46 V to only provide 5.95 mA cm−2. The estimated onset potential of Pt1–NiMnO3 (1.82 V) is also inferior to that of Pt/C (1.93 V) and NiMnO3 (2.07 V). The Tafel slope of Pt1–NiMnO3 is found to be 0.19 V dec−1 which is lower than that of Pt/C (0.196 V dec−1) and NiMnO3 (0.28 V dec−1), demonstrating its faster OER kinetics, Fig. 6b. Also, the Tafel slope of Pt1–NiMnO3 at a current density of 1 mA cm−2 was lower than that of NiMnO3 and Pt/C (0.325 V dec−1) (Fig. S1b†). Additionally, the accelerated i–t durability test for 5 h at 1.82 V of Pt1–NiMnO3 revealed its higher stability than NiMnO3 and Pt/C catalysts (Fig. S2b†). The Nyquist impedance plots revealed that the resistance of Pt1–NiMnO3 is significantly inferior to that of Pt/C and NiMnO3, implying its higher conductivity and enhanced OER kinetics, Fig. 6c.
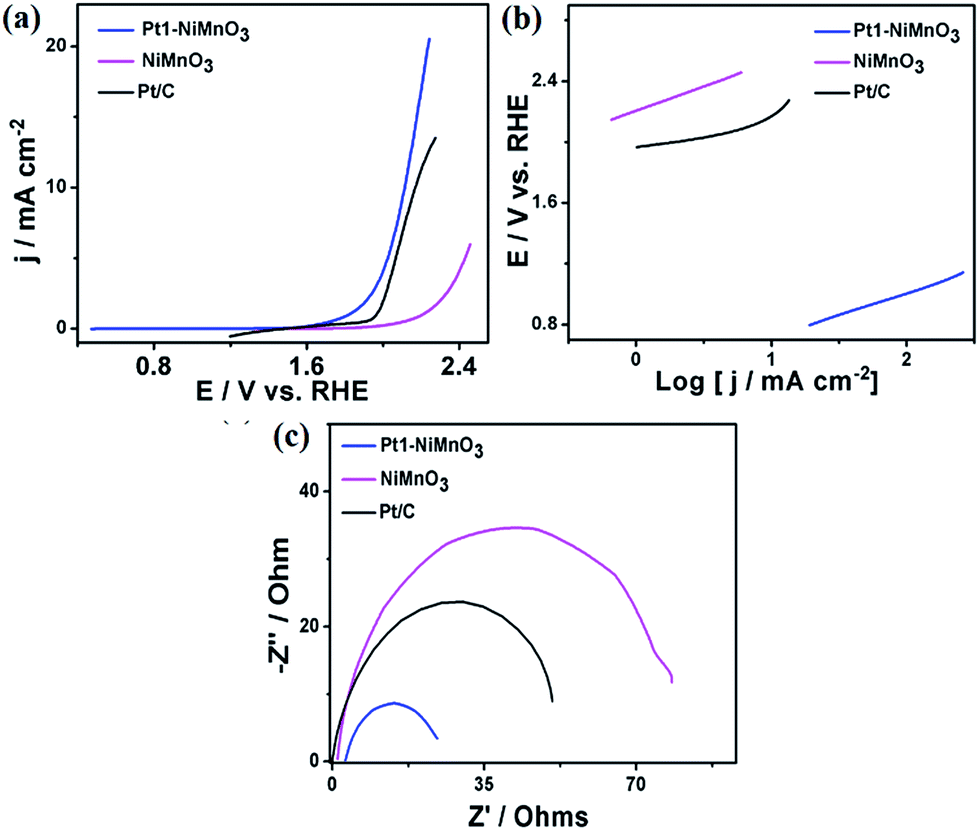 | ||
| Fig. 6 (a) LSV curves of Pt1–NiMnO3 and NiMnO3 benchmarked in PBS (pH = 7.4) at a scan rate of 5 mV s−1, (b) Tafel plots, and (c) Nyquist impedance plots. | ||
Fig. 7a shows the LSV curves of Pt1–NiMnO3 benchmarked in H2SO4 relative to commercial Pt/C and NiMnO3. The maximum current density of Pt1–NiMnO3 (18.2 mA cm−2) measured at 2.18 V is 10.4 times higher than that of Pt/C (1.74 V) and 22.74 times higher than that of NiMnO3 (0.8 mA cm−2). Pt1–NiMnO3 showed an onset potential of 1.8 V which is significantly lower than that of Pt/C (2.06 V) and NiMnO3 (2.23 V). Meanwhile, Pt1–NiMnO3 consumed only an ηOER of 0.9 V to provide 10 mA cm−2 which is lower than that of Pt/C (1.13 V), while NiMnO3 exhausted 2.37 V to merely yield a current density of 3.46 mA cm−2. The durability i–t tests further indicated the superior durability of Pt1–NiMnO3 compared to Pt/C and NiMnO3 (Fig. S2c†). The Tafel slope of Pt1–NiMnO3 (0.2 V dec−1) is lower than that of Pt/C (0.22 V dec−1) and NiMnO3 (0.33 V dec−1), demonstrating its ability to facilitate OER kinetics, Fig. 7b. This is also in addition to the lower Tafel slope of Pt1–NiMnO3 at a current density of 1 mA cm−2 compared to Pt/C and NiMnO3 (Fig. S1c†). This may be due to the lower resistance of Pt1–NiMnO3 than that of Pt/C and NiMnO3 as reflected by the Nyquist impedance plots (Fig. 7c).
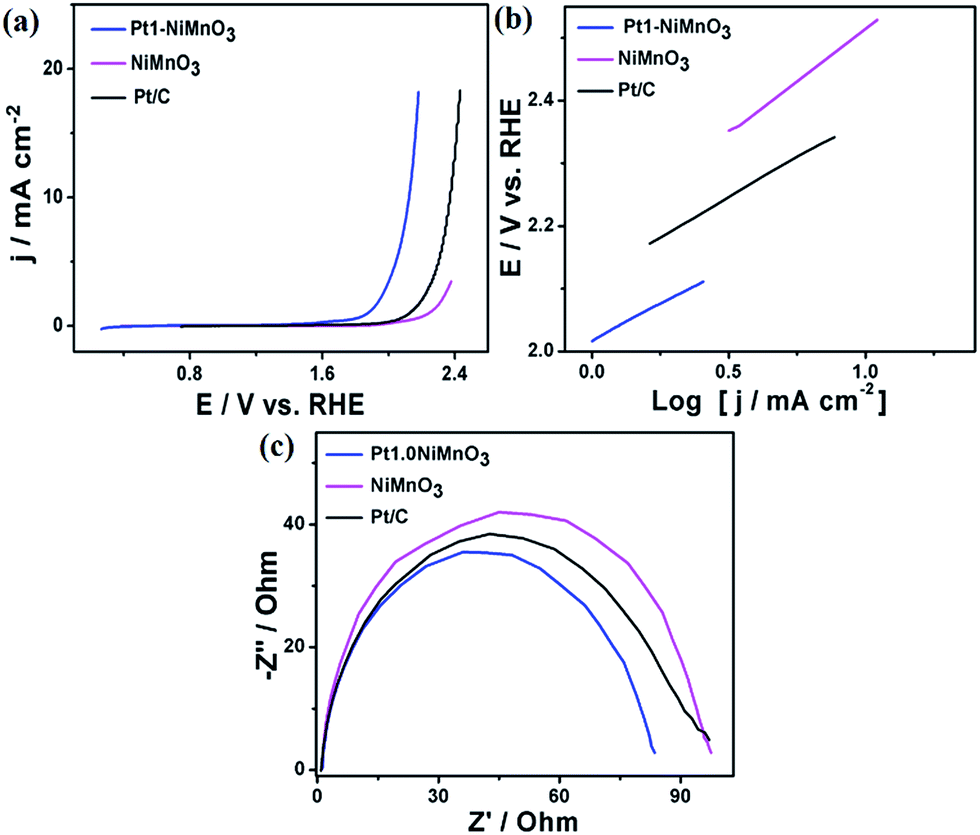 | ||
| Fig. 7 (a) LSV curves of Pt1–NiMnO3 and NiMnO3 benchmarked in H2SO4 (0.5 M) at a scan rate of 5 mV s−1, (b) Tafel plots, and (c) Nyquist impedance plots. | ||
The results ascertained clearly warrant the superior OER activity and durability of Pt1–NiMnO3 at all pH values relative to NiMnO3 owing to the substantial catalytic effect of Pt.12,13,21 In particular, the size, morphology, and composition are the predominant factors behind the enhancement of the catalytic performance. Pt1–NiMnO3, with its small nanosizes, can be well-dispersed, which led to an increase in the number of available catalytic sites on its surface along with decreasing the binding energies of O2 molecules.12,13 Moreover, Pt1–NiMnO3 with its anisotropic nanostructure and high surface area not only entails various surface corners but also has less tendency to aggregate and affords various accessible active sites for O2 molecules.
Another important factor behind the enhancement in the electrocatalytic performance is the integration of Pt into the highly stable NiMnO3.13 This led to a decrease in its d-band level, facilitating O–O bond splitting at lower potentials, and lessening of the binding energies of the reaction intermediates (e.g., HOO− and HO−) in addition to protecting the active catalytic sites from blocking. Moreover, merging Pt within the Ni/Mn lattice stabilizes them against dissolution and transformation into another valence state in different electrolytes.12,13,61
Conclusion
In summary, Pt–NiMnO3 NCs with different compositions are synthesized via a citrate-gel method. Incorporation of Pt into the spinel structure of NiMnO3 has a significant effect on the particle size of the resulting Pt–NiMnO3 NCs as well as their catalytic performance towards the OER. The particle size decreases with increasing the Pt-content, whereas the OER activity and durability increase. Pt1–NiMnO3 exhibits superior OER activity and durability at different pH values relative to its counterparts Pt0.5–NiMnO3, Pt0.1–NiMnO3, NiMnO3 NCs, and commercial Pt/C. The ηOER values at 10 mA cm−2 of Pt1–NiMnO3 are found to be 0.44, 0.88, and 0.9 V in 1.0 M KOH, PBS, and H2SO4, respectively. The presented work opens new frontiers in the fabrication of perovskite/noble metal hybrids as efficient nanocatalysts for the OER.Acknowledgements
This work was made possible by NPRP Grant no. NPRP 7-485-1-091 from the Qatar National Research Fund (a member of the Qatar Foundation). The statements made herein are solely the responsibility of the authors.References
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- K. Eid, H. Wang, V. Malgras, Z. A. Alothman, Y. Yamauchi and L. Wang, J. Phys. Chem. C, 2015, 119, 19947–19953 CAS.
- S. Lu, K. Eid, W. Li, X. Cao, Y. Pan, J. Guo, L. Wang, H. Wang and H. Gu, Sci. Rep., 2016, 6, 26196 CrossRef CAS PubMed.
- S. Lu, K. Eid, M. Lin, L. Wang, H. Wang and H. Gu, J. Mater. Chem. A, 2016, 4, 10508–10513 CAS.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 9, 9266–9291 CrossRef PubMed.
- K. Neyerlin, G. Bugosh, R. Forgie, Z. Liu and P. Strasser, J. Electrochem. Soc., 2009, 156, B363–B369 CrossRef CAS.
- R. Forgie, G. Bugosh, K. Neyerlin, Z. Liu and P. Strasser, Electrochem. Solid-State Lett., 2010, 13, B36–B39 CrossRef CAS.
- Y. Shen, A. C. Lua, J. Xi and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 3464–3472 CAS.
- H. Jin, K. W. Lee, N. T. Khi, H. An, J. Park, H. Baik, J. Kim, H. Yang and K. Lee, Small, 2015, 11, 4462–4468 CrossRef CAS PubMed.
- J. Greeley, I. Stephens, A. Bondarenko, T. P. Johansson, H. A. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897–3901 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- Y. Zhu, W. Zhou and Z. Shao, Small, 2017, 13, 1603793 CrossRef PubMed.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150–7155 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- C. Zhang, B. Wang, X. Shen, J. Liu, X. Kong, S. S. Chuang, D. Yang, A. Dong and Z. Peng, Nano Energy, 2016, 30, 503–510 CrossRef CAS.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. Stephens and I. Chorkendorff, Chem. Sci., 2015, 6, 190–196 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- N. M. AlYami, A. P. LaGrow, K. S. Joya, J. Hwang, K. Katsiev, D. H. Anjum, Y. Losovyj, L. Sinatra, J. Y. Kim and O. M. Bakr, Phys. Chem. Chem. Phys., 2016, 18, 16169–16178 RSC.
- F. Jiao and H. Frei, Angew. Chem., 2009, 121, 1873–1876 CrossRef.
- R. E. Rettew, N. K. Allam and F. M. Alamgir, ACS Appl. Mater. Interfaces, 2011, 3, 147–151 CAS.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- C. G. Read, J. F. Callejas, C. F. Holder and R. E. Schaak, ACS Appl. Mater. Interfaces, 2016, 8, 12798–12803 CAS.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 12, 4661–4672 CrossRef.
- Y. Wu, G. D. Li, Y. Liu, L. Yang, X. Lian, T. Asefa and X. Zou, Adv. Funct. Mater., 2016, 26, 4999 CrossRef.
- E. Detsi, J. B. Cook, B. K. Lesel, C. L. Turner, Y.-L. Liang, S. Robbennolt and S. H. Tolbert, Energy Environ. Sci., 2016, 9, 540–549 CAS.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- Z. Chen, C. X. Kronawitter, Y.-W. Yeh, X. Yang, P. Zhao, N. Yao and B. E. Koel, J. Mater. Chem. A, 2017, 5, 842–850 CAS.
- B. Weng, F. Xu, C. Wang, W. Meng, C. R. Grice and Y. Yan, Energy Environ. Sci., 2017, 10, 121–128 CAS.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- M. Al-Hoshan, J. Singh, A. Al-Mayouf, A. Al-Suhybani and M. Shaddad, Int. J. Electrochem. Sci., 2012, 7, 4959–4973 CAS.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 CAS.
- S.-C. M. O. T. Film, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef PubMed.
- Y. Qiu, L. Xin and W. Li, Langmuir, 2014, 30, 7893–7901 CrossRef CAS PubMed.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- C. Zhang, M. Shao, L. Zhou, Z. Li, K. Xiao and M. Wei, ACS Appl. Mater. Interfaces, 2016, 8, 33697–33703 CAS.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- J. Hao, Y. Liu, H. Shen, W. Li, J. Li, Y. Li and Q. Chen, J. Mater. Sci.: Mater. Electron., 2016, 27, 6598–6605 CrossRef CAS.
- P. Sun, H. Yi, T. Peng, Y. Jing, R. Wang, H. Wang and X. Wang, J. Power Sources, 2017, 341, 27–35 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- A. C. Thenuwara, E. B. Cerkez, S. L. Shumlas, N. H. Attanayake, I. G. McKendry, L. Frazer, E. Borguet, Q. Kang, R. C. Remsing and M. L. Klein, Angew. Chem., Int. Ed., 2016, 22, 10381–10385 CrossRef PubMed.
- P. W. Menezes, A. Indra, O. Levy, K. Kailasam, V. Gutkin, J. Pfrommer and M. Driess, Chem. Commun., 2015, 51, 5005–5008 RSC.
- D. Hong, Y. Yamada, A. Nomura and S. Fukuzumi, Phys. Chem. Chem. Phys., 2013, 15, 19125–19128 RSC.
- J. Tian, N. Cheng, Q. Liu, X. Sun, Y. He and A. M. Asiri, J. Mater. Chem. A, 2015, 3, 20056–20059 CAS.
- X. He, F. Yin, Y. Li, H. Wang, J. Chen, Y. Wang and B. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 26740–26757 CAS.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- M. Ledendecker, S. Krick Calderón, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., 2015, 127, 12538–12542 CrossRef.
- J. B. Gerken, J. G. McAlpin, J. Y. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- W. Sun, K. Qiao, J.-y. Liu, L.-m. Cao, X.-q. Gong and J. Yang, ACS Comb. Sci., 2016, 18, 195–202 CrossRef CAS PubMed.
- S. Giri, D. Ghosh and C. K. Das, Dalton Trans., 2013, 42, 14361–14364 RSC.
- T. Rajesh, A. K. Rajarajan, C. S. Gopinath and R. N. Devi, J. Phys. Chem. C, 2012, 116, 9526–9532 CAS.
- W.-Y. Xia, N. Li, Q.-Y. Li, K.-H. Ye and C.-W. Xu, Sci. Rep., 2016, 6, 23398 CrossRef CAS PubMed.
- H. Shi and G. Zhao, J. Phys. Chem. C, 2014, 118, 25939–25946 CAS.
- J. Yu, Y. Zhong, W. Zhou and Z. Shao, J. Power Sources, 2017, 338, 26–33 CrossRef CAS.
- W. Hu, Q. Wang, S. Wu and Y. Huang, J. Mater. Chem. A, 2016, 4, 16920–16927 CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 CAS.
- K. Eid, H. Wang, Y. Yamauchi and L. Wang, Chem.–Asian J., 2016, 6, 1388–1393 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00111h |
| ‡ Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |