
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectrocatalytic water treatment systems: degradation, kinetics and intermediate products studies of sulfamethoxazole on a TiO2–exfoliated graphite electrode
Moses G. Peleyejuab,
Eseoghene H. Umukoroa,
Luthando Tshwenyaab,
Richard Moutloaliab,
Jonathan O. Babalolac and
Omotayo A. Arotiba *abd
*abd
aDepartment of Applied Chemistry, University of Johannesburg, Doornfontein 2028, South Africa. E-mail: oarotiba@uj.ac.za
bDST/Mintek Nanotechnology Innovation Centre, University of Johannesburg, South Africa
cDepartment of Chemistry, University of Ibadan, Nigeria
dCentre for Nanomaterials Science Research, University of Johannesburg, South Africa
First published on 21st August 2017
Abstract
Sulfamethoxazole is an antibacterial agent which is commonly prescribed for the treatment of infections in humans and animals. The detection of this drug in the aqueous environment has raised considerable health concerns. Herein, we report the photoelectrocatalytic degradation of sulfamethoxazole at a TiO2–exfoliated graphite (TiO2–EG) anode. The TiO2–EG nanocomposite, synthesised by sol–gel and microwave methods, was characterised by XRD, Raman and FTIR spectroscopies, SEM and TEM. The cyclic voltammograms of the fabricated electrodes were obtained in [Fe(CN)6]3− redox probe. Concentration abatement of the antibiotic was monitored using a UV-vis spectrophotometer and the possible intermediates were investigated using LCMS. After 6 h of the photoelectrocatalytic process, almost 100% of the drug had been degraded and a 90% COD decay was achieved. The photoelectrocatalytic degradation of sulfamethoxazole entailed γ-, β-, δ- and ε-cleavages, hydroxylation and rings opening. The outcome of this study shows that the EG–TiO2 anode can be applied for the photoelectrocatalytic remediation of water contaminated by pharmaceuticals.
1. Introduction
Antibiotics have been and are being used in the treatment of a broad range of bacteria-causing infections, saving humans and animals from pain, distress and death. In recent time, however, there has been a growing concern over the proliferation of antibiotic-resistant bacteria and it is believed that the presence of antibacterial agents in the environment has contributed to the emergence of these bacteria strains.1,2 A number of these substances are not easily biodegradable and thus they persist in the environment. The detection of antibiotics in surface and ground waters is well documented and sulfamethoxazole (SMX), a sulfonamide, is among the most frequently detected.3–7 SMX finds a wide application in human and veterinary medicine for treatment of infections.8 Incomplete metabolism in the body and ineffective treatment of wastewater from pharmaceutical industries may result in the accumulation of this substance and its metabolites in the environment.There has been a large number of investigations on the destruction of organic pollutants via electrochemical approach.9–12 In particular, direct or indirect oxidation involving the generation of hydroxyl radicals at a feasible potential has been shown to be promising for the remediation of water contaminated by organic substances.13,14 The development of photoelectrochemical process to enhance oxidation of organic pollutants at the anode renders this approach more effective and more attractive. A typical photoelectrochemical cell is equipped with an anode that is photoactive; such that the generation of the powerful hydroxyl radicals, which react unselectively with many recalcitrant organic pollutants, is maximised in the presence of light. In photoelectrocatalytic system, the challenges often associated with the traditional photocatalytic process are minimised. For instance, the problem of electrons and holes recombination15 is reduced as the electrons generated are driven away via the external circuit.16,17 Similarly, recovering of the photocatalyst from the suspension after treatment is not required as it is immobilised/localised on a substrate. The synergistic benefit of electrical energy and solar energy also makes the photoelectrocatalytic method the more suitable for oxidising organic contaminants in wastewater.
Photocatalytic semiconductors have been immobilised onto conductive substrates and employed as photoanodes for treating water polluted by organic substances. In a recent report by Li et al., a phosphate modified BiOCl photocatalyst was immobilised onto FTO glass and the resulting photoanode was employed for water oxidation and methyl orange degradation.18 Similarly, Zeng et al. prepared a film of WO3 nanoplates on FTO glass and applied the anode for the photoelectrochemical degradation of methylene blue.19 In another study by Lin and co-workers, Bi2WO6 film was obtained on ITO glass and the electrode was utilised for the photo-assisted anodic oxidation of rhodamine B.20 TiO2 is one of the most explored photoactive materials in many photocatalytic applications.21 It has received considerable attention as a choice material for the preparation of anodes for use in photoelectrochemical processes. TiO2 is preferred because of its high photocatalytic activity, chemical stability, low cost and non-toxicity. In a report by Su et al., TiO2 photoanode was prepared by treating Ti foil in a solution of titanium(IV)chloride and nitric acid. The prepared electrode was used for the degradation of sulfamethoxazole in the presence of UV light, and it was reported to be effective for the degradation of the pharmaceutical especially in the chloride supported cell.17 In another investigation by Kondalkar et al., TiO2 was immobilised onto FTO glass substrate by dip coating and the photoanode reportedly showed good electrocatalytic activity towards the oxidation of the antibiotic, cefotaxime, under UV irradiation.22 Also, Wei et al. prepared a film of TiO2/g-C3N4 hybrid heterostructure on ITO glass by surface hybridisation and dip-coating approach. The obtained anode reportedly exhibited good photoelectrocatalytic property towards phenol oxidation.23
Efforts have been made to trap TiO2 nanoparticles in the pores of certain carbon materials. For instance, a few reports have emerged on the use of graphite rods and expanded graphite as supports for the immobilisation of TiO2. In these studies, the obtained photoanodes were used for the degradation of phenols and dyes.24–27 In particular, the porous and the compressible nature of exfoliated graphite were advantageous for the trapping of the photocatalyst. In addition, factors such as low cost, ease of preparation, and high conductivity also encouraged the use of graphite-based supports for TiO2 photoanode. The impressive electron transport property of exfoliated graphite is desirable for channeling away the photogenerated electrons,28 consequently, the lifespan of the charges can be prolonged. In essence, this promotes the production of the powerful hydroxyl radicals which are required for the oxidation of the organic compounds in water.
In this work, a nanocomposite of TiO2 and exfoliated graphite (EG) was prepared via sol–gel and microwave techniques. The composite material was fabricated into electrode and employed for the photoelectrochemical oxidation of sulfamethoxazole for the first time, to the best of our knowledge. The extent of the degradation of the antibiotic was monitored with UV-visible spectroscopy and chemical oxygen demand analysis. The possible degradation products were investigated using liquid chromatography coupled with mass spectrometry.
2. Experimental
2.1. Reagents and materials
Natural graphite flakes, nitric acid, sulfuric acid, n-butanol, titanium butoxide, potassium ferricyanide, potassium nitrate, sodium sulfate, sodium chloride and sulfamethoxazole were purchased from Sigma-Aldrich South Africa and used as obtained. All solutions were prepared with deionised water.2.2. Preparation of EG and TiO2–EG
Preparation of EG was done according to a method reported earlier.29 Firstly, sieving of natural graphite flakes was achieved by 300 micron sieve. The uniformly sized graphite flakes were then dispersed in a mixture of nitric acid and sulfuric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v). The mixture was kept at ambient conditions for 24 h, after which the resulting acid intercalated material was washed with copious deionised water until a near neutral pH was achieved. The material was then air dried and subsequently subjected to a thermal treatment at 800 °C for 60 s. The obtained product is a puffed up material with a large volume-to-mass ratio.
:3 v/v). The mixture was kept at ambient conditions for 24 h, after which the resulting acid intercalated material was washed with copious deionised water until a near neutral pH was achieved. The material was then air dried and subsequently subjected to a thermal treatment at 800 °C for 60 s. The obtained product is a puffed up material with a large volume-to-mass ratio.
In a typical preparation of the nanocomposite of EG and TiO2, 2 g of EG was dispersed in 30 mL of n-butanol, and the mixture was stirred magnetically for 5 min. Thereafter, 6 mL of titanium butoxide was added to the mixture, and the hydrolysis of the titanium butoxide was achieved by drop-wise addition of 5 mL of water to the mixture while stirring vigorously. The sol formed was subsequently transferred into a reaction vessel and placed in Anton Paar microwave synthesis reactor operated at 600 W for 10 min. The material obtained was calcined at 400 °C for 2 h. Pristine TiO2 was prepared following the same procedure except that EG was not added.
2.3. Characterisation
X-ray diffraction (XRD) patterns were obtained on Rigaku Smartlab X-ray diffractometer (USA) with Cu Kα radiation. Raman analyses were performed on Witec alpha300 R confocal Raman microscope (Germany). Electron images were obtained on scanning electron microscope (TESCAN, Vega3 XMU, Czech Republic) and transmission electron microscope (JEM 2100 TEM, 200 kV, Japan). UV-vis spectra of the aliquots of SMX withdrawn from the reaction cell at fixed time intervals were recorded on Agilent Cary 60 spectrophotometer (Malaysia) and the chemical oxygen demand was determined on HACH DR3900 spectrophotometer. SMX degradation intermediates were identified using Shimadzu LCMS 8030 (triple quad) equipped with a Shimadzu LC-30AD Nexera Liquid Chromatography, a Shimadzu SIL-30 AC Nexera autosampler, and a Shimadzu CTO-20 AC Prominence Column Oven. The analytical column was a C18 column (Shimadzu 2 μm, 2.1 mm × 100 mm) which was maintained at 40 °C. The flow rate was 0.2 mL min−1 and injection volume was 5 μL, the mobile phases were acetonitrile and 0.1% formic acid.2.4. Fabrication of electrodes
The as-prepared TiO2–EG was compressed into 15.0 mm pellets at high pressure using a hydraulic press. A clean copper wire of diameter 1.0 mm and resistivity 1.673 μΩ cm was coiled at one end to form a flat surface, conductive silver glue was applied onto this surface and the pellet was subsequently placed. It was then left to air dry at room temperature for 30 minutes. The edges of the pellets were then covered with non-conductive epoxy resin and the electrode was then placed in a glass rod and sealed with the resin and Teflon tape, exposing only the electrode surface and the terminal copper wire.2.5. Electrochemical and photoelectrochemical experiments
Electrochemical experiments were carried out on a computer-controlled potentiostat/galvanostat (autolab, PGSTAT 302N model). Three electrode configuration was used with EG (or TiO2–EG), platinum foil and Ag/AgCl (3 M KCl) as working, auxiliary and reference electrodes respectively. The diameter of the working electrode was 1.5 cm. Cyclic voltammograms and photocurrent responses were obtained in a 5 mM (KCl supported) solution of ferricyanide. Electrochemical and photoelectrochemical oxidation of sulfamethoxazole was carried out in an undivided cell (with quartz window) using chronopotentiometry. Typically, a 75 mL of 25 mg L−1 SMX prepared in 0.1 M Na2SO4 was electrolysed in an experiment and the electrolytic solution was being magnetically stirred during the course of electrolysis. The pH of the SMX solution was maintained at 6.3 prior to degradation. Simulated sunlight was from a solar simulator, Oriel LCA-100 equipped with a 100 W xenon lamp and an Air Mass 1.5 Global filter which produces 100 mW cm−2. The surface of the electrode was made to align with the light source and the distance between the electrochemical cell and the lamp was 10 cm.3. Results and discussions
3.1. X-ray diffraction, Raman spectroscopy and FTIR spectroscopy
The XRD patterns of TiO2 and TiO2–EG are shown in Fig. 1a, TiO2 gave sharp and well defined peaks at 25.3, 37.8, 47.9, 54.5, 62.7, 69.5 and 75.0° which correspond to (101), (004), (200), (211), (204), (220) and (215) crystal lattice planes of anatase TiO2 respectively (JCPDS no. 21-1272). A new peak at 26.4° can be observed in the spectrum of the nanocomposite material, this reflection peak corresponds to (002) crystal plane of expanded graphite.29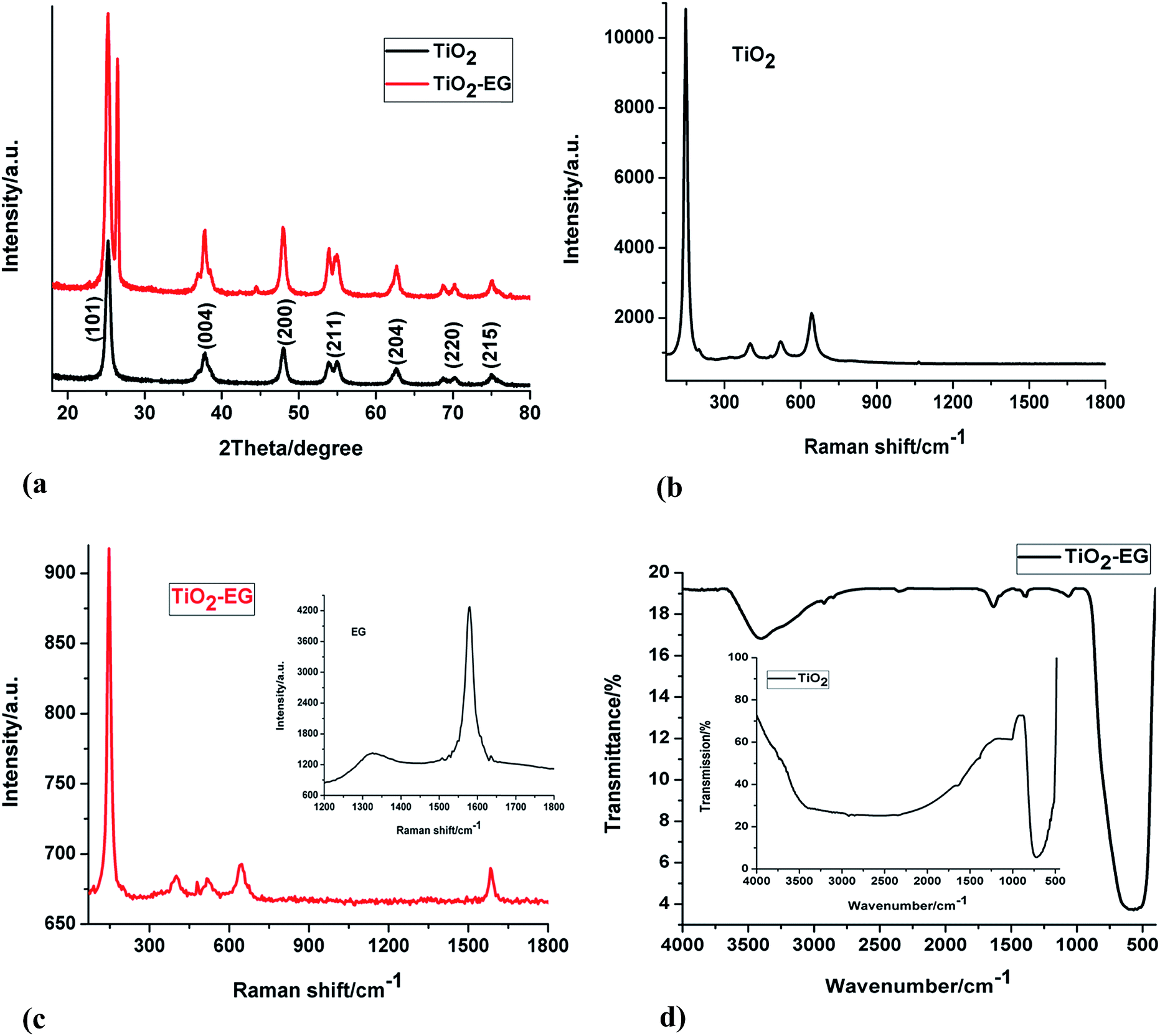 | ||
| Fig. 1 (a) XRD patterns of TiO2 and TiO2–EG (b) Raman spectrum of pristine TiO2 (c) Raman spectra of TiO2–EG and EG (inset) (d) FTIR spectra of TiO2–EG and TiO2 (inset). | ||
Raman analyses of the materials were carried out to investigate the structural features of the electrode materials. Fig. 1b and c display the Raman spectra of TiO2 and TiO2–EG. The characteristic bands of anatase TiO2 can be seen at around 146, 398, 521 and 643 cm−1 corresponding to Eg, B1g, A1g and Eg Raman lattice vibrations of the metal oxide semiconductor respectively.30–32 The spectrum of the nanocomposite shows the signature peaks of TiO2 which are present in the spectrum of the pristine material, while a new peak centred at 1589 cm−1 can also be seen. This new peak is the G band of the EG material which arises from the vibration of sp2 carbon atoms. The D band which is due to defect in the lattice structure of the graphitic material has a very low intensity and therefore cannot be clearly seen in the spectrum of the composite material, it is however present in the spectrum of the pristine EG shown in the inset.
The FTIR spectra of TiO2 and TiO2–EG can be seen in Fig. 1d, the peak at around 550 cm−1 in the spectrum of pristine TiO2 can be attributed to the vibration of the Ti–O bonds in the TiO2 lattice. In the spectrum of the composite material, peaks at 1634 and 1068 cm−1 can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O–C stretch of the EG. Also, the weak bands at 2924 and 2854 cm−1 are due to C–H stretch. The broad peak at 3408 cm−1 can be assigned to the vibration of hydroxyl groups of adsorbed water.
C and C–O–C stretch of the EG. Also, the weak bands at 2924 and 2854 cm−1 are due to C–H stretch. The broad peak at 3408 cm−1 can be assigned to the vibration of hydroxyl groups of adsorbed water.
3.2. Morphologies of electrode materials
The electron micrographs of TiO2 and TiO2–EG are presented in Fig. 2. From the SEM image of TiO2, it can be reasonably assumed that the nanoparticles are uniform-sized. The TEM image reveals that there is agglomeration of the particles, this can also be deduced from the SEM image as the particles are closely situated. In the TEM image of the TiO2–EG, however, the TiO2 particles are seen to sit on the graphite sheets, and agglomeration of the particles are minimal. Also, the SEM image of TiO2–EG shows that the graphite layers are decorated by the TiO2 particles. | ||
| Fig. 2 (A) SEM image of TiO2 (B) SEM image of TiO2–EG (C) TEM images of TiO2 and TiO2–EG and (D) TEM image of TiO2–EG. | ||
3.3. Cyclic voltammetry at EG and TiO2–EG electrodes
Cyclic voltammetry is a widely used technique which offers qualitative information on electrochemical reactions. The voltammograms obtained from the cyclic voltammetric studies of the electrodes carried out using potassium ferricyanide as a redox probe are presented in Fig. 3. The separation between the peak potentials for a reversible couple (at 298 K) is given by| ΔEp = 0.059/n |
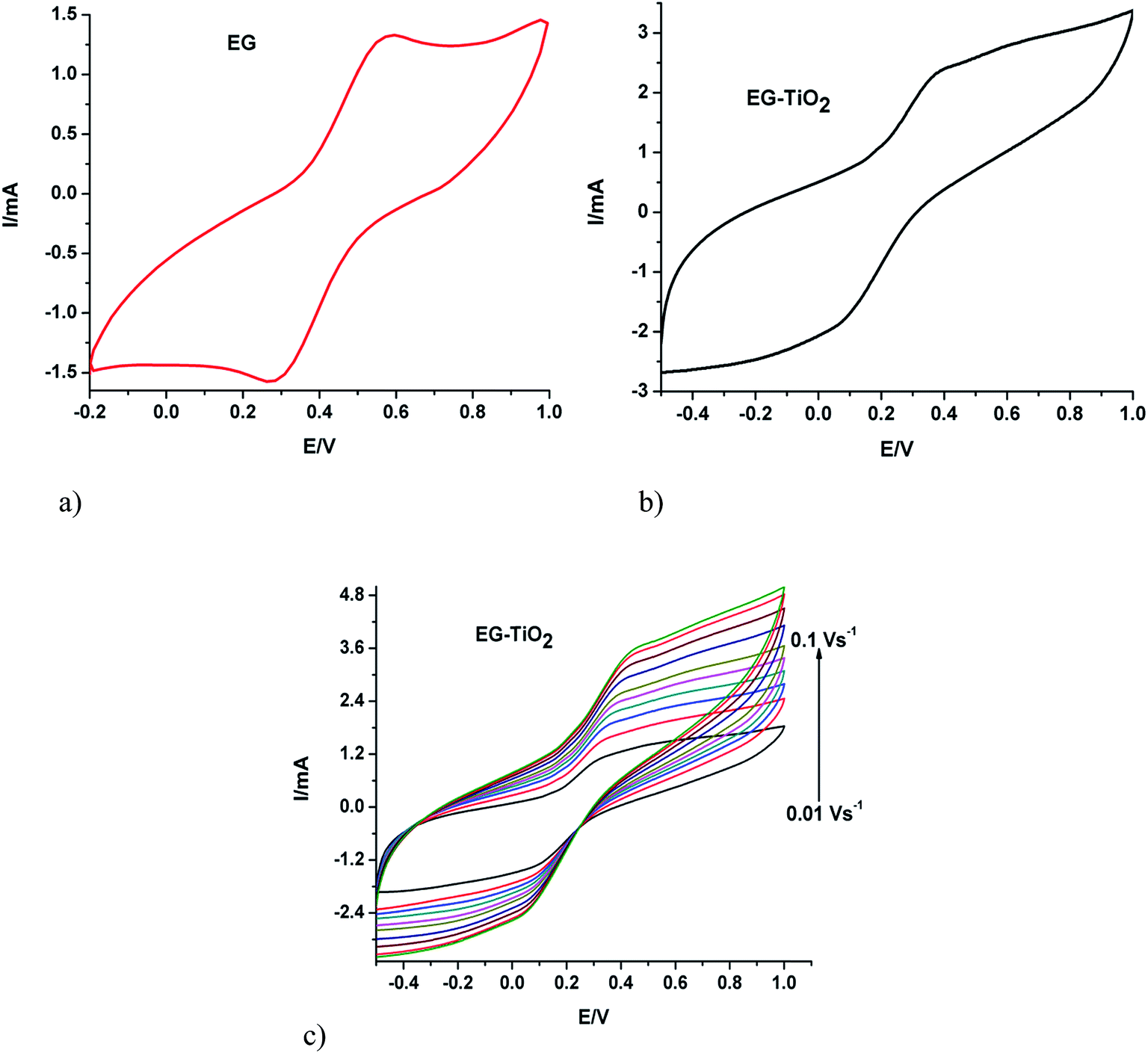 | ||
| Fig. 3 Cyclic voltammograms of (a) EG and (b) EG–TiO2 recorded at a scan rate of 0.05 V s−1 and (c) EG–TiO2 at different scan rates in 5 mM ferricyanide solution. | ||
3.4. Degradation of SMX at EG and TiO2–EG electrodes
Oxidation of the analyte was monitored on UV-vis spectrophotometer at its wavelength of maximum absorption (λmax = 257 nm). In both electrochemical and photoelectrochemical processes at the TiO2–EG electrode, there was a gradual but very significant reduction (with time) in the absorbance of the antibiotic at the λmax (Fig. 4a). This confirmed its degradation in the processes. However, a much higher degradation efficiency was obtained in the photo-assisted process (Fig. 4b). It is also noteworthy that the composite electrode exhibited a higher removal efficiency of the analyte than the pristine electrode in the electrochemical process (Fig. 4c). The improved degradation obtained in the photoelectrochemical process at the TiO2 based electrode can be attributed to the photocatalytic behaviour of the semiconductor and the combined effect of electrical energy and solar energy which are beneficial for generating the hydroxyl radicals required to destroy the pollutant. Upon irradiation with light, the photo-generated holes in TiO2 reacts with water to produce hydroxyl radicals, this powerful oxidant attacks and degrades the pollutant molecules until they are mineralised. Similarly, the holes also act as oxidant, attacking the organic specie until it is completely broken down. Furthermore, when the applied potential is sufficiently high, oxidation of water on the surface of the anode to form the hydroxyl radicals takes place. The enhanced performance of EG–TiO2 compared to EG in the electrochemical process can be related to the higher electro-active surface area of the EG–TiO2 which provides for greater number of sites for the oxidation of water to generate the oxidant. In addition, since direct electron transfer from the contaminant to the anode often occurs simultaneously with the indirect oxidation,33,34 a larger electro-active surface area favours the oxidation of SMX at the anode. This observation is in agreement with the results of the cyclic voltammetry experiments in which higher current was obtained at the composite electrode. | ||
| Fig. 4 (a) UV-vis spectra of oxidised SMX solution and normalised plots of concentration abatement for (b) electrochemical and photoelectrocatalytic degradation of SMX at TiO2–EG photoelectrode (c) electrochemical degradation of SMX at EG and TiO2–EG anodes (experiments carried out at pH 6.3, j = 10 mA cm−2, [SMX]0 = 25 mg L−1, supporting electrolyte = Na2SO4) (d) current-dependence of SMX degradation (e) effect of bulk solution pH on SMX degradation. | ||
The influence of current density on the degradation process was investigated. Oxidation of the analyte was achieved at the initial current density of 7 mA cm−2. There was however improvement in the process when the value was increased to 10 mA cm−2. Further increase to 13 mA cm−2 did not lead to appreciable increase in the degradation efficiency (Fig. 4d). This trend suggests that the oxidation of SMX is mainly achieved by holes and hydroxyl radicals in the photoelectrochemical process. It is also believed that the anodic potential at 10 mA cm−2 is optimal to prevent recombination of photogenerated charges and that further increase in potential is unutilised.
The effect of pH of the bulk solution on the degradation of SMX was studied. It can be observed that the rate of oxidation is higher at acidic pH (Fig. 4e), with the rate at pH 3 being slightly higher than the rate at pH 6. The pKa1 and pKa2 values of SMX are 1.7 and 5.6 respectively.35 Below and above these values SMX is either positively or negatively charged. The proportion of cationic or anionic form of SMX present in a solution at a time depends on the pH of the solution. At pH 3, SMX molecules are expected to be in neutral form and since the degradation efficiency at this pH is higher than at the other two pH values, it can be said that the degradation of the analyte in this process is favoured when the molecules are uncharged. The difference between the rates at pH 3 and pH 6 is not very large, thus it can be asserted that there is still a considerable amount of the neutral molecules at pH 6. Furthermore, although the degradation of SMX at pH 6.3 is a little slower, the pH of the electrolytic solution can still be maintained at 6.3 since it is milder.
3.5. Kinetics of SMX degradation, COD decay and current efficiency at TiO2–EG photoanode
Hydroxyl radicals mediated degradation of organic contaminants can be described by the Langmuir–Hinshelwood model, specifically the simplified pseudo-first order equation can be used to examine the kinetics in the degradation process. The equation is given as:| lnC/C0 = −kt |
C0/C against t presented a good linearity and the rate constant, k of the reaction was calculated from slope of the line. The apparent rate constant obtained for the degradation process carried out at a current density of 10 mA cm−2 and pH 6.3 was 5.74 × 10−3 min−1. This value of k is smaller compared to that obtained in a related study by Su et al., however, when factors such as electrode surface area, supporting electrolyte are taken into consideration, the rate of SMX decay in this work can be said to be appreciably good.
An efficient wastewater treatment technique is expected to lead to a significant reduction in the chemical oxygen demand (COD) of the water at a reasonable energy input. The removal of COD in this investigation was calculated using the relations:
| COD decay (%) = (COD0 − CODt)/COD0 × 100% |
Using the COD approach, the current efficiency for the degradation of SMX was calculated using the following equation:36
| Total current efficiency (TCE) = FV × (COD0 − CODt)/8IΔt |
487 C mol−1), V is the volume of the electrolytic solution (in litre), I is the current (A) and t is the electrolysis time (s). The TCE calculated for the degradation of SMX at optimum conditions was 0.674. This value indicates that a fairly high proportion of the electrical energy applied was utilised for the decay of the contaminant.
3.6. Identification of intermediate products during the degradation of SMX
LCMS analysis of standard sample and aliquots of degraded SMX solution were carried out. The chromatograms obtained are in Fig. 5. The major aromatic intermediates identified are m/z = 94, 157, 172. These species can result from the γ-, β-, δ- and ε-cleavages of the sulfamethoxazole molecules.37 The peak at m/z = 94 can be considered as protonated aniline, it has been reported in a number of studies dealing with oxidation of SMX by hydroxyl radicals.3,37–39 It can be thought to be formed by the attack of hydroxyl radicals on sulfonated moiety, leading to release of SO42−.38 The formation of a product having m/z = 157 has been attributed to the cleavage of the S–N bond in the parent molecule,40 and the m/z = 172 may be a sulfanilamide or ionic form of sulfanilic acid resulting from ε-cleavage.37,41 The peaks at m/z = 270 and 286 have been indicated to result from the mono- and di-hydroxylated products of SMX.42,43 It can therefore be reasonably suggested that the photoelectrocatalytic oxidation of SMX is by cleavage of the S–N bond and hydroxylation and opening of ring systems in the molecule. A schematic representation of the degradation route of SMX is presented in Fig. 6.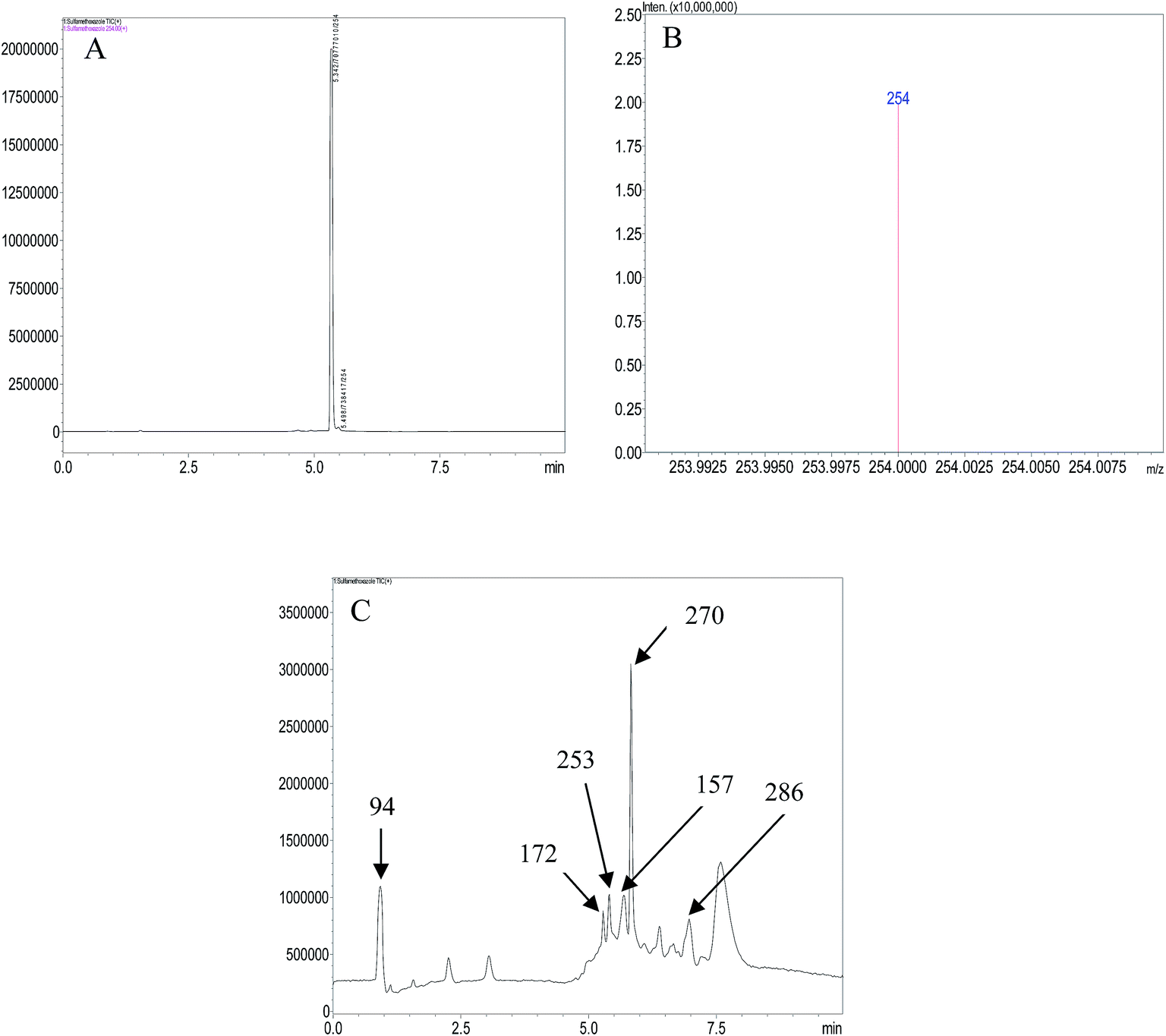 | ||
| Fig. 5 (A) Chromatogram of SMX solution (B) mass spectrum of SMX solution (C) chromatogram of degraded SMX solution. | ||
 | ||
| Fig. 6 Proposed degradation route of SMX by photoelectrochemical process. | ||
4. Conclusion
The applicability of TiO2–EG electrode for the oxidation of SMX via photoelectrocatalytic process was demonstrated in this study. The performance of the photoanode was enhanced in the presence of simulated sunlight, with a fourfold increase in the removal efficiency of the contaminant. The degradation of SMX at the electrode was dependent on pH of the solution and applied current, pH 6.3 and current density 10 mA cm−2 were taken as optimum. After 6 h of the process at the optimum conditions, 90% of the COD of the solution was removed. LC-MS analysis of the degraded SMX revealed the formation of aniline and other by-products which are believed to result from the interaction of the photoelectrochemically generated hydroxyl radicals and electrons with the parent molecule. Given the ease of preparation, low cost and performance of the TiO2–EG anode, it can be further explored for the removal of pharmaceuticals from wastewater.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial supports from the following institutions in South Africa are gratefully acknowledged: Faculty of Science, University of Johannesburg; DST/Mintek Nanotechnology Innovation Centre, University of Johannesburg; Centre for Nanomaterials Science Research, University of Johannesburg; National Research Foundation of South Africa (CPRR Grant number: 98887) and Water Research Commission of South Africa (Grant number: K5/2567). The authors would like to thank Ms Vallerie Muckoya and Dr Patrick Njobeh for assistance with LC-MS.References
- D. Lucas, M. Badia-Fabregat, T. Vicent, G. Caminal, S. Rodríguez-Mozaz, J. Balcazar and D. Barcelo, Chemosphere, 2016, 152, 301–308 CrossRef CAS PubMed.
- C. Stange, J. Sidhu, A. Tiehm and S. Toze, Int. J. Hyg. Environ. Health, 2016, 219, 823–831 CrossRef CAS PubMed.
- S. Hussain, S. Gul, J. R. Steter, D. W. Miwa and A. J. Motheo, Environ. Sci. Pollut. Res., 2015, 22, 15004–15015 CrossRef CAS PubMed.
- N. Voulvoulis, D. Barceló and P. Verlicchi, in Pharmaceuticals in the Environment, 2015, pp. 120–179 Search PubMed.
- B. E. Haggard, J. M. Galloway, W. R. Green and M. T. Meyer, J. Environ. Qual., 2006, 35, 1078–1087 CrossRef CAS PubMed.
- A. K. Brown and C. S. Wong, J. Chromatogr. A, 2016, 1471, 34–44 CrossRef CAS PubMed.
- P. Rodríguez-Escales and X. Sanchez-Vila, Water Res., 2016, 105, 540–550 CrossRef PubMed.
- L. Xu, G. Wang, F. Ma, Y. Zhao, N. Lu, Y. Guo and X. Yang, Appl. Surf. Sci., 2012, 258, 7039–7046 CrossRef CAS.
- F. C. Moreira, R. A. Boaventura, E. Brillas and V. J. Vilar, Appl. Catal., B, 2017, 202, 217–261 CrossRef CAS.
- E. Brillas and C. A. Martínez-Huitle, Appl. Catal., B, 2015, 166, 603–643 CrossRef.
- X. He, Z. Chai, F. Li, C. Zhang, D. Li, J. Li and J. Hu, J. Chem. Technol. Biotechnol., 2013, 88, 1568–1575 CrossRef CAS.
- B. Ntsendwana, B. B. Mamba, S. Sampath and O. A. Arotiba, RSC Adv., 2013, 3, 24473–24483 RSC.
- B. P. Chaplin, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 CAS.
- M. Turabik, N. Oturan, B. Gözmen and M. A. Oturan, Environ. Sci. Pollut. Res., 2014, 21, 8387–8397 CrossRef CAS PubMed.
- D. Zhou, Z. Chen, Q. Yang, C. Shen, G. Tang, S. Zhao, J. Zhang, D. Chen, Q. Wei and X. Dong, ChemCatChem, 2016, 8, 3064–3073 CrossRef CAS.
- E. H. Umukoro, M. G. Peleyeju, J. C. Ngila and O. A. Arotiba, Chem. Eng. J., 2017, 317, 290–301 CrossRef CAS.
- Y.-f. Su, G.-B. Wang, D. T. F. Kuo, M.-l. Chang and Y.-h. Shih, Appl. Catal., B, 2016, 186, 184–192 CrossRef CAS.
- Z. Li, Y. Qu, K. Hu, M. Humayun, S. Chen and L. Jing, Appl. Catal., B, 2017, 203, 355–362 CrossRef CAS.
- Q. Zeng, J. Li, J. Bai, X. Li, L. Xia and B. Zhou, Appl. Catal., B, 2017, 202, 388–396 CrossRef CAS.
- J. Li, X. Zhang, Z. Ai, F. Jia, L. Zhang and J. Lin, J. Phys. Chem. C, 2007, 111, 6832–6836 CAS.
- S. Sarkar, R. Das, H. Choi and C. Bhattacharjee, RSC Adv., 2014, 4, 57250–57266 RSC.
- V. V. Kondalkar, S. S. Mali, R. M. Mane, P. Dandge, S. Choudhury, C. K. Hong, P. S. Patil, S. R. Patil, J. H. Kim and P. N. Bhosale, Ind. Eng. Chem. Res., 2014, 53, 18152–18162 CrossRef CAS.
- Z. Wei, F. Liang, Y. Liu, W. Luo, J. Wang, W. Yao and Y. Zhu, Appl. Catal., B, 2017, 201, 600–606 CrossRef CAS.
- G. Palmisano, V. Loddo, H. H. El Nazer, S. Yurdakal, V. Augugliaro, R. Ciriminna and M. Pagliaro, Chem. Eng. J., 2009, 155, 339–346 CrossRef CAS.
- D. Li, J. Jia, Y. Zhang, N. Wang, X. Guo and X. Yu, J. Hazard. Mater., 2016, 315, 1–10 CrossRef CAS PubMed.
- B. Ntsendwana, S. Sampath, B. Mamba and O. Arotiba, Photochem. Photobiol. Sci., 2013, 12, 1091–1102 CAS.
- X. Yu, Y. Zhang and X. Cheng, Electrochim. Acta, 2014, 137, 668–675 CrossRef CAS.
- T. Ndlovu, A. T. Kuvarega, O. A. Arotiba, S. Sampath, R. W. Krause and B. B. Mamba, Appl. Surf. Sci., 2014, 300, 159–164 CrossRef CAS.
- M. Peleyeju, E. Umukoro, J. Babalola and O. Arotiba, Electrocatalysis, 2016, 7, 132–139 CrossRef CAS.
- F. Hardcastle, J. Arkansas Acad. Sci., 2011, 65, 43–48 CAS.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- G. Zhu, H. Yin, C. Yang, H. Cui, Z. Wang, J. Xu, T. Lin and F. Huang, ChemCatChem, 2015, 7, 2614–2619 CrossRef CAS.
- J.-F. Zhi, H.-B. Wang, T. Nakashima, T. N. Rao and A. Fujishima, J. Phys. Chem. B, 2003, 107, 13389–13395 CrossRef CAS.
- A. Donaghue and B. P. Chaplin, Environ. Sci. Technol., 2013, 47, 12391–12399 CrossRef CAS PubMed.
- H. Chen, B. Gao, H. Li and L. Q. Ma, J. Contam. Hydrol., 2011, 126, 29–36 CrossRef CAS PubMed.
- M. Zhou, Q. Dai, L. Lei, C. a. Ma and D. Wang, Environ. Sci. Technol., 2005, 39, 363–370 CrossRef CAS PubMed.
- K. P. de Amorim, L. L. Romualdo and L. S. Andrade, Sep. Purif. Technol., 2013, 120, 319–327 CrossRef CAS.
- G. Liu, X. Li, B. Han, L. Chen, L. Zhu and L. C. Campos, J. Hazard. Mater., 2017, 322, 461–468 CrossRef CAS PubMed.
- H. Y. Kim, T.-H. Kim, S. M. Cha and S. Yu, Chem. Eng. J., 2017, 313, 556–566 CrossRef CAS.
- W. Zhu, F. Sun, R. Goei and Y. Zhou, Appl. Catal., B, 2017, 207, 93–102 CrossRef CAS.
- L. Hu, P. M. Flanders, P. L. Miller and T. J. Strathmann, Water Res., 2007, 41, 2612–2626 CrossRef CAS PubMed.
- E. Ioannidou, Z. Frontistis, M. Antonopoulou, D. Venieri, I. Konstantinou, D. I. Kondarides and D. Mantzavinos, Chem. Eng. J., 2017, 318, 143–152 CrossRef CAS.
- J. Radjenovic and M. Petrovic, J. Hazard. Mater., 2017, 333, 242–249 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |