DOI:
10.1039/C7RA07239B
(Paper)
RSC Adv., 2017,
7, 40581-40590
Upgrading of palmitic acid over MOF catalysts in supercritical fluid of n-hexane†
Received
30th June 2017
, Accepted 30th July 2017
First published on 21st August 2017
Abstract
The addition of phosphotungstic acid (PTA) to the synthesis mixture of PdCu@FeIII–MOF-5 yields the direct encapsulation of PTA inside the MOF structure (i.e. PTA@PdCu@FeIII–MOF-5) through a facile solvothermal approach. The deoxygenation reaction of palmitic acid has been investigated over PdCu@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 under a hydrogen atmosphere in the supercritical fluid (SCF) of n-hexane. The results showed that palmitic acid can be converted completely at 240 °C on PTA@PdCu@FeIII–MOF-5 with a high selectivity of hexadecane. Owning to the improvement of acidity of the MOF catalyst by the encapsulation of PTA inside the hollow octahedral nanostructures of PdCu@FeIII–MOF-5, the selectivity for hexadecane over the PTA@PdCu@FeIII–MOF-5 catalyst is higher than that over PdCu@FeIII–MOF-5. The excellent performance in the catalytic hydrodeoxygenation (HDO) of palmitic acid is associated with the synergistic effect between yolk–shell PTA@PdCu@FeIII–MOF-5 nanostructures and SCF medium.
1. Introduction
Concerns about the environment and rising costs of fossil fuels have stimulated the search for renewable supplements to fossil fuels. Biomass and its derivatives can provide the sustainable production of liquid fuels and valuable chemicals.1–4 Algae is mainly composed of proteins, lipids and polysaccharides. Only lipids in algae can be used by traditional extraction esterification methods for the preparation of biodiesel,1 while thermo-chemical conversion technology can realize the usage of all components of algae. The resulting liquid product can be called bio-oil.2 Algae-derived biofuel is regarded as the only current renewable source of oil that could meet the global demand for transport fuels and high added valued chemicals.3,4 However, large amounts of oxygen-containing organic compounds exist in the algae-based bio-oil and lead to polymerization, low calorific value, poor stability and strong corrosion.4,5 As a result, the bio-oil cannot be directly used as transportation fuels without upgrading C16–C18 long-chain fatty acids (the main components exist in algal bio-oil).6,7 Catalytic cracking, catalytic esterification and HDO are the main methods for upgrading bio-oil. In particular, the HDO method, which can fundamentally overcome the disadvantage of high oxygen content in bio-oil, has become the focus of research recently. However, some shortages still exist in the HDO of algal-based bio-oil. The catalyst efficiency is relatively low as the large molecules like C16–C18 long-chain fatty acids are hard to spread to the active sites in the pore of the catalyst in conventional medium. In spite of low hydrogen solubility in traditional reaction medium, the high temperature needed in hydrogenation reactions further reduces the hydrogen solubility. Yet the hydrogen dissolvability remains limited even under 13–18 MPa. The high hydrogen pressure restricted the gas–liquid mass transfer process as well as decreasing the safety of the reaction. The aggregation of reactants due to slow diffusion caused carbon deposit on the activity sites leading to the deactivation of catalyst.
The key parameters affect the activity and product selectivity in porous catalysts are adsorption/desorption and pore-transport.8,9 Due to the unique tunable properties (e.g. diffusivity, transport ability), SCFs have with a broad range of applications and dramatically out-performed traditional solvents.10–13 SCFs had been utilized as reaction medium for the hydrogenation upgrading of model compounds recently.14–18 Supercritical methanol technology has proved to be effective for the upgrading of low-boiling fraction of bio-oil.14 The upgrading of crude bio-oil was conducted in supercritical ethanol by Dang et al.15 Hydroprocessing of model bio-oil compound p-cresol in both water and supercritical n-heptane indicated that the reaction were mass-transfer controlled in water while in supercritical n-heptane the mass-transfer limitations were eliminated due to the complete H2 miscibility and enhanced pore diffusivities.16 Deoxygenation of the model bio-oil compound nonanal over Pt/Al2O3 in supercritical n-hexane at 300 °C and a pressure range of 0.7–8 MPa showed a high solubility of H2, and obtained 60% conversion and 40% selectivity to C8 and C9 hydrocarbons.17 The upgrading of bio-oil prepared from hydrothermal liquefaction of cornstalks over Ni/ZrO2 in supercritical cyclohexane under 573 K and 5 MPa H2 obtained 81.6% yield of hydrocarbons with an excellent quality.18 The studies above are based on the model compounds in bio-oil comes from terrestrial biomass. The catalytic HDO of model compounds in algal-based bio-oil like palmitic acid in SCF media has not yet been reported. The properties of SCFs enable a good compatibility of the reactants hydrogen and macromolecules like C16 long-chain fatty acids in SCF and more likely to form a single phase. The pore-transport of the reactants to the active sites of catalyst was promoted. Meanwhile, the products can be removed timely, thus alleviating the gas–liquid mass transfer limit existed in HDO reaction. Avoiding high-pressure hydrogen the security was also improved.9
MOFs as a unique class of porous materials are widely applied to catalysis, separation, gas storage and molecular recognition19,20 and are typically characterized by large surface areas, tunable and uniform pore structures, and interconnected three-dimensional cavities accessible through small pore windows.21–23 These characteristics enable MOFs to confine nanoparticles (NPs) e.g. polyoxometalates (POMs) and metal nanoparticles (MNPs) like Au, Pd, Pt and Cu etc.21 Interaction among the incorporated MNPs were impeded, thus avoiding agglomeration and detachment of the MNPs, whereas the smooth diffusion of reactants into and out of MOFs were guaranteed.21 The hollow cage of MOFs could facilitate the diffusion of substrates onto the internal metal surface as well as the desorption of products.24,25 Recently, MOFs used for catalytic hydrogenation reaction had been systematically explored.26–31 Zhao et al. showed that MOFs with special sandwich nanostructures had an excellent catalytic performance for the hydrogenation of α,β-unsaturated aldehydes.28 Pd@MOF-5 were proved to be a moderately active catalyst for the hydrogenation of cyclooctene.27 The hydrogenation of 2,3,5-trimethylbenzoquinone using Pd28@UiO-66-NH2 has been investigated in the literature.29 The ultrasmall Pd NPs encaged in amine-functionalized UiO-66 in HDO of vanillin in water indicated that 100% conversion of vanillin with exclusive selectivity for 2-methoxy-4-methylphenol were achieved under mild conditions.30 The heterometallic MOF systems bearing two metals e.g. the yolk–shell PdCu@FeIII–MOF-5 nanostructures were successfully designed though a facile solvothermal method and were found to show highly improved catalytic performance in the liquid-phase selective hydrogenation of 1-chloro-2-nitrobenzene relative to the pure yolk.31,32 In short, MOFs encapsulated with MNPs had displayed remarkable catalytic hydrogenation performance due to the unique porous structure and hydrogen storage capacity. POMs consisted of heteropolyanions are complex Brønsted acids and can be encapsulated within the pores of MOF.33 Studies of many POMs based MOFs have been reported.34 Incorporating the acid functionality of PTA in MIL-101 improved the acidity of MOFs,35,36 thereby contributing to the dehydration of carbohydrates.37 Encapsulating MOFs with POMs makes them excellent candidates as heterogeneous catalysts.38 The hydrogenation of long-chain fatty acid by MOFs has been rarely reported.
Exploiting SCF as the reaction medium and MOFs with both microporous and mesoporous structures as the catalyst are supposed to conquer the above existed deficiencies about catalytic HDO of algal-based bio-oil. In this study, palmitic acid is selected as the model compound of algal-based bio-oil. The encapsulation of PTA to the synthesis mixture of PdCu@FeIII–MOF-5 obtained PTA@PdCu@FeIII–MOF-5 through one-pot solvothermal approach.24 The as-prepared yolk–shell PTA@PdCu@FeIII–MOF-5 are used for the catalytic HDO reaction of palmitic acid with the SCF of n-hexane (Tc = 234.5 °C, Pc = 3 MPa) as reaction medium. The remarkable performance of the catalytic HDO reactions attributes to the excellent solubility and diffusibility of SCF media, the high hydrogen storage capacity and special pore structure of the MOF catalysts.39–41
2. Experimental
2.1 Materials
Palmitic acid (99%), hexadecanol (≥99.5%), hexadecanal (≥99.5%), hexadecane (≥99.5%), pentadecane (≥99.5%), N,N-dimethylformamide (DMF) (≥99.5%), ethanol (≥99.5%), Pd(acac)2, Cu(acac)2, Fe(acac)3, n-hexane and PTA were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Zn(NO3)2·6H2O, H2BDC and PVP (Mw = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were bought from Shanghai Chemical Reagents Co., Ltd. (Shanghai, China). All reagents were used as-received without further purification.
000) were bought from Shanghai Chemical Reagents Co., Ltd. (Shanghai, China). All reagents were used as-received without further purification.
2.2 Synthesis of catalysts
2.2.1 PTA@FeIII–MOF-5 hollow octahedral nanostructures. FeIII–MOF-5 encapsulating PTA (donated as PTA@FeIII–MOF-5) was synthesized using a facile solvothermal method.32 Typically, 60 mg Fe(acac)3, 46.4 mg Zn(NO3)2·6H2O, 9.6 mg H2BDC, 9.6 mg PTA and 200 mg PVP were dissolved in 25.6 mL DMF–ethanol mixture (v/v = 5:3) under magnetic stirring for 10 min at room temperature. The resulting homogeneous solution was transferred to a 40 mL Teflon-lined stainless-steel autoclave. After heating the sealed vessel at 100 °C for 6 h it was cooled to room temperature. Separation and purification of the products via centrifugation was the same as the treating of PTA@MOF-5.
2.2.2 PTA@PdCu@FeIII–MOF-5 hollow nanostructures. Briefly, a 2 mL nanoparticles solution of desired concentration and 200 mg PVP were mixed with 23.6 mL DMF–ethanol mixture (v/v = 5:3) under magnetic stirring for 10 minutes at room temperature. Then, 60 mg Fe(acac)3, 46.4 mg Zn(NO3)2·6H2O, 9.6 mg H2BDC and 9.6 mg PTA were put into the above solution under magnetic stirring for another 10 min. The resulting solution was transferred to a 40 mL Teflon-lined stainless-steel autoclave. The sealed vessel was then heated at 100 °C for 6 h before it was cooled to room temperature. Separation and purification of the products was the same as described above.MOF-5 (Fig. S1 of the ESI data†), PTA@MOF-5 (Fig. S2 of the ESI data†), FeIII–MOF-5 (Fig. S3 of the ESI data†), PdCu@FeIII–MOF-5 (Fig. S4 of the ESI data†), Pd NPs@FeIII–MOF-5 (Fig. S5 of the ESI data†), Au NPs@FeIII–MOF-5 (Fig. S6 of the ESI data†), PdCu dendrites (Fig. S7 of the ESI data†), Pd NPs (Fig. S8 of the ESI data†) and Au NPs (Fig. S9 and S12 of the ESI data†) were synthesized according the previous reports.32,42
2.3 Experiments and analysis of the products
The catalytic hydrogenation experiments of palmitic acid were conducted in a 100 mL batch autoclave heated with an external electrical furnace. And the temperature was measured by a thermocouple and controlled at ±1 °C. In a typical experiment, palmitic acid (0.3 g) dissolved in n-hexane (30 mL) and the catalyst (0.1 g) were added into the reactor, before pressurizing with appropriate amount of H2 the autoclave was purged with H2 (0.2 MPa) for three times to remove air at ambient temperature. The products were confirmed by GC-MS (Agilent 6820 N series) and quantitatively analyzed by the gas chromatograph (GC) equipped with a GsBP-Inowax (30 m × 0.32 mm, 0.25 Micron) column and flame ionization detector (FID) with He (10 mL min−1) as carrier gas and injector temperature 320 °C.
3. Results and discussion
3.1 Catalysts characterization
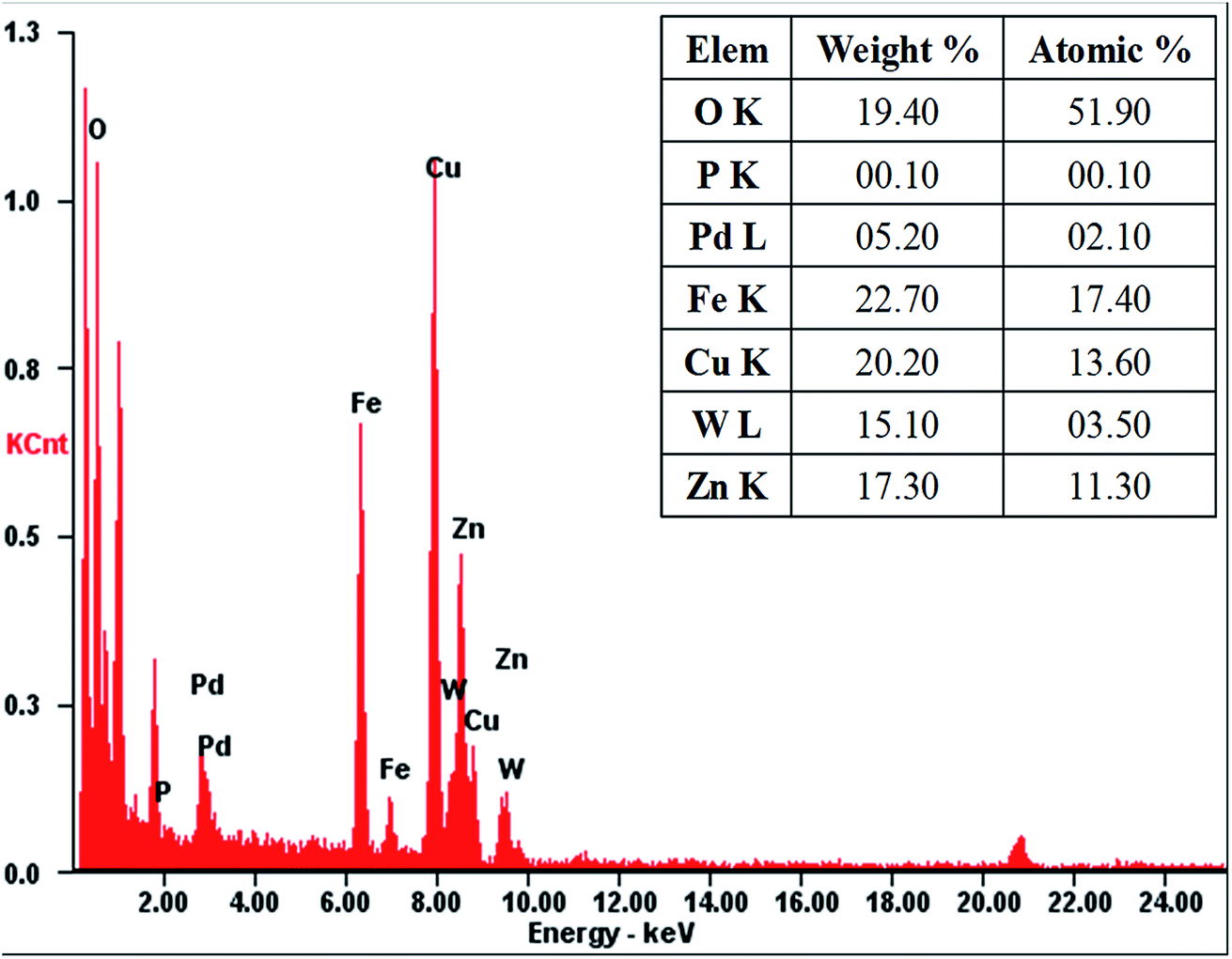
The morphology and structural features of as-prepared PTA@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 hollow nanospheres were characterized by transmission electron microscopy (TEM). As exhibited in Fig. S2,† PTA@MOF-5 remains a roughly spherical shape with an average diameter of 30 nm compared with that of MOF-5 hollow nanospheres 135 nm (ref. 32) (Fig. S1†). PTA@FeIII–MOF-5 showed a hollow octahedral nanostructure as displayed in Fig. 1 without distinct change comparing with FeIII–MOF-5 (Fig. S3†). Nevertheless, PTA@PdCu@FeIII–MOF-5 indicated a polyhedron structure as shown in Fig. 3 attributing to the encapsulation of PTA in PdCu@FeIII–MOF-5 (Fig. S4†). Energy-dispersive X-ray spectroscopy (EDS) analysis manifested that the elements P and W distributed throughout PTA@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 homogeneously as shown in Fig. 2 and 4 respectively. ICP-OES confirmed that the concentration of the element Pd, Cu and W was 9.7 mM L−1, 10.3 mM L−1 and 0.3 mM L−1 respectively.
 |
| | Fig. 1 TEM image of PTA@FeIII–MOF-5. | |
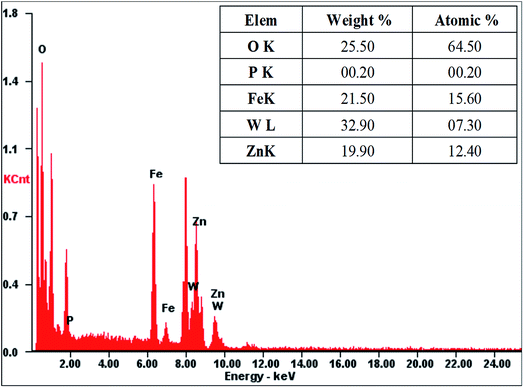 |
| | Fig. 2 EDS analysis of PTA@FeIII–MOF-5. | |
 |
| | Fig. 3 TEM image of PTA@PdCu@FeIII–MOF-5. | |
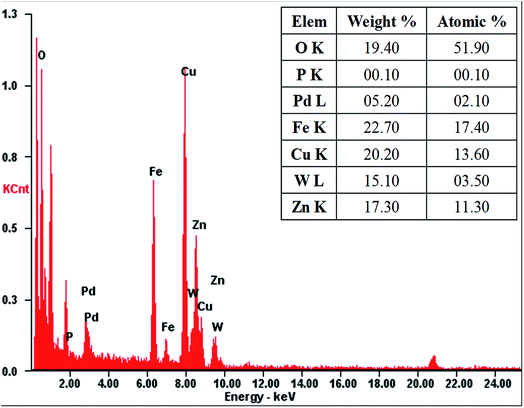 |
| | Fig. 4 EDS analysis of PTA@PdCu@FeIII–MOF-5. | |
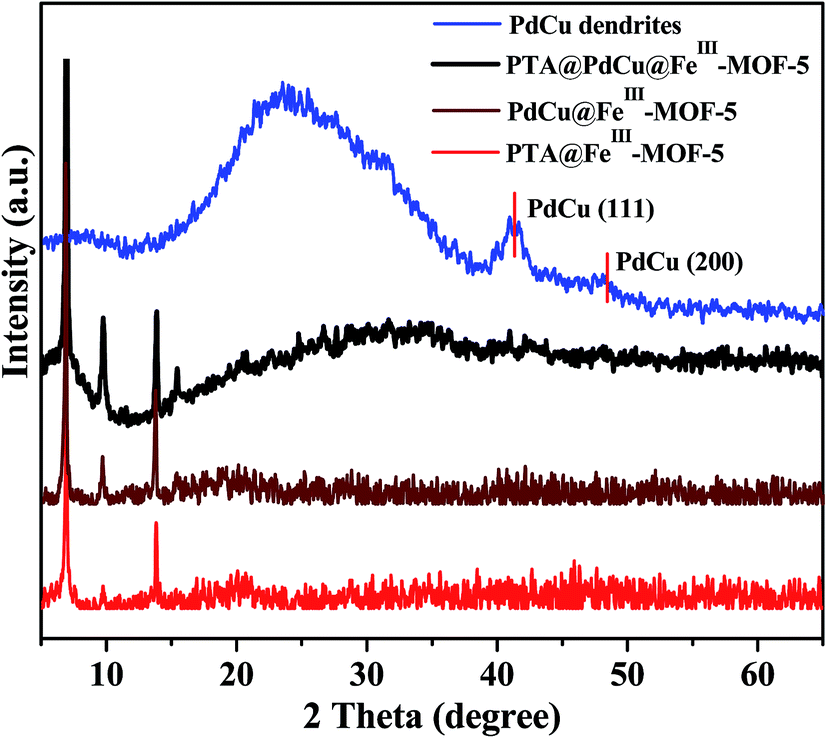
XRD analysis of PTA@PdCu@FeIII–MOF-5, PdCu@FeIII–MOF-5, PTA@FeIII–MOF-5 and PdCu dendrites were displayed in Fig. 5. XRD patterns of PTA@FeIII–MOF-5, PdCu@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 are almost identical to that of FeIII–MOF-5 (Fig. S10†), respectively. This confirms that PTA should be encapsulated into FeIII–MOF-5 without disturbing the MOF structure. Moreover, no patterns belonging to PTA crystals were observed suggesting that PTA is highly dispersed in the cage of FeIII–MOF-5 by small clusters.43 The peaks at 41.5° and 48° of PdCu dendrites coincided with the (111) and (200) peak of PdCu alloys (JCPDS 48-1551),42,44 respectively. The element P, W, Pd and Cu were not detected in X-ray diffraction patterns of PTA@FeIII–MOF-5, PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5, indicating a good confinement effect of FeIII–MOF-5 hollow octahedral nanostructure for PdCu dendrites and PTA.
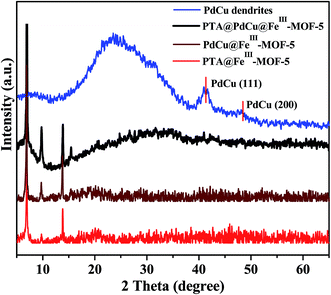 |
| | Fig. 5 XRD analysis of PTA@PdCu@FeIII–MOF-5, PdCu@FeIII–MOF-5, PTA@FeIII–MOF-5 and PdCu dendrites. | |
Acidic properties of PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5 characterized by NH3-TPD were shown in Fig. 6. The distinct peak of PTA@PdCu@FeIII–MOF-5 observed in the range of 150–250 °C corresponded to the weak acid sites.45,46 Compared to PdCu@FeIII–MOF-5 the acidic property of PTA@PdCu@FeIII–MOF-5 was considerably pronounced after the encapsulation of PTA.
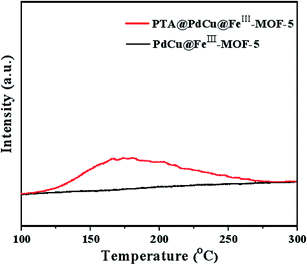 |
| | Fig. 6 NH3-TPD analysis of PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5. | |
TG and DTG analysis of the as-obtained catalysts in nitrogen were shown in Fig. 7. Weight losses below 150 °C are associated with the loss of hygroscopic water by dehydration. In the temperature range 200–300 °C, the weight losses are attributed to the solvent liberation.47 Above 400 °C, weight losses can be attributed to the thermal decomposition of MOF structures. The TG and DTG analysis demonstrated that PTA@PdCu@FeIII–MOF-5 polyhedral nanostructures did not exhibit significant weight loss up to at least 350 °C, and indicated a good thermal stability below 350 °C of both PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5.
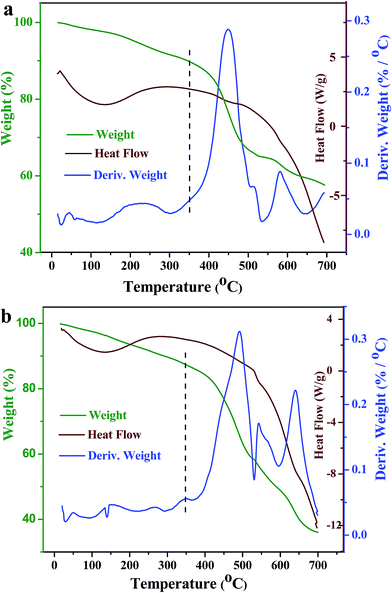 |
| | Fig. 7 Thermogravimetric (TG) analysis and derivative thermogravimetric (DTG) (rate of weight loss) of (a) the yolk–shell PTA@PdCu@FeIII–MOF-5 and (b) PdCu@FeIII–MOF-5 in nitrogen. Heating rate 10 °C min−1 from room temperature to 700 °C. | |
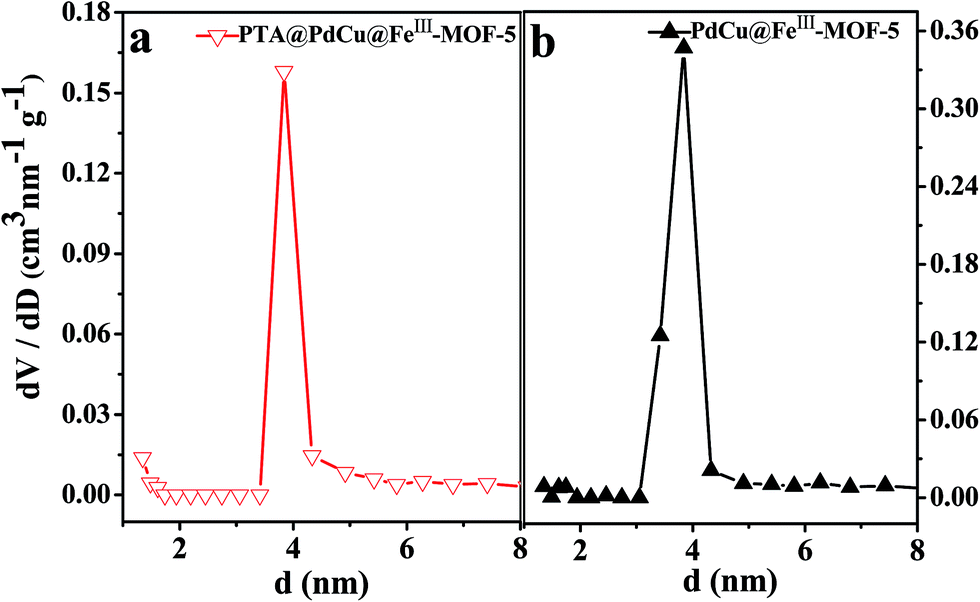
The permanent porosity of the as-obtained MOFs was confirmed by pore size distribution measurements and nitrogen adsorption–desorption isotherms as shown in Fig. 8 and 9 respectively. As shown in Fig. 9, both PdCu@FeIII–MOF-5 and PdCu@PTA@FeIII–MOF-5 exhibited a typical type IV sorption isotherm, indicating mesoporosity of the MOFs.48 Additionally, it was observed that nitrogen uptake displays an obvious increase at a relatively low pressure (P/P0 < 0.01), suggesting the presence of inherent micropores in these hollow nanocages.32,49 The pore size distribution calculated from the BJH method using the desorption branch in Fig. 8 showed that the shells of PdCu@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 mainly contain mesopores with diameters of 3.83 nm and 3.84 nm respectively. Due to the mesopores and micropores of the yolk–shell MOFs, PdCu@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 shows a BET surface area of 240.3 m2 g−1 and 188.3 m2 g−1 respectively. The pore volumes of PdCu@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 are 0.40 and 0.17 cm3 g−1, respectively. The encapsulation of PTA inside PdCu@FeIII–MOF-5 structure resulted in the decrease of pore volume and BET surface area of PTA@PdCu@FeIII–MOF-5. The voids between coordination polymer aggregated in the shells presumably formed the mesopores of the catalysts.50
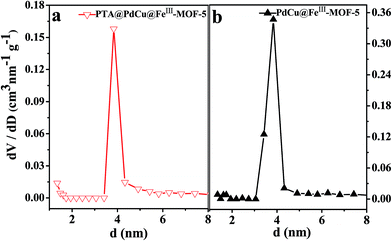 |
| | Fig. 8 Pore size distribution of PTA@PdCu@FeIII–MOF-5 (a) and PdCu@FeIII–MOF-5 (b) calculated from the BJH method using desorption branch. | |
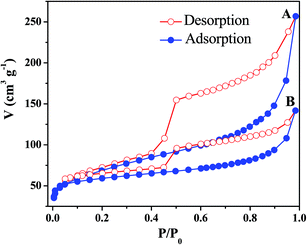 |
| | Fig. 9 Nitrogen adsorption–desorption isotherms of PdCu@FeIII–MOF-5 (A) and PTA@PdCu@FeIII–MOF-5 (B) measured at 77 K. | |
As shown in Fig. 10, the FT-IR spectra of FeIII–MOF-5 and PdCu@FeIII–MOF-5 hollow octahedral nanostructures verify the coordination of the carboxylate groups to metal ions, as evidenced by a red shift in the carboxylate stretching frequency to 1575 cm−1 from 1680 cm−1 for uncoordinated H2BDC, which is consistent with the previous report.32,51 Meanwhile, the carboxylate stretching frequency of PTA@FeIII–MOF-5 and PTA@PdCu@FeIII–MOF-5 exhibited unconspicuous shift compared with FeIII–MOF-5. And the XPS spectra of PTA@FeIII–MOF-5 hollow nanostructures (Fig. S11†) coincides with that of FeIII–MOF-5.32 Both TEM, FT-IR and XPS spectra analysis confirmed that PTA were encapsulated inside the FeIII–MOF-5 structures and the structure of PTA@FeIII–MOF-5 maintained approximately the same with that of FeIII–MOF-5.
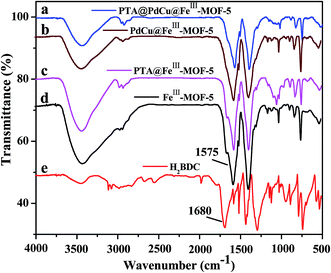 |
| | Fig. 10 FT-IR spectra of as-synthesized PTA@PdCu@FeIII–MOF-5 (a), PdCu@FeIII–MOF-5 (b), PTA@FeIII–MOF-5 (c), FeIII–MOF-5 (d) and H2BDC (e). | |
3.2 Catalytic HDO of palmitic acid
3.2.1 Effect of different as-prepared catalysts. The catalytic HDO of palmitic acid on PTA@PdCu@FeIII–MOF-5, PdCu@FeIII–MOF-5, Pd NPs@FeIII–MOF-5, Au NPs@FeIII–MOF-5, PdCu dendrites, Pd NPs and Au NPs were indicated in Table 1. The SCF of n-hexane can be obtained at 240 °C and under the initial 2 MPa H2, hence 240 °C could be selected as the optimum reaction temperature. It can be concluded from Table 1 that the catalytic activity of PdCu@FeIII–MOF-5, Pd NPs@FeIII–MOF-5 and Au NPs@FeIII–MOF-5 is remarkably superior to FeIII–MOF-5, PTA@FeIII–MOF-5, PdCu dendrites, Pd NPs and Au NPs respectively. It can be concluded from the entry 2, 3, 4 and entry 5 that metal active sites played a key role in catalytic HDO of palmitic acid. Moreover, the catalytic efficiency of the yolk–shell catalysts for the HDO reactions were increased remarkably attributing to the high hydrogen storage capacity of MOFs and the immobilization and dispersion effect of MOF for MNPs. Notably, the catalytic hydrogenation performance of bimetallic MOF system PdCu@FeIII–MOF-5 has an advantage over monometal MOF system i.e. Pd NPs@FeIII–MOF-5 and Au NPs@FeIII–MOF-5. The products of catalytic HDO of palmitic acid mainly contained hexadecanal, hexadecanol and hexadecane. The catalytic HDO of palmitic acid over PdCu@FeIII–MOF-5 mainly produced hexadecanol. Compared to PdCu@FeIII–MOF-5, the products distribution manifested that the selectivity of hexadecane increased significantly on PTA@PdCu@FeIII–MOF-5 due to the enhancement of acidity of the catalyst. Compared to the catalyst exploited to the HDO of palmitic acid in the previous work,7,46 the yolk–shell PTA@PdCu@FeIII–MOF-5 exhibited a considerable advantage as can be seen in Table S1.†
Table 1 HDO of palmitic acid over different as-prepared catalystsa
| Cat. |
Conv./% |
Selectivity/% |
| Hexadecanal |
Hexadecanol |
Hexadecane |
Pentadecane |
| Reaction conditions: solvent 30 mL n-hexane, reactant palmitic acid 0.3 g, temperature 240 °C, catalyst 0.1 g, initial H2 pressure 2 MPa, reaction time 2 h. |
| PTA@PdCu@FeIII–MOF-5 |
100 |
0 |
5.3 |
90.6 |
4.1 |
| PdCu@FeIII–MOF-5 |
100 |
0.2 |
74 |
24.8 |
1 |
| Pd NPs@FeIII–MOF-5 |
82.1 |
5.2 |
76.2 |
18.1 |
0.5 |
| Au NPs@FeIII–MOF-5 |
70.5 |
9.8 |
71.1 |
19.1 |
0 |
| FeIII–MOF-5 |
3.6 |
19.1 |
80.9 |
0 |
0 |
| PTA@FeIII–MOF-5 |
3.9 |
18.6 |
81.4 |
0 |
0 |
| PdCu dendrites |
25.7 |
20.1 |
60.3 |
19.6 |
0 |
| Pd NPs |
13.2 |
25.3 |
65 |
9.7 |
0 |
| Au NPs |
10.5 |
29.2 |
64.3 |
6.5 |
0 |
3.2.2 Effect of different reaction media. Table 2 showed the catalytic HDO of palmitic acid over PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5 in supercritical n-hexane and conventional n-decane media respectively. As shown, the performance of the catalysts in SCF of n-hexane is superior to that in conventional n-decane media, the conversion of palmitic acid in SCF of n-hexane is about 15% higher than that in decane due to the good compatibility of the hydrogen and palmitic acid of SCF. The enhancement of acidity of PTA@PdCu@FeIII–MOF-5 contributed to the dehydration of hexadecanol to produce hexadecane. With the temperature increasing to 240 °C the final pressure of SCF system of n-hexane is approximately 0.3 MPa lower than that of traditional n-decane system as a result of the good compatibility of the hydrogen of SCF of n-hexane and high hydrogen storage capacity of MOF materials.
Table 2 Catalytic HDO of palmitic acid in SCF of n-hexane and conventional n-decane mediaa
| Media |
Cat. |
Conv./% |
Selectivity/% |
| Hexadecanal |
Hexadecanol |
Hexadecane |
Pentadecane |
| Reaction conditions: solvent n-hexane 30 mL, reactant palmitic acid 0.3 g, temperature 240 °C, catalyst 0.1 g, initial H2 pressure 2 MPa, reaction time 2 h. |
| n-Hexane |
PTA@PdCu@FeIII–MOF-5 |
100 |
0 |
5.3 |
90.6 |
4.1 |
| PdCu@FeIII–MOF-5 |
100 |
0.2 |
74 |
24.8 |
1 |
| n-Decane |
PTA@PdCu@FeIII–MOF-5 |
85.1 |
0.6 |
5.9 |
90.4 |
3.1 |
| PdCu@FeIII–MOF-5 |
84.6 |
0.1 |
74.9 |
24.2 |
0.8 |
3.3 Catalytic HDO of palmitic acid over PTA@PdCu@FeIII–MOF-5 in n-hexane
3.3.1 Effect of reaction temperatures. Generally, reaction temperature directly affects the reaction rate and equilibrium.52 As listed in Table 3, catalytic reaction over PTA@PdCu@FeIII–MOF-5 at different reaction temperature showed that the conversion was relatively low at 180 °C, but increased dramatically to 100% at 240 °C (the supercritical state of n-hexane). Analysis of product distribution revealed that when the temperature increased to 240 °C, 93.7% selectivity of n-hexadecane was achieved. Further elevating the temperature to 260 °C, the selectivity of hexadecane almost constant. Large amounts of hexadecanol could be obtained when conducting the reaction below 200 °C indicating that the dehydration of the hexadecanol has the highest temperature barrier in the whole reaction. The selectivity of the decarbonylation product pentadecane increased with a very small extent from 0% at 180 °C to 5.9% at 260 °C. And the hydrocracking products were not detected during the reaction. Indicating that HDO is the main pathway. PdCu@FeIII–MOF-5 were also investigated as the catalyst as displayed in Table 3, the conversion increased from 78.1% at 200 °C to 99.8% at 240 °C, the selectivity of hexadecanol increased from 67.2% at 200 °C to 74% at 240 °C, then slightly decreased to 65.3% at 260 °C. Whereas the selectivity of hexadecane was relatively low that increased from 11.6% at 200 °C to 33.2% at 260 °C as a result of the inacidity of PdCu@FeIII–MOF-5. The encapsulation of PTA inside the yolk–shell composites enhanced the acidity of PTA@PdCu@FeIII–MOF-5, thus, contributing to the dehydration of hexadecanol.53 The different acidity of PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5 elucidated the different products distribution of HDO of palmitic acid.
Table 3 Catalytic HDO of palmitic acid over PTA@PdCu@FeIII–MOF-5 and PdCu@FeIII–MOF-5 in n-hexanea
| Cat. |
T/°C |
Conv./% |
Selectivity/% |
| Hexadecanal |
Hexadecanol |
Hexadecane |
Pentadecane |
| Reaction conditions: solvent 30 mL n-hexane, reactant palmitic acid 0.3 g, catalyst 0.1 g, initial H2 pressure 2 MPa, reaction time 2 h. |
| PTA@PdCu@FeIII–MOF-5 |
180 |
55.8 |
25.1 |
48.2 |
26.7 |
0 |
| 200 |
86.5 |
10.3 |
63.2 |
31.4 |
1.1 |
| 220 |
99.8 |
0 |
31.1 |
66.6 |
2.3 |
| 240 |
99.9 |
0 |
2.3 |
93.6 |
4.1 |
| 260 |
100 |
0 |
0.4 |
93.7 |
5.9 |
| PdCu@FeIII–MOF-5 |
200 |
78.1 |
21.2 |
67.2 |
11.6 |
0 |
| 220 |
96.5 |
15.3 |
70 |
18.5 |
0.2 |
| 240 |
99.8 |
0 |
74 |
25 |
1 |
| 260 |
100 |
0 |
65.3 |
33.2 |
1.5 |
3.3.2 Effect of hydrogen pressure. Initial hydrogen pressure was an important parameter that affected the HDO rate of palmitic acid. The pressure variation experiments were investigated using PTA@PdCu@FeIII–MOF-5 as a catalyst under 240 °C as shown in Fig. 11. As expected, with the initial H2 pressure increased from 0 to 2.5 MPa the conversion of palmitic acid increased from 5.5% to 100% and the selectivity of hexadecane increased from 0 to 96.7%. And the selectivity of hexadecanal and hexadecanol increased first from 0 to 28% and 31% then decreased to 0 and 2.1% respectively. While the selectivity of the decarbonylation product n-pentadecane was very low (from 0 at 0 MPa to 1.2% at 2.5 MPa). Hydrogen participated in converting hexadecanal into hexadecanol, the branching step in the reaction. Increasing the amount of H2 facilitates the equilibrium toward hexadecanol accelerating the pathway to n-hexadecane. Confirming that the main route is hydrogenation–dehydration–hydrogenation reactions to generate hexadecane without carbon loss. The result indicated that higher hydrogen pressure facilitates the HDO pathway over PTA@PdCu@FeIII–MOF-5 coinciding with previous reports.54–56
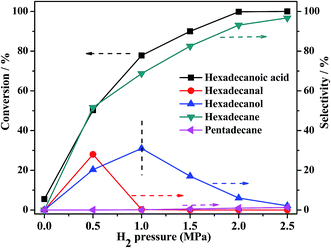 |
| | Fig. 11 Effect of H2 pressure on palmitic acid conversion and selectivity to hydrogenation products over PTA@PdCu@FeIII–MOF-5. Reaction conditions: solvent n-hexane 30 mL, palmitic acid 0.3 g, catalyst 0.1 g, reaction temperature 240 °C, reaction time 2 h. | |
3.3.3 Effect of reaction time. For the study of reaction mechanism, the effect of reaction time on the catalytic HDO of palmitic acid was also studied as depicted in Fig. 12. The conversion of palmitic acid reached almost 100% after reacting for 2 h. Fig. 12 reveals that the selectivity of hexadecanol gradually increased from 38% at 0.5 h to 51% at 1 h, and then gradually decreased to 0% at 3 h, along with that of hexadecanal decreased consecutively from 34% at 0.5 h to 0% at 3 h. Whereas that of hexadecane increased continuously to 99.8% at 3 h. And the selectivity of pentadecane remained very low during the reaction. It is observed that palmitic acid exhausted earlier than hexadecanol. A fast hydrogenation reduction of palmitic acid to hexadecanal proceeded firstly, immediately following by the hydrogenation of hexadecanal to generate hexadecanol. Then the main product hexadecane was produced by slow dehydration of hexadecanol. The reduction rate of hexadecanal to hexadecanol was confirmed to be faster than the production rate of hexadecanal over PTA@PdCu@FeIII–MOF-5.
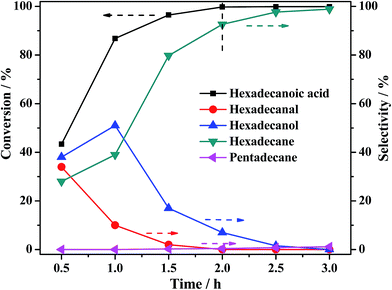 |
| | Fig. 12 Effect of reaction time on palmitic acid conversion and the selectivity of hydrogenation products over PTA@PdCu@FeIII–MOF-5. Reaction conditions: solvent n-hexane 30 mL, palmitic acid 0.3 g, catalyst 0.1 g, initial H2 pressure 2 MPa, reaction temperature 240 °C. | |
4. Mechanism study
Hexadecanal was placed to the reaction conditions to confirm that hexadecanol was generated from the hydrogenation of hexadecanal. As shown in Fig. 13, the selectivity of hexadecanol gradually increased from 49% at 0.5 h to 55% at 1 h and then decreased to 2.2% at 3 h, meanwhile the selectivity of the HDO product hexadecane increased from 51% to 99%, indicating that HDO was the main pathway for the conversion of hexadecanal over PTA@PdCu@FeIII–MOF-5. Conversely, the selectivity of the decarbonylation product (pentadecane) was less than 1%. This suggests that hexadecanol obtained from hexadecanal is through a hydrogenation mechanism as displays in Scheme 1. The (de)hydrogenation between hexadecanol and hexadecanal is reversible. Hexadecane was generated from the hydrogenation of hexadecene that came from the dehydration of hexadecanol.
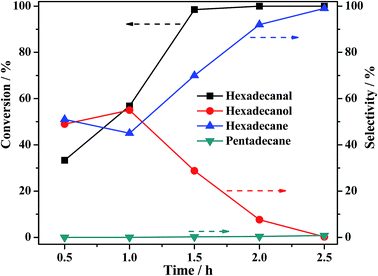 |
| | Fig. 13 Effect of reaction time on hexadecanal conversion and the selectivity of hydrogenation products over PTA@PdCu@FeIII–MOF-5. Reaction conditions: solvent n-hexane 30 mL, palmitic acid 0.3 g, catalyst 0.1 g, initial H2 pressure 2 MPa, reaction temperature 240 °C. | |
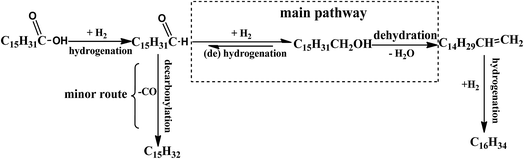 |
| | Scheme 1 Proposed hydrogenation process for palmitic acid over PTA@PdCu@FeIII–MOF-5. | |
5. Conclusions
A series of well-defined MOF hollow nanostructures encapsulated with PTA (PTA@MOF-5, PTA@FeIII–MOF-5, PTA@PdCu@FeIII–MOF-5) were successfully synthesized by a facile solvothermal approach. It is demonstrated that the performance of the catalysts in SCF of n-hexane is superior to that in the conventional media. Selective hydrogenation results of palmitic acid indicate that the yolk–shell PTA@PdCu@FeIII–MOF-5 nanostructures is in favor of achieving excellent catalytic performance as compared to the pure noble-metal nanocrystals and other catalysts used in the previous work. The acidic property of PTA@PdCu@FeIII–MOF-5 was greatly pronounced compared to PdCu@FeIII–MOF-5, resulting in the high selectivity of hexadecane. The high performance in the catalytic HDO of palmitic acid is associated with the synergistic effect between yolk–shell PTA@PdCu@FeIII–MOF-5 nanostructures and SCF. The present work demonstrated a new green catalytic process towards the hydrogenation upgrading of algal-based bio-oil and a great potential in catalyzing organic reactions.
Acknowledgements
The authors are grateful for the financial supports from the National Natural Science Foundation of China (No. 21576155, 21376136 and No. 21376140), Research Project of Guangdong Provincial Department of Science and Technology Department (No. 2015B020215004).
References
- P. Collet, L. Lardon, A. Hélias, S. Bricout, I. Lombaert-Valot, B. Perrier, O. Lépine, J.-P. Steyer and O. Bernard, Renewable Energy, 2014, 71, 525–533 CrossRef CAS
 .
. - Z. Shuping, W. Yulong, Y. Mingde, I. Kaleem, L. Chun and J. Tong, Energy, 2010, 35, 5406–5411 CrossRef .
- E. P. Bennion, D. M. Ginosar, J. Moses, F. Agblevor and J. C. Quinn, Appl. Energy, 2015, 154, 1062–1071 CrossRef CAS .
- Y. Chen, Y. Wu, D. Hua, C. Li, M. P. Harold, J. Wang and M. Yang, RSC Adv., 2015, 5, 18673–18701 RSC .
- P. Duan and P. E. Savage, Bioresour. Technol., 2011, 102, 1899–1906 CrossRef CAS PubMed .
- S. Zou, Y. Wu, M. Yang, C. Li and J. Tong, Energy Environ. Sci., 2010, 3, 1073–1078 CAS .
- R. Ding, Y. Wu, Y. Chen, J. Liang, J. Liu and M. Yang, Chem. Eng. Sci., 2015, 135, 517–525 CrossRef CAS .
- A. Baiker, Chem. Rev., 1999, 99, 453–474 CrossRef CAS PubMed .
- B. Subramaniam, Appl. Catal., A, 2001, 212, 199–213 CrossRef CAS .
- P. Pollet, E. A. Davey, E. E. Ureña-Benavides, C. A. Eckert and C. L. Liotta, Green Chem., 2014, 16, 1034 RSC .
- M. Tsang, G. Philippot, C. Aymonier and G. Sonnemann, Green Chem., 2016, 18, 4924–4933 RSC .
- Z. Sun, G. Bottari and K. Barta, Green Chem., 2015, 17, 5172–5181 RSC .
- D. Verma, R. Insyani, Y.-W. Suh, S. M. Kim, S. K. Kim and J. Kim, Green Chem., 2017, 19, 1969–1982 RSC .
- W. Li, C. Pan, Q. Zhang, Z. Liu, J. Peng, P. Chen, H. Lou and X. Zheng, Bioresour. Technol., 2011, 102, 4884–4889 CrossRef CAS PubMed .
- Q. Dang, Z. Luo, J. Zhang, J. Wang, W. Chen and Y. Yang, Fuel, 2013, 103, 683–692 CrossRef CAS .
- H. Wan, R. V. Chaudhari and B. Subramaniam, Top. Catal., 2012, 55, 129–139 CrossRef CAS .
- J. W. Ford, R. V. Chaudhari and B. Subramaniam, Ind. Eng. Chem. Res., 2010, 49, 10852–10858 CrossRef CAS .
- W. Shi, Y. Gao, S. Song and Y. Zhao, Ind. Eng. Chem. Res., 2014, 53, 11557–11565 CrossRef CAS .
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520–524 CrossRef CAS .
- A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494–3495 CrossRef CAS PubMed .
- H. Liu, L. Chang, C. Bai, L. Chen, R. Luque and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5019–5023 CrossRef CAS PubMed .
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC .
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed .
- Z. Zhang, Y. Chen, S. He, J. Zhang, X. Xu, Y. Yang, F. Nosheen, F. Saleem, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 126, 12725–12729 CrossRef .
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889–10893 CrossRef CAS PubMed .
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS .
- S. Hermes, M. K. Schroter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed .
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed .
- D.-L. Chen, S. Wu, P. Yang, S. He, L. Dou and F.-F. Wang, J. Phys. Chem. C, 2017, 121, 8857–8863 CAS .
- F. Zhang, S. Zheng, Q. Xiao, Y. Zhong, W. Zhu, A. Lin and M. Samy El-Shall, Green Chem., 2016, 18, 2900–2908 RSC .
- X. Xu, Z. Zhang and X. Wang, Adv. Mater., 2015, 27, 5365–5371 CrossRef CAS PubMed .
- Z. Zhang, Y. Chen, X. Xu, J. Zhang, G. Xiang, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 429–433 CrossRef CAS PubMed .
- R. Canioni, C. Roch-Marchal, F. Sécheresse, P. Horcajada, C. Serre, M. Hardi-Dan, G. Férey, J.-M. Greneche, F. Lefebvre and J.-S. Chang, J. Mater. Chem., 2011, 21, 1226–1233 RSC .
- R. Yu, X.-F. Kuang, X.-Y. Wu, C.-Z. Lu and J. P. Donahue, Coord. Chem. Rev., 2009, 253, 2872–2890 CrossRef CAS .
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS .
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC .
- Y. Zhang, V. Degirmenci, C. Li and E. J. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed .
- D. Farrusseng, Metal–organic frameworks: applications from catalysis to gas storage, John Wiley & Sons, 2011 Search PubMed .
- B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 117, 4823–4827 CrossRef .
- B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS PubMed .
- A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494–3495 CrossRef CAS PubMed .
- B. Xu, Z. Zhang and X. Wang, Nanoscale, 2013, 5, 4495–4505 RSC .
- J. Juan-Alcañiz, E. V. Ramos-Fernandez, U. Lafont, J. Gascon and F. Kapteijn, J. Catal., 2010, 269, 229–241 CrossRef .
- L. Wang, J.-J. Zhai, K. Jiang, J.-Q. Wang and W.-B. Cai, Int. J. Hydrogen Energy, 2015, 40, 1726–1734 CrossRef CAS .
- L. Vilcocq, R. Koerin, A. Cabiac, C. Especel, S. Lacombe and D. Duprez, Appl. Catal., B, 2014, 148–149, 499–508 CrossRef CAS .
- Y. Duan, Y. Wu, Q. Zhang, R. Ding, Y. Chen, J. Liu and M. Yang, J. Mol. Catal. A: Chem., 2015, 398, 72–78 CrossRef CAS .
- S.-L. Zhong, R. Xu, L.-F. Zhang, W.-G. Qu, G.-Q. Gao, X.-L. Wu and A.-W. Xu, J. Mater. Chem., 2011, 21, 16574–16580 RSC .
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and Á. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS .
- Y. Y. Karabach, M. F. C. Guedes da Silva, M. N. Kopylovich, B. Gil-Hernandez, J. Sanchiz, A. M. Kirillov and A. J. Pombeiro, Inorg. Chem., 2010, 49, 11096–11105 CrossRef CAS PubMed .
- S.-L. Zhong, R. Xu, L.-F. Zhang, W.-G. Qu, G.-Q. Gao, X.-L. Wu and A.-W. Xu, J. Mater. Chem., 2011, 21, 16574–16580 RSC .
- W. Cho, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2008, 130, 16943–16946 CrossRef CAS PubMed .
- X. Fang, Z. Yin, H. Wang, J. Li, X. Liang, J. Kang and B. He, J. Catal., 2015, 329, 187–194 CrossRef CAS .
- Y. Zhang, V. Degirmenci, C. Li and E. J. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed .
- K. Kandel, J. W. Anderegg, N. C. Nelson, U. Chaudhary and I. I. Slowing, J. Catal., 2014, 314, 142–148 CrossRef CAS .
- L. Boda, G. Onyestyák, H. Solt, F. Lónyi, J. Valyon and A. Thernesz, Appl. Catal., A, 2010, 374, 158–169 CrossRef CAS .
- R. Ding, Y. Wu, Y. Chen, H. Chen, J. Wang, Y. Shi and M. Yang, Catal. Sci. Technol., 2016, 6, 2065–2076 CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07239b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab and
Mingde Yanga
*ab and
Mingde Yanga