
A biocatalytic approach towards the stereoselective synthesis of protected inositols†
L. H.
Sayer
,
G. J.
Florence
* and
T. K.
Smith
*
Biomedical Science Research Complex, Schools of Chemistry and Biology, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: gjf1@st-andrews.ac.uk; tks1@st-andrews.ac.uk
First published on 12th December 2016
Abstract
A biocatalytic flow system utilising Trypanosoma bruceiL-myo-inositol 1-phosphate synthase has been developed to prepare significant quantities of L-myo-inositol 1-phosphate. Using this highly valuable biochemical, we have demonstrated its utility as a viable starting material in a drastically improved and shortened synthesis of a key intermediate in the synthesis of the biologically important myo-inositol derivatives. This biotransformation allows rapid and stereospecific inositol derivatives to be synthesized, on a larger scale than ever before.
Introduction
myo-Inositol is ubiquitous in nature and can be found as the structural core of a diverse range of biological metabolites, including inositol phosphates, phosphatidylinositols and their phosphates, glycosylphosphatidylinositol (GPI) anchors1 and mycothiol.2 Many of these compounds have important and fundamental biological functions, including cellular signalling,3 protein anchoring1 and pathogen virulence.4 Given their essential function in living systems, they have been the focus of significant research in both academia and industry.The stereo-controlled synthesis of protected myo-inositol derivatives presents a number of obstacles and challenges, often requiring lengthy synthetic sequences, which suffer significant material loss as a consequence.5myo-Inositol chemistry faces the regioselectivity issues associated with classical carbohydrate chemistry, but the situation is further complicated by the inherent symmetry of the myo-inositol core and the requirement to break the symmetry plane to access the desired enantiomer. In order to overcome the complex enantioselectivity issues it is generally necessary to perform a chemical or enzymatic resolution, which further detriments the yield of synthetic routes.
The eukaryotic de novo biosynthesis of myo-inositol (3), shown in Scheme 1, involves the sequential action of two enzymes: L-myo-inositol 1-phosphate synthase (INO1, E.C.5.5.1.4) and myo-inositol monophosphatase (IMPase, E.C.3.1.3.25).6 The rate-limiting step of this process is the conversion of D-glucose 6-phosphate (GP, 1) to L-myo-inositol 1-phosphate (IP, 2) by INO1, which employs nicotinamide adenine dinucleotide (NAD+) as a cofactor. A biocatalytic approach using INO1 would facilitate direct access to enantiopure IP (2).7 This is an attractive target due to its high commercial value (€196 mg−1, Cayman Chemical), but also due to the potential for its use as a starting point in the syntheses of numerous myo-inositol derivatives with significantly reduced step counts. Using selective protection strategies we envisaged the ability to gain access to any protection pattern around the myo-inositol core. A clear advantage of the present approach would be the removal of bottleneck resolution step (50% max yield) as IP (2) is produced directly and would provide a unique and previously unavailable chiral pool starting material. However, the use of IP for chemical synthesis is not without challenge and will require an extensive study of protecting group conditions compatible with the phosphate or it's corresponding phosphate ester.
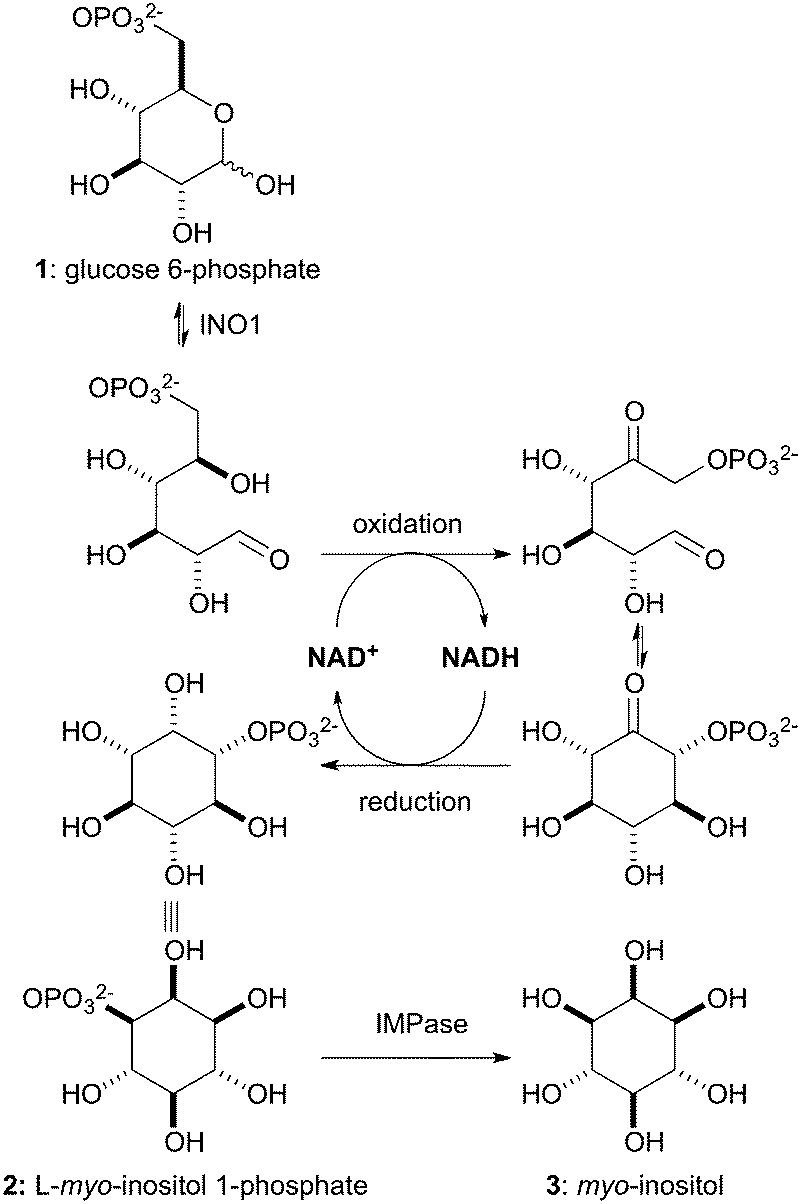 | ||
| Scheme 1 Biosynthesis of myo-inositol (3). | ||
In considering the use of an INO1 enzyme for biocatalysis we identified several key facets. Firstly, the cofactor, NAD+, is both reduced and reoxidised within the reaction, so there is no net change. This allows a much lower amount of NAD+ to be used which would be the most expensive component of the system, lowering the potential need for co-factor regeneration in situ. The starting material, GP (1), is relatively inexpensive (€29 g−1, Sigma Aldrich) compared to the IP product. Once established as viable, this system should be able to incorporate a commercial hexokinase upstream to access GP (1) from glucose. Biocatalytic production of IP (2) has previously been accomplished using whole cell fermentation of recombinant S. cerevisiae INO1 (ref. 8) and isolated recombinant INO1 enzyme preparations.9,10 This work clearly demonstrates the potential for the biocatalytic production of IP, however within an industrial setting these methods may fail to provide an efficient and economical route to IP (2).
The protozoan parasite Trypanosoma brucei relies upon the de novo synthesis of myo-inositol for its survival.11,12 The T. brucei INO1 (TbINO1) has been cloned and overexpressed in Escherichia coli using a pBAD TA vector, which encodes a C-terminal hexa-His tag.11 The recombinant TbINO1 protein was shown to be catalytically active as part of a coupled assay developed for a drug discovery programme, whereby the inhibition of TbINO1 activity was assessed by the phosphate released by produced IP via an IMPase. Saturation kinetic studies showed TbINO1 to have an apparent Km of 0.58 mM and Vmax of 0.25 mole per hour with respect to [GP (1)]. The specific activity of recombinant TbINO1 was determined to be 756 U mg−1 at 37 °C where 1 U is the amount of enzyme catalysing the formation of 1 nmole of IP (2) per minute at 37 °C.11 The specific activity of recombinant TbINO1 is approximately 10-fold higher than that of human recombinant INO1 (70 U mg−1).13 The protein proved relatively stable operating at an optimum temperature of 37 °C. The reaction was conducted on a <1 mg scale in tris(hydroxymethyl)aminoethane acetate buffer at pH 8.0 and 37 °C. The reaction solution contained 5 mM GP (1), 1 mM NAD+, 2 mM ammonium acetate, 1 mM dithiothreitol (DTT) and 5 μg of TbINO1 in a total volume of 150 μL. Enzyme activity was dependent upon NH4+ ions, with activity reduced to 10 U mg−1 in their absence. TbINO1 has been shown to furnish complete conversion to IP (2), which is important when considering the optimization and scale up of this process. It not only increases efficiency, but also avoids potential difficulties in the separation of IP (2) from any remaining GP (1) due to their similarly high polarity. TbINO1, therefore, looks like an ideal candidate for use in the biocatalytic production of IP (2).
Results and discussion
TbINO1 expression, purification and assay
Recombinant TbINO1 was expressed as previously detailed14 and purified by Ni2+-affinity chromatography, eluting with an increasing gradient of imidazole in 20 mM ammonium bicarbonate (pH 8.5) and 250 mM NaCl. The protein was dialysed against a solution of 20 mM ammonium bicarbonate (pH 8.5), 50 mM NaCl and 5 mM DTT, and stored with 20% glycerol at −80 °C. This process yielded the recombinant TbINO1 enzyme in a yield of 75 mg L−1 of culture broth. Conversion of TbINO1 reactions was determined colourimetrically, using the established IMPase assay.11Batch scale and flow optimisation
In assessing the original TbINO1 assay conditions for batch reactions,14 we sought to exchange the buffer from tris(hydroxymethyl)aminoethane acetate to ammonium bicarbonate. This change facilitates the direct isolation of IP due to the volatility of ammonium bicarbonate, while also reducing the number of components in the reaction solution but maintaining ammonium ion concentration. Initial experiments were run in the ammonium bicarbonate buffer on a 0.4 mg scale in a total volume of 150 μL. Optimisation to a conversion of >95% was achieved with the reaction containing 10 mM GP (1), 2 mM NAD+, 1 mM DTT, 50 mM ammonium bicarbonate (pH 8.5) and 87 μg TbINO1 at 37 °C overnight. Conversion was determined by an IMPase assay using the malachite green method of Itaya and Ui to determine free phosphate released.15 Scaling up to 10 mg scale was accomplished by increasing TbINO1 to 300 μg and the total reaction volume to 600 μL and provided quantitative conversions. The concentrations of NAD+, DTT and ammonium bicarbonate (pH 8.5) were maintained at the original levels.Attempts to further increase the scale of the batch reaction beyond a 10 mg scale proved less satisfactory. On 100 mg GP (1) scale a maximum conversion of 76% was achieved after six days using a reaction system containing 4 mM NAD+, 1 mM DTT, 50 mM ammonium bicarbonate (pH 8.5) and 300 μg TbINO1 in a total volume of 1 mL. In assessing the various factors that prevented full conversion, we observed the yellowing of the reaction mixture to be consistent with the breakdown of NAD+ over extended time periods in basic reaction medium.16 Furthermore, additional NAD+ dosing did not restore enzyme activity and no further GP conversion was observed. Furthermore, the abrupt halt to reaction progress after six days coincided with precipitation of denatured TbINO1. Thus for the batch reaction to be viable full conversion must be achieved within this time frame. Efforts to improve upon the 76% conversion including enzyme redosing, GP concentration variation and dosing, failed to deliver and the lack of full conversion on this larger batch scale was attributed to an equilibrium affect between GP and IP. Thus, it was concluded that the batch reaction would not prove viable for scale up of the process given the requirements defined at the outset.
Conducting the biocatalysis in-flow presents many advantages over an in-batch system.17 In general, flow systems are more readily amenable to scale up than batch reaction processes. Immobilisation of enzymes tends to improve their stability and aids enzyme recycling. Additionally, product purification is further simplified with high conversion rates. The use of a flow system for TbINO1 is particularly attractive because it would resolve many of the issues encountered within batch process, in other words an equilibrium between substrate and product that would be detrimental would not be encountered. An immobilized TbINO1 system set up that passes a low concentration of GP (1) at an optimal flow rate would ensure full conversion to IP (2) and overcome any issues associated with unfavourable product inhibition at equilibrium. Once protein activity drops the reaction can be stopped and fractions containing only fully converted product can be collated for isolation.
In these proof of concept studies, our recombinant TbINO1 was immobilized directly on a Ni2+-charged IMAC Sepharose™ Fast Flow 6 column taking advantage of the C-terminal hexa His-tag. This resin, designed for protein purification though not optimal, has a loading capacity of up to 40 mg mL−1 of resin and provided a suitable enzyme support to perform preliminary studies. Initially a 1 mL column was loaded with purified TbINO1 by passing a solution at 25 mL h−1.
A reaction solution containing 5 mM GP (1), 2 mM NAD+, 1 mM β-mercaptoethanol and 50 mM ammonium bicarbonate (pH 8.5) could then be passed through the system (Fig. 1). The immobilized TbINO1 was able to achieve an 81% conversion in one pass at a flow rate of 2.5 mL h−1 at 37 °C. The product was collected and acidified to pH <2 using concentrated hydrochloric acid, and following lyophilisation furnished IP, in its free acid form, as a white solid containing traces of NAD+/NADH. Dissolution in MeOH and filtration to remove the sparingly soluble NAD/NADH, followed by concentration gave the final product. However initial attempts to chemically modify the IP (2) were hampered by Ni2+-ions that had leached from the immobilised TbINO1 column. This issue was resolved by the incorporation of a Ni2+-free column to scavenge leached Ni2+ ions.
 | ||
| Fig. 1 Reaction set up. The TbINO1 loaded Ni2+-charged column was placed within a column oven to enable the reaction to be conducted at a constant 37 °C, and a peristaltic pump was used to control the movement of liquid through the system. | ||
Following this initial success the stationary phase volume was increased to a 5 mL column to accommodate additional TbINO1 (up to 200 mg enzyme) to allow for a higher concentration of GP (1) to be processed under flow conditions and enable reaction monitoring via1H NMR analysis. A reaction solution containing 18 mM GP (1), 1 mM NAD+, 1 mM β-mercaptoethanol and 50 mM ammonium bicarbonate (pH 8.5) can be passed through the system and achieve full conversion to IP (2) in one pass. After 36 hours the TbINO1 begins to significantly lose activity and was unable to fully convert the substrate (Fig. 2). At this flow rate, and amount of enzyme (∼200 mg), the system produced 420 mg of IP (2) in 36 hours. The increased level of conversion in the flow system can be attributed to the increased amount of enzyme immobilized on the Sephadex support avoiding the problems of protein denaturation and co-factor decomposition which plagued the homogeneous batch reaction variant. Furthermore the lack of newly formed product (IP) interacting with the enzyme allowing an unfavourable equilibrium between GP and IP is significantly reduced, thus, near full conversion is achieved under flow conditions.
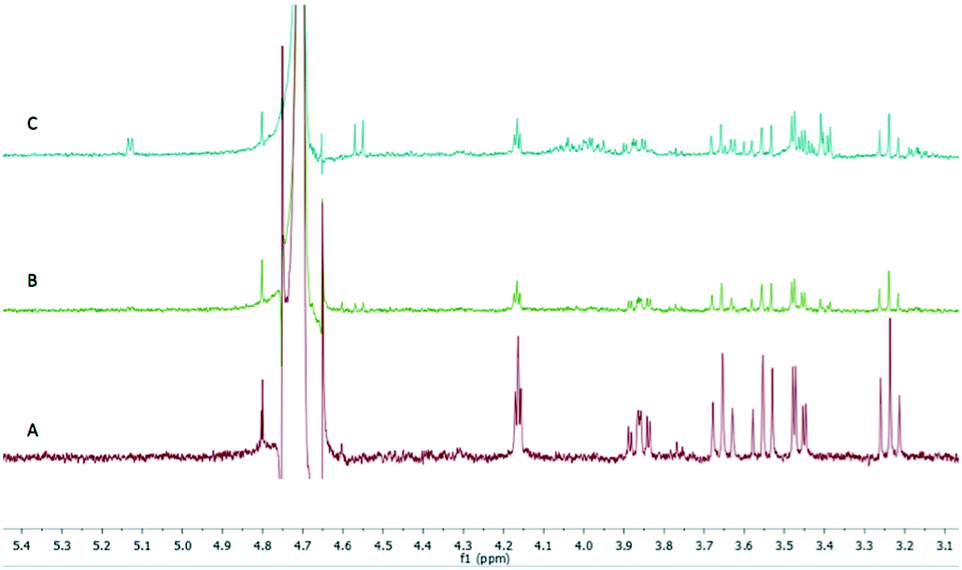 | ||
| Fig. 2 Reaction progress monitored by 1H NMR spectroscopy. Reaction was conducted at 37 °C with a flow rate of 2.5 mL h−1 and the solution contained 18 mM GP (1), 1 mM NAD+, 1 mM β-mercaptoethanol and 50 mM ammonium bicarbonate (pH 8.5). A: 0 to 20 h fractions, B: 20 to 30 h fraction, C: 30 to 40 h fraction. | ||
Further processing and chemical derivatisation of L-myo-inositol-1-phosphate (2)
Having established access to IP (2) we sought to explore its utility as a potential starting material in chemical synthesis. Our first priority was to effectively cap the sensitive phosphate moiety in 2. Not unsurprisingly there is little precedent for this transformation, however Griffin et al. used diazomethane for the selective methylation of simpler phosphates over alcohols.18 Following this precedent we opted for benzylation of the phosphate with phenyldiazomethane (Scheme 2). Thus, treatment of a high dilution solution of IP (2) in MeOH with freshly prepared ethereal phenyldiazomethane provide the bis-benzyl phosphate ester derivative 4 in 97% yield as a colourless solid after lyophilisation.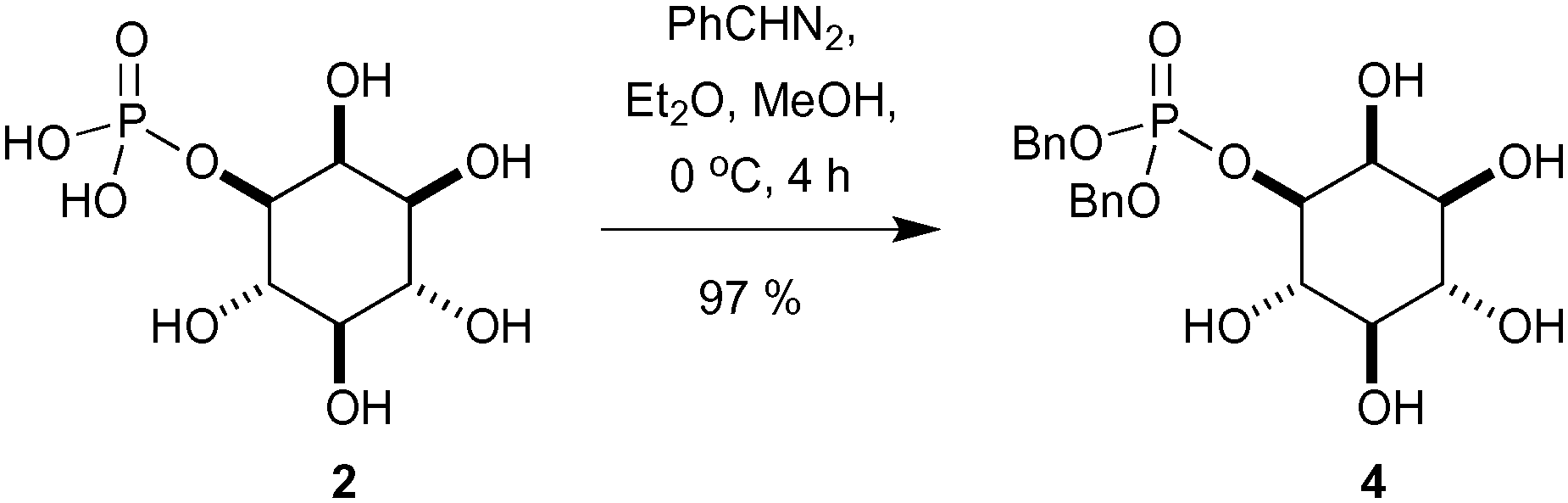 | ||
| Scheme 2 Selective phosphate benzylation using phenyldiazomethane. | ||
In order to demonstrate the viability of 4 as a building block for the synthesis of inositol derivatives we identified the pentaacetate 5 as suitable protected inositol intermediate, as shown in Scheme 3. This intermediate in either of its enantiomeric forms has been used in the synthesis of mycothiol 6 by Steenkamp et al.,19 and in the preparation of biotinylated chemical probes 7 by Otsuka et al.20 Mycothiol is the major low molecular weight thiol used in most actinomycetes cells to reduce toxic oxidants and maintain an intracellular reducing environment.21–24Mycobacterium tuberculosis, contains some of the highest levels of mycothiol,19 and it is hypothesized that this organism could become vulnerable to therapeutics and other stress factors if the biosynthesis or functions of mycothiol are interrupted.25 Therefore, mycothiol and analogues thereof are important targets that could lead to urgently required novel therapeutic treatments for tuberculosis.26
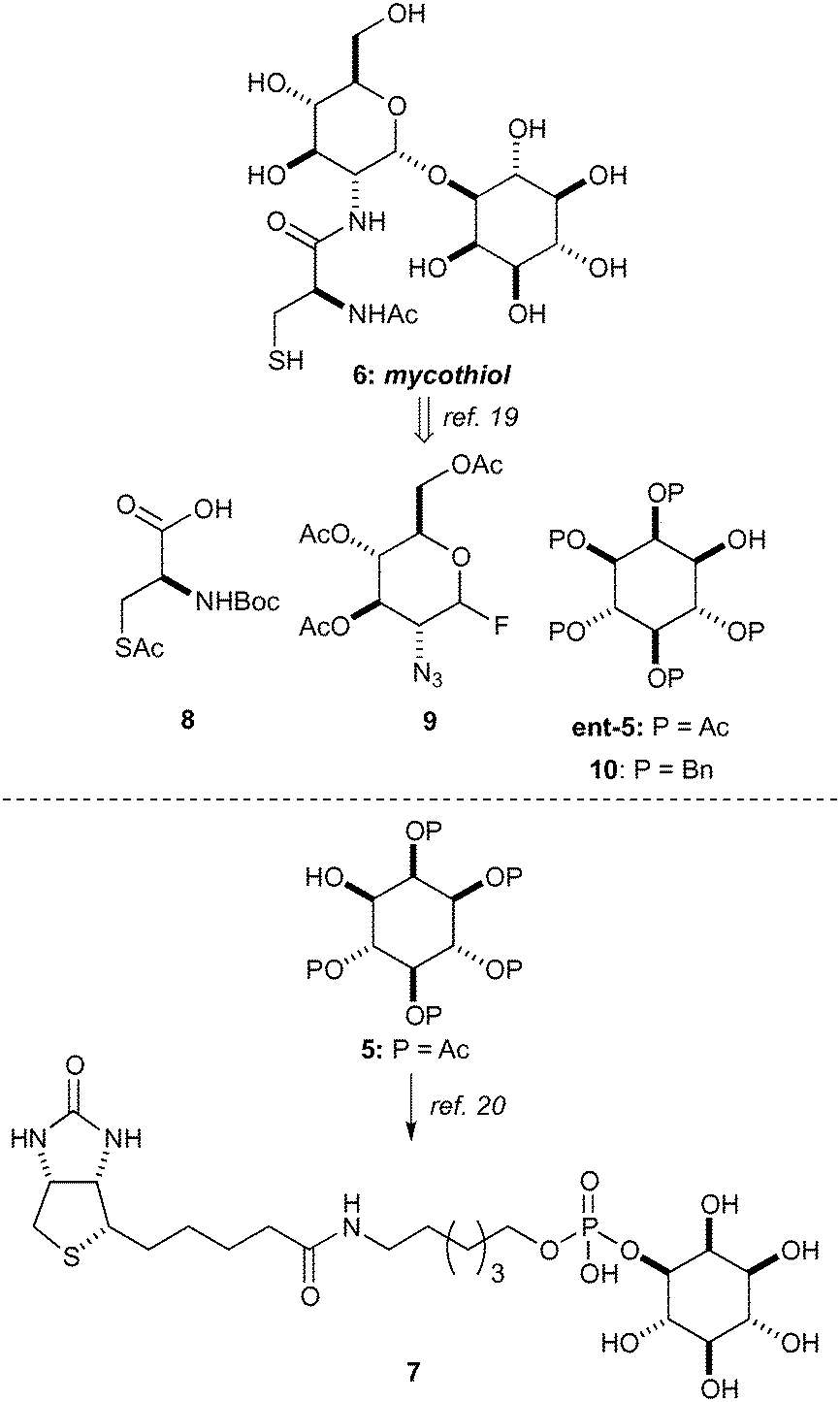 | ||
| Scheme 3 Applications of inositol pentaacetate 5 in synthesis. | ||
Synthetic routes to mycothiol (6) and derivatives adhere to similar synthetic strategies (Scheme 3) involving a protected cysteine (8), a glucosamine donor (9) and an enantiopure myo-inositol acceptor, such as ent-5 or 10. Synthetic routes have relied upon classical manipulations of myo-inositol with the shortest route to date of ent-5 requiring seven steps proceeding with a 7% overall yield.19 From our dibenzylated phosphate derivative 4 we sought to rapidly access the pentaacetate 5 (Scheme 4),27 which could be used in the synthesis of an unnatural analogue (11) and that has previously proven to be valuable in the study of mycothiol biosynthesis.19
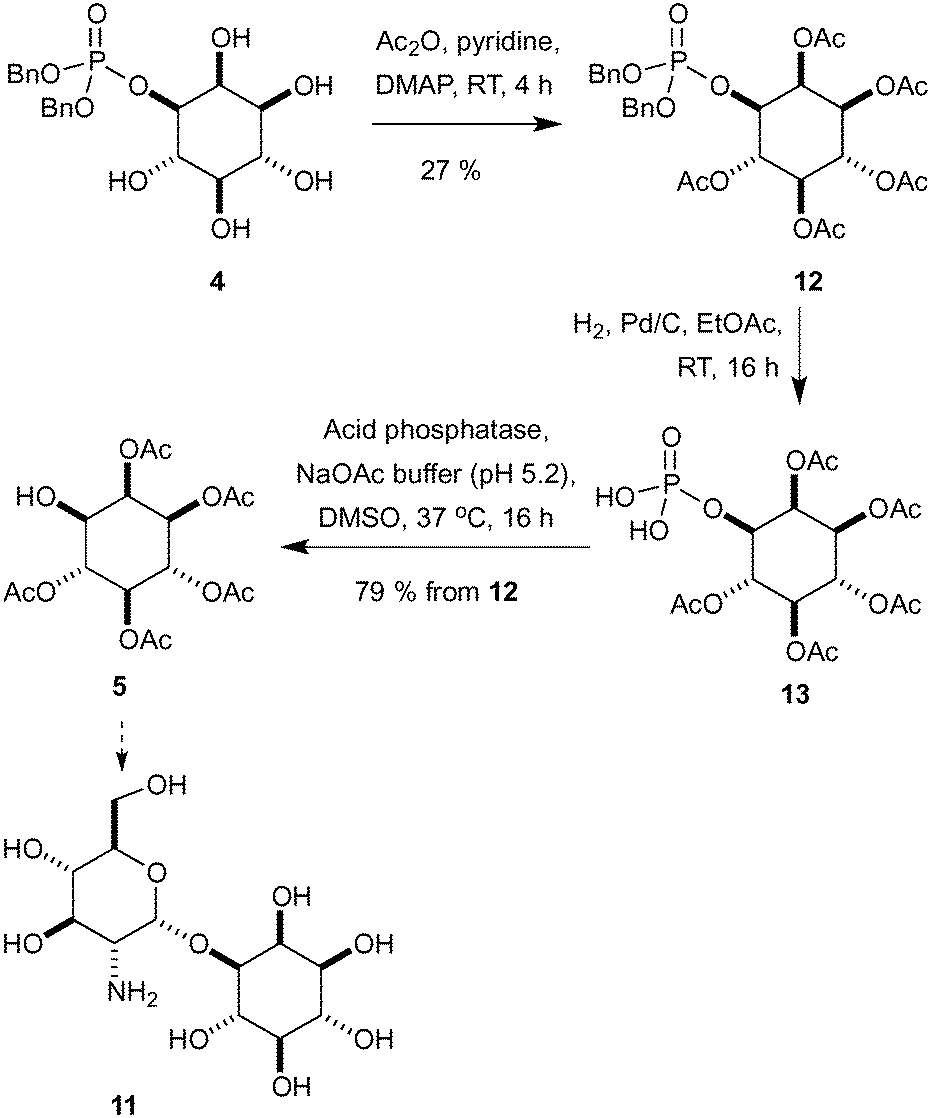 | ||
| Scheme 4 Synthesis of inositol pentaacetate derivative 5. | ||
Starting with peracetylation with acetic anhydride, pyridine and DMAP at room temperature provided 12 in a modest 27% yield (Scheme 4).28 The removal of the phosphate was now required and a variety of direct acid hydrolysis conditions were screened without success including aqueous HF and HCl/MeOH. To overcome this problem, the benzyl groups were removed by hydrogenolysis and the crude intermediate 13 was treated directly with an acid phosphatase to produce desired alcohol 5. This route produced the desired peracetylated inositol intermediate 5 in four steps and 21% yield from L-myo-inositol 1-phosphate (2), an improvement over those present within the literature.19,20,27
Conclusions
We have developed a novel biocatalytic flow process that can produce L-myo-inositol 1-phosphate (2) at a rate of roughly 12 mg h−1. The recombinant TbINO1 is stable under the reaction conditions for roughly 36 hours, allowing access to 430 mg of the highly desirable product in one preparation. We have demonstrated the convenience of using enantiopure L-myo-inositol 1-phosphate (2) as a starting point in the synthesis of myo-inositol derivatives, furnishing enantiomerically pure pentaacetate 5 in four steps, a vast improvement over literature methods. We aim to achieve different protection patterns about the myo-inositol core, in order to gain access to and drastically reduce routes to other important myo-inositol derivatives. The biocatalytic TbINO1 flow system is not fully optimised at this stage requiring further investigation to determine the best method to immobilise TbINO1 and will be reported on in due course. However these studies clearly demonstrate the potential of a biocatalytic approach in addressing the complexities of inositol synthesis.Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rcf, 20 minutes, 4 °C). The supernatant was filtered (0.45 μm) prior to loading onto a freshly charged, new 5 mL Ni2+-Sepharose column. The column was washed with 25 mL of a solution containing 10 mM imidazole, 50 mM ammonium bicarbonate (pH 8.5) and 1 mM 2-mercaptoethanol to elute the unwanted cell extracts. The column was finally washed with a solution containing 50 mM ammonium bicarbonate (pH 8.5) and 1 mM 2-mercaptoethanol, before fitting a Sepharose column in series that had been previously stripped of any Ni2+ ions by washing with 50 mM EDTA solution.
000 rcf, 20 minutes, 4 °C). The supernatant was filtered (0.45 μm) prior to loading onto a freshly charged, new 5 mL Ni2+-Sepharose column. The column was washed with 25 mL of a solution containing 10 mM imidazole, 50 mM ammonium bicarbonate (pH 8.5) and 1 mM 2-mercaptoethanol to elute the unwanted cell extracts. The column was finally washed with a solution containing 50 mM ammonium bicarbonate (pH 8.5) and 1 mM 2-mercaptoethanol, before fitting a Sepharose column in series that had been previously stripped of any Ni2+ ions by washing with 50 mM EDTA solution.
Acknowledgements
We thank the EPSRC and the University of St Andrews for PhD studentship funding. We thank the EPSRC National Mass Spectroscopy Service for mass spectroscopic analysis.Notes and references
- M. A. Ferguson, S. W. Homans, R. A. Dwek and T. W. Rademacher, Science, 1988, 239, 753–759 CAS.
- S. Sakuda, Z. Y. Zhou and Y. Yamada, Biosci., Biotechnol., Biochem., 1994, 58, 1347–1348 CrossRef CAS PubMed.
- M. J. Berridge and R. F. Irvine, Nature, 1989, 341, 197–205 CrossRef CAS PubMed.
- G. L. Newton, N. Buchmeier and R. C. Fahey, Microbiol. Mol. Biol. Rev., 2008, 72(3), 471–494 CrossRef CAS PubMed.
- M. S. Shashidhar, K. M. Sureshan, T. Praveen and T. Das, Chem. Rev., 2003, 103, 4477–4503 CrossRef PubMed.
- F. A. Loewus and M. W. Loewus, Annu. Rev. Plant Physiol., 1983, 34, 137–161 CrossRef CAS.
- (a) C. J. Hamilton, Nat. Prod. Rep., 2004, 21, 365–385 RSC; (b) A. S. Rowan and C. J. Hamilton, Nat. Prod. Rep., 2006, 23, 412–443 RSC.
- C. A. Hansen, A. B. Dean, K. M. Draths and J. W. Frost, J. Am. Chem. Soc., 1999, 3799–3800 CrossRef CAS.
- L. Chen, C. Zhou, H. Yang and M. F. Roberts, Biochemistry, 2000, 12415–12423 CrossRef CAS.
- M. W. Vetting, P. A. Frantom and J. S. Blanchard, J. Biol. Chem., 2008, 283, 15834–15844 CrossRef CAS PubMed.
- K. L. Martin and T. K. Smith, Mol. Microbiol., 2006, 61, 89–105 CrossRef CAS PubMed.
- (a) T. K. Smith, A. Crossman, J. S. Brimacombe and M. A. Ferguson, EMBO J., 2004, 23, 4701–4708 CrossRef CAS PubMed; (b) K. Nagamune, T. Nozaki, Y. Maeda, K. Ohishi, T. Fukuma, T. Hara, R. T. Schwarz, C. Sütterlin, R. Brun, H. Riezman and T. Kinoshita, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10336–10341 CrossRef CAS PubMed; (c) T. Chang, K. G. Milne, M. L. S. Güther, T. K. Smith and M. J. Ferguson, J. Biol. Chem., 2002, 277, 50176–50182 CrossRef CAS PubMed.
- S. Ju, G. Shaltiel, A. Shamir, G. Agam and M. L. Greenberg, J. Biol. Chem., 2004, 279, 21759–21765 CrossRef CAS PubMed.
- A. González-Salgado, M. Steinmann, L. L. Major, E. Sigel, J.-L. Reymond, T. K. Smith and P. Bütikofer, Eukaryotic Cell, 2015, 14(6), 616–624 CrossRef PubMed.
- K. Itaya and M. Ui, Clin. Chim. Acta, 1966, 14, 361–366 CrossRef CAS.
- C. H. Wong and G. M. Whitesides, Enzymes in Synthetic Organic Chemistry, Elsevier Science Ltd, 1st edn, 1994 Search PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- B. E. Griffin, J. A. Haines and C. B. Reese, Biochim. Biophys. Acta, 1967, 142(2), 536–538 CrossRef CAS.
- M. A. Jardine, H. S. C. Spies, C. N. Nkambule, D. W. Gammon and D. J. Steenkamp, Bioorg. Med. Chem., 2002, 10, 875–881 CrossRef CAS PubMed.
- K. Anraku, T. Inoue, K. Sugimoto, T. Morii, Y. Mori, Y. Okamoto and M. Otsuka, Org. Biomol. Chem., 2008, 6, 1822–1830 CAS.
- V. K. Jothivasan and C. J. Hamilton, Nat. Prod. Rep., 2008, 25, 1091–1117 RSC.
- M. Rawat and Y. Av-Gay, FEMS Microbiol. Rev., 2007, 31, 278–292 CrossRef CAS PubMed.
- G. L. Newton, R. C. Fahey, G. Cohen and Y. Aharonowitz, J. Bacteriol., 1993, 175, 2734–2742 CrossRef CAS PubMed.
- G. L. Newton, K. Arnold, M. S. Price, C. Sherill, S. B. Delcardayre, Y. Aharonowitz, G. Cohen, J. Davies, R. C. Fahey and C. Davis, J. Bacteriol., 1996, 178, 1990–1995 CrossRef CAS PubMed.
- S. Knapp, S. Gonzalez, D. S. Myers, L. L. Eckman and C. A. Bewley, Org. Lett., 2002, 4, 4337–4339 CrossRef CAS PubMed.
- S. W. Riordan, J. J. Field, H. M. Corkran, N. Dasyam, B. L. Stocker, M. S. Timmer, J. E. Harvey and P. H. Teesdale-Spittle, Bioorg. Med. Chem. Lett., 2015, 25, 2152–2155 CrossRef CAS PubMed.
- V. I. Shivets, B. A. Klyashchitskii, A. E. Stepanov and R. P. Evstigneeva, Tetrahedron, 1973, 29, 331–340 CrossRef.
- The derivatisation of 5 under a variety of peracetylation conditions was studied (Ac2O/pyr; Ac2O/I2; Ac2O, pH 10.5, DMF or DMSO). In all cases the C1-phosphate ester was prone to migration resulting in low conversions and isolated yields. Further studies are now focussed on the assessment of alternative protecting group manipulations compatible with the presence of the phosphate ester in 4.
Footnote |
| † Electronic supplementary information (ESI) available: For details of general experimental procedures, synthetic procedures and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c6re00175k |
| This journal is © The Royal Society of Chemistry 2017 |