
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactivity-guided mixed synthesis and evaluation of α-alkenyl-γ and δ-lactone derivatives as potential fungicidal agents†
Yong-Ling Wua,
Yan-Qing Gaoab,
De-Long Wanga,
Chen-Quan Zhonga,
Jun-Tao Feng *ab and
Xing Zhangab
*ab and
Xing Zhangab
aResearch and Development Center of Biorational Pesticide, Northwest A&F University, Yangling 712100, Shaanxi, China. E-mail: fengjt@nwsuaf.edu.cn; Fax: +86-29-87092122; Tel: +86-29-87092122
bResearch Center of Biopesticide Technology & Engineering, Yangling 712100, Shaanxi, China
First published on 15th December 2017
Abstract
In view of the great antifungal activities of sesquiterpene lactones and natural product Tulipalin A, 52 derivatives derived from α-methylene-γ-butyrolactone substructures were synthesized to study antifungal activities. In vitro and in vivo antifungal activity results revealed that compounds 2-25, which contain a γ-butyrolactone scaffold and cinnamic aldehyde moiety, have greater potent fungicidal activity than other compounds. The preliminary structure–activity relationships (SARs) demonstrated that compounds with electron-withdrawing groups and small steric hindrance would have more desirable potency. Meanwhile, the quantitative structure–activity relationship (QSAR) model (R2 = 0.947, F = 65.77, and S2 = 0.0028) revealed a convincing correlation of antifungal activity against B. cinerea with molecular structures of title compounds. The present study provided a more detailed insight into the antifungal activity of the α-methylene-γ-butyrolactone substructure, which provided a potential expectation for the exploration of α-alkenyl-γ-butyrolactone structures in agriculture.
Introduction
In recent years, fungicide resistance and food security problems have becoming increasingly serious.1,2 Since it is difficult to find appropriate fungal-specific targets of fungal cells, the discovery of a new mode of action or novel fungicidal agents based on novel scaffolds from natural products has become important to overcome the chemical resistance problem. In fact, natural products with structural modification have been broadly used for discovering and developing pesticides in the market today, due to the structural diversities, environmental compatibilities, easy biodegradation, and lower environmental and mammalian toxicity characteristics.3–6 In addition, discovering and screening candidates with antifungal activity from thousands of natural products is virtually and economically impossible, while employing a bioactivity-guided mixed synthesis method would reduce the problems with cost and time requirements for screening.A wide variety of structures of sesquiterpene lactones (STLs) have a diversity of biological activities, such as cytotoxic, anticancer, antifungal, anti-inflammatory, and antimicrobial properties.7–10 In fact, α-methylene-γ-lactone and cyclopentenone structures reacting with the sulfhydryl moieties of enzymes or functional proteins via the Michael type addition among numerous natural products are the important chemical scaffolds of STLs.11,12 In our previous work, carabrone and its alcohol analogue carabrol, two known sesquiterpene lactones isolated from the fruits of Carpesium macrocephalum, were found to possess potent antifungal activity in a protective manner which blocked the early penetrative infection process of pathogenic spores.13 The study of the structure–activity relationships (SARs) of carabrone derivates also revealed that the α-methylene-γ-butyrolactone core boosts their antifungal potency.14,15 At the same time, Tulipalin A and B, two natural pharmacophores, both of which contain a methylene group, exerted great antimicrobial activities.16 Therefore, as shown in Fig. 1, the bioactivity of these compounds guided us to continue to seek new analogous α-methylene-γ-lactone bearing templates with potent fungicidal efficacy.
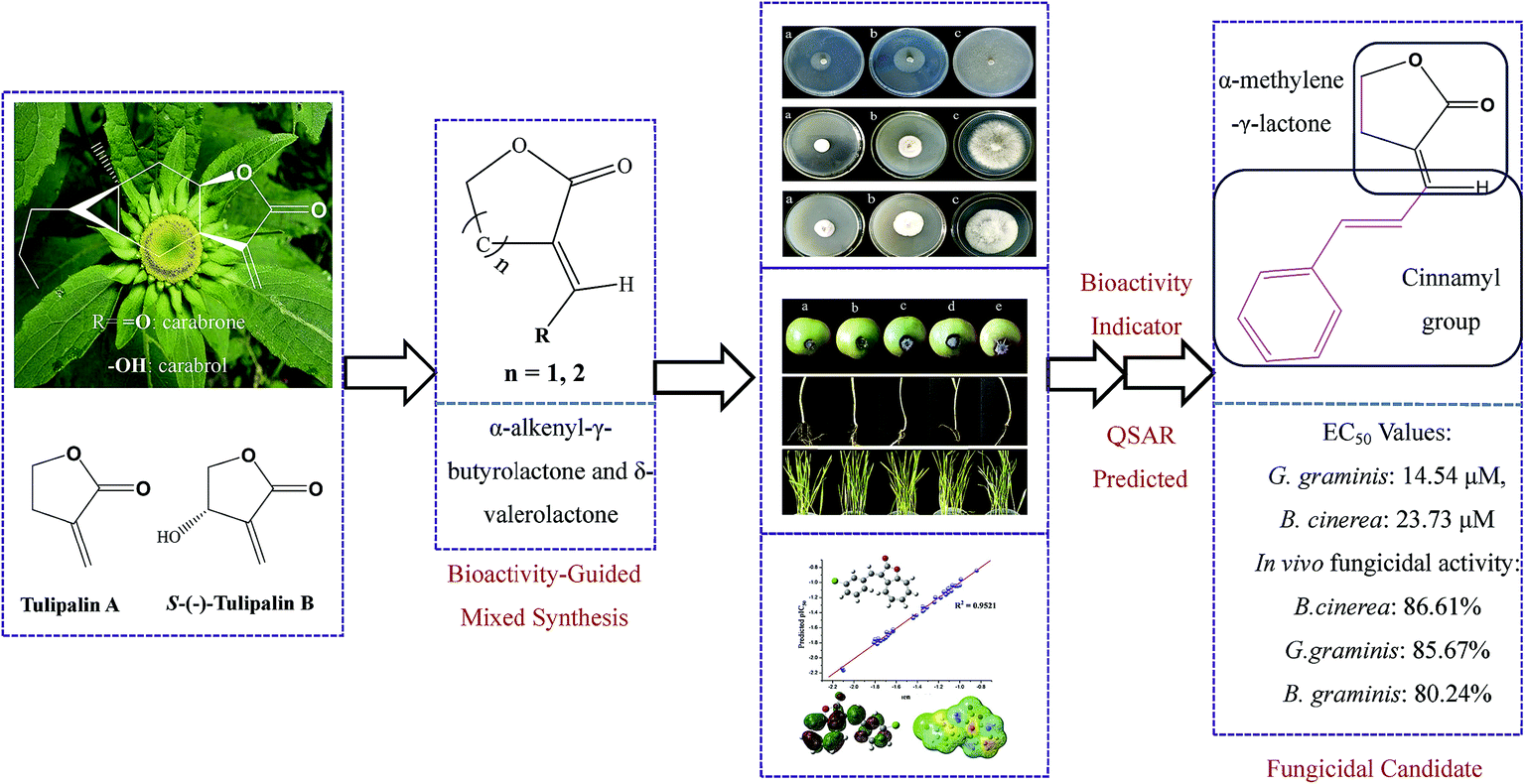 | ||
| Fig. 1 The design strategy to find promising fungicidal candidates. | ||
α-Alkenyl-γ-butyrolactone derivates with a chain-ring have been demonstrated to reduce their cytotoxicity.17 Moreover, little is known about the effect of the lactone ring conformation and constraints on the bio-activity of α-methylene-γ-lactone. Thus this prompted us to design and synthesize a series of α-alkenyl-γ-butyrolactone and δ-valerolactone derivates. Since the in vitro antifungal activity of fungicides does not always match the in vivo activity, i.e. in compounds with high in vitro antifungal activity, the in vivo activities were also tested. At the same time, SAR and constructed QSAR models were built to elaborate the relationship between structural features of the activity and mechanism actions.18,19 The objective of this article was to seek an efficient candidate based on bioactivity-guided mixed synthesis.
Experimental
Chemicals and instruments
Chemicals and reagents used in this research were of analytical grade (purchased from Aladdin Industrial Inc., Shanghai, China); all solvents were dried and redistilled before use. Analytical thin-layer chromatography (TLC) was performed on silica gel GF254. Column chromatographic (CC) purification was carried out using silica gel (200–300 mesh), which was obtained from Qingdao Haiyang Chemical Co., Ltd. (Qingdao, China). The melting points of these synthetic derivatives were determined on an X-5 apparatus and uncorrected, and the apparatus was purchased from Beijing Tech. Nuclear magnetic resonance spectra (NMR) were obtained on a Bruker Avance 500 MHz instrument. HR-MS (ESI) was observed using a Bruker Apex-Ultra 7.0 T spectrometer. The reaction progress was monitored by thin-layer chromatography on silica gel GF-254 with detection by UV light.Synthesis of compound α-alkenyl-γ-butyrolactone
As shown in Scheme 1, to a cooled (0 °C) solution of γ-butyrolactone (5.0 g, 58 mmol) and benzaldehyde (5.85 g, 55.1 mmol) in dry benzene (75 mL) was added KOtBu (7.82 g, 69.7 mmol) portionwise.20,21 After the addition, the thick orange solution was stirred at room temp for 6 h. The mixture was acidified with dilute H2SO4 (aq) and extracted with Et2O (3 × 30 mL). The organic layer was dried (MgSO4) and evaporated under reduced pressure. The residue was purified using flash column chromatography (silica gel, EtOAc/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) to yield 2 (5.4 g, 56%) as a white solid. The data of compound 2 is shown as follows.
:5) to yield 2 (5.4 g, 56%) as a white solid. The data of compound 2 is shown as follows.
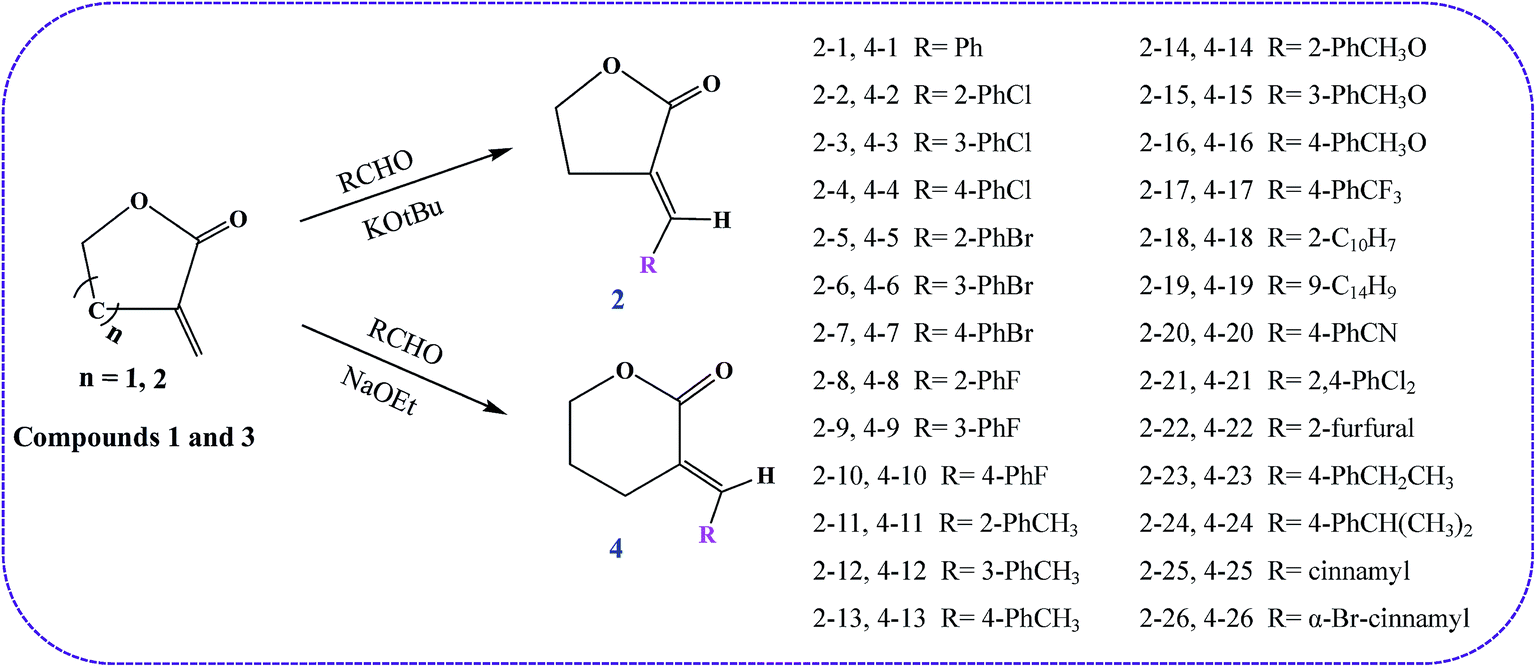 | ||
| Scheme 1 Synthesis route of the target compounds 2-1–26 and 4-1–26. | ||
135.20, 132.41, 131.68, 130.80, 129.98, 129.36, 127.23, 126.63, 68.01, 26.62. HR-MS (ESI): m/z calcd for C11H9ClO2 ([M + Na]+) 208.0291, found 231.0183.Synthesis of compound α-alkenyl-δ-valerolactone
A solution of aromatic aldehyde (5 mmol), δ-valerolactone (5 mmol) in anhydrous methyl alcohol was added dropwise to a methyl alcohol solution of NaOEt (10 mmol) under ice-cooling. The mixture was stirred at 0–10 °C for 3 hours. TLC was used to monitor whether the reaction was completed. Thereafter, the mixture was diluted with EtOAc, and acidified with dilute H2SO4 (aq). The organic layer was then washed with NaHCO3 solution and water, dried with MgSO4 and removed under reduced pressure to give the crude product which was purified by column chromatography with petroleum ether–EtOAc (10:1) to afford compound 4. The structures of all compounds were confirmed by 1H and 13C NMR and HR-MS analyses. The synthesis route is shown in Scheme 1 and the data of compound 4 is shown as follows.
Biological assays
In vitro antifungal assay
According to the mycelium linear growth rate method reported previously,22–24 the in vitro antifungal activities of compounds against five strains of plant pathogenic fungi were tested.The compounds with higher initial activities against B. cinerea and G. graminis var. tritici were selected to assay 50% inhibition concentration values (IC50). In order to get a series of stock solutions, the test compound (2 mM) was diluted with 5% DMSO aqueous solution by a double-fold dilution method. Each stock solution (10 mL) was mixed with the autoclaved PDA medium (190 mL) to provide a set of mediums with different concentrations of the test compound (3.125, 6.25, 12.5, 25, 50 and 100 μM). The culture medium containing only 0.25% DMSO was used as a blank control, and Tulipalin A, carabrone and carbendazim were used as positive controls. Each test was performed in triplicate. Antifungal toxicity regression equations and IC50 values were established according to the method previously reported.14
In vivo antifungal assay
The stock solution of the test compound was diluted with water containing 0.1% tween-80 to 200 μg mL−1.

Building and validation of the QSAR model
Building and validation of the QSAR model was carried out with a common procedure. Briefly, using the Gaussian 03W package of programs (Gaussian Inc.), the optimal conformers of the title compounds with the lowest energy were computed at the DFT/6-31G (d) level.27 Meanwhile, the most stable configurations of the compounds were generated and the corresponding “.log” and “.chk” files were gained. Afterwards, the calculated results were transformed into a form compatible with CODESSA 2.7.15 using Ampac 9.1.3.28,29 At last, all of the molecular descriptors involved in these compounds were calculated by CODESSA 2.7.15, and the heuristic analysis method was used to build the QSAR model, which determines the most significant structural features for antifungal activity against B. cinerea. During the model development process, the squared correction coefficient (R2), the squared standard error of the estimates (S2), and the Fisher significance ratio (F) were used to clarify the standards of statics. Moreover, the tested IC50 values were converted into the corresponding plogIC50 values and used as dependent variables to get better linear regression. The quality of the final model was ensured using internal validation and the “leave-one-out” cross-validation methods.30,31
Results and discussion
Chemistry
The synthetic routes of the title compounds are outlined in Scheme 1. To evaluate the essentiality of the substituent on the exocyclic carbon–carbon double bond of γ-lactone and δ-lactone, we synthesized a series of α-methylene-γ-lactone and α-methylene-δ-lactone compounds. γ-Butyrolactone and δ-valerolactone compounds were reacted with different substituent aldehydes, carried out in an ice-cooling bath under KOtBu or NaOEt catalysts. With these concise and stereospecific methods, the stereochemistry of the exocyclic double bond was exclusively E in moderate yields. It is worth noting that the yield of the γ-butyrolactone derivates (44–61%) was obviously higher than that of the δ-valerolactone derivates (41–47%), which may be due to the ring tension of the former lactone compounds being lower than that of the latter, and results in the γ-butyrolactone derivates tending to be more stable.Antifungal activity and SARs
The in vitro screening results of the title compounds for preliminary antifungal activities against five pathogenic fungi (Botrytis cinerea, Colletotrichum lagenarium, Phytophthora capsici, Gaeumannomyces graminis var. tritici, and Sclerotinia sclerotiorum, Fusarium graminearum) at 100 μg mL−1 are listed in Table 1 and Fig. 2. The results showed that most of the compounds exhibited certain to great inhibition activity against each of the fungi at 100 μg mL−1 (inhibition rate = 46.0–95.8%). Particularly, the antifungal activity of compounds 2-3–4, 2-7–10, 2-17, 2-17–19, 2-21, 2-25–26, 4-8–10, 4-17, 4-21, and 4-25–26 was more potent than that of Tulipalin A and equal or higher than that of the natural product carabrone. Furthermore, from the results we can see that all the derivatives showed higher activity against B. cinerea and G. graminis (inhibition rate > 55%) compared to the other three pathogenic fungi.| Compound | Average inhibition rate (%) (100 μg mL−1; 72 h) | ||||||
|---|---|---|---|---|---|---|---|
| No. | R | B. c. | C. l. | G. g. | S. s. | F. g. | |
| a B. c., Botrytis cinerea; C. l., Colletotrichum lagenarium; G. g., Gaeumannomyces graminis var. tritici; S. s., Sclerotinia sclerotiorum; F. g., Fusarium graminearum. | |||||||
| 1 | 2-1 | Ph | 81.4 ± 0.7 | 76.7 ± 0.3 | 81.2 ± 0.8 | 63.2 ± 0.4 | 72.5 ± 0.2 |
| 2 | 2-2 | 2-PhCl | 81.4 ± 0.5 | 77.9 ± 0.1 | 83.6 ± 0.4 | 68.7 ± 0.3 | 75.6 ± 0.4 |
| 3 | 2-3 | 3-PhCl | 82.7 ± 0.8 | 77.6 ± 0.5 | 84.1 ± 0.2 | 69.1 ± 0.2 | 76.3 ± 0.5 |
| 4 | 2-4 | 4-PhCl | 85.6 ± 0.8 | 80.1 ± 0.6 | 86.8 ± 0.2 | 72.3 ± 0.4 | 76.0 ± 0.1 |
| 5 | 2-5 | 2-PhBr | 78.5 ± 0.2 | 72.3 ± 0.3 | 81.6 ± 0.7 | 63.6 ± 0.5 | 68.5 ± 0.3 |
| 6 | 2-6 | 3-PhBr | 79.1 ± 0.7 | 74.9 ± 0.8 | 80.2 ± 0.4 | 65.4 ± 0.7 | 70.9 ± 0.5 |
| 7 | 2-7 | 4-PhBr | 82.6 ± 0.5 | 78.6 ± 0.3 | 83.2 ± 0.5 | 66.3 ± 0.2 | 71.2 ± 0.5 |
| 8 | 2-8 | 2-PhF | 86.3 ± 0.1 | 80.5 ± 0.5 | 91.7 ± 0.3 | 72.4 ± 0.5 | 77.3 ± 0.4 |
| 9 | 2-9 | 3-PhF | 92.6 ± 0.3 | 87.5 ± 0.5 | 93.3 ± 0.5 | 75.5 ± 0.7 | 80.8 ± 0.6 |
| 10 | 2-10 | 4-PhF | 95.8 ± 0.4 | 90.4 ± 0.5 | 95.1 ± 0.2 | 76.2 ± 0.1 | 82.2 ± 0.1 |
| 11 | 2-11 | 2-PhCH3 | 73.3 ± 0.6 | 68.3 ± 0.2 | 75.5 ± 0.3 | 58.6 ± 0.3 | 63.6 ± 0.8 |
| 12 | 2-12 | 3-PhCH3 | 75.6 ± 0.3 | 70.8 ± 0.5 | 73.6 ± 0.4 | 56.2 ± 0.2 | 61.4 ± 0.3 |
| 13 | 2-13 | 4-PhCH3 | 77.6 ± 0.8 | 81.2 ± 0.1 | 77.6 ± 0.4 | 58.6 ± 0.3 | 64.5 ± 0.2 |
| 14 | 2-14 | 2-PhCH3O | 69.7 ± 0.8 | 64.5 ± 0.3 | 71.2 ± 0.1 | 53.4 ± 0.3 | 60.6 ± 0.5 |
| 15 | 2-15 | 3-PhCH3O | 72.6 ± 0.5 | 68.1 ± 0.4 | 76.6 ± 0.6 | 52.4 ± 0.7 | 56.6 ± 0.7 |
| 16 | 2-16 | 4-PhCH3O | 73.4 ± 0.2 | 69.8 ± 0.2 | 74.4 ± 0.3 | 55.3 ± 0.9 | 61.5 ± 0.4 |
| 17 | 2-17 | 4-PhCF3 | 96.1 ± 0.7 | 91.3 ± 0.6 | 93.9 ± 0.5 | 79.8 ± 0.7 | 83.1 ± 0.5 |
| 18 | 2-18 | 2-C10H7 | 85.2 ± 0.3 | 80.5 ± 0.7 | 84.4 ± 0.3 | 71.3 ± 0.6 | 75.5 ± 0.2 |
| 19 | 2-19 | 9-C14H9 | 87.1 ± 0.8 | 81.3 ± 0.7 | 84.8 ± 0.6 | 72.2 ± 0.3 | 68.9 ± 0.4 |
| 20 | 2-20 | 4-PhCN | 75.2 ± 0.8 | 71.3 ± 0.1 | 76.2 ± 0.7 | 65.1 ± 0.3 | 62.3 ± 0.8 |
| 21 | 2-21 | 2,4-PhCl2 | 88.2 ± 0.7 | 82.6 ± 0.5 | 88.4 ± 0.3 | 74.5 ± 0.7 | 78.2 ± 0.7 |
| 22 | 2-22 | 2-Furfural | 81.6 ± 0.4 | 75.3 ± 0.3 | 73.6 ± 0.6 | 69.3 ± 0.8 | 73.5 ± 0.3 |
| 23 | 2-23 | 4-PhCH2CH3 | 72.4 ± 0.6 | 65.2 ± 0.8 | 71.2 ± 0.3 | 57.2 ± 0.4 | 63.2 ± 0.4 |
| 24 | 2-24 | 4-PhCH(CH3)2 | 69.7 ± 0.7 | 63.4 ± 0.1 | 71.2 ± 0.3 | 55.6 ± 0.2 | 60.0 ± 0.1 |
| 25 | 2-25 | Cinnamyl | 92.2 ± 0.5 | 87.2 ± 0.6 | 92.3 ± 0.7 | 75.6 ± 0.3 | 82.9 ± 0.4 |
| 26 | 2-26 | α-Br-cinnamyl | 96.0 ± 0.9 | 88.3 ± 0.9 | 92.8 ± 0.6 | 78.2 ± 0.7 | 83.1 ± 0.7 |
| 27 | 4-1 | Ph | 79.2 ± 0.4 | 73.4 ± 0.8 | 77.7 ± 0.3 | 56.2 ± 0.1 | 62.2 ± 0.8 |
| 28 | 4-2 | 2-PhCl | 78.4 ± 0.8 | 74.3 ± 0.2 | 75.5 ± 0.4 | 61.8 ± 0.4 | 66.6 ± 0.3 |
| 29 | 4-3 | 3-PhCl | 79.5 ± 0.8 | 74.4 ± 0.8 | 74.2 ± 0.3 | 61.3 ± 0.1 | 67.3 ± 0.2 |
| 30 | 4-4 | 4-PhCl | 80.6 ± 0.4 | 75.4 ± 0.7 | 82.1 ± 0.5 | 68.2 ± 0.5 | 74.8 ± 0.9 |
| 31 | 4-5 | 2-PhBr | 76.7 ± 0.9 | 70.7 ± 0.8 | 76.8 ± 0.9 | 58.3 ± 0.2 | 64.2 ± 0.5 |
| 32 | 4-6 | 3-PhBr | 77.2 ± 0.4 | 72.5 ± 0.3 | 77.4 ± 0.5 | 61.8 ± 0.7 | 66.3 ± 0.8 |
| 33 | 4-7 | 4-PhBr | 80.6 ± 0.5 | 74.1 ± 0.3 | 75.3 ± 0.2 | 61.4 ± 0.5 | 68.2 ± 0.7 |
| 34 | 4-8 | 2-PhF | 83.1 ± 0.3 | 78.2 ± 0.1 | 85.2 ± 0.6 | 68.3 ± 0.2 | 80.8 ± 0.3 |
| 35 | 4-9 | 3-PhF | 86.4 ± 0.8 | 81.5 ± 0.3 | 87.1 ± 0.5 | 70.1 ± 0.4 | 76.3 ± 0.1 |
| 36 | 4-10 | 4-PhF | 89.5 ± 0.6 | 83.4 ± 0.8 | 89.5 ± 0.4 | 72.6 ± 0.8 | 77.6 ± 0.2 |
| 37 | 4-11 | 2-PhCH3 | 69.9 ± 0.3 | 63.1 ± 0.2 | 68.7 ± 0.3 | 53.5 ± 0.3 | 59.2 ± 0.4 |
| 38 | 4-12 | 3-PhCH3 | 71.2 ± 0.3 | 66.5 ± 0.7 | 83.6 ± 0.2 | 51.1 ± 0.3 | 57.3 ± 0.7 |
| 39 | 4-13 | 4-PhCH3 | 74.1 ± 0.6 | 68.3 ± 0.7 | 72.3 ± 0.4 | 52.7 ± 0.6 | 58.8 ± 0.4 |
| 40 | 4-14 | 2-PhCH3O | 65.7 ± 0.6 | 64.6 ± 0.4 | 55.2 ± 0.5 | 48.8 ± 0.3 | 54.1 ± 0.6 |
| 41 | 4-15 | 3-PhCH3O | 68.2 ± 0.3 | 66.8 ± 0.2 | 66.5 ± 0.9 | 46.0 ± 0.9 | 48.4 ± 0.3 |
| 42 | 4-16 | 4-PhCH3O | 69.2 ± 0.5 | 63.7 ± 0.4 | 62.0 ± 0.6 | 50.4 ± 0.3 | 51.6 ± 0.7 |
| 43 | 4-17 | 4-PhCF3 | 91.4 ± 0.7 | 87.9 ± 0.6 | 87.3 ± 0.1 | 74.2 ± 0.1 | 75.2 ± 0.6 |
| 44 | 4-18 | 2-C10H7 | 80.2 ± 0.8 | 74.1 ± 0.8 | 78.4 ± 0.2 | 66.6 ± 0.7 | 71.1 ± 0.2 |
| 45 | 4-19 | 9-C14H9 | 81.6 ± 0.9 | 78.5 ± 0.3 | 77.5 ± 0.1 | 65.4 ± 0.3 | 70.3 ± 0.8 |
| 46 | 4-20 | 4-PhCN | 69.5 ± 0.5 | 61.8 ± 0.5 | 78.7 ± 0.2 | 61.2 ± 0.1 | 63.6 ± 0.6 |
| 47 | 4-21 | 2,4-PhCl2 | 83.4 ± 0.5 | 78.9 ± 0.6 | 83.6 ± 0.9 | 69.6 ± 0.5 | 75.4 ± 0.9 |
| 48 | 4-22 | 2-Furfural | 76.5 ± 0.8 | 72.5 ± 0.3 | 65.6 ± 0.2 | 63.4 ± 0.8 | 69.2 ± 0.6 |
| 49 | 4-23 | 4-PhCH2CH3 | 70.8 ± 0.2 | 65.4 ± 0.4 | 63.2 ± 0.4 | 52.5 ± 0.4 | 56.5 ± 0.8 |
| 50 | 4-24 | 4-PhCH(CH3)2 | 68.2 ± 0.6 | 62.2 ± 0.6 | 55.3 ± 0.5 | 50.6 ± 0.9 | 53.6 ± 0.3 |
| 51 | 4-25 | Cinnamyl | 85.0 ± 0.3 | 80.4 ± 0.2 | 87.6 ± 0.5 | 71.1 ± 0.4 | 77.3 ± 0.5 |
| 52 | 4-26 | α-Br-cinnamyl | 86.6 ± 0.2 | 83.1 ± 0.2 | 86.2 ± 0.4 | 72.6 ± 0.3 | 80.1 ± 0.7 |
| 53 | Carabrone | 82.2 ± 0.7 | 75.6 ± 0.7 | 86.9 ± 0.7 | 71.3 ± 0.7 | 68.2 ± 0.7 | |
| 54 | Tulipalin A | 64.9 ± 0.7 | 57.3 ± 0.7 | 68.3 ± 0.7 | 59.2 ± 0.7 | 56.5 ± 0.7 | |
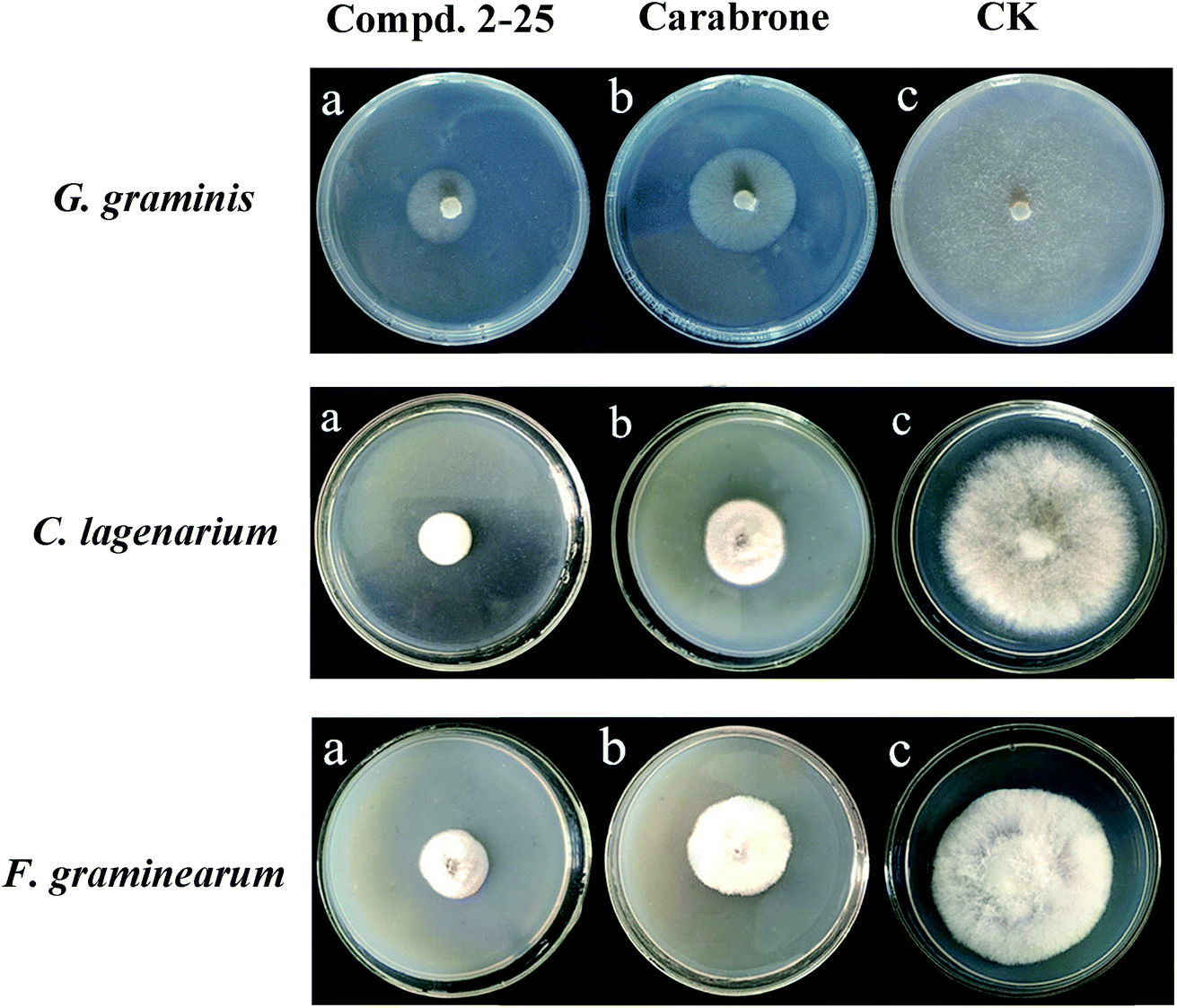 | ||
| Fig. 2 In vitro antifungal activity of compounds against Gaeumannomyces graminis var. tritici, Colletotrichum lagenarium and Fusarium graminearum at 100 μg mL−1. | ||
According to these preliminary results, the half maximal inhibitory concentration (IC50) of all the derivatives against G. graminis and B. cinerea was tested by the mycelial growth inhibitory rate method. It is notable that most of the γ-butyrolactone derivates (IC50 = 14.54–134.56 μM) showed a superior activity against G. graminis compared to the δ-valerolactone derivates (IC50 = 24.45–154.88 μM), which might be plausibly ascribed to the intrinsic biological feature of the lactone ring and the fact that the γ-butyrolactone derivates bear conformation core size for receptor binding sites; on the contrary, the δ-valerolactone derivates disrupt the bioactive conformation. In order to investigate the SARs of the substituents on the lactone ring, different kinds and positions of substituent were introduced, and G. graminis was selected for SAR research for all compounds. According to the data in Table 2, we can construct SARs from the preliminary conclusions. The compounds 2-2–10 and 4-2–10 containing a halogen atom (-PhF, -PhCl, and -PhBr) exhibited obviously potent antifungal activity compared to the -PhCH3O and -PhCH3 derivatives, which maybe due to the former derivates containing the electron-withdrawing group being beneficial to antifungal activity. In general, the activity of the compounds follows: F > Cl > Br > CH3 > CH3O. Remarkably, 2-17 displayed an IC50 value of 14.78 μM, an involved the -PhCF3 electron-withdrawing substituent, the value was higher than that of the isopropyl (2-24, IC50 = 123.75 μM) and ethyl substituent (2-23, IC50 = 104.21 μM). In contrast with 2-17, disubstitution of the benzylidene ring with a 2,4-PhCl2 substituent did not improve activity, as seen from compound 2-21 (IC50 = 33.85 μM). A plausible explanation on such a decrease assumes that the steric effect interferes with receptor binding when a Cl atom is on the ortho-position.
| R | Compd | G. graminis | B. cinerea | Compd | G. graminis | B. cinerea | ||
|---|---|---|---|---|---|---|---|---|
| IC50, μM | IC50, μM | pIC50c | IC50, μM | IC50, μM | pIC50c | |||
| a All half maximal inhibitory concentration (IC50) values are presented as the mean ± SD (n = 3), μM.b Commercial fungicide, carbendazim was used as the positive control.c pIC50, the tested IC50 values were converted into the corresponding plogIC50 values. |
||||||||
| Ph | 2-1 | 88.21 | 94.96 | −1.98 | 4-1 | 102.41 | 119.26 | −2.08 |
| 2-PhCl | 2-2 | 45.45 | 63.50 | −1.80 | 4-2 | 64.98 | 82.11 | −1.91 |
| 3-PhCl | 2-3 | 34.87 | 50.14 | −1.70 | 4-3 | 51.20 | 79.00 | −1.90 |
| 4-PhCl | 2-4 | 21.54 | 46.39 | −1.67 | 4-4 | 48.58 | 66.44 | −1.82 |
| 2-PhBr | 2-5 | 46.65 | 66.80 | −1.83 | 4-5 | 66.73 | 83.58 | −1.92 |
| 3-PhBr | 2-6 | 41.76 | 58.54 | −1.77 | 4-6 | 64.54 | 78.47 | −1.89 |
| 4-PhBr | 2-7 | 33.64 | 50.92 | −1.71 | 4-7 | 51.41 | 70.12 | −1.85 |
| 2-PhF | 2-8 | 45.47 | 49.31 | −1.69 | 4-8 | 62.35 | 79.83 | −1.90 |
| 3-PhF | 2-9 | 28.52 | 39.26 | −1.59 | 4-9 | 48.55 | 74.35 | −1.87 |
| 4-PhF | 2-10 | 20.15 | 28.33 | −1.45 | 4-10 | 40.05 | 66.78 | −1.82 |
| 2-PhCH3 | 2-11 | 122.13 | 136.06 | −2.13 | 4-11 | 124.38 | 165.43 | −2.22 |
| 3-PhCH3 | 2-12 | 98.47 | 127.45 | −2.11 | 4-12 | 103.47 | 147.27 | −2.17 |
| 4-PhCH3 | 2-13 | 78.77 | 107.72 | −2.03 | 4-13 | 96.24 | 130.25 | −2.11 |
| 2-PhCH3O | 2-14 | 134.54 | 145.29 | −2.16 | 4-14 | 154.88 | 175.22 | −2.24 |
| 3-PhCH3O | 2-15 | 115.64 | 138.43 | −2.14 | 4-15 | 126.37 | 173.16 | −2.24 |
| 4-PhCH3O | 2-16 | 104.12 | 131.62 | −2.12 | 4-16 | 123.24 | 153.30 | −2.19 |
| 4-PhCF3 | 2-17 | 14.78 | 21.65 | −1.34 | 4-17 | 31.38 | 56.00 | −1.75 |
| 2-C10H7 | 2-18 | 47.85 | 57.88 | −1.76 | 4-18 | 75.25 | 89.55 | −1.95 |
| 9-C14H9 | 2-19 | 50.54 | 39.84 | −1.60 | 4-19 | 70.74 | 60.88 | −1.78 |
| 4-PhCN | 2-20 | 87.76 | 106.80 | −2.03 | 4-20 | 98.54 | 135.50 | −2.13 |
| 2,4-PhCl2 | 2-21 | 33.85 | 42.77 | −1.63 | 4-21 | 55.98 | 65.04 | −1.81 |
| 2-Furfural | 2-22 | 98.74 | 113.69 | −2.06 | 4-22 | 114.36 | 133.45 | −2.13 |
| 4-PhCH2CH3 | 2-23 | 104.21 | 145.63 | −2.16 | 4-23 | 148.47 | 163.54 | −2.21 |
| 4-PhCH(CH3)2 | 2-24 | 123.75 | 152.10 | −2.18 | 4-24 | 142.55 | 176.65 | −2.25 |
| Cinnamyl | 2-25 | 14.54 | 23.73 | −1.38 | 4-25 | 24.45 | 38.95 | −1.84 |
| α-Br-cinnamyl | 2-26 | 16.44 | 21.12 | −1.32 | 4-26 | 27.02 | 46.17 | −1.66 |
| Tulipalin A | 58.27 | 67.86 | — | Carabrone | 28.54 | 25.74 | — | |
| Carbendazimb | 7.84 | 8.34 | — | — | — | — | — | |
As we can see, the steric effect also played an essential role for the antifungal activity. For the title compounds, substitution at the para-position was strongly favored over the corresponding meta- and/or ortho-positions, which was verified between the derivatives 2-2–16 and 4-2–16, and the derivatives 2-2 (2-PhCl, IC50 = 45.45 μM) and 2-5 (2-PhBr, IC50 = 46.65 μM) were less potent than the corresponding derivatives 2-4 (4-PhCl, IC50 = 21.54 μM) and 2-7 (4-PhBr, IC50 = 33.64 μM). Meanwhile, with the bulky aryl groups, the derivatives (2-18–19 and 4-18–19) bearing a 2′-naphthyl or a 9′-anthryl group have no obvious increase in activity (IC50 = 47.85, 50.54, 75.25, 70.74 μM). Moreover, isopropyl with a bulky substituent was unfavorable to the activity. Taken together, these results could demonstrate the importance of substituents with a proper size, the para-position and the electron-withdrawing characteristic. Compared with aromatic substituent compounds 2-1 and 4-1 (IC50 = 88.21, 102.41 μM), a small decrease in activity was observed for compounds 2-22 and 4-22 (IC50 = 98.74, 114.36 μM) with the 2-furfural substituent. Unexpectedly, the cinnamic aldehyde compounds 2-25–26 and 4-25–26 (IC50 = 14.54, 16.44, 24.45, and 27.02 μM) showed higher antifungal activity toward G. graminis than other compounds and the natural product carabrone (IC50 = 28.54 μM), and derivatives 2-25 also displayed approximately 2-fold more activity than carbendazim (IC50 = 7.84 μM). This phenomenon may be due to the cinnamic aldehyde structure have some antifungal capability.
In vivo fungicidal bioactivity
In order to further confirm the antifungal activity of these title compounds, the in vivo fungicidal activities (protective effect) of the 14 promising derivatives were tested against B. cinerea on tomato fruits and against G. graminis and B. graminis on wheat seedlings.As can be seen in Table 3 and Fig. 3, the inhibitory rates of compounds 2-10 and 2-25 exceed 80% against all three fungi at 200 μg mL−1, meanwhile, compound 2-25 is equivalent with commercial fungicide (carbendazim or triadimefon). Compounds 2-17 and 4-7 also showed great activity, and their protective inhibitory rates were higher than 70%. The above results indicated that the in vivo fungicidal activities in this research were aligned with in vitro activities; moreover, the in vivo fungicidal activities were better than the in vitro activity.
| No. | Compounds (200 μg mL−1) | Against B. cinerea (%) | Against G. graminis (%) | Against B. graminis (%) |
|---|---|---|---|---|
| a Values are the means of three replicates; letters a–d represent a significant difference at p = 0.05; commercial fungicide, triadimefon and carbendazim were used as the positive control. | ||||
| 1 | 2-2 | 52.21d | 57.65d | 56.36d |
| 2 | 2-10 | 82.43ab | 83.59a | 81.67a |
| 3 | 2-13 | 61.23cd | 59.65d | 52.34d |
| 4 | 2-17 | 78.65b | 81.54ab | 78.65ab |
| 5 | 2-19 | 56.54d | 41.68e | 42.52e |
| 6 | 2-21 | 55.65d | 66.58c | 64.38c |
| 7 | 2-23 | 66.51c | 48.68e | 51.25d |
| 8 | 2-25 | 86.61a | 85.67a | 80.24a |
| 9 | 4-1 | 56.42d | 52.24d | 46.75e |
| 10 | 4-7 | 77.85b | 70.36b | 73.51b |
| 11 | 4-15 | 56.24d | 62.87c | 46.27 |
| 12 | 4-18 | 68.57c | 66.34c | 59.68d |
| 13 | 4-20 | 66.33c | 62.24c | 57.46d |
| 14 | 4-25 | 80.47ab | 76.39b | 79.38ab |
| 15 | Tulipalin A | 54.21d | 58.36d | 42.94e |
| 16 | Carabrone | 70.36bc | 80.24ab | 73.58b |
| 17 | Triadimefon | — | 87.29a | 82.39a |
| 18 | Carbendazim | 89.54a | — | — |
 | ||
| Fig. 3 In vivo antifungal activity of compounds against Botrytis cinerea, Gaeumannomyces graminis var. tritici, and Blumeria graminis at 200 μg mL−1. aCommercial fungicide; carbendazim was used as the B. cinerea positive control and triadimefon was used as the G. graminis and B. graminis positive control. | ||
QSAR study and antifungal activity against B. cinerea
Conformer optimization and minimum energy calculations are the essential procedures in the construction of a QSAR model. In this paper, 52 γ-butyrolactone and δ-valerolactone compounds were used as samples and 5 groups of descriptors were obtained. Heuristic regression was chosen to acquire a QSAR model with satisfactory values of R2, F, and S2, which were used to establish a relationship between antifungal activity and molecular descriptors.The number of descriptors was achieved by the “breaking point” rule, and according to the t values in the test, the descriptors were obtained. Meanwhile, the numbers of samples and descriptors also meet the equation 3D ≤ S − 3 (S means the number of samples; D means the number of descriptors). At last, the final 5-descriptor model was generated. In the ESI,† the five significant descriptors, the “breaking point” rule figure and their values are listed.
The fabricated QSAR model equation has five descriptors presented in a grading down way based on the importance of the statistical analysis, as shown in Table 4. According to this optimized model, the comparison chart of the predictive and practical activity of the 52 compounds was exhibited in Fig. 4. Compared with experimental pIC50 (negative logIC50), we could conclude that the generated model was reliable. The final QSAR model with 5 descriptors can be described as
| pIC50 = −4.6536 + 5.4325 × qOmax. − 3.1128 × no + 2.5461 × MAOEP + 0.0665 × μc − 3.5452 × qmax., N = 52, R2 = 0.947, F = 65.77, S2 = 0.0028 |
| Descriptor no. | X | ±ΔX | t-Text | Descriptor |
|---|---|---|---|---|
| a Max. net atomic charge for an O atom.b Number of occupied electronic levels of atoms.c Max. atomic orbital electronic population.d Total point-charge composition of the molecular dipole.e Max. net atomic charge. | ||||
| 0 | −4.6536 | 3.2135 × 10−1 | 6.6525 | Intercept |
| 1 | 5.4325 | 3.4576 × 10−1 | −2.7665 | qOmax.a |
| 2 | 3.1128 | 5.7687 | −1.6532 | nob |
| 3 | 2.5461 | 1.0893 | 2.6675 | MAOEPc |
| 4 | 6.6523 × 10−2 | 3.2254 × 10−2 | 1.9878 | μcd |
| 5 | 3.5452 | 2.2153 × 10−1 | 2.6673 | qmax.e |
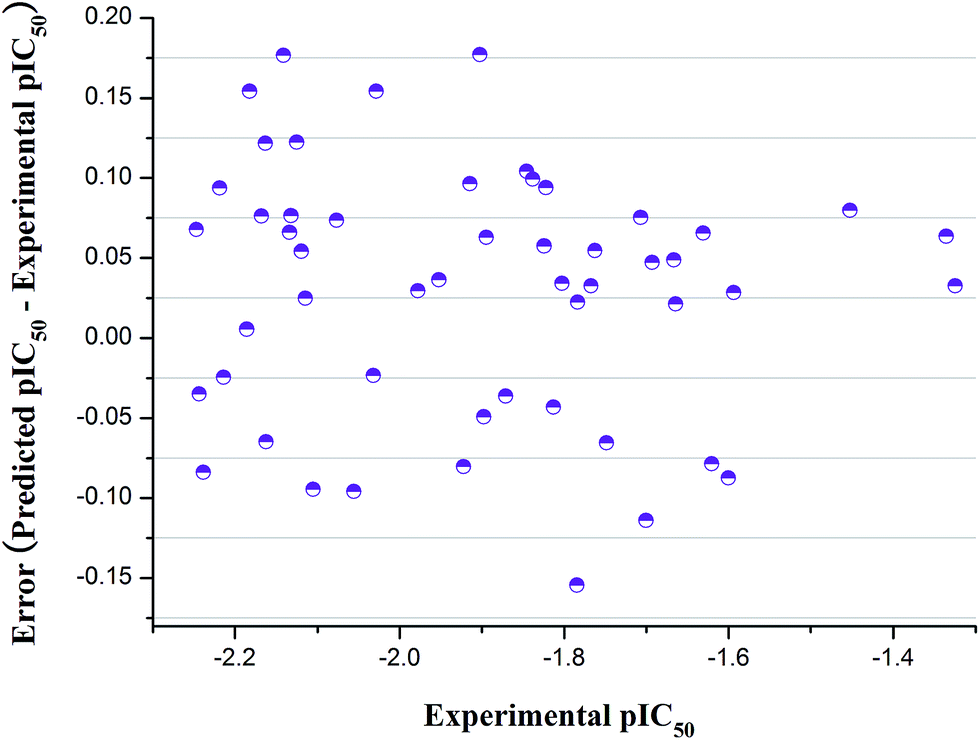 | ||
| Fig. 4 Graphic of pEC50 values versus respective errors. | ||
Internal validation and “leave-one-out” cross-validation methods were carried out to validate the established QSAR model.32 Internal validation results were presented in the ESI.† The RTraining2 and RTest2 were within 5% for all three sets, and the average values of RTraining2 and RTest2 approached the overall R2 value. So, the obtained QSAR model indicated the predictive power of 3-fold cross-validation. In the “leave-one-out” method, every fourth compound 1, 5, 9, etc., was an external test set, and the others were the training set. The R2 values of the training set and test set were close, and the QSAR model acquired in this study was available.
By explaining the descriptors of this model, we could gain some insight into the structural features and antifungal activity. The 1st and 5th descriptors obtained in this study were the max. net atomic charge for an O atom and the max. net atomic charge. These two descriptors belonged to electrostatic descriptors and they reflect the charge distribution of the molecules as shown in the contour maps indicating the occupied electronic levels of compounds 2-4 and 4-17 (Fig. 5), which represent the geometric mean of atomic electronegativities.33,34 According to the frontier molecular orbital theory (FMO) of chemical reactivity, the formation of a transition state is due to an interaction between the frontier orbitals (HOMO and LUMO) of the reacting species. So the electrostatic properties of an O atom and max. net atomic charge were important elements for the antifungal activity of the title compounds.
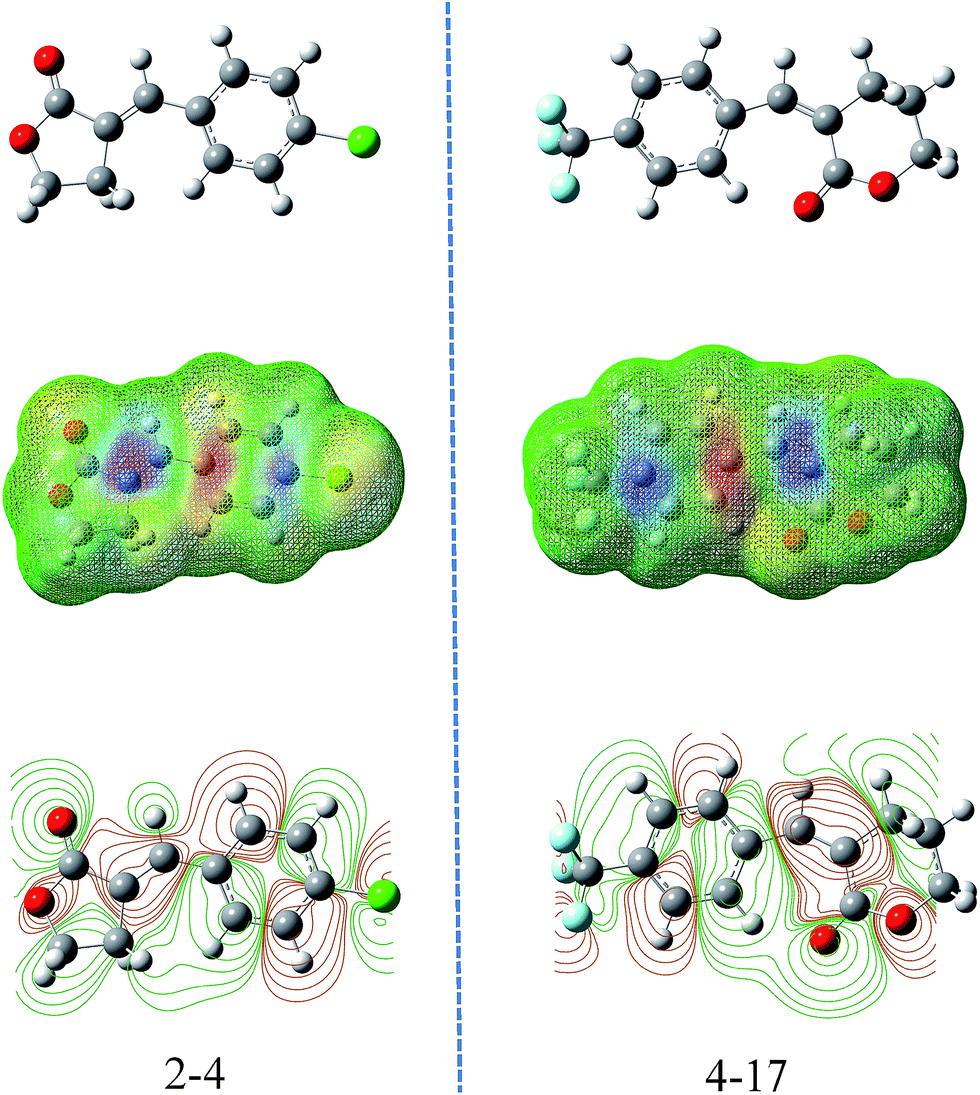 | ||
| Fig. 5 Molecular electrostatic potential and contour maps of compounds 2-4 and 4-17. The green parts represent positive molecular orbitals, and the red parts represent negative molecular orbitals. | ||
The second and third most important descriptors obtained in this model were the maximum atomic orbital electronic population and the number of occupied electronic levels of atoms. The former is an important descriptor to reflect the nucleophilicity of the molecule, which is directly in contact with the molecular nucleophilic capacity and describes the susceptibility of the molecule to electrophilic attack.35 The number of occupied electronic levels of atoms belongs to quantum-chemical descriptors, which therefore act directly on the quantum chemically calculated charge distribution in the molecules and describes the molecular polar interactions. Meanwhile, as the result of the of electron activity difference between the atoms, the optimized geometries and permanent polarization are shown in the molecular electrostatic potential map simultaneously (Fig. 5), and we can see that the exocyclic carbon–carbon double bond exhibits greater negative electrostatic potential which easily occurred in the nucleophilic reaction.36,37 Actually, α-methylene-γ-butyrolactone derivatives with an electrophilic α,β-unsaturated carbonyl system (Michael acceptor), which had higher electron deficiency, could react with biological nucleophiles.38,39
The fifth important descriptor was the total point-charge component of the molecular dipole (μc), which measured the hydrophilic/lipophilic property of the compounds.40–42 The appropriate μc value illustrated the penetration ability of the molecules to the cell as well as interaction with the action target to a large extent. As shown in Fig. 6, the optimized geometries and charge distribution on the atoms of compounds 2-4, 2-9, 2-13, 4-17, 4-21 and 4-24 were demonstrated simultaneously. It was illustrated that, after lactone ring modification with aromatics, the hydrophilic and lipophilic property of the compounds was regulated, therefore the μc value was an essential factor in adjusting the antifungal activity.
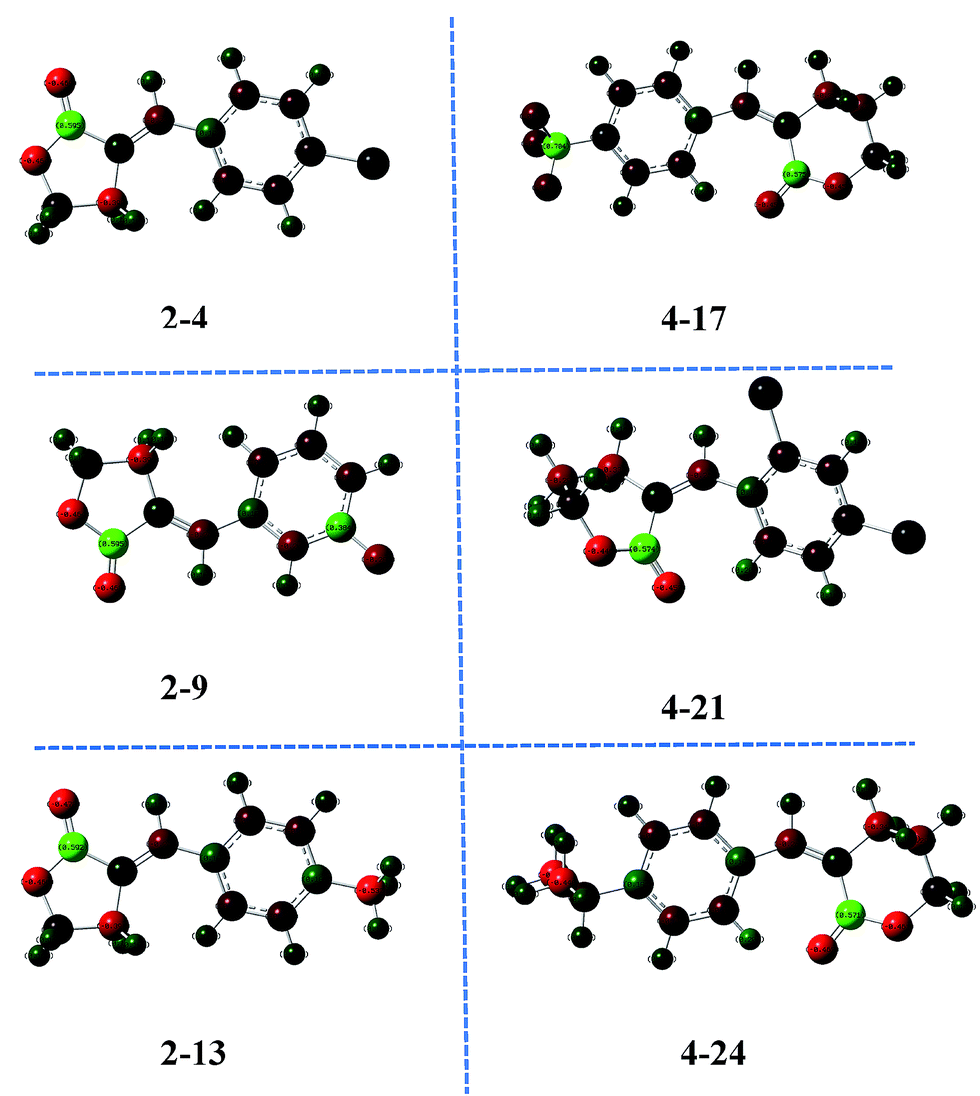 | ||
| Fig. 6 Optimized geometries and charge distribution of compounds 2-4, 2-9, 2-13, 4-17, 4-21, and 4-24. | ||
Conclusion
Inspired by the bioactivity of sesquiterpene lactones and the natural product Tulipalin A, a series of α-methylene-γ-lactone and α-methylene-δ-lactone compounds were prepared through structural modification. The antifungal activity of all compounds against G. graminis and B. cinerea were evaluated, and the γ-lactone derivatives exhibited higher antifungal activity than the δ-lactone derivatives. Particularly, compound 2-25 connected with a cinnamic aldehyde structure showed superior in vitro and in vivo fungicide activities. The SAR indicated that electron-withdrawing groups and steric effects played important roles simultaneously. Moreover, the QSAR model (R2 = 0.947, F = 65.77, and S2 = 0.0028) indicated that the net atomic charges, nucleophilicity and hydrophilic/lipophilic property of the compounds are the most important in this research. In view of these results, we know more about the antifungal activity of α-methylene-γ-lactone substructures, especially that compound 2-25 contains a γ-butyrolactone scaffold and cinnamic aldehyde moiety and has the potential to be a fungicide candidate.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We greatly appreciate the funding support for this research provided by the National Natural Science Foundation of China (No. 31471800). We also thank the Kunming Institute of Botany, Chinese Academy of Science for the HR-MS spectral data.References
- M. Ishtiaq, M. A. Saleem and M. Razaq, Crop Prot., 2012, 33, 13–20 CrossRef CAS.
- P. K. Anderson, A. A. Cunningham, N. G. Patel, F. J. Morales, P. R. Epstein and P. Daszak, Trends Ecol. Evol., 2004, 19, 535–544 CrossRef PubMed.
- F. E. Dayan, C. L. Cantrell and S. O. Duke, Bioorg. Med. Chem., 2009, 17, 4022–4034 CrossRef CAS PubMed.
- C. F. Nising, S. Hillebrand and L. Rodefeld, Chem. Commun., 2011, 47, 4062–4073 RSC.
- L. G. Copping and B. P. Khambay, Pest Manage. Sci., 2015, 56, 649–650 CrossRef.
- L. G. Copping, Crop Prot., 1997, 16, 491–492 Search PubMed.
- A. K. Picman, Biochem. Syst. Ecol., 1986, 14, 255–281 CrossRef CAS.
- M. F. Neerman, Int. J. Aromather., 2003, 13, 114–120 CrossRef.
- G. Lyss, A. Knorre, T. J. Schmidt, H. L. Pahl and I. Merfort, J. Biol. Chem., 1998, 273, 33508–33516 CrossRef CAS PubMed.
- T. J. Schmidt, Curr. Org. Chem., 1999, 3, 577–608 CAS.
- A. K. Picman, E. Rodriguez and C. H. N. Towers, Chem.-Biol. Interact., 1979, 28, 83–90 CrossRef CAS PubMed.
- P. M. Bork, M. L. Schmitz, M. Kuhnt, C. Escher and M. Heinrich, FEBS Lett., 1997, 402, 85–90 CrossRef CAS PubMed.
- J. Feng, H. Wang, S. Ren, J. He and X. Zhang, J. Agric. Food Chem., 2012, 60, 3817–3823 CrossRef CAS PubMed.
- J. Feng, D. Wang, Y. Wu, H. Yan and X. Zhang, Bioorg. Med. Chem. Lett., 2013, 23, 4393–4397 CrossRef CAS PubMed.
- Y. Wu, D. Wang, E. Guo, S. Song, J. Feng and X. Zhang, Bioorg. Med. Chem. Lett., 2017, 27, 1284–1290 CrossRef CAS PubMed.
- J. Lee, J. Lee, J. Lim, S. Sim and D. Park, J. Med. Plants Res., 2008, 2, 059–065 Search PubMed.
- T. Janecki, E. Błaszczyk, K. Studzian, A. Janecka, U. Krajewska and M. Rozalski, J. Med. Chem., 2005, 48, 3516–3521 CrossRef CAS PubMed.
- I. G. Shibi, L. Aswathy, R. S. Jisha, V. H. Masand, A. Divyachandran and J. M. Gajbhiye, Eur. J. Med. Chem., 2015, 77, 9–23 CAS.
- C. Hansch and R. P. Verma, Eur. J. Med. Chem., 2009, 44, 260–273 CrossRef CAS PubMed.
- A. K. Mahalingam, A. Linda, K. E. Jenny, W. Johan, K. Jacob, T. Unge, W. Hans, M. Larhed and H. Anders, J. Med. Chem., 2010, 53, 607–615 CrossRef CAS PubMed.
- Y. Ru Meng and Y. D. Guan, Chin. Chem. Lett., 2002, 13, 1039–1042 Search PubMed.
- S. Brase, A. Encinas, J. Keck and C. F. Nising, Chem. Rev., 2009, 109, 3903–3990 CrossRef CAS PubMed.
- M. Wang, Q. Zhang, Q. Ren, X. Kong, L. Wang, H. Wang, J. Xu and Y. Guo, J. Agric. Food Chem., 2014, 62, 10945–10953 CrossRef CAS PubMed.
- L. Fang, M. Wang, S. Gou, X. Liu, H. Zhang and F. Cao, J. Med. Chem., 2014, 57, 1116–1120 CrossRef CAS PubMed.
- R. P. John, R. D. Tyagi, D. Prévost, S. K. Brar, S. Pouleur and R. Y. Surampalli, Crop Prot., 2010, 29, 1452–1459 CrossRef.
- F. Chen, M. Wang, Y. Zheng, J. Luo, X. Yang and X. Wang, World J. Microbiol. Biotechnol., 2010, 26, 675–684 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin and J. C. Burant, Gaussian 03, Gaussian, Inc., Wallingford, CT, USA, 2004 Search PubMed.
- A. Katritsky, M. Karelson, V. S. Lobanov, R. Dennington, T. A. Keith and R. D. A. T. Keith, Codessa 2.7.15, Semichem, Inc., Shawnee, KS, USA, 2004 Search PubMed.
- M. J. S. Dewar, A. J. Holder, I. Roy, D. Dennington, D. A. Liotard, D. G. Truhlar, T. A. Keith, J. M. Millam and C. D. Harris, AMPAC 9.3.1, Semichem, Inc., Shawnee, KS, USA, 2004 Search PubMed.
- Y. Gao, X. Tian, J. Li, S. Shang, Z. Song and M. Shen, ACS Sustainable Chem. Eng., 2016, 4, 2741–2747 CrossRef CAS.
- J. Li, X. Tian, Y. Gao, S. Shang, J. Feng and X. Zhang, RSC Adv., 2015, 5, 66947–66955 RSC.
- A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. Varoli, N. Calonghi and R. H. Shoemaker, J. Med. Chem., 2008, 51, 7508–7513 CrossRef CAS PubMed.
- A. Andreani, A. Locatelli, A. Leoni, M. Rambaldi, R. Morigi, R. Bossa, M. Chiericozzi, A. Fraccari and I. Galatulas, Eur. J. Med. Chem., 1997, 32, 919–924 CrossRef CAS.
- J. Li, Y. Gao, S. Shang, X. Rao, J. Song and Z. Wang, RSC Adv., 2014, 4, 58190–58199 RSC.
- R. Kumar, A. Kumar, S. Jain and D. Kaushik, Eur. J. Med. Chem., 2011, 46, 3543–3550 CrossRef CAS PubMed.
- M. Witschel, Bioorg. Med. Chem., 2009, 17, 4221–4229 CrossRef CAS PubMed.
- L. Zhao and S. Feng, J. Colloid Interface Sci., 2007, 274, 55–68 CrossRef PubMed.
- A. Cho, C. E. Song, S. K. Lee, W. S. Shin and E. Lim, J. Mater. Sci., 2016, 51, 6770–6780 CrossRef CAS.
- D. Sharma, B. Narasimhan, P. Kumar and A. Jalbout, Eur. J. Med. Chem., 2009, 44, 1119–1127 CrossRef CAS PubMed.
- P. Sharma, A. Kumar, S. Upadhyay, V. Sahu and J. Singh, Eur. J. Med. Chem., 2009, 44, 251–259 CrossRef CAS PubMed.
- K. Kalani, D. Yadav, F. Khan, S. Srivastava and N. Suri, J. Mol. Model., 2012, 18, 3389–3413 CrossRef CAS PubMed.
- T. J. Schmidt, A. M. M. Nour, S. A. Khalid, M. Kaiser and R. Brun, Molecules, 2009, 14, 2062–2076 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12471f |
| This journal is © The Royal Society of Chemistry 2017 |