
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight- and heat-triggered reversible luminescent materials based on polysiloxanes with anthracene groups†
Dongdong Han,
Hang Lu,
Wensi Li,
Yonghao Li and
Shengyu Feng *
*
Key Laboratory of Special Functional Aggregated Materials, Key Laboratory of Colloid and Interface Chemistry (Shandong University), Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: fsy@sdu.edu.cn
First published on 15th December 2017
Abstract
In this study, reversible silicone elastomers were successfully developed by light-triggered dimerization and heat depolymerization of the anthryl groups. Polysiloxanes with anthryl groups were prepared from poly(aminopropylmethylsiloxane) (PAPMS) with electron-donating (9-anthracenylmethyl acrylate) and electron-withdrawing (anthracene-9-carboxylic acid) units. The cross-linking networks were formed with the via 4π–4π photo-cycloadditions of the anthryl groups upon the UV light excitation (365 nm). 9-Anthracenylmethyl acrylate or anthracene-9-carboxylic acid efficiently dimerized through the photodimerization of the anthryl groups in the organic solvents, which was proven by UV-vis spectra, NMR spectra, and LC/MS. The covalent bonds between pendant anthryl groups were cleaved after heating at 120 °C. Furthermore, repeatable dimerization–depolymerization conversion was confirmed. In addition, for the first time, we found that the sunlight can also initiate the cycloaddition, which was “greener” and more environment-friendly. The green luminescence was observed from the PAPMS-1 film instead of the quenching effect caused by aggregation after the cycloaddition. Thus, a colorful UV-light emitting diode (LED) cell was obtained by coating the films on the commercially available LED cell.
Introduction
Silicone elastomers are widely used as safe and durable materials for domestic and industrial applications because of their resistance to weathering, high and low temperatures, radiation, and fire.1–5 However, traditional silicones are normally thermoset materials, which are prepared by three main methods: (1) Pt-catalyzed hydrosilylation between Si–H and vinyl groups; (2) condensation reaction between Si-OR and –OH; (3) free-radical cross-linking.6 Recently, many classical reactions such as thiol–ene “click” reactions,7 Michael addition,8 and [2 + 2] photo-cycloaddition9 were utilized to develop self-healing, thermo-sensitive, fluorescent, or reversible silicone elastomers. For example, Brook et al. prepared a thermo-sensitive silicone elastomer via [2 + 2] photo-cycloaddition of coumarin. Coumarin is UV-sensitive and can undergo a reversible reaction upon UV irradiation.9 In addition, Brook et al. prepared a self-associating silicone elastomer based on azide/alkyne cycloaddition reactions.10 In our group, highly sensitive stimuli-responsive polysiloxanes (SPSis) were prepared via a facile, efficient, and catalyst-free aza-Michael addition of poly(aminopropylmethylsiloxane) (PAPMS) with N-isopropylacrylamide.11 However, these methods have some drawbacks, including potential explosiveness of the azide monomers, the unpleasant smell of thiol–enes, the high costs of the catalysts, and the inherent difficulties of the [2 + 2] photo-cross-linking reactions. Thus, it is desirable to develop a simple, efficient, and catalyst-free system to prepare functional and cross-linked silicones.In spite of many synthetic methods used to produce silicone materials, there are few reports about photo-cross-linking reactions without a photo-initiator or catalysts. Anthracene is commonly used in photo-responsive compounds, which can form covalent bonds through photo-cycloaddition.12–22 Anthracene groups undergo [4 + 4] photo-dimerization when irradiated by UV light (λ > 300 nm) and can be reversed to the original monomers via exposure to a higher energy UV light (λ < 300 nm) (Scheme 1)23 or heating to 100–130 °C.24,25 Xie et al. successfully obtained shape-memory materials by dangling photoresponsive anthracene groups from different polymer chains.23,26,27 However, there are few reports on reversible silicone elastomers based on the anthracene groups.
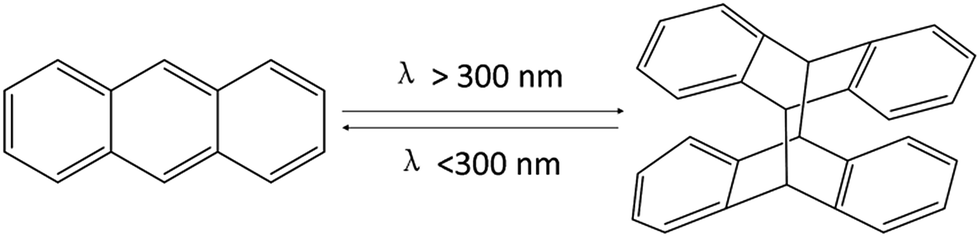 | ||
| Scheme 1 Reversible photo-dimerization reaction of anthracene groups.23 | ||
Based on the above discussion, we supposed that the stimuli or environmentally responsive silicone elastomers could be obtained from the reaction of polysiloxanes and the anthracene groups.
In this study, we appended the anthryl groups to polysiloxanes with different anthracene derivatives and conducted a comprehensive examination on the reversible conversion process with heat. It is well-known that the dimerization or dissociation rate of anthryl dimers depends on the substituents on the anthracene ring.28 Thus, we synthesized silicone elastomers by combining poly(aminopropylmethylsiloxane) (PAPMS) with two derivatives of 9-substituted anthracene, in which one was linked with an electron-donating group (9-anthracenylmethyl acrylate, PAPMS-1) and the other was linked with an electron-withdrawing substituent (anthracene-9-carboxylic acid, PAPMS-2). The prepolymer of PAPMS-1 was successfully prepared from 9-anthracenylmethyl acrylate and PAPMS through an aza-Michael reaction, which is operated at mild reaction conditions, has a high efficiency, does not require a catalyst, and produces no by-products. However, the prepolymer of PAPMS-2 was prepared via “salt-forming vulcanization”6 by neutralizing PAPMS with anthracene-9-carboxylic acids to yield an ion-association complex. Then, light- and heat-triggered reversible silicone elastomers were prepared via [4 + 4] photo-dimerization of the pendant anthryl groups of the prepolymers. The cross-linked network was formed among the polymer chains after the polymer solution was treated with UV light (365 nm).
Sunlight-induced photo-dimerization was applied, for the first time, in the anthracene system, thus making it a “greener” and more environment-friendly process. As expected, dimerization efficiency of anthryl groups was remarkable when the sunlight was applied in the PAPMS-1 system. The covalent bonds formed via dimerization of the pendant anthryl groups could be reversed when the polymer was heated at 120 °C. We found that the PAPMS-1 film could emit green light under UV irradiation, which made the composites suitable for optoelectronic devices applications, such as light emitting diodes (LEDs). When the polymer solution was coated on the commercially available UV-LED cells and cured in situ, the colorful LED cells could be fabricated in an economical and effective way.
Experimental
Materials
Octamethylcyclotetrasiloxane (D4) and 3-aminopropyl-methyl-diethoxysilane were obtained as the commercial products and used directly. Dichloromethane, acetic acid, petroleum, ethanol, toluene, trichloromethane, and methanol were obtained from Tianjin Fuyu Chemical Co., Ltd. 9-Anthraldehyde, acryloyl chloride and anthracene-9-carboxylic acid were supplied by Energy Chemical Technology Co., Ltd. Sodium borohydride was provided by the Tianjin Kermel Chemical Reagent Co., Ltd.Synthesis
1H NMR (400 MHz, CDCl3): δ 8.48 (s, 1H), 8.43 (d, J = 9.0 Hz, 2H), 8.04 (d, J = 8.4 Hz, 2H), 7.64–7.53 (m, 2H), 7.49 (dd, J = 7.9, 7.0 Hz, 2H), 5.69 (s, 2H).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. A yellow solid (4.6 g) was obtained after vacuum drying (yield 81%).
:1) as an eluent. A yellow solid (4.6 g) was obtained after vacuum drying (yield 81%).1H-NMR (400 MHz, CDCl3): δ 8.55 (s, 1H), 8.39 (d, J = 9.0 Hz, 2H), 8.07 (d, J = 8.4 Hz, 2H), 7.66–7.57 (m, 2H), 7.57–7.46 (m, 2H), 6.45 (dd, J = 17.3, 1.4 Hz, 1H), 6.27 (s, 2H), 6.16 (dd, J = 17.3, 10.4 Hz, 1H), 5.83 (dd, J = 10.4, 1.4 Hz, 1H).
1H NMR (400 MHz, CDCl3) δ 2.67 (dd, J = 15.1, 8.1 Hz, 2H), 1.58–1.25 (m, 4H), 0.60–0.46 (m, 2H), 0.25–0.10 (m, 73H).
Preparation of the elastomers PAPMS-1 and PAPMS-2
For preparation of PAPMS-1, as shown in Scheme 2, 9-anthracenylmethyl acrylate and PAPMS, with a certain mole ratio, were added to a flask. The mixture was dissolved in the mixed solvents of methanol (1 mL) and toluene (10 mL). Then, the resultant mixture was refluxed and stirred at 45 °C. The FT-IR spectra of the mixture are presented in the ESI (Fig. S1†). As shown in Fig. S1,† the double bond disappeared after 8 h, which indicated that the aza-Michael reaction was completed. The mixture (PAPMS containing anthryl groups) was stirred in an ice bath under 365 nm UV-irradiation for 1 h. Then, most of the solvent was removed through distillation under reduced pressure till the resultant mixture could still be fluid. The thick precursor was placed in a Teflon mold and then the remaining solvent was slowly volatilized at room temperature.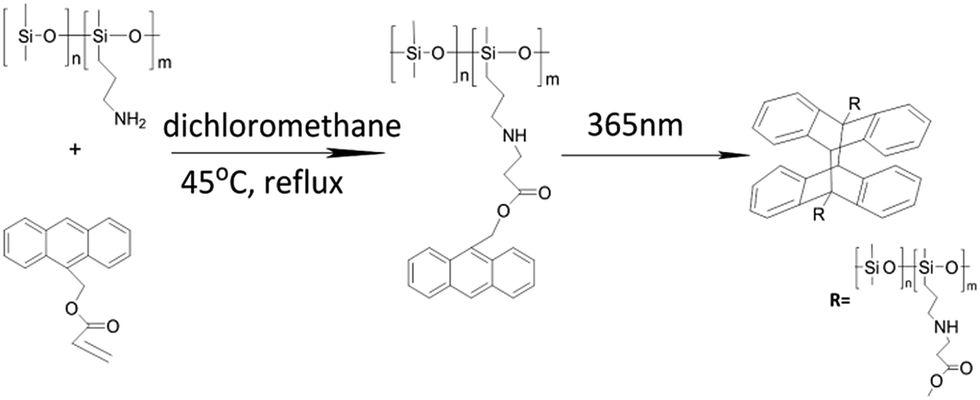 | ||
| Scheme 2 Synthesis route of PAPMS-1. | ||
In addition, for the first time, sunlight-induced photo-dimerization was attempted for anthryl silicone elastomers. A mixture of 9-anthracenylmethyl acrylate and PAPMS was irradiated by sunlight for 3 days instead of UV light. Then, most of the solvent was removed through distillation under reduced pressure till the resulting mixture could still be fluid. The thick precursor was placed into a Teflon mold, and then the remaining solvent was slowly volatilized at room temperature.
PAPMS-2 was prepared via “salt-forming vulcanization” by neutralizing PAPMS with anthracene-9-carboxylic acids to yield an ion-association complex. Typically, anthracene-9-carboxylic acids and PAPMS, in a certain mole ratio, were added to a flask. The mixture was dissolved in the mixed solvents of methanol (1 mL) and toluene (10 mL). The mixture was stirred in an ice bath upon UV-irradiation (365 nm). Fig. S2† depicts the IR spectra of anthracene-9-carboxylic acids, PAPMS, and the PAPMS-2 elastomer. The results confirmed the formation of an ion-association complex. In PAPMS-2, –COOH and –NH2 groups disappeared completely. Therefore, the new absorption band was observed at 1570 cm−1, which was caused by the asymmetric deformation of –COO−. This result inferred the complex formation of –COO− with –NH3+. The mixture was stirred in an ice bath under UV-irradiation at 365 nm. Then, most of the mixed-solvent was removed under vacuum till the resulting mixture can still be fluid. The thick precursor was poured into a Teflon mold and the rest of the solvent evaporated naturally.
Characterization and measurements
The dimerization of the anthracene groups was accomplished by irradiating with UV light from a Spectroline Model SB-100P/FA lamp (365 nm, 100 W). The UV intensity was set at 4500 mW cm−2 at a distance of 38 cm. The proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a Bruker AVANCE 400 spectrometer at 25 °C using CDCl3 (or CD3OD) as the solvent and without tetramethylsilane as an interior label. The molecular weights were determined by an Agilent HP1100 LC-Applied Biosystems API 4000 TQ mass spectrometer (LC/MS). The luminescence (excitation and emission) spectra of the samples were determined with a Hitachi F-7000 fluorescence spectrophotometer using a monochromated Xe lamp as an excitation source. Both excitation and emission slits were set at 5 nm. The ultraviolet absorption (UV) spectra in dichloromethane (or toluene) solution were detected using a Beijing TU-1901 double beam UV-vis spectrophotometer. Thermogravimetric analysis (TGA) was performed under N2 using a TA SDTQ600 over a temperature range from room temperature to 800 °C with a heating rate of 10 °C min−1. The scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800 (7 kV). The samples were cut and coated with a thin layer of platinum before the investigation.Results and discussion
Anthracene groups undergo dimerization ([4 + 4] cycloaddition reaction) when irradiated by UV (λ > 300 nm).26 In this study, the photo-cycloaddition between the anthracene groups was accomplished through UV-irradiation at 365 nm. In order to confirm the effectiveness of dimerization under the UV light, the control experiments were performed using 9-anthracenylmethyl acrylate or anthracene-9-carboxylic acid under the same conditions. The final products were characterized by UV-vis spectrometry, 1H-NMR, and mass spectrometry. The 1H-NMR spectra of the dimers are presented in Fig. 1. Compared to the spectrum of the monomer of before irradiation, few new peaks are observed in the spectrum of the resultant dimer (after the UV irradiation). For 9-anthracenylmethyl acrylate, the peaks k and l are observed at 7.0 and 6.8 ppm, respectively, after the UV-irradiation (Fig. 1a), which matched with the structure of a dimer. This result confirmed the occurrence of the dimerization reaction. For anthracene-9-carboxylic acid, the peaks k and l are located at 7.0 and 6.9 ppm, respectively, after the UV-irradiation (Fig. 1c), which showed the abundant changes in the anthracene unit signals compared with 9-anthracenylmethyl acrylate after the photo-irradiation. The results are primarily related to the substituents on the anthracene rings. However, the proton peaks of the monomer still existed after the irradiation, which indicated that the dimerization reaction was incomplete. The mass spectra also showed a dimer peak and a monomer peak in the final product after the irradiation as shown in Fig. S3 and S4.† The results obtained from the 1H-NMR and mass spectra demonstrated that only small amount of the monomer can undergo a cycloaddition reaction upon UV irradiation at 365 nm.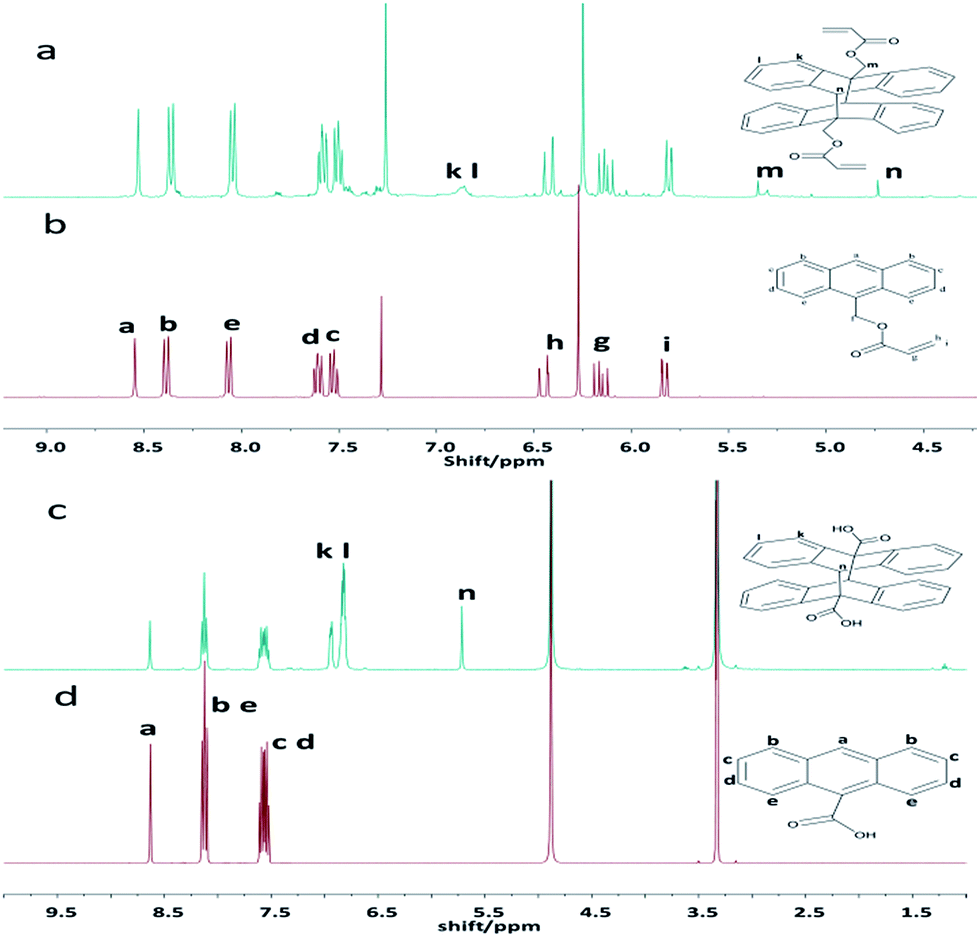 | ||
| Fig. 1 1H-NMR spectra of the anthracene derivatives after ((a) and (c)) and before ((b) and (d)) the [4 + 4] photo-cycloaddition of 9-anthracenylmethyl acrylate and anthracene-9-carboxylic acid, respectively, upon a UV irradiation at 365 nm for 1 h. | ||
The main absorption peak of anthracene in the UV-vis spectra is between 300 and 400 nm. Upon dimerization at the 9 and 10 positions of the anthracene moieties, the conjugated π-system was broken, which led to a depletion of the characteristic UV absorption bands of the anthracene groups. As shown in Fig. 2, the absorption intensity decreased upon the UV-irradiation, which indicated that the cycloaddition had occurred. In addition, the progress of the dimerization reaction was estimated by the proportion of the decrease in the absorbance compared to its original value before the irradiation, using the equation (A0 − A)/A0, where A0 and A represent the absorption intensity at 368 nm before and after the irradiation, respectively.29 It is well-known that the rate of this dimerization depends on the substituents of the anthracene ring and the rate of reaction with the electron-withdrawing substituents is higher than that with the electron-donating substituents. As shown in Fig. 2, the efficiency of dimerization of the monomers was 25% and 35%, respectively. The results achieved by UV-vis spectrometry, 1H-NMR, and mass spectrometry proved that the dimerization reaction easily occurs when the substituent on the anthracene ring is an electron-withdrawing group.
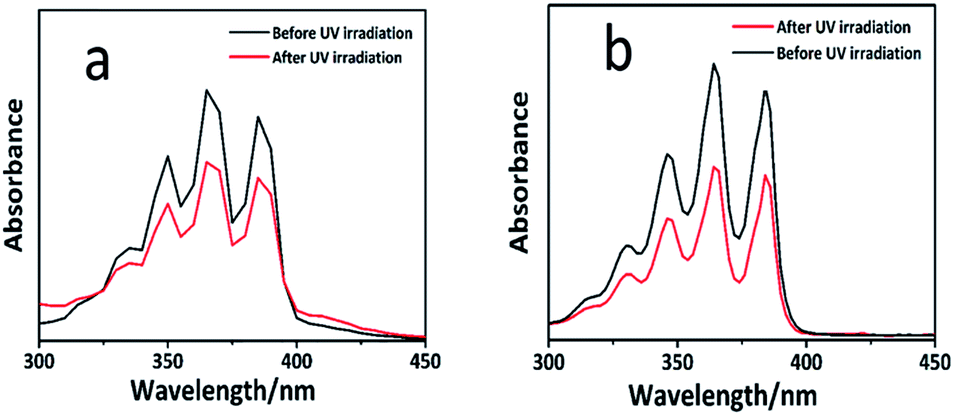 | ||
| Fig. 2 (a) UV-vis spectra of 9-anthracenylmethyl acrylate before (top) and after (bottom) irradiation (365 nm); (b) UV-vis spectra of anthracene-9-carboxylic acid before (top) and after (bottom) the irradiation (365 nm). | ||
The photo-dimerization reaction ([4 + 4] photo-cycloaddition) in the polymer solution can also be determined by UV-vis spectroscopy. Fig. S5† shows the UV-vis spectra of the PAPMS-1 solution and the PAPMS-2 solution before and after the UV-irradiation. Although the absorption intensity of both polymer solutions decreased after the UV-irradiation, the absorption of the PAPMS-2 solution decreased more than that of the PAPMS-1 solution, which was primarily attributed to the substituents of the anthracene rings. The absorption intensity of the polymer solution was related to the exposure time as illustrated in Fig. 3. The polymer solution before the irradiation has the strongest absorption intensity in the range of 300–400 nm. The intensity of the absorption peak decreased with the increase in the time of irradiation, which indicated that the efficiency of the photo-dimerization increased with the increase in the exposure time (insets in Fig. 3). For the same irradiation time, the absorption intensity of PAPMS-2 decreased more than that of the PAPMS-1. Our data have further confirmed that the efficiency of photo-dimerization is higher if the substituent on the anthracene ring is an electron-withdrawing group.
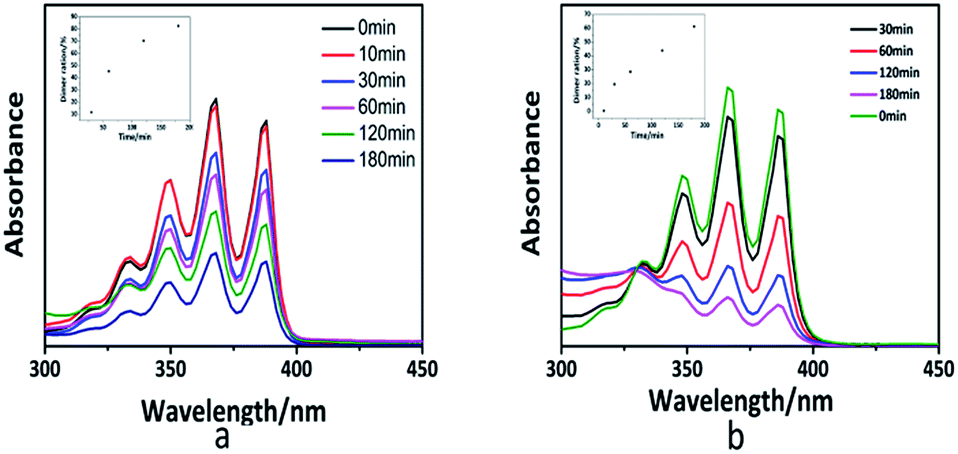 | ||
| Fig. 3 UV-vis spectra and dimer ration (insets) of the PAPMS-1 solution (a) and PAPMS-2 solution (b) with different exposure times (365 nm). | ||
Polysiloxanes containing pendant anthryl groups are expected to show an ascendancy of the controllable reversibility and reactivity as well as avoid the formation of by-products. The reversibility of dimerization is favorable for the degradation and the recovery of the materials. In the previous reports, the cleavage of the photo-dimers can be either optically triggered by short-wavelength UV irradiation (254 nm) or thermally-induced bond scission.30,31 However, compared with the thermal degradation, the UV irradiation may cause some side reactions, which limited the use of this reversible system. In order to investigate the reversible formation of the photo-cross-linking network, a heat-triggered de-cross-linking route with a thermal annealing step at 120 °C was designed. UV-vis spectroscopy was utilized to monitor the reversible dimerization of the anthracene groups. The light- (λ = 365 nm, 60 min) and heat-triggered (120 °C) reaction, denoted as a reversible cycle, was operated. As illustrated in Fig. 4a and b, the absorption intensity of the PAPMS-1 solution and the PAPMS-2 solution decreased after the irradiation, but almost recovered to the original value after the thermal process. These results indicated that some of the anthryl groups could form dimers after the UV irradiation and the dimers were cleaved to form the anthryl groups again after the thermal treatment. Nevertheless, more time was needed for the PAPMS-1 solution for de-cross-linking (120 °C, 240 min) compared to that for the PAPMS-2 solution (120 °C, 180 min). In addition, we detected the reversible property of the films as shown in Fig. S6.† The results also revealed the dimerization upon UV-irradiation at 365 nm and the bond cleavage of the anthracene dimers by heat treatment (120 °C). Furthermore, the model experiment was performed. As shown in Fig. 4d, the prepolymer PAPMS-2 was a viscous liquid at room temperature, whereas it was solidified when exposed to a 365 nm light. It is surprising that PAPMS-2 appeared to be a liquid when heated at 120 °C and changed into the solid sample when irradiated at 365 nm. In addition, the reversible cycle (Fig. S7†) of PAPMS-1 was longer than that of PAPMS-2. It is reported that the rate of this dissociation, which is caused by anthracene dimers that dissociate cleanly at the elevated temperatures through a diradical transition state, depends on the substituents on the anthracene ring.32 As expected, PAPMS-2 could be easily depolymerized than PAPMS-1 because the anthracene in PAPMS-2 was linked to an electron-withdrawing group.
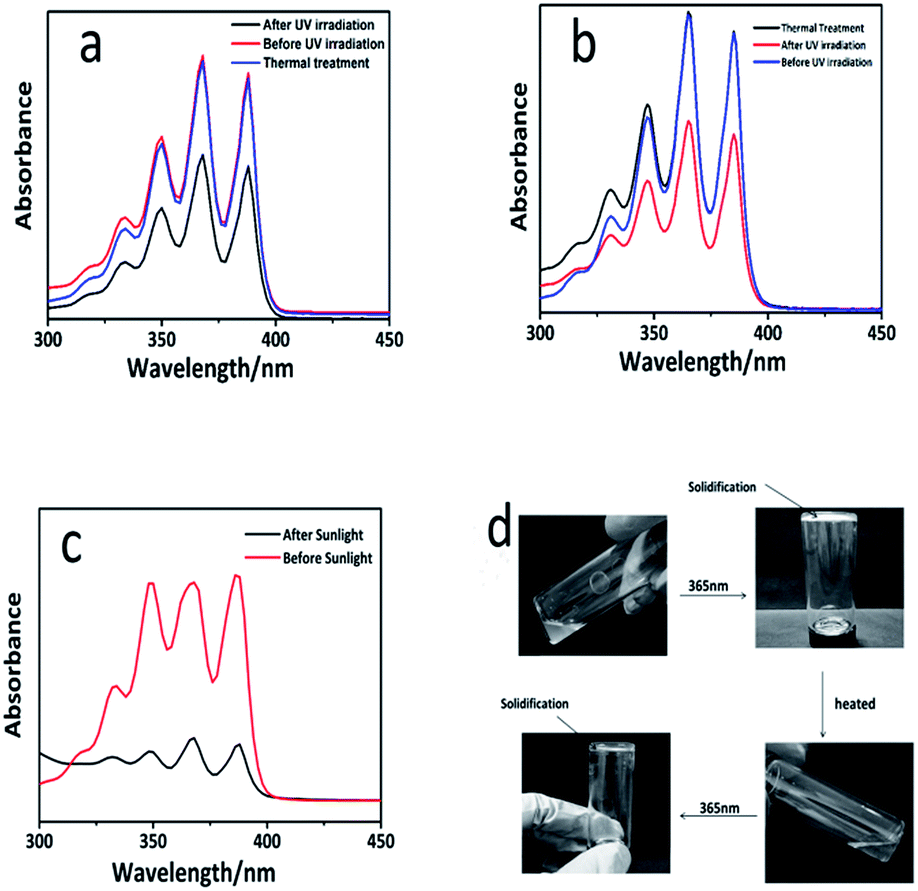 | ||
| Fig. 4 (a) UV-vis spectra of the PAPMS-1 solution in different processes; (b) UV-vis spectra of PAPMS-2 solution in different processes; (c) UV-vis spectra of the anthracene modified polysiloxane solution before and after the irradiation (sunlight); (d) illustrations of PAPMS-2 at 365 nm upon repeating irradiation and heating cycles. | ||
Sunlight is a green energy source, which includes light of various wavelengths; when it comes to the application in chemistry, sunlight is primarily used in the field of photocatalysis. Moreover, some studies have indicated that some reactions can be triggered by the ultraviolet ray of sunlight. For example, silicone elastomer could be prepared via thiol–ene click reaction induced by sunlight.33 It is an advantage that some reactions can be induced by sunlight because they are “greener” and do not need excessive power as compared to the conventional reactions triggered by a UV light. In this study, the sunlight was applied, for the first time, to trigger the dimerization reaction of the anthryl group. PAPMS-1 was chosen as an example to design the experiment. The prepolymer PAPMS-1 was prepared and the same amount of the PAPMS-1 solution was diluted to 10 mL in two colorimetric tubes. One tube was placed under the sunlight and the other was placed indoors. After three days, the samples were tested by UV-vis spectroscopy as illustrated in Fig. 4c. The absorption intensity of the anthracene groups decreased after being irradiated by sunlight, which proved that the dimerization reaction had occurred. Based on these data, it can be concluded that our study can produce the potential self-repair materials by exposure to sunlight.
Luminescent properties of the films
Anthracene is commonly used in fluorophores that are present in the fluorescent materials and its solution can emit a strong blue light under the UV light irradiation. Simultaneously, anthracene is the exact fetch of most of the fluorescent materials that exhibit aggregation fluorescence quenching (ACQ) effect. They can emit a strong blue light in the solution, but their fluorescence intensity would weaken or even quench in the high-concentration solution or solid. We were surprised to find that PAPMS-1 emitted green fluorescence under a UV light. Thus, we investigated the fluorescence properties of PAPMS-1. It could be observed from Fig. 5a that the fluorescence behavior of PAPMS-1 was similar to that of anthracene. PAPMS-1 can emit a strong blue light in the solution, while the fluorescence intensity weakened or even quenched in the high-concentration solution or the polymer film. Though PAPMS-1 appears like a solid solution, the anthryl group was aggregated due to π–π stacking. In such case, the fluorescence intensity of the film was much lower than that of the solution. The maximum emission wavelength of the PAPMS-1 film was red shifted to 520 nm compared with that in the solution (450 nm). Simultaneously, the cured film was green under a UV light as observed by naked eyes. In PAPMS-1, the anthryl groups were in a dispersed state in the solution. However, they were aggregated owing to π–π stacking in the films, which led to the red shift of the maximum emission wavelength and exhibited different colors of the fluorescence between the polymer solution and the films. Using the CIE 1931 RGB colorimetric system, the bright emissions of the cured film and the polymer solution can be easily observed by the naked eyes upon excitation with a wavelength of 398 nm as shown in Fig. 5b.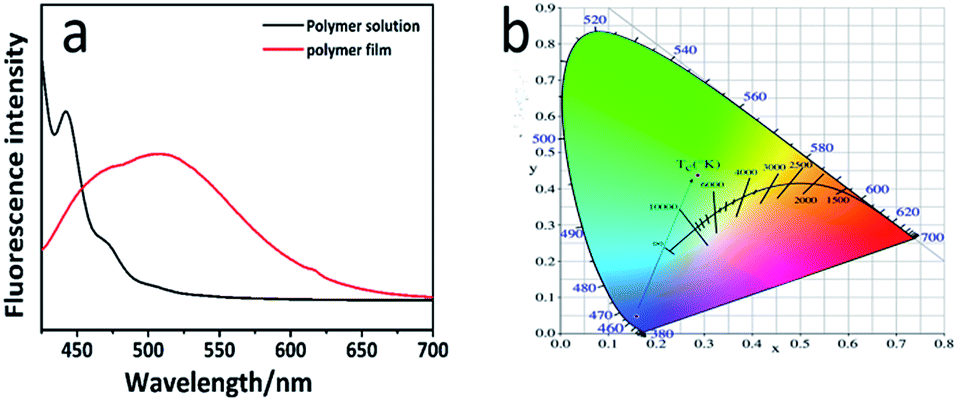 | ||
| Fig. 5 (a) Fluorescence spectra of the PAPMS-1 film and its solution under UV-irradiation at a 365 nm wavelength (excitation at 398 nm); (b) emission colors of the films in the CIE 1931 chromaticity diagram; CIE color coordinates: polymer solution (0.1588, 0.0478) and cured film (0.3133, 0.421). | ||
Interestingly, the fluorescence intensity of the film changed after the irradiation. As shown in Fig. 6, the fluorescence intensity of the PAPMS-1 film increased after the UV-irradiation, which was different from the ACQ effect. The fluorescence emission reduced due to π–π stacking in the PAPMS-1 film. The UV-triggered dimerization occurred between the anthracene groups after the irradiation. The π–π stacking was broken and the aggregated state of the anthracene groups was changed. Thus, the ACQ effect was weakened and the emission increased.
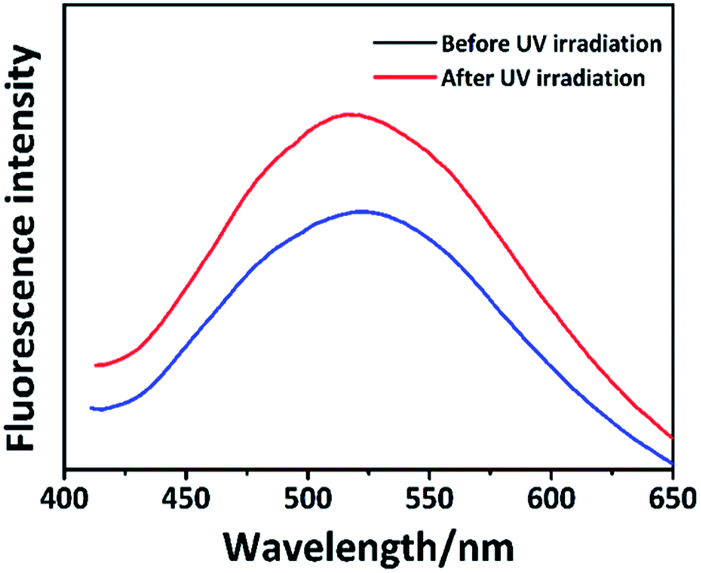 | ||
| Fig. 6 Fluorescence spectra of the untreated and treated PAPMS-1 film upon UV-irradiation. | ||
Coating on a commercial available UV-LED light
The potential application on the commercially available LED cell was determined because of the unique luminescent performance of the PAPMS-1 film. When the PAPMS-1 film was coated on a flat substrate exposed to a UV lamp, a green luminescence was observed even with the naked eyes as illustrated in Fig. 7a. PAPMS-1 has an excellent film-forming ability and can be easily coated on any round-shaped object. Thus, the reactant mixture containing 9-anthracenylmethyl acrylate grafted polysiloxane was also coated onto a commercially available UV-LED cell instead of casting on the flat substrate. Thus, the colorful UV-light emitting diode (LED) cell can be obtained through a facile and efficient method, in which the commercially available LED cell was coated with the transparent luminescent films from the solution medium, followed by in situ curing. As shown in Fig. 7b, a bright green light was observed. We can speculate that if the fluorescent molecular species were changed or the excitation wavelength was adjusted, the change in the emission colors of the LED light could be observed. Therefore, different sorts of colorful LED lights can be obtained via the similar approaches. In addition, the PAPMS-1 film exhibited an excellent thermal stability, which was proven by thermogravimetric analysis (TGA) under N2 (Fig. S8†). The microphase separation of the PAPMS-1 film was not observed by the scanning electronic microscopy (SEM) as shown in the Fig. S9.† | ||
| Fig. 7 (a) Images of the flat substrate coated with the luminescent film under UV irradiation (365 nm); (b) images of a commercially available UV-LED cell (left) and a UV-LED coated with the luminescent film (right). | ||
Conclusion
The synthesis of novel anthracene-containing polysiloxane via aza-Micheal or neutralization reaction and irradiation under a UV light is described in this paper. It is evident that the photo-cross-linking process is almost reversible, which is proven by the data from the absorption intensity of the UV spectra obtained when the target products were heated. In particular, sunlight can be used instead of UV light to trigger the reaction and it was applied for the first time here in the anthracene dimerization. Moreover, a sunlight-induced reaction is beneficial for the environment. The green luminescent films based on polysiloxane and the anthracene group were developed and possessed the excellent film properties. These transparent films could be coated on a UV-LED cell, which makes them ideal candidates, particularly for fabricating devices used in displaying and lighting in an environment-friendly way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21774070), the National Science Foundation of Shandong Province (ZR2015BQ008), and Shandong Special Fund for Independent Innovation and Achievements transformation (No. 2014ZZCX01101).References
- Y. Zuo, Z. Gou and C. Zhang, Macromol. Rapid Commun., 2016, 37(15), 1052–1059 CrossRef CAS PubMed.
- C. Erbil, E. Kazancıoğlu and N. Uyanık, Eur. Polym. J., 2004, 40(6), 1145–1154 CrossRef CAS.
- R. P. Eckberg, Ultraviolet light curable acrylic functional silicone compositions, US Pat., US4348454, 1982.
- S. Matsubara, M. Yoshioka and K. J. Utimoto, Chem. Lett., 1994, 5(5), 827–830 CrossRef.
- G. Zhiming, Y. Zuo and S. Feng, RSC Adv., 2016, 6(77), 73140–73147 RSC.
- H. Lu and S. Feng, J. Polym. Sci., Part A: Polym. Chem., 2017, 55(5), 903–911 CrossRef CAS.
- Y. Zuo, Z. Gou and J. Cao, et al., RSC Adv., 2016, 6(51), 45193–45201 RSC.
- Y. Yue, Y. Liang, H. Wang, L. Feng, S. Feng and H. Lu, Photochem. Photobiol., 2013, 89, 5–13 CrossRef CAS PubMed.
- A. S. Fawcett, T. C. Hughes, L. Zepeda-Velazquez and M. A. Brook, Macromolecules, 2015, 48(18), 6499–6507 CrossRef CAS.
- A. S. Fawcett and M. A. Brook, Macromolecules, 2014, 47(5), 1656–1663 CrossRef CAS.
- S. Li and S. Feng, RSC Adv., 2016, 6, 99414–99421 RSC.
- T. Bartz, M. Klapper, K. Müllen and R. C. Schulz, Polym. Int., 1993, 31, 153–162 CrossRef CAS.
- M. Coursan, J. P. Desvergne and A. Deffieux, Macromol. Chem. Phys., 1996, 197, 1599–1608 CrossRef CAS.
- J. T. Goldbach, T. P. Russell and J. Penelle, Macromolecules, 2002, 35, 4271–4276 CrossRef CAS.
- S. Paul, S. Stein, W. Knoll and K. Müllen, Acta Polym., 1996, 47, 92–98 CrossRef CAS.
- L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Adv. Funct. Mater., 2008, 18, 3315–3322 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- H. Wang, F. Liu, R. C. Helgeson and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 655–659 CrossRef CAS PubMed.
- J. F. Xu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS PubMed.
- L. Lopez-Vilanova, I. Martínez, T. Corrales and F. Catalina, Eur. Polym. J., 2014, 56, 69–76 CrossRef CAS.
- X. A. Trinh, J. Fukuda, Y. Adachi, H. Nakanishi and T. Norisuye, Macromolecules, 2007, 40, 5566–5574 CrossRef CAS.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7725 RSC.
- H. Xie, C. Y. Cheng and X. Y. Deng, et al., ACS Appl. Mater. Interfaces, 2016, 8, 9431–9439 CAS.
- S. Radl, M. Kreimer, T. Griesser, A. Oesterreicher, A. Moser, W. Kern and S. Schlogl, Polymer, 2015, 80, 76–87 CrossRef CAS.
- S. Bringmann, R. Brodbeck, R. Hartmann and C. Schafer, Org. Biomol. Chem., 2011, 9(21), 7491–7499 CAS.
- H. Xie, C. Y. Cheng and X. Y. Deng, et al., Macromolecules, 2017, 50, 5155–5164 CrossRef CAS.
- H. Xie, C. Y. Cheng and L. Du, et al., Macromolecules, 2016, 49, 3845–3855 CrossRef CAS.
- J. Van Damme, L. laminck and G. Van Assche, et al., Tetrahedron, 2016, 72(29), 4303–4311 CrossRef CAS.
- T. Yamamoto, S. Yagyu and Y. J. Tezuka, J. Am. Chem. Soc., 2016, 138, 3904–3911 CrossRef CAS PubMed.
- H. D. Becker, Unimolecular photochemistry of anthracenes, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- S. Tazuke and N. Hayashi, Functionality and structure of polymers, J. Polym. Sci., Polym. Chem. Ed., 1978, 16, 2729–2739 CrossRef CAS.
- J. Van Damme, O. van den Berg and J. Brancart, et al., Macromolecules, 2017, 50(5), 1930–1938 CrossRef CAS.
- Y. Zuo, J. Cao and S. Feng, Adv. Funct. Mater., 2015, 25, 2754–2762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12201b |
| This journal is © The Royal Society of Chemistry 2017 |