
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic Fe3O4@silica sulfuric acid nanoparticles promoted regioselective protection/deprotection of alcohols with dihydropyran under solvent-free conditions†
Kalyani
Rajkumari
,
Juri
Kalita
,
Diparjun
Das
and
Samuel Lalthazuala
Rokhum
 *
*
Department of Chemistry, National Institute of Technology, Silchar-788010, Assam, India. E-mail: rokhum@che.nits.ac.in; Fax: +91 3842 224797; Tel: +91 3842 242915
First published on 15th December 2017
Abstract
Protection (and deprotection) of hydroxyl groups via tetrahydropyranylation was carried out effectively using a catalytic amount of Fe3O4 supported silica sulphuric acid nanoparticles (Fe3O4@SiO2@SO3H) under solvent-free conditions. The synthesized nanocatalyst was characterized by XRD, TEM, FT-IR etc. A wide range of tetrahydropyranylated alcohol derivatives were synthesized using this heterogeneous magnetic nanocatalyst within 10–20 min with high yields. In addition, tetrahydropyranyl ethers could also be deprotected to the parent alcoholic compounds in the presence of MeOH using the same catalyst. After completion of the reactions, the catalyst was easily separated from the reaction medium using an external magnet, which ameliorated the overall synthetic process. The catalyst was recovered and reused for five successive reactions without any appreciable loss in its activity. Mild reactions conditions, operational simplicity, solvent free conditions, high selectivity, easy recyclability of the magnetic nanocatalyst, and high yields can be considered as the advantageous features of our procedure.
1. Introduction
During the synthesis of an organic compound, the protection (and deprotection) of the free hydroxyl group in a multifunctional alcohol substrate or intermediate is one of the most frequently used strategies.1,2 For this type of functional manipulation, etherification of the hydroxyl group using 3,4-dihydro-2H-pyran (DHP) is recognized as the most popular and easy protocol. This is because of its several attractive advantages over other protecting reagents, which include ease of its preparation, stability of the corresponding tetrahydropyranyl ethers towards various reaction conditions and use of reagents such as strong bases, hydrides, Grignard reagents and other organometallic reagents.3–5 Moreover, THP ethers can easily be converted to various derivatives, such as sulfides, halides,6 esters,7 cyanides and carbonyl compounds8 employing specific methods, which makes them ideal protecting reagents for tetrahydropyranylation.Enumerable catalysts including both homogeneous and heterogeneous catalysts have been reported to date, which have been proposed to catalyze tetrahydropyranylation of free hydroxyl groups. Taking into account the advantages of heterogeneous catalysts over homogeneous catalysts in terms of separation and recoverability, several effective heterogeneous catalysts have been reported in recent years.9,10 However a serious disadvantage of heterogeneous catalysts is that their efficiency gets restricted due to the limited surface area.11 This drawback can be overcome by a nanoporous catalyst support due to its “plenty of room at the bottom” nature, which can also be considered as a bridge between heterogeneous and homogenous catalysts.12
In recent decades, magnetic nanoparticles (MNPs) have gained enormous attention since they can be easily separated from the reaction medium by an external magnet, preventing the use of column chromatography, avoiding loss of product, enhancing recyclability and thus ameliorating the overall synthesis process.13 Magnetic Fe3O4 has been investigated the most due to its potential magnetic nature although bare iron oxide nanoparticles have several limitations, which include immediate aggregation into large clusters and rapid exothermic reactions with oxygen.14,15 However surface modification by forming a silica coating over the MNP surface can make it stable, inert, non-toxic, resistant under catalytic conditions as well as improve the surface functionality due to the availability of abundant silanol groups (–SiOH), which act as a support for immobilization of the SO3H function. Owing to the unique properties of magnetic nanoparticles, sulphuric acid adsorbed on silica coating MNPs core–shell structures (Fe3O4@SiO2@SO3H) have been utilized as a catalyst in various organic transformations, such as Biginelli reaction,16 synthesis of amidoalkyl naphthols17 and protection–deprotection of hydroxyl groups using hexamethyldisilazane (HMDS).18 In this communication, we report tetrahydropyranylation/depyranylation of alcohols with sulphuric acid adsorbed on a silica coating MNPs core–shell structure under solvent-free conditions, which makes it free from the hazardous effects of organic toxic solvents, offering a green and eco-friendly approach.
2. Experimental methodology
2.1 Materials and methods
All alcohols, DHP (3,4-dihydro-2H-pyran), silica gel for TLC (Thin Layer Chromatography) and column chromatography were of analytical grade and purchased from SpectroChem. TEOS (tetraethyl orthosilicate) was purchased from Sigma Aldrich, India. The chemicals were used without further purification. The solvent used were of extra pure grade, purchased from Merck India. Double distilled deionized water was used for the synthesis of magnetic nanoparticles.For characterization of the catalyst, high resolution transmission electron microscopy (HRTEM) was conducted using an electron microscope JEM-2100, 200 kV, JEOL (SAIF-NEHU, Shillong). The samples were dispersed in ethanol, DCM (dichloromethane) and then treated ultrasonically in order to disperse individual particles over a copper grid. Powder X-Ray Diffraction (XRD) patterns were obtained on an X'Pert Pro PANalytical diffractometer (SAIF-GU, Guwahati) under the following conditions: K-Alpha 1 wavelength (λ = 1.54056 Å), K-Alpha 2 wavelength (λ = 1.54439 Å), generator voltage of 40 kV, a tube current of 35 mA and the count time of 0.5 s per 0.02° in the range of 5°–90° with a copper anode. FTIR spectra were recorded on a Nicolet 6700, Nicolet Continuum FTIR Microscope (IASST, Guwahati). NMR spectra were recorded on Bruker Avance II, 400 MHz (SAIF NEHU, Shillong).
2.2 Preparation of catalysts
 | ||
| Scheme 1 Schematic diagram for synthesis of Fe3O4@silica sulfonic acid nanocatalyst. | ||
2.3 General procedure for the tetrahydropyranylation of alcohols using Fe3O4@silica sulfonic acid (Fe3O4@SiO2@SO3H)
A mixture of alcohol (1 mmol), DHP (1.2 mmol) and Fe3O4@silica sulfonic acid catalyst (0.15 g, 20 mol% of substrate) was stirred in a small reaction vessel at room temperature. The reaction progress was monitored by thin layer chromatography. After completion of the reaction, ethyl acetate (10 mL) was added to the solution and the catalyst was separated by an external magnet. The solution was concentrated under reduced pressure and the product was passed through a column of silica gel eluting with hexane/ethyl acetate (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain pure THP ether with high purity. The catalyst was washed several times with ethyl acetate and dried under vacuum (24 mmHg) at 60 °C for 24 h.
:1) to obtain pure THP ether with high purity. The catalyst was washed several times with ethyl acetate and dried under vacuum (24 mmHg) at 60 °C for 24 h.
2.4 General procedure for the deprotection of alcohols
THP ether (1 mmol) and Fe3O4@silica sulfonic acid catalyst (20 mol% of substrate) were stirred in methanol (0.5 mL) at room temperature for 30 min. The cleavage of THP ethers was monitored by TLC. After completion of the reaction, the catalyst was separated with the help of an external magnet and washed with ethyl acetate. The solution was concentrated under reduced pressure to obtain the corresponding pure hydroxyl compound in a quantitative yield.3. Result and discussion
3.1 Catalyst characterization
The synthesized Fe3O4@silica sulfonic acid nanoparticles were characterized by Fourier Transform Infrared (FT-IR) spectroscopy, X-Ray Diffraction and Transmission Electron Microscopy (TEM). | ||
| Fig. 1 FT IR spectra of (a) Fe3O4, (b) Fe3O4@SiO2 and (c) Fe3O4@silica sulfonic acid. | ||
The absorption peak at approximately 579 cm−1 (Fig. 1(a)) corresponds to the stretching vibration of the Fe–O bond17 and the adsorption of the silica coating on the magnetite surface was indicated by the band near 1031 and 893 cm−1, which are assigned to the Si–O stretching vibration (Fig. 1(b)). In addition, successful sulfonic acid functionalization of the silica layer on Fe3O4 surface was evidenced by the absorption bands at 1076 cm−1 and 1136 cm−1 related to the stretching of the S–O bonds. A peak appeared at about 3402 cm−1 due to the stretching of the OH groups in the SO3H moiety (Fig. 1(c)). These FT-IR spectra provided evidence of the formation of a silica shell onto the surface of Fe3O4 and the acid functionalization of the silica shell.17,18
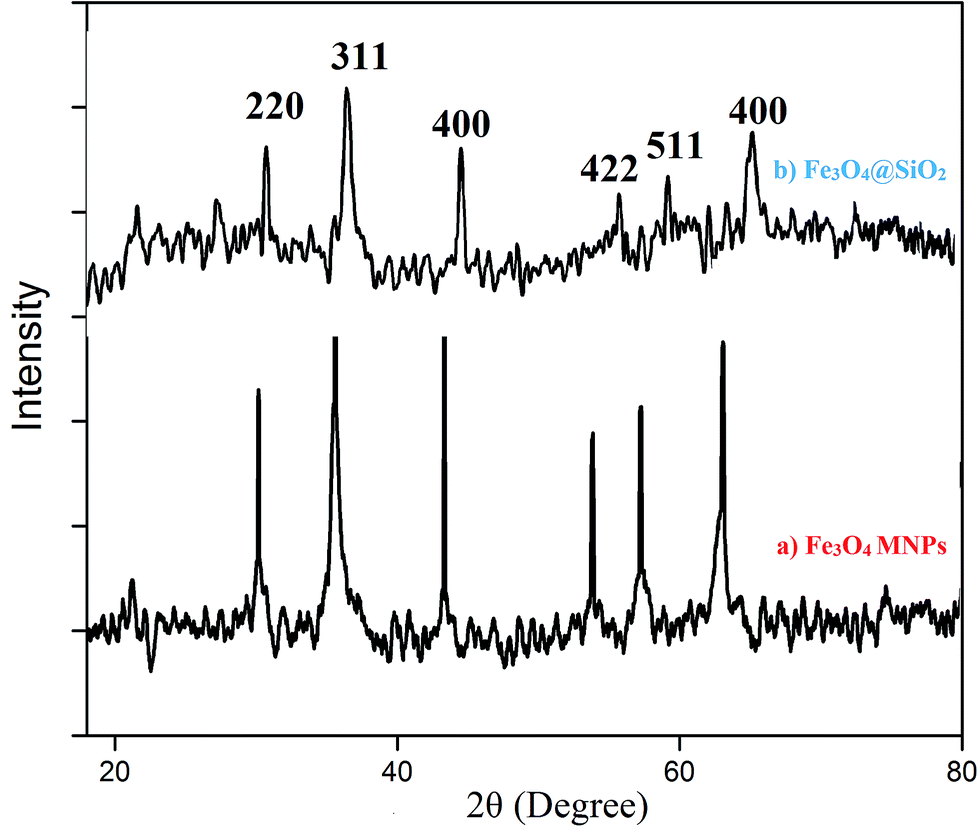 | ||
| Fig. 2 XRD spectra of (a) Fe3O4, (b) Fe3O4@silica sulfonic acid. | ||
 | ||
| Fig. 3 (a) HRTEM images of Fe3O4 nanoparticles, (b) HRTEM images of Fe3O4@SiO2, (c) HRTEM images of Fe3O4@SiO2@SO3H, (d) ED pattern of Fe3O4@SiO2@SO3H. | ||
The fine coating of silica over the Fe3O4 nanoparticles can be observed from the TEM images shown in Fig. 3(b) and (c), which indicates the retention of the silica coating even after the sulfonic acid functionalization on the silica surface.17,19
Further, the SAED (Selected Area Electron Diffraction) pattern (Fig. 3(d)) of the Fe3O4 nanoparticles confirms that the sample is crystalline and also confirms our conclusions from the X-ray diffraction pattern.
3.2 Protection and deprotection of alcohols
In continuation of our ongoing research interest, we applied the synthesized catalyst in tetrahydropyranylation of various alcohol substituents. Initially, we tried it with benzyl alcohol and, to our delight; protection of alcohol was successful under solvent-free conditions within a very short time of 1 h only. First, we took an equimolar mixture of benzyl alcohol (1 mmol) and DHP (1 mmol) and 10 mol% of the nanocatalyst and stirred the mixture for 1 h. After stirring for 1 h, we observed only slight formation of the product as indicated by TLC. Surprisingly, this did not increase even after stirring for another 1 h. Then, we added additional DHP (0.2 mmol) and found that the reaction was completed within the next 1 h. Inspired by these findings, our next evident target was to optimize the amount of catalyst. To our satisfaction, we found that 20 mol% of the nanocatalyst was optimum for the protection of alcohols (Table 1, entry 4).| Entry | Catalyst amount (mol%) | Time (min) | Yield (%) |
|---|---|---|---|
| a Reaction conditions: benzyl alcohol (1 mmol), DHP (1.2 mmol), room temperature, solvent-free condition. | |||
| 1 | 5 | 150 | 65 |
| 2 | 10 | 120 | 85 |
| 3 | 15 | 55 | 90 |
| 4 | 20 | 45 | 95 |
| 5 | 25 | 45 | 95 |
Encouraged by the result and the advantage of using magnetic nanocatalysts over soluble homogeneous catalysts, we carried out the conversion of various alcohols into their protected products as described in Scheme 2.
 | ||
| Scheme 2 Tetrahydropyranylation of alcohols using 20 mol% of nanocatalyst. | ||
With the optimum reaction condition in hand, we carried out the tetrahydropyranylation of a variety of primary, secondary, tertiary, benzylic and allylic alcohols which were smoothly converted to the corresponding desired products in good to excellent yields at room temperature. It was observed that benzylic alcohols substituted with electron-donating groups are highly reactive under our reaction condition, giving the corresponding protected alcohols in excellent yields (Table 2, entries 2 and 3). However, reactions of benzyl alcohols substituted with electron-withdrawing groups were, to some extent, slower as compared to their electron-donating counterparts (Table 2, entry 5). Although the reaction rates of aliphatic alcohols were lower as compared to the benzylic ones, they were easily converted into their corresponding products in high yields. It was observed that aliphatic alcohols with larger chain lengths take more time for completion than those with smaller chain lengths (Table 2, entries 7, 8, and 9). However, secondary and tertiary alcohols took longer time for complete conversion (Table 2, entries 11 and 12). Allyl alcohols (Table 2, entries 13 and 14) were converted into the corresponding protected alcohols leaving the olefinic bonds intact.



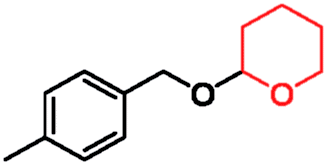




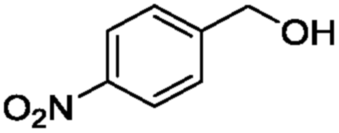



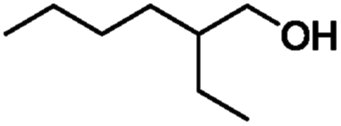












In order to gain more insights into the selectivity of our method, we studied various competitive reactions between structurally different alcohols in binary mixtures as shown in Table 3. This study reveals that benzylic alcohols can be converted into their protected products with excellent selectivity in the presence of aliphatic alcohols (Table 3, entry 1). Interestingly, benzylic alcohols were converted quantitatively into their protected alcohols, while the secondary alcohol remained intact (Table 3, entry 2).
| Entry | Alcohols | Products | Time (min) | % Conversion |
|---|---|---|---|---|
| a Reaction condition: alcohol (1 mmol), DHP (1.2 mmol), catalyst (20 mol%), room temperature, solvent-free conditions. | ||||
| 1 |

|

|
45 | 95 |

|

|
5 | ||
| 2 |
|

|
45 | 100 |

|

|
0 | ||
| 3 |

|
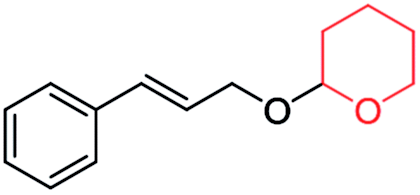
|
60 | 94 |

|

|
6 | ||
| 4 |

|

|
60 | 100 |

|

|
0 | ||
Allyl alcohols also reacted faster as compared to their aliphatic counterparts, affording the allyl products in high yield (Table 3, entry 3). Similarly, primary alcohols can also be converted to their corresponding THP ether in the presence of tertiary ones with complete selectivity (Table 3, entry 4).
The mechanism proposed for the tetrahydropyranylation of alcohols catalyzed by Fe3O4@SiO2@SO3H is shown in Scheme 3.
 | ||
| Scheme 3 Proposed mechanism for the tetrahydropyranylation of alcohols using Fe3O4@SiO2@SO3H nanocatalyst. | ||
3.3 Recycling of the catalyst
The recyclability of the magnetic nanocatalyst was investigated with consecutive tetrahydropyranylation reactions using different substrates. Fig. 4 depicts the results of consecutive runs performed by reusing the catalyst under our optimal reaction conditions. After each catalytic run, the catalyst was recovered by an external magnet, washed with ethyl acetate and then dried at 100 °C in Abderhalden apparatus under reduced pressure overnight before being used again in a new reaction. We demonstrated that no depreciations in the catalytic performance were observed in all the test reactions even after five catalytic cycles. In addition, no difference in the IR spectrum of the catalyst was observed after these repeated cycles (Table 4).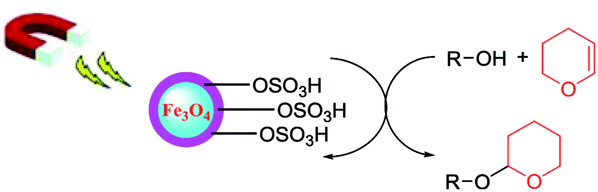 | ||
| Fig. 4 Recyclability test of nanocatalyst for protection/deprotection of alcohols. | ||
| Entry | Products | Run/yield % | |||||
|---|---|---|---|---|---|---|---|
| a Reaction condition: alcohol (1 mmol), DHP (1.2 mmol), catalyst (20 mol%), room temperature. | |||||||
| 1 |
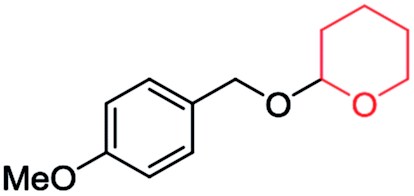
|
Run | 1 | 2 | 3 | 4 | 5 |
| Yield (%) | 96 | 96 | 94 | 94 | 93 | ||
| 2 |

|
Run | 1 | 2 | 3 | 4 | 5 |
| Yield (%) | 90 | 89 | 89 | 89 | 88 | ||
| 3 |

|
Run | 1 | 2 | 3 | 4 | 5 |
| Yield (%) | 88 | 88 | 86 | 86 | 84 | ||
3.4 Deprotection of THP ether
The deprotection of THP ethers was investigated by changing the solvent system. We found that the addition of methanol serves as an efficient deprotecting reagent for THP ethers in the presence of Fe3O4@silica sulfonic acid as a catalyst at room temperature to provide the corresponding free alcohols in an excellent yield of 90–95% (Table 5) irrespective of the structural variations. It is interesting to note that the deprotection of all the compounds reported in the present study was completed within 30 min. The proposed mechanism for the deprotection of THP ether is shown in Scheme 4.
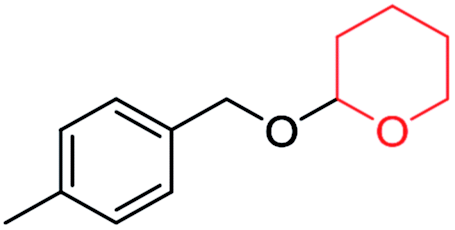


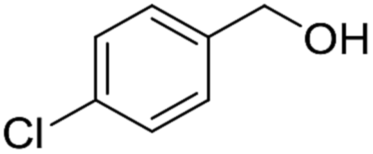
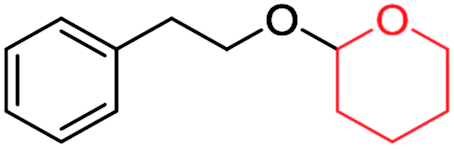
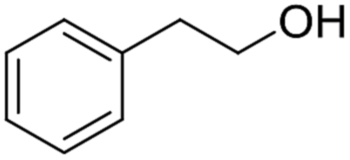

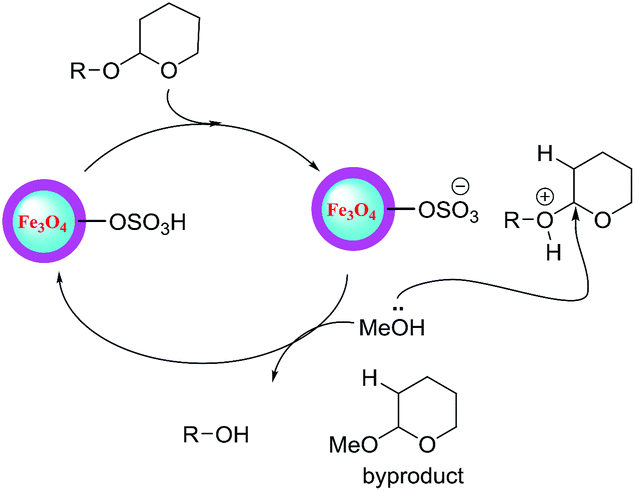 | ||
| Scheme 4 Plausible mechanism for the deprotection/dehydropyranylation of THP ether. | ||
4. Conclusions
Protection/deprotection is one of the most frequently applied synthetic strategies by organic chemists; discovery of more green and efficient heterogeneous catalysts for these reactions has been a fundamental necessity. The tetrahydropyranylation of alcohols using our reported magnetic Fe3O4@silica sulfonic acid nanocatalyst offers a green, mild, less toxic, stable, and solvent-free process of protection/deprotection reaction with easy recoverability of the catalyst. In short, our method is highly economical and environmentally benign.5. Spectral data
5.1 2-((4-Methylbenzyl)oxy)-tetrahydro-2H-pyran (Table 2, entry 2)
1H-NMR (400 MHz, CDCl3, TMS): δ 7.27 (d, J = 8 Hz, 2H), 7.18 (d, J = 8 Hz, 2H), 4.79–4.71 (m, 2H), 4.50 (d, J = 12 Hz, 1H), 3.95–3.92 (m, 1H), 3.58–3.55 (m, 1H), 2.36 (s, 3H), 1.91–1.67 (m, 1H), 1.58–1.54 (m, 5H); 13C-NMR (100 MHz, CDCl3, TMS): δ 137.28, 135.34, 129.16, 128.09, 97.64, 68.27, 62.17, 30.71, 25.64, 21.29, 19.49.5.2 2-(4-Methoxybenzyloxy)-tetrahydro-2H-pyran (Table 2, entry 3)
1H-NMR (400 MHz, CDCl3, TMS): δ 7.26–7.28 (d, J = 8 Hz, 2H), 6.84–6.86 (d, J = 8 Hz, 2H), 4.66–4.71 (m, 2H), 4.43 (d, J = 4 Hz, 1H), 3.90 (t, J = 4 Hz, 1H), 3.74 (s, 3H), 3.53 (t, J = 4 Hz, 1H), 1.49–1.83 (m, 6H); 13C-NMR (100 MHz, CDCl3, TMS): δ 159.24, 130.37, 129.56, 113.81, 97.49, 68.53, 62.11, 55.21, 30.70, 25.61, 25.57, 19.50.5.3 Tetrahydro-2-(phenethyloxy)-2H-pyran (Table 2, entry 6)
1H-NMR (400 MHz, CDCl3, TMS): δ 7.13–7.25 (m, 4H), 4.56 (t, J = 3.6 Hz, 1H), 3.38–3.95 (m, 4H), 2.88 (t, J = 7.2 Hz, 2H), 1.42–1.82 (m, 7H); 13C-NMR (100 MHz, CDCl3, TMS): δ 139.15, 128.76, 126.25, 98.614, 68.291, 62.8, 39.38, 36.446, 30.712, 25.546, 19.63.5.4 2-(2-Ethylhexyloxy)-tetrahydro-2H-pyran (Table 2, entry 7)
1H-NMR (400 MHz, CDCl3, TMS): δ 4.51–4.52 (t, J = 4 Hz, 1H), 3.58–3.81 (m, 2H), 3.18–3.56 (m, 2H), 1.48–1.77 (m, 7H), 1.24–1.29 (m, 8H), 0.82–0.86 (m, 6H); 13C-NMR (100 MHz, CDCl3, TMS): δ 99.04, 98.93, 70.4, 62.08, 39.73, 39.66, 30.79, 30.61, 29.17, 29.13, 25.62, 23.94, 23.14, 19.57, 14.13, 11.17, 11.06.5.5 2-(8-Methylnonyloxy)-tetrahydro-2H-pyran (Table 2, entry 8)
1H-NMR (400 MHz, CDCl3, TMS): δ 4.572 (s, 1H), 3.72–3.88 (m, 2H), 3.49 (d, J = 12 Hz, 2H), 1.40–1.84 (m, 19H), 1.07–1.33 (m, 10H); 13C-NMR (100 MHz, CDCl3, TMS): δ 19.45, 22.74, 25.26, 26.68, 27.14, 27.80, 29.24, 29.81, 30.62, 41.04, 62.13, 67.38, 98.47.5.6 2-(Octyloxy)-tetrahydro-2H-pyran (Table 2, entry 9)
1H-NMR (400 MHz, CDCl3, TMS): δ 4.54 (t, J = 4 Hz, 1H), 3.83–3.70 (m, 1H), 3.69–3.66 (m, 1H), 3.51–3.47 (m, 1H), 3.35–3.32 (m, 1H), 1.78–1.55 (m, 1H), 1.53–1.51 (m, 1H), 1.51–1.48 (m, 6H), 1.32–1.23 (m, 11H), 0.85 (t, J = 8 Hz, 3H); 13C-NMR (100 MHz, CDCl3, TMS): δ 98.87, 67.74, 62.34, 31.91, 30.84, 29.82, 29.53, 26.32, 25.58, 22.73, 19.74, 14.16.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge SERB, New Delhi for financial support (Grant No. SB/FT/CS-103/2013 and SB/EMEQ-076/2014).References
- B. Kumar, M. A. Aga, D. Mukherjee, S. S. Chimni and S. C. Taneja, Tetrahedron Lett., 2009, 50, 6236 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, in Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 2007 Search PubMed.
- D. B. G. Williams, S. B. Simelane, M. Lawton and H. H. Kinfe, Tetrahedron, 2010, 66, 4573 CrossRef CAS.
- M. Miyashita, A. Yoshikoshi and P. A. Grieco, J. Org. Chem., 1977, 42, 3774 CrossRef.
- T. Sato, J. Otera and H. Nozaki, J. Org. Chem., 1990, 55, 4770 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and H. Hazarkhani, Tetrahedron Lett., 2002, 43, 7139 CrossRef CAS.
- S. Kim and W. J. Lee, Synth. Commun., 1986, 16, 659 CrossRef CAS.
- B. Akhlaghinia, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1783 CrossRef CAS.
- M. Yadegari and M. Moghadam, Appl. Organomet. Chem., 2016, 30, 872 CrossRef CAS.
- B. Karimi and M. Khalkhali, J. Mol. Catal. A: Chem., 2005, 232, 113 CrossRef CAS.
- R. A. Sheldon and R. S. Downing, Appl. Catal., A, 1999, 189, 163 CrossRef CAS.
- B. F. G. Johnson, Top. Catal., 2003, 24, 147 CrossRef CAS.
- A. K. Rathi, R. Zboril, R. S. Varma and M. B. Gawande, in Magnetite (Ferrites)-Supported Nano-Catalysts: Sustainable Applications in Organic Transformations, ACS Symposium Series, ACS, Washington DC, 2016, ch. 2, p. 39 Search PubMed.
- L. M. Liz-Marzin and P. Mulvaney, J. Phys. Chem. B, 2003, 107, 7312 CrossRef.
- O. B. Miguel, P. Tartaj, M. P. Morales, P. Bonville, U. G. Schindler, X. Q. Zhao and S. V. Verdaguer, Small, 2006, 2, 1476 CrossRef PubMed.
- B. Dam, A. Kumar Pal and A. Gupta, Synth. Commun., 2016, 46, 275 CrossRef CAS.
- J. Safari and Z. Zarnegar, J. Mol. Catal. A: Chem., 2013, 379, 269 CrossRef CAS.
- A. Rostami, A. Ghorbani-Choghamarani, B. Tahmasbi, F. Sharifi, Y. Navasi and D. Moradi, J. Saudi Chem. Soc., 2017, 21, 399 CrossRef CAS.
- A. R. Kiasat and J. Davarpanah, J. Mol. Catal. A: Chem., 2013, 373, 46 CrossRef CAS.
- H. Naeimi and S. Mohamadabadi, Dalton Trans., 2014, 43, 12967 RSC.
- F. Nemati, M. M. Heravi and R. Saeedirad, Chin. J. Catal., 2012, 33, 1825 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of synthesized compounds. See DOI: 10.1039/c7ra12458a |
| This journal is © The Royal Society of Chemistry 2017 |