
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast synthesis of amides from ethyl salicylate under microwave radiation in a solvent-free system†
Rafaely N. Lima a,
Valdenizia R. Silvab,
Luciano de S. Santosb,
Daniel P. Bezerrab,
Milena B. P. Soaresb and
André L. M. Porto*a
a,
Valdenizia R. Silvab,
Luciano de S. Santosb,
Daniel P. Bezerrab,
Milena B. P. Soaresb and
André L. M. Porto*a
aLaboratório de Química Orgânica e Biocatálise, Instituto de Química de São Carlos, Universidade de São Paulo, Av. João Dagnone, 1100, Ed. Química Ambiental, Santa Angelina, 13563-120, São Carlos, SP, Brazil. E-mail: almporto@iqsc.usp.br; Fax: +55 16 33739952; Tel: +55 16 33738103
bFundação Oswaldo Cruz, Centro de Pesquisas Gonçalo Moniz, R. Waldemar Falcão, 121, Candeal, 40296-710, Salvador – BA, Brazil
First published on 15th December 2017
Abstract
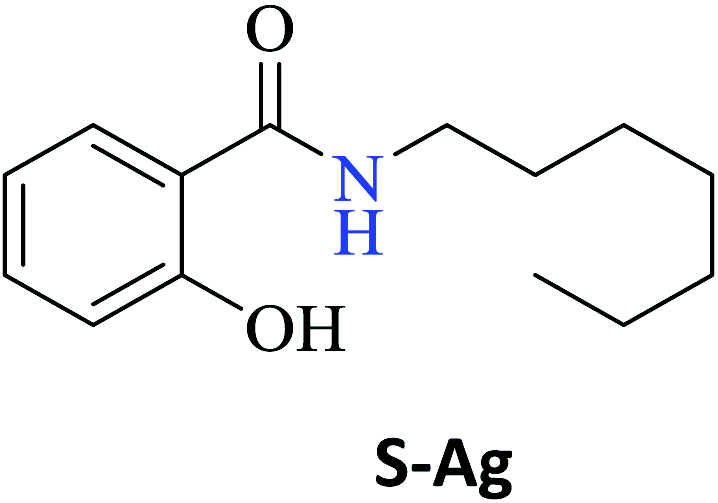
In this study, amide bond formation, one of the most important reactions in organic chemistry, it was evaluated using ethyl salicylate and ten different primary amines. Under the optimized experimental conditions, i.e. 60 °C, hexane, phenylboronic acid-PBA (15 mol%), boric acid-BA (15 mol%) or without catalyst-WC, using a hot-plate for 24 h, amides were obtained in excellent isolated yields (WC, 77–94% to S-Aa-Ad; PBA, 11–94% to S-Ae-Aj; and BA, 28–90% to S-Ae-Aj). The reaction employing CAL-B also permitted a moderate conversion for the production of amides S-Ae-Aj (3–42%). However, in our efforts to reduce the amide synthesis time (24 h), the reactions were performed in the presence of microwave-MW radiation using a free-solvent system [60 °C, PBA (15 mol%) or WC], which reduced the time of the reaction by 32-fold (45 min) and afforded nine amides (S-Aa-Ah and S-Aj) in 80–99% isolated yield and S-Ai in 23% yield. A cytotoxicity assay demonstrated that the amide S-Ag was capable of inhibiting four human tumor cell lines (MCF-7, HCT116, HepG2, and HL-60) with an IC50 ranging from 8.68 to 17.57 μg mL−1. In this study, MW radiation provided an attractive way for faster reactions, improved yields, and cleaner reactions, as well as the synthesis of amide S-Ag with cytotoxic activity.
Introduction
Salicylic acid (SA) is a phenolic compound and member of the β-hydroxy acid family, which is widely found in many plants to induce a disease resistance response.1 It has also been widely used as a key ingredient in many skin-care products for the treatment of acne, wrinkles, warts, exfoliation, and aging;2–4 however, the use of SA in these formulations has been limited due to strong irritation of skin caused by it.5To minimize or improve the activity of SA, novel methodologies or analogues compounds have been developed or synthesized. For example, due to its low solubility in water, microcapsules containing SA have been developed, which show controlled release and improved antibacterial activity against Staphylococcus aureus, Staphylococcus epidermidis, and Propionibacterium acnes.5
Aspirin (acetylsalicylic acid, ASA), an ester product obtained from SA, is a drug used worldwide due to its analgesic, anti-thermal, and anti-inflammatory activities.6,7 Investigations have demonstrated that aspirin plus dipyridamole and individual ASA present antithrombotic activity after ischemic stroke.8,9 Salicylanilides display activity against tuberculosis and cytotoxicity to multi-drug resistant bacteria.10 SA conjugated to hydroquinone (SAH) displayed better activity for the treatment of melasma through chemical peeling as compared to its isolated compounds, and SAH had higher lipophilicity and skin absorption.11
Fatty amide derivatives of SA also show a high affinity for stratum corneum and present safer topical application since they do not metabolize and enter into systemic circulation, accumulating in the uppermost layer of skin.12,13 Lipophilic hydroxy acid derivatives of SA reduced the number of non-inflammatory lesions of comedonal acne by up to 55.6% as compared to salicylic acid, which reduced it by 48.5%;14 SA amides also present antioxidant activity and HIV-1 integrase inhibition,15,16 same as observed with 3-keto SA chalcones, exhibiting HIV-1 integrase inhibition.17
Therefore, SA and its analogues (amides, esters, fatty amides, and fatty acid) present a large number of biological activities and have been largely studied in academic, medicinal, and pharmaceutical areas. Thus, it is important to synthesize amides from SA for applications in biotechnology, agriculture, and medicine according to Green Chemistry principles using facile procedures and eco-friendly methodologies with lower amounts of waste, solvent, and time. Microwave irradiation has become a very usual technique in organic synthesis, improving products yields and decreasing reaction times; moreover, when it is used under solvent-free conditions, it results in cleaner reactions.18–20
Herein, we report a facile, rapid, high yielding, and eco-friendly solvent-free synthesis of amide derivatives from ethyl salicylate under microwave radiation in the absence or presence of lipase obtained from Candida antarctica (CAL-B) and phenylboronic acid or boric acid as catalysts. The amides synthesized were also submitted to an in vitro cytotoxicity assay using four human tumor cells lines (breast adenocarcinoma, colon carcinoma, hepatocellular carcinoma, and promyelocytic leukemia).
Results and discussion
Aminolysis reactions of ethyl salicylate using Candida antarctica lipase and phenylboronic acid
To investigate the synthetic reactions of amides, ethyl salicylate ES (50 mg, 0.30 mmol) and 2-aminoethan-1-ol Aa (0.30 mmol) have been used during the reaction optimization process, and the optimized conditions are as follows: initial temperature at 45 °C, 3 mL of hexane as a solvent, and immobilized commercial CAL-B (50 mg). In this assay, it was observed that CAL-B did not catalyze the aminolysis reaction after 24 h. Upon increasing the amount of amine Aa to 3 equivalents (0.90 mmol), amide S-Aa was produced in 59% yield after 24 h. However, a similar yield (57%) without the use of the enzyme was obtained. These reactions showed that the use of CAL-B did not efficiently catalyze the reaction after 24 h. However, upon prolonging the reaction time to 48 h, 79% conversion was obtained in the presence of CAL-B, whereas 62% conversion was obtained without CAL-B. In this case, the enzyme showed a moderate catalytic effect in the reaction.To optimize the production of amide S-Aa, the reaction was carried out at 45 °C using 3 equivalents of amine Aa (0.90 mmol) while decreasing the amount of solvent to 1 mL of hexane. The conversion obtained in this assay was 74% in the presence of CAL-B (50 mg) and 75% without CAL-B. Similar results were obtained after 48 h when the same conditions were used: the amide S-Aa was obtained with 87% conversion in the presence of CAL-B and with 79% conversion without CAL-B. Again, it was observed that CAL-B had a modest effect on the reaction yield.
In accordance with the previous experiments, the difference in conversion after 24 h or 48 h in the presence or absence of CAL-B was not significant when 3 equivalents of amine (0.90 mmol) and 1 mL of hexane were used. However, optimization of the reaction conditions was significant since it was not necessary to add the enzyme to achieve a high yield of amide S-Aa at 45 °C after 48 h using 3 equiv. of amine and 1 mL of hexane (79%).
In addition, the aminolysis reaction of ES (0.3 mmol) and amine Aa (0.90 mmol) in hexane (1 mL) without CAL-B at 60 °C produced a 93% conversion of S-Aa after 24 h (Table 1). 2-Hydroxy-N-(2-hydroxyethyl)benzamide S-Aa was isolated (92% yield) and characterized using GC-MS, IR, and NMR analysis.
|
||||||
|---|---|---|---|---|---|---|
| T (°C) | Hexane (mL) | Ratio ES![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Aa :Aa |
24 h | 48 h | ||
| c (%) amide S-Aa with CAL-B | c (%) amide S-Aa without CAL-B | c (%) amide S-Aa with CAL-B | c (%) amide S-Aa with CAL-B | |||
| a Performed in duplicate.b Isolated yield. c = conversion determined by the GC-FID analysis. | ||||||
| 45 | 3 | (1:1) |
0 | 0 | — | — |
| 3 | (1:3) |
57 | 59 | 62 | 79 | |
| 1 | (1:1) |
26 | 29 | 41 | 53 | |
| 1 | (1:3) |
75 | 74 | 79 | 87 | |
| 60 | 3 | (1:3) |
59 | 68 | 93 | 94 |
| 1 | (1:3) |
93 (92)b | 94 | |||
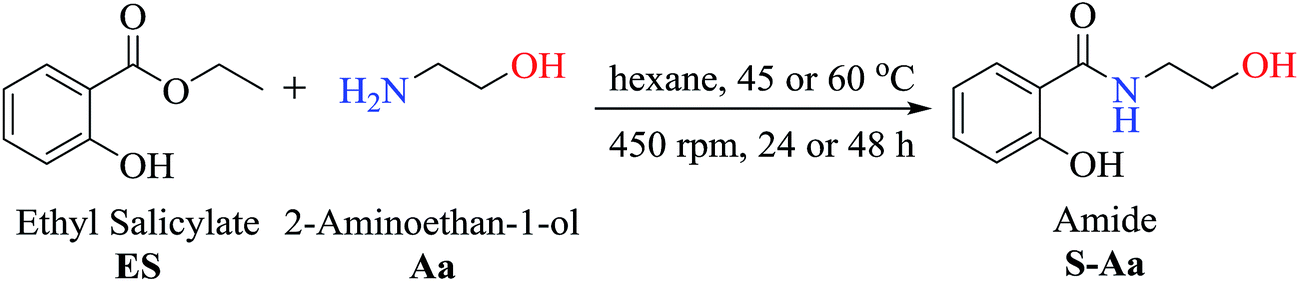
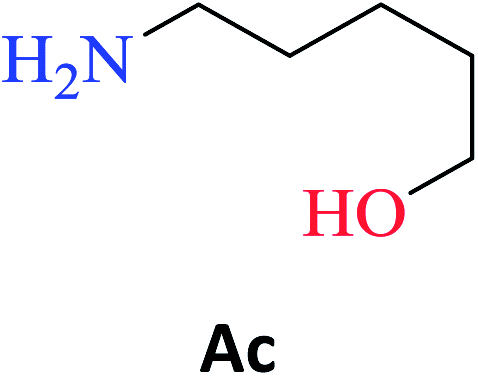
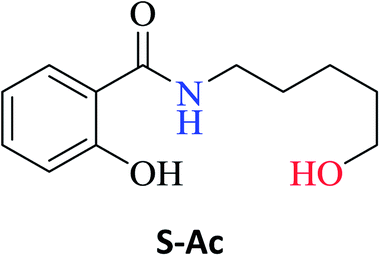
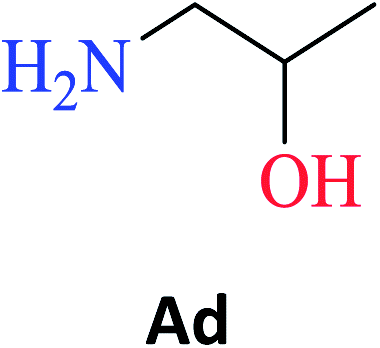
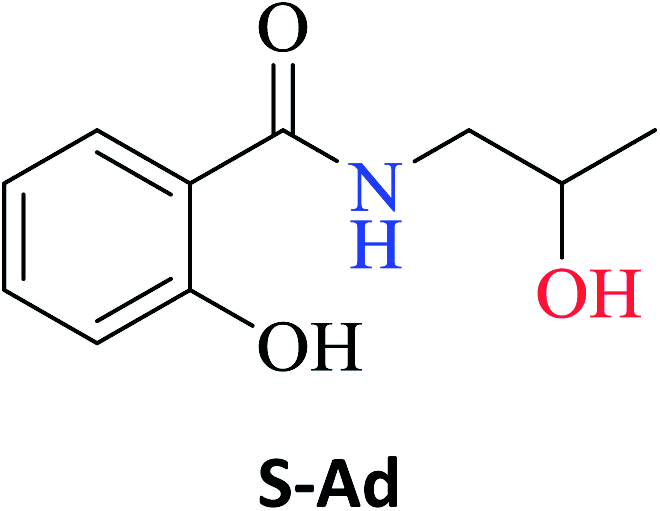
The optimized reaction conditions were used to realize the reaction between ethyl salicylate ES (0.30 mmol) and nine different amines Ab-Aj (0.90 mmol, 60 °C, 1 mL of hexane, 450 rpm, and 24 h). High isolated yields were obtained for 2-hydroxy-N-(3-hydroxypropyl)benzamide S-Ab (94%), 2-hydroxy-N-(5-hydroxypentyl)benzamide S-Ac (84%), and 2-hydroxy-N-(2-hydroxypropyl)benzamide S-Ad (77%), as reported in Table 2.
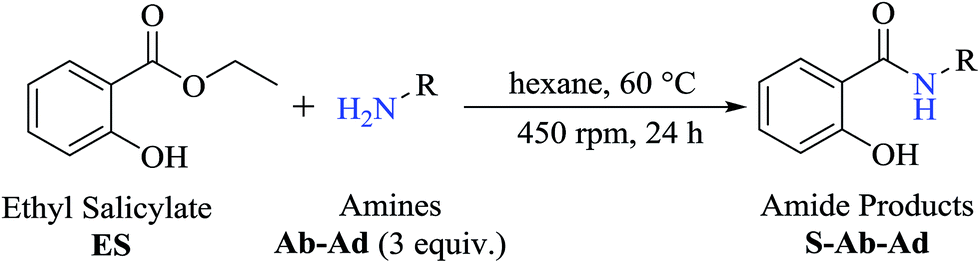
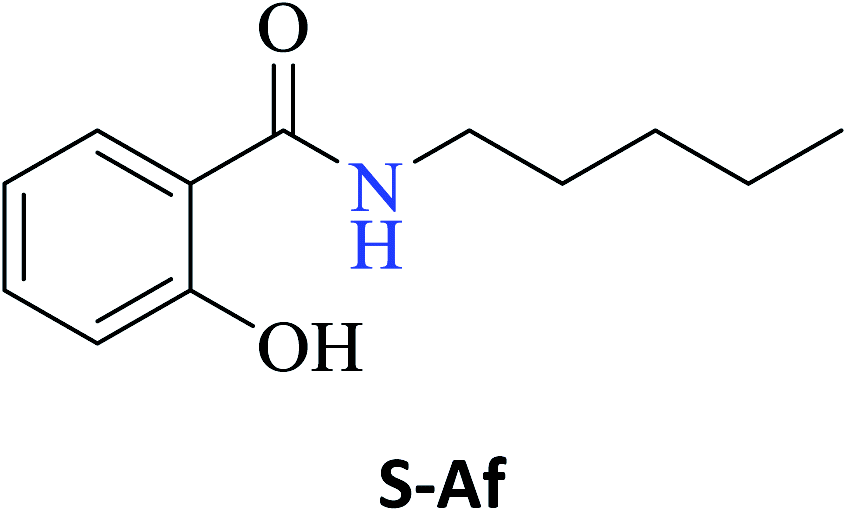
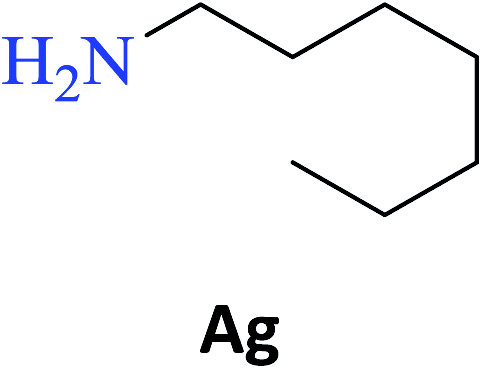
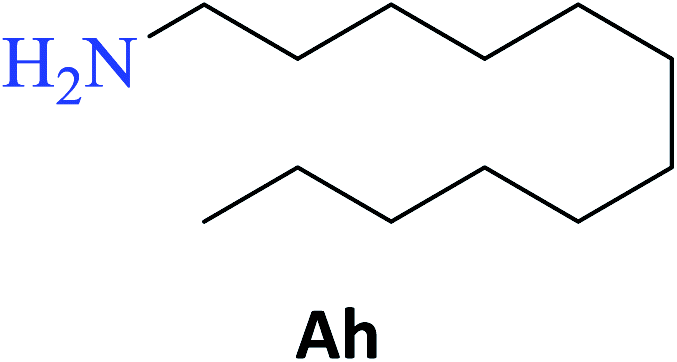
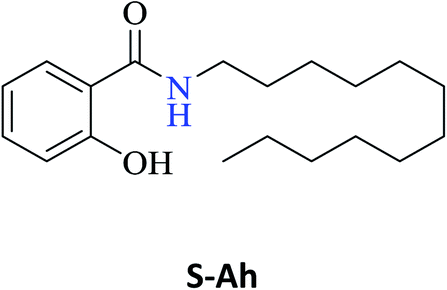
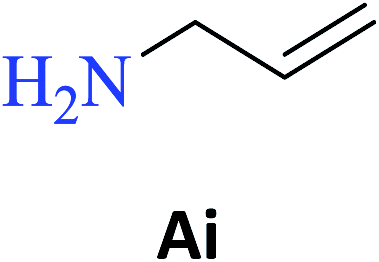
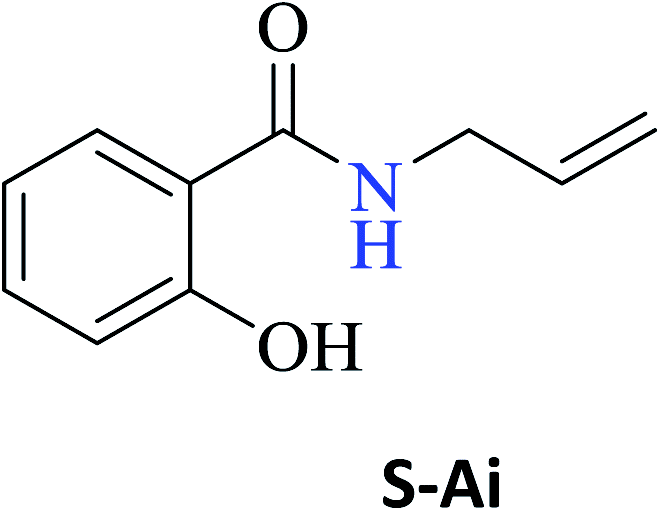
Interestingly, it was observed that for the amides N-butyl-2-hydroxybenzamide S-Ae, 2-hydroxy-N-pentylbenzamide S-Af, N-heptyl-2-hydroxybenzamide S-Ag, N-allyl-2-hydroxybenzamide S-Ai, and N-benzyl-2-hydroxybenzamide S-Aj, the aminolysis reactions did not occur without the use of CAL-B as a catalyst with the exception of amide N-dodecyl-2-hydroxybenzamide S-Ah, which was formed with only 6% conversion.
The reaction conversions (3–42%) observed in the preparation of amides S-Ae to S-Aj using CAL-B showed the moderate catalytic effect of the lipase (Table 3).
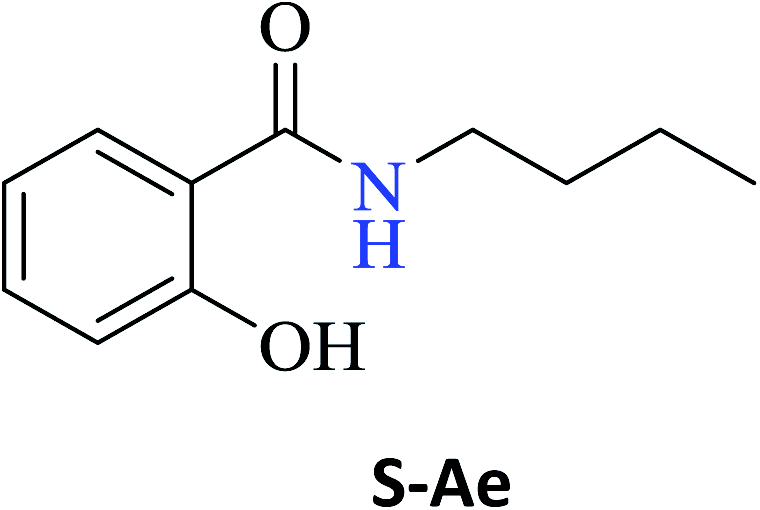
To obtain the amides S-Ae-Aj in better yields, phenylboronic acid (PBA) was used as a catalyst. The reaction to produce S-Ae was carried out using 5 mol%, 10 mol%, and 15 mol% PBA for 24 h. It was observed that 15% PBA was able to catalyze the production of amide S-Ae with 95% conversion (determined by GC-FID), and a 79% isolated yield was obtained via flash column chromatography using silica gel. Lower yields of the amide S-Ae were obtained using 10 mol% PBA (yield = 70%) and 5 mol% PBA (yield = 60%) (Scheme 1). Following the procedure described, the amides S-Ae-Aj were analyzed by GC-FID and isolated, and the yields were obtained (Table 3).
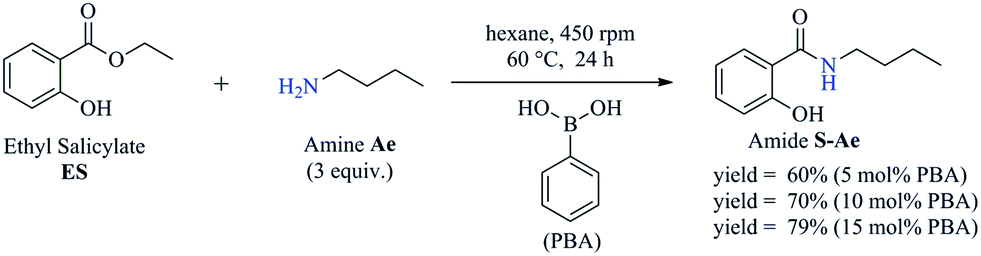 | ||
| Scheme 1 The synthesis of amide S-Ae via the aminolysis reaction of n-butylamine and ES in the presence of PBA as a catalyst. | ||
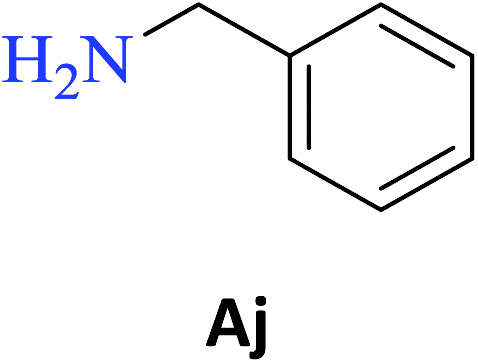
In the presence of PBA, ethyl salicylate, ES, was substantially converted to its respective amides in high isolated yields after 24 h (S-Af, 86%; S–Ag, 91%; and S-Ah, 90%). However, the amides S-Ai and S-Aj were obtained in only 11% and 63% yields, respectively (Table 3).
As phenylboronic acid was observed to be a good catalyst in the aminolysis reactions using ethyl salicylate ES and mono-functionalized amines (Ae-Aj), boric acid (BA, H3BO3), as a cheaper and non-toxic catalyst, was used with the aim to reduce the reaction cost.
Upon carrying out the reaction using BA under the same reaction conditions (60 °C, 15 mol%, 450 rpm, 1 mL of hexane, and 24 h) used with phenylboronic acid, it was observed that by increasing the amine carbon chain (Ae < Af < Ag < Ah), inferior yields were obtained as compared to the case of PBA (S-Ae, 53%; S-Af, 77%; S-Ag, 86%; and S-Ah, 90%).
The observed effect was attributed to the lower solubility of BA in hexane as compared to that of PBA, which reduced the catalytic effect of BA and consequently the production of amides. On the other hand, both catalysts have the same effect on the increased yields, which were observed in the order of S-Ae < S-Af < S-Ag < S-Ah (BA and PBA).
PBA can also infer some π electronic repulsion or interaction with the amines Ai and Aj, which have a similar π-bond component causing a reduction in the amide conversion (S-Ai, 11% and S-Aj, 63%) as compared to BA (S-Ai, 28% and S-Aj, 77%) with the yields being greater than those obtained with PBA. Thus, both catalysts allowed high isolated yields for the synthesis of several amide derivatives of ethyl salicylate ES.
Fig. 1 shows an adapted proposal21–23 for the catalytic cycle using phenylboronic acid as the catalyst to perform the aminolysis reactions between ethyl salicylate ES and the amines Ae-Aj providing the amides S-Ae-Aj, respectively. The catalytic cycle starts with the formation of a cyclic intermediate between PBA and ethyl salicylate ES. This intermediate activates the carbonyl group of ES via hydrogen bonding; this facilitates amine attack, consequent formation of the amide, and regeneration of PBA.
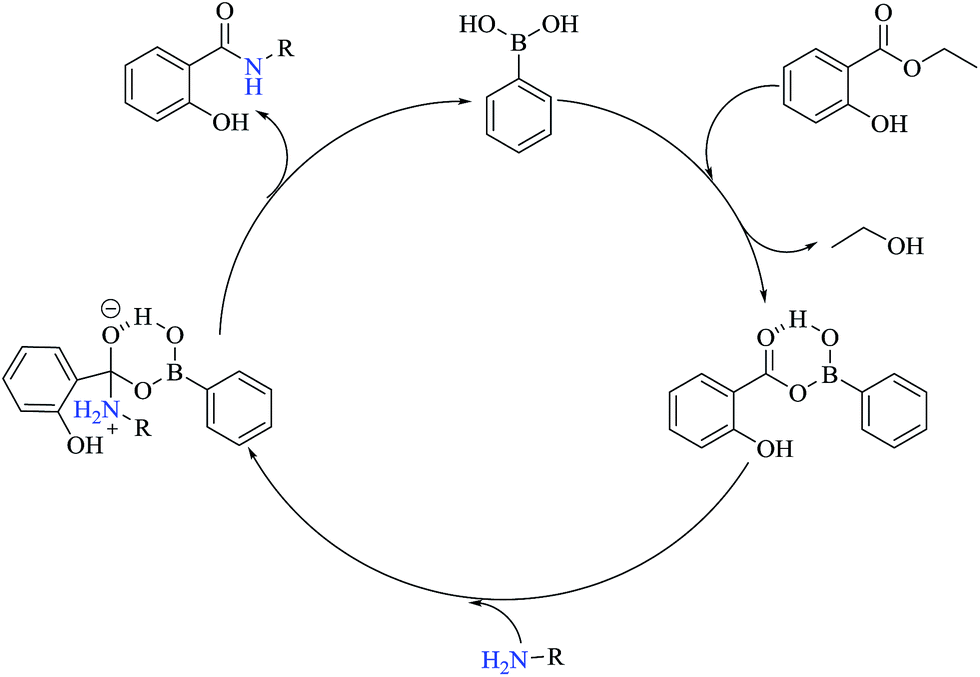 | ||
| Fig. 1 The proposed catalytic cycle of the aminolysis reaction of ethyl salicylate ES with different amines catalyzed by PBA. | ||
As PBA showed good catalytic effect for the production of amides from mono-functionalized amines, it was also used for bi-functionalized amines, Aa-Ad, in the aminolysis reactions with ES. The results showed that PBA was not able to catalyze the reactions for the production of S-Aa (93%), S-Ab (94%), S-Ac (84%), and S-Ad (77%) since the results obtained were very similar to those achieved in the absence of the catalyst after 24 h of reaction. These results can be explained through the intramolecular interaction observed with amino-alcohols between the N–H and O–H functional groups that forms a cyclic conformation, which does not occur with mono-functionalized amines. These interactions can cause steric hindrance and electronic effects that make it difficult for the catalyst to react and accelerate the reaction.24
Aminolysis reactions of ethyl salicylate ES and amines Aa-Aj under microwave radiation
To decrease the reaction time obtained previously using a hot plate at 24 hours, the aminolysis reaction was conducted under microwave radiation to accelerate the reaction. As hexane is the best solvent for the aminolysis reactions with ethyl salicylate ES, as reported in the previous studies and according to the literature,25 it does not present a good effect when applied under MW; thus, the aminolysis reactions in this study have been carried out using a solvent-free system.The reaction under MW using ethyl salicylate ES and 2-aminoethan-2-ol Aa (3 equiv.) in a solvent-free system at 60 °C was followed by thin layer chromatography-CCD, and after 45 min, the ES was totally consumed; this decreased the reaction time determined earlier by conventional heating in hexane (24 h) by 32-fold.
According to the high performance of the aminolysis reaction under MW, the amides S-Aa-Aj were synthesized using this methodology. All products were obtained in excellent yield (80–99%), except for amide S-Ai (23%). The low yield of the amide S-Ai can be explained by the low vapor pressure and boiling point (53 °C) of the amine Ai. When submitted to a system at 60 °C, it can be evaporated into the atmosphere even if the reaction has been performed using a reflux apparatus.
As observed in Table 4, several amides were obtained in high isolated yields (from 80 to 99%) under MW radiation, and the results were slightly higher than those obtained upon heating on a hot-plate (Table 3). The exception was S-Ai (23%) although its yield was double of that obtained with conventional heating on a hot plate (11%).
|
|
|---|---|
| Amide products | Yielda (%) |
| a Isolated yield.b Without catalyst.c 15 mol% PBA. | |
| 2-Hydroxy-N-(2-hydroxyethyl)benzamide (S-Aa) | 99b |
| 2-Hydroxy-N-(3-hydroxypropyl)benzamide (S-Ab) | 91b |
| 2-Hydroxy-N-(5-hydroxypentyl)benzamide (S-Ac) | 87b |
| 2-Hydroxy-N-(2-hydroxypropyl)benzamide (S-Ad) | 92b |
| N-Butyl-2-hydroxybenzamide (S-Ae) | 80c |
| 2-Hydroxy-N-pentylbenzamide (S-Af) | 94c |
| N-Heptyl-2-hydroxybenzamide (S-Ag) | 97c |
| N-Dodecyl-2-hydroxybenzamide (S-Ah) | 97c |
| N-Allyl-2-hydroxybenzamide (S-Ai) | 23c |
| N-Benzyl-2-hydroxybenzamide (S-Aj) | 86c |

Finally, due to the good yields obtained in the aminolysis reaction using the optimized procedure with 3 equivalents of amine under MW radiation, the reaction was investigated using 1 equivalent of amine Aa. In this case, the reaction to produce S-Aa was then scaled up to 1 mmol and carried out at 100 °C for 45 min and 2 h. Under these conditions, S-Aa was obtained in 75% and 80% yields. Therefore, it was concluded that even upon increasing the temperature (100 °C) and prolonging the reaction time to 2 h, the yield did not increase (only 5%); this confirmed the need to use an excess amount of amine to achieve a better reaction performance and yield.
MW irradiation has been used for the aminolysis reactions without the use of an organic solvent, and good isolated yields have been obtained; however, these methodologies still employ tosyl chloride as the condensing agent26 or high temperatures (150 °C),18,27 which results in a less eco-friendly methodology.
Others previous studies have described the synthesis of the amide S-Aa from methyl salicylate without the use of an organic solvent at high temperatures (210–215 °C).28 A similar procedure was also used to produce the amides S-Aa, S-Ab, and S-Ac at 170 °C.29,30 An one-pot system using 2-chlorobenzoyl chloride and dichloromethane (step-one, 5 h) and KOH, CuI, 1,10-phenanthroline, and water at 100 °C (step-two, 10 h) also produced the amide S-Aa.31
Amide S-Ae was previously obtained using a solution of phenol in toluene, BCl3, and n-butylisocyanate after 12 h of reaction.32 The amide S-Af was synthetized using N-butyl-o-iodobenzamide, LiOH, CuBr, and 1,10-phenanthroline in DMSO at 100 °C after 12 h.33 The amide S-Aj was synthetized using salicyloyl chloride in dichloromethane at 0 °C and TEA (t.a., overnight).34
Greener conditions were applied to produce the amide S-Ag using salicylic acid and 10% CAL-B at 75 °C (18 h) and amide S-Ah using ethyl salicylate and 5 g mol−1 of CAL-B without the use of a solvent at 65 °C (20 h).12,33 The amide S-Ai was obtained from salicylic acid using silica gel 60 (230–400 mesh) under microwave radiation (200 W, 130 °C, and 20 min).36 As observed, most of these reactions required temperatures between 100 °C and 215 °C, time in the range from 12 to 20 h, complex catalysts, and chloride or iodine reagents. Milder, greener, and faster reactions were obtained using CAL-B (65–75 °C, 18–20 h)12,33 and MW irradiation (200 W, 130 °C, 20 min).36 These conditions are superior (temperature or time) when compared with those of the procedure reported by us in this study (17 W, 60 °C, 15 mol% PBA or BA, without a solvent, and 45 min).
Antitumor activity
Table 5 shows the IC50 data obtained for the amide series S-Aa-Aj synthesized from ethyl salicylate. As shown in Table 5, only amide S-Ag exhibited good cytotoxic activity for the four-tumor cell lines (MCF-7, HCT116, HepG2, and HL-60) investigated herein. The best IC50 observed was obtained for MCF-7 cells (8.68 μg mL−1). The amide S-Ag was also able to inhibit HepG2, HL-60, and HCT116 cells with the IC50 values of 15.56 μg mL−1, 16.30 μg mL−1, and 17.57 μg mL−1, respectively. The other amides presented IC50 values above 25 μg mL−1 and thus were not considered to have a promising cytotoxic activity.37,38| Amides | Human tumor cell lines | |||
|---|---|---|---|---|
| MCF7 (μg mL−1) | HCT116 (μg mL−1) | HepG2 (μg mL−1) | HL-60 (μg mL−1) | |
| a MCF7 (human breast adenocarcinoma), HCT116 (human colon carcinoma), HepG2 (human hepatocellular carcinoma), and HL-60 (human promyelocytic leukemia). Doxorubicin (DOX) was used as a positive control. ND = not determined. | ||||
| S-Aa | >25 | >25 | >25 | >25 |
| S-Ab | >25 | >25 | >25 | >25 |
| S-Ac | >25 | >25 | >25 | >25 |
| S-Ad | ND | >25 | >25 | >25 |
| S-Ae | >25 | >25 | >25 | >25 |
| S-Af | >25 | >25 | >25 | >25 |
| S-Ag | 8.68 | 17.57 | 15.56 | 16.30 |
| S-Ah | >25 | >25 | >25 | >25 |
| S-Ai | >25 | >25 | >25 | >25 |
| S-Aj | ND | >25 | >25 | >25 |
| DOX | 1.78 | 0.11 | 0.07 | 0.05 |
Conclusion
After employing three different catalysts for the synthesis of ten amides S-Aa-Aj from ethyl salicylate ES, it was observed that CAL-B showed a moderate effect for catalyzing the aminolysis reactions (3–42%), especially for the mono-functionalized amines Ae-Aj using a hot-plate and hexane as a solvent. The reactions with phenylboronic acid (63–91%; S-Ae-Ah, S-Aj) and boric acid (53–90%; S-Ae-Ah, S-Aj) as catalysts resulted in high isolated yields, except for S-Ai (11% and 23%, respectively). The use of microwave radiation in a solvent-free system allowed the preparation of amides in higher yields (80–99%; S-Aa-Ah, S-Aj) than those obtained using a hot plate (63–94%; S-Aa-Ah, S-Aj), and a 32-fold reduction in the reaction time as compared to the case of conventional heating (24 h) was observed. Via the results obtained, it was possible to verify the great technological potential of microwave radiation to improve the reaction time in a solvent-free system and the capacity of the catalysts based on boron to improve the synthesis of amides, e.g. amide S-Ag that presented cytotoxic activity in the presence of four tumor cell lines.Experimental
Material and methods
Lipase obtained from Candida antarctica (CAL-B), expressed in Aspergillus niger and immobilized on acrylic resin, deuterated chloroform (CDCl3-99.8% with 0.5% TMS), deuterated methanol (CD3OD, 99.9%), 2-aminoethan-2-ol Aa (≥99%), 3-aminopropan-1-ol Ab (99%), 5-aminopentan-1-ol Ac (95%), DL-1-aminopropan-2-ol Ad (≥90%), butan-1-amine Ae (99%), pentan-1-amine Af (≥99%), heptan-1-amine Ag (97%), dodecan-1-amine Ah (98%), prop-2-en-1-amine Ai (99%), phenylmethanamine Aj (≥99%), ethyl salicylate (99%), and phenylboronic acid (95%) and boric acid (99.5%) were purchased from Sigma Aldrich. Hexane was purchased from Synth, ethyl acetate (EtOAc) was obtained from NEON, and silica gel C18 on TLC (Thin Layer Chromatography) was obtained from ALUGRAM® XTra SIL G/UV245. All reagents were used without further purification.GC-FID analysis of the aminolysis reactions
The analyses of the aminolysis reactions were performed using a gas chromatograph (GC2010, Shimadzu Corporation, Japan) equipped with an AOC-20i (Shimadzu) automatic injection sampler and flame ionization detector (FID). The ionization process was carried out using nitrogen gas (30 mL min−1) and the flame formed from hydrogen 5.0 (40 mL min−1) and synthetic air (400 mL min−1). The separations were performed on a fused silica capillary column (DB-5MS 5%-phenyl–95%-dimethylpolysiloxane 30 m × 0.25 mm internal diameter, 0.25 mm film thickness) in a stream of nitrogen 5.0 of 0.95 mL min−1. The injector temperature was 230 °C, and 1.0 μL of the sample was used. The oven temperature setting started from 100 °C and increased at 7 °C min−1 until 300 °C, and this temperature was maintained for 5 min. The total time of the analysis was 33.57 min.Biocatalytic synthesis of the amide S-Aa
In a 5 mL vial, ethyl salicylate ES (50 mg, 0.30 mmol), 2-aminoethan-1-ol Aa (0.30 or 0.90 mmol), Candida antarctica lipase (50 mg), and hexane (1 or 3 mL) were added. The vial was kept under magnetic stirring (450 rpm) on a hot-plate (IKA®/RCT basic) at 45 °C or 60 °C. The samples (200 μL) were obtained, diluted with hexane in a 5 mL volumetric flask, and analyzed by GC-FID after 24 and 48 hours. The yield was calculated based on an analytical curve (R2 = 0.9952) obtained by the GC-FID analysis of a stock solution (670 ppm, 33.5 mg/50 mL) and dilutions (570, 470, 370, 170, 70, and 28.7 ppm) (ESI†). Reactions without CAL-B were also performed and analyzed by GC-FID.Synthesis of the amides S-Aa-Ad
In a 5 mL vial, ethyl salicylate ES (50 mg, 0.30 mmol), amine Aa-Ad (0.90 mmol), and hexane (1 mL) were added. The reaction mixtures were kept under magnetic stirring (450 rpm) on a hot-plate (IKA®/RCT basic) at 60 °C for 24 h. To minimize the solvent cost, the amides were not subjected to an extraction process. After the reaction, the amides were isolated using preparative thin layer chromatography. For the amides S-Aa-Ad, 90% EtOAc and 10% hexane were used as the eluent for the isolation and purification of the products.Synthesis of the amides S-Ae-Aj using phenylboronic acid and boric acid as catalysts
In a 5 mL flask, ethyl salicylate ES (50 mg, 0.30 mmol), butan-1-amine Ae (0.90 mmol), hexane (1 mL), and the organic catalyst phenylboronic acid (PBA, 5 mol%, 10 mol% and 15 mol%) or boric acid (BA, 15 mol%) were added, and the reaction was conducted at 60 °C for 24 h. After the purification process, the best aminolysis reaction condition was applied to 6 different amines (Ae-Aj). After the reaction, the amides were isolated using thin layer chromatography. For the amides S-Ai and S-Aj, a mixture of 90% hexane and 10% EtOAc was used as the eluent, and for S-Ae-Ah, a mixture of 80% hexane and 20% EtOAc was used as the eluent. The control reactions did not detect production of the amides S-Ae-Aj.Synthesis of the amides S-Aa to S-Ad under microwave radiation without the use of a catalyst and using phenylboronic acid for the amides S-Ae to S-Aj
In a 5 mL round-bottomed flask, ES (50 mg, 0.30 mmol) and the amines Aa-Ad (0.90 mmol) were added. For the S-Ae-Aj synthesis, ES, the amines Ae to Aj, and 15 mol% of PBA were used. For the production of the amides S-Aa-Ad, the use of a catalyst was not necessary. The reaction was carried out without the addition of an organic solvent and subjected to microwave radiation (17 W, 60 °C, 45 min). To avoid the use of excess amine, the reactions were also carried out using ES and the amine Aa in equivalent proportions (1 mmol) at 100 °C for 45 min and 2 h.Gas chromatography-mass spectrometry (GC-MS)
The reactions analyses were conducted using a gas chromatograph (GC-MS-QP2010 Ultra Shimadzu Corporation, Japan) equipped with an auto-sampler injection AOC-20i (Shimadzu). An electron capture detector was used with the electronic impact of 70 eV, a scanning speed of 1.666, and a scan interval of 0.30 fragments per s; the fragments were detected from 40 to 500 Da. The separations were performed using a fused silica capillary column (DB-5MS 5% phenyl–95%–dimethylpolysiloxane, 30 m × 0.25 mm internal diameter, and 0.25 mm film thickness) in a stream of helium 5.0 at 0.95 mL min−1. The injector temperature, oven temperature, rate increase, amount of sample, and total time analysis were the same as presented in the GC-FID analysis (see the ESI†).Fourier transform infrared (FTIR) analysis
FTIR spectra were obtained using a Shimadzu IRAffinity-1 spectrometer. The analyses were performed using a thin film formed by the application of diluted samples (CHCl3) on silicon plates. The transmittance was expressed in cm−1 for the bands observed between 4000 and 400 cm−1.Nuclear magnetic resonance (NMR)
NMR spectra were obtained using an Agilent apparatus operated at 500 MHz for 1H and 126 MHz for 13C. The samples were solubilized in CDCl3 or CD3OD, and the chemical shifts were expressed in ppm relative to an internal standard, TMS. Coupling constants (J) are reported in hertz (Hz). The analyses were conducted using 15–20 mg of isolated amides. The acquisition time for 1H analysis was 4.0894 s at a sweep width of 8012.82 Hz, and for 13C analysis, the acquisition time was 1.0486 s at a sweep width of 31250.00 Hz.
Cytotoxicity assay
The samples (S-Aa-Aj) were diluted in sterile and pure DMSO (5 mg mL−1) and tested at concentrations ranging from 0.19 to 25 μg mL−1. To evaluate the cytotoxicity of the amides S-Aa-Aj, an Alamar blue test was performed after 72 h of exposure with the test substances.Alamar blue, identified as resazurin,40 is a fluorescent/colorimetric indicator with redox properties. As a tetrazolium salt, Alamar blue is reduced in proliferating cells. Its oxidized form is blue (non-fluorescent/non-viable cell), and the reduced form is pink (fluorescent/viable cell). The reduction of Alamar blue reflects cell proliferation. It was initially used to indicate cell growth and/or viability in monitoring lymphocyte proliferation,41 and currently has several applications. Initially, the cells were plated in 96-well plates (100 μL per well of a solution of 0.3 × 106 cells per mL for cells in suspension and 0.7 × 105 cells per mL for adhered cells). After 24 hours of incubation, amides S-Aa-Aj (previously dissolved in DMSO) were added to the human tumor cells lines and incubated at 37 °C for 72 h. Doxorubicin was used as a positive control. The negative control received the same amount of DMSO. Then, four hours (twenty-four hours for PBMC) before the end of the incubation period, 20 μL of a stock solution (0.312 mg mL−1) of Alamar blue (resazurin) was added to each well. The absorbance was measured at the wavelength of 570 nm (reduced) and 595 nm (oxidized) using a plate reader.41 The percentage of inhibition was calculated, the percentage of inhibition × log of the concentration was calculated, and its IC50 was determined from the non-linear regression using the Prisma version 5.0 program (GraphPad Software).
Cells
Tumor cells, MCF-7 (human breast adenocarcinoma), HCT116 (human colon carcinoma), HepG2 (human hepatocellular carcinoma), and HL-60 (human promyelocytic leukemia), were obtained from the American Type Culture Collection (ATCC)42 and used in the assays. The tumor cells were cultured in cell culture bottles (75 cm3, 250 mL volume) in media comprising RPMI 1640 and supplemented with 10% fetal bovine serum. The tumor cells were maintained in incubators under a 5% CO2 atmosphere at 37 °C. Cellular growth was monitored daily using an inversion microscope. The medium was changed whenever the cell growth reached the necessary confluence for nutrient renewal. For maintenance of the adhered cells, trypsin (0.25%) was used for the cells to escape from the walls of the bottles. Cell cultures showed microplasma negatives, as judged by placement with Hoechst (Mycoplasma Stain Kit, Cat. MYC1, Sigma-Aldrich, St. Louis, MO, USA).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
RNL is thankful for the fellowship provided by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). ALMP thanks the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/Proc. 301987/2013-0) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP/Proc. 2014/18257-0) for financial support.References
- C. M. Pieterse and L. C. van Loon, Trends Plant Sci., 1999, 4, 52–58 CrossRef PubMed
.
- E. Merinville, A. J. Byrne, A. V. Rawlings, A. J. Muggleton and A. C. Laloeuf, J. Cosmet. Dermatol., 2010, 9, 174–184 CrossRef PubMed
- H.-S. Lee and I.-H. Kim, Dermatol. Surg., 2003, 29, 1196–1199 Search PubMed
- J. A. Zeichner, Journal of Clinical and Aesthetic Dermatology, 2016, 9, 40–43 Search PubMed
- Z. Yang, Y. Fang and H. Ji, Chin. J. Chem. Eng., 2016, 24, 421–426 CrossRef CAS
- Aspirina® – Aspirina, http://www.aspirina.com.br/pt/produtos/aspirina/, (accessed February 22, 2017).
- R. Menegatti, C. Alberto and M. Fraga, Quim. Nova Esc., 2001, 3, 16–22 Search PubMed
- T. Esprit and S. Group, Lancet, 2006, 367, 1665–1673 CrossRef
- H. C. Diener, L. Cunha, C. Forbes, J. Sivenius, P. Smets and A. Lowenthal, J. Neurol. Sci., 1996, 143, 1–13 CrossRef CAS PubMed
- G. Paraskevopoulos, S. Monteiro, R. Vosátka, M. Krátký, L. Navrátilová, F. Trejtnar, J. Stolaříková and J. Vinšová, Bioorg. Med. Chem., 2017, 25, 1524–1532 CrossRef CAS PubMed
- P. W. Hsieh, I. A. Aljuffali, C. L. Fang, S. H. Chang and J. Y. Fang, J. Dermatol. Sci., 2014, 76, 120–131 CrossRef CAS PubMed
- A. M. Mouad, D. Taupin, L. Lehr, F. Yvergnaux and A. L. M. Porto, J. Mol. Catal. B: Enzym., 2016, 126, 64–68 CrossRef CAS
- Y. D. Yan, J. H. Sung, D. W. Lee, J. S. Kim, E. M. Jeon, D. D. Kim, D. W. Kim, J. O. Kim, M. G. Piao, D. X. Li, C. S. Yong and H. G. Choi, Int. J. Pharm., 2011, 419, 154–160 CrossRef CAS PubMed
- A. Levesque, I. Hamzavi, S. Seite, A. Rougier and R. Bissonnette, J. Cosmet. Dermatol., 2011, 10, 174–178 CrossRef PubMed
- N. M. Storozhok, N. P. Medyanik, A. P. Krysin, S. A. Krekov and V. E. Borisenko, Russ. J. Org. Chem., 2013, 49, 1031–1034 CrossRef CAS
- N. M. Storozhok, N. P. Medyanik, P. Krysin, I. P. Pozdnyakov and S. Krekov, Kinet. Catal., 2012, 53, 162–171 CrossRef CAS
- H. Sharma, S. Patil, T. W. Sanchez, N. Neamati, R. F. Schinazi and J. K. Buolamwini, Bioorg. Med. Chem., 2011, 19, 2030–2045 CrossRef CAS PubMed
- L. Perreux, A. Loupy and F. Volatron, Tetrahedron, 2002, 58, 2155–2162 CrossRef CAS
- R. S. Varma and K. P. Naicker, Tetrahedron Lett., 1999, 40, 6177–6180 CrossRef CAS
- L. Perreux, A. Loupy and M. Delmotte, Tetrahedron, 2003, 59, 2185–2189 CrossRef CAS
- F. Yun, C. Cheng, J. Zhang, J. Li, X. Liu, R. Xie, P. Tang and Q. Yuan, Synthesis, 2017, 49, 1583–1596 CrossRef CAS
- K. Ishihara and Y. Lu, Chem. Sci., 2016, 7, 1276–1280 RSC
- G. Arce, G. Carrau, A. Bellomo and D. Gonzalez, World J. Chem. Educ., 2015, 3, 27–29 CrossRef CAS
- R. N. Lima and A. L. M. Porto, Catal. Commun., 2017, 100, 157–163 CrossRef CAS
- A. M. Sanseverino, Quim. Nova, 2002, 25, 660–667 CrossRef
- A. Khalafi-Nezhad, A. Parhami, M. N. S. Rad and A. Zarea, Tetrahedron Lett., 2005, 46, 6879–6882 CrossRef CAS
- L. Perreux, A. Loupy and M. Delmotte, Tetrahedron, 2003, 59, 2185–2189 CrossRef CAS
- A. P. Phillips and R. Baltzly, J. Am. Chem. Soc., 1947, 69, 200–204 CrossRef CAS PubMed
- D. S. C. Black and M. J. Wade, Aust. J. Chem., 1972, 25, 1797–1810 CrossRef CAS
- F. Benedini, G. Bertolini, R. Cereda, G. Donà, G. Gromo, S. Levi, J. Mizrahi and A. Sala, J. Med. Chem., 1995, 38, 130–136 CrossRef CAS PubMed
- S. J. Balkrishna and S. Kumar, Synthesis, 2012, 44, 1417–1426 CrossRef CAS
- O. Piccolo, L. Filippini, L. Tinucci, E. Valoti and A. Citterio, Tetrahedron, 1986, 885, 885–891 CrossRef
- X. Chen, W. Hao and Y. Liu, Org. Biomol. Chem., 2017, 3, 1–28 Search PubMed
- Y. A. Ibrahim, H. Behbehani, M. R. Ibrahim and R. N. Malhas, Tetrahedron, 2003, 59, 7273–7282 CrossRef CAS
- P. Kaushik, N. A. Shakil, J. Kumar and B. B. Singh, Synth. Commun., 2015, 45, 579–587 CAS
- A. Ojeda-porras, A. Hernández-santana and D. Gamba, Green Chem., 2015, 5, 1–44 CrossRef
- M. Suffness and J. M. Pezzuto, in Methods in Plant Biochemistry: Assays for Bioactivity, ed. K. Hostettmann, Academic Press, London, 1990, pp. 71–133 Search PubMed
- J. Boik, Natural compounds in cancer therapy, Oregon Medical Press, Minnesota, USA, 2001 Search PubMed
- A. M. Mouad, D. Taupin, L. Lehr, F. Yvergnaux and A. L. M. Porto, J. Mol. Catal. B: Enzym., 2016, 126, 64–68 CrossRef CAS
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef
- S. Ansar Ahmed, R. M. Gogal and J. E. Walsh, J. Immunol. Methods, 1994, 170, 211–224 CrossRef CAS
- U. Boulevard and V. 20110 U. Manassas, American Type Culture Collection (ATCC), https://www.atcc.org/.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopic spectra, GC-MS analysis, mass spectra and infrared analysis. See DOI: 10.1039/c7ra11434f |
| This journal is © The Royal Society of Chemistry 2017 |