
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step purification and oriented attachment of protein A on silica and graphene oxide nanoparticles using sortase-mediated immobilization
Seyed Mehdi Qafariab,
Gholamreza Ahmadian*a and
Mehdi Mohammadi *b
*b
aSystems Biotechnology Department, Institute of Industrial and Environmental Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran. E-mail: ahmadian@nigeb.ac.ir; Fax: +98 21 44787399; Tel: +98 21 44787143
bBioprocess Engineering Department, Institute of Industrial and Environmental Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), P. O. Box: 14965/161, Tehran, Iran. E-mail: M.mohammadi@nigeb.ac.ir; Fax: +98 21 44787399; Tel: +98 21 44580461
First published on 12th December 2017
Abstract
Site-specific immobilization of proteins is a major challenge due to the large number of nucleophilic groups on the surface of polypeptides. Here we present the use of the specificity of the sortase A transpeptidation reaction to covalent and oriented attachment of protein A as a model protein through the short C-terminal linker region on chemically modified silica and graphene oxide (GO) nanoparticles. Two types of nucleophile donor groups (–(glycine)5–NH2 and –CH2–CH2–NH2) were introduced to the surface of both carriers and successful functionalization were confirmed by FT-IR, TG and XRD analysis. Immobilization of protein A with LPxTG domain was performed with mild conditions providing single step purification and immobilization of the target protein. To address the effects of nucleophile and carrier on the functional properties of the immobilized protein, biological activity tests were conducted by the use of the immobilized preparations in Human Immunoglobulin G (IgG) purification followed by SDS-PAGE analysis. We found that amine nucleophile of ethylenediamine on the surface of GO nanoparticles is more effective than pentaglycine which is widely reported as a good substrate for sortase-mediated ligation.
1. Introduction
One of the four major classes of macromolecules in organisms is proteins. Each kind of protein has its own unique sequence of amino acids and three-dimensional structure. Therefore, they usually act with high efficiency and high specificity. Regarding these high-valued characteristics, they can be utilized in biotechnological processes both on the laboratory and industrial scales.1 Despite the great advantages of using proteins in biotechnological processes, they have a high cost of isolation and purification from the reaction media. Immobilization of proteins on a solid support is an efficient way in order to make these processes more cost effective as the immobilized proteins can be easily recovered and used in the continues rounds of the reaction.2 Protein functions normally depend on immobilization approach which can be classified into five main groups: adsorption,3 covalent bonding,4 entrapment,5 polymerization6 and encapsulation.7 Among them, covalent attachment of proteins on a solid carrier is the most robust approach which is usually preferred when leaching of protein from the support is the main concern.8,9 From other point of view, protein immobilization can be categorized in random and oriented/site-directed immobilization.10 In random immobilization, proteins are bonded through side chain amino groups to the surface of carrier from different sites, thus exposing various orientation of the immobilized protein to the surrounding environment.11 Therefore, the protein native function can be affected by this unpredictable orientation. Although this non-specific covalent linkage can be used for the cases with relaxed orientation requirements, in the case where homogeneity is needed, these methods may not be suitable for protein immobilization. To bring negative effect of immobilization down, several researches report the use of oriented immobilization of proteins on activated supports.12 In these methods, proteins are bonded to the functional groups of a carrier from a unique site, providing identical orientation of proteins to environment. In fact, oriented immobilization of proteins via a particular amino acid permits proteins to be arranged in a more controlled style. Furthermore, as the reaction site of protein-carrier linkage is selectable, immobilization has very little or no effect on native functions of the target protein.13 There are several methodologies to perform oriented immobilization which can be categorized into two main groups.In the first approach, a proper sequence is introduced into the structure of target protein e.g. capture protein. The capture part (e.g. cutinase and avidin) fused to target protein, can recognize its motif on carrier.14–16 Oriented immobilization by intein is the other example of this category in which intein splits into two part, C-intein and N-intein. One of this two part is fussed to target protein and other one is bonded to carrier. When C-intein and N-intein come together, intein is activated and self-spliced with out-coming intein lead to immobilization of target protein on carrier.17 The second group of oriented immobilization is based on introducing a single amino acid in a spatial site of the target protein and subsequent process for immobilization of this protein from the selected site.18–22
For example, Shi and co-workers have reported site directed immobilization of testis-specific protease 50 by introducing azide groups into its structure followed by the use of click chemistry approach for immobilization of the protein on alkyne activated carrier.20 Staudinger ligation, has been also used for site directed immobilization of ribonuclease S′. In this methodology, un-natural azide groups of a genetically modified protein have been used to react with phosphine moieties of a solid carrier.23 Diels–Alder cycloaddition, thiol–ene additions and oxime reaction are the other approaches that are frequently used for the oriented immobilization of proteins on properly activated supports.24,25
Chemoenzymatic approach for covalent immobilization is another technique that can be used for site specific conjugation of proteins on solid supports. This methodology is based on utilization of the inherent chemoselectivity of enzymes. In this context, Sortase A (SrtA) is a high selective and well documented example that catalyzes a transpeptidation reaction known as “sortagging”. SrtA as a cysteine transpeptidase enzyme (EC 3.4.22.70) naturally located at the plasma membrane and facilitates covalently anchoring a variety of proteins to the cell wall.26
SrtA recognizes substrates with LPxTG domain and catalyzes the cleavage of the amide bond between the threonine (T) and glycine (G). The rest of protein from glycine till the end of the protein is released in environment and acyl–enzyme intermediate (thioester) is created through cysteine 184 (active site).27–29 The free carboxyl group of the thioester intermediate then undergoes nucleophilic attack by an amino group of a multiglycine substrate (e.g. pentaglycine in S. aureus) and the enzyme is recycled for the next round of reaction. SrtA has high specificity for LPxTG sequence and the free amino group of the multiglycine substrate. However, it has been well-documented that all natural amino acid residues except Cys and Trp can be used as the amino group donor.30 Recent studies have shown that SrtA with potent substrate specificity provides a robust and gentle approach for the oriented and covalent immobilization of proteins.31
In this regard, Raeeszadeh-Sarmazdeh and co-workers have attached GFP and mCherry on the gold surface by sortase-mediated immobilization.32 In other report, a fibronectin-binding protein has been immobilized on CM5 chips by this methodology. Immobilization of T-cell adapter protein on agarose beads with the aim of SrtA has also been reported.33
In this research, we took the advantage of ability of S. aureus to oriented immobilization of Protein A (SpA) as a model protein. Silica and graphene nanoparticles after functionalization with two different linkers were used as solid supports for immobilization of the model protein. Sortase-mediated immobilization also permitted a single step purification and immobilization of protein A. To the best of our knowledge this is the first report on oriented immobilization of a protein by using SrtA on the surface of silica and graphene nano-sheets. SpA is a virulence factor which is located in S. aureus cell wall and is able to bind Fc immunoglobulins (spatially IgG) of the most of mammalians.34 Therefore, it can be used for the purification of immunoglobulin or Fc conjugated protein from serum or other proteins. Furthermore, the presence of LPxTG motif in its structure makes it suitable to use as a model protein for the sortase-mediated immobilization on different carriers.
2. Material and methods
2.1. Materials
Pentaglycine was from CHEM-IMPEX INT'L INC COMPANY. Sodium bicarbonate, Fmoc (N-hydroxysuccinimide ester), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and piperidine were from sigma. N-[3-(Trimethoxysilyl)propyl]ethylenediamine, diethylamide and silica gel (70–230 mesh, 63–200 μm) were purchased from Merck. Graphene oxide nano particles was obtained from Neutrino Port. Co (5–18 nm, IP ≃ 1.6). Other reagents and solvents were of analytical or HPLC grade. PCR amplification was carried out with a thermal cycler (PTC-150 Mini Cycler (MJResearch)). Fourier transform infrared spectra (FT-IR) were recorded on a Bomen FT-IR-MB-series instrument with a KBr pellet technique. Thermogravimetry and differential thermal analysis (TGA and DTA) were carried out from 10 °C to 800 °C at a heating rate of 20 °C min−1 in air atmosphere using a STA 503M system from Bahr GmbH, Germany. XRD analysis was carried out 5–80° with step size 2°, time/step: 20 s at room temperature and atmosphere using X-ray Diffraction X'Pert PRO MPD, Poland.2.2. Methods
Plasmid pET-26-SpA4 containing just 5 IgG-binding domain of SpA gene, previously cloned and was supplemented from NIGEB collection. The plasmid was transformed into E. coli BL21 DE3 by heat shock methods. It was expressed and purified as the same as SrtAΔ59 procedure; except that culture media contain 50 μg mL−1 Kanamycin as a selection marker.
The resulting silica–(Gly)5–Fmoc particles were collected by filtration and washed thoroughly with acetone. Then, the silica–(Gly)5–Fmoc particles were added to 100 mL of 20% piperidine and incubated for 30 min. Unprotected silica–pentaglycine particles were filtered and washed twice with distilled water and stored at 4 °C.
3. Results and discussion
3.1. Preparation of modified silica gel
It is well-documented that in the sortase-mediated strategies, the highest performance obtains when the amine nucleophile was bonded to at least one sp3 carbon atom (–CH2–NH2).28 To provide this condition for sortase-mediated immobilization of SpA, two different functionalization methods were used (Fig. 1). In the first approach, the amine group was introduced to the surface of silica by the reaction of hydroxyl group of silica surface and N-[3-(trimethoxysilyl)propyl]ethylenediamine. In this way, the surface of carrier is modified with amine moieties having the needed domain. | ||
| Fig. 1 A schematic illustration of chemical modification of silica particles for oriented immobilization of protein A. | ||
In the second approach, the amine functional groups were subjected to further modification with pentaglycine through EDC-based coupling chemistry. To prevent tandem reactions between two molecules of the pentaglycine, at first, the amine group of pentaglycine was protected with Fmoc and subsequent activation of carboxylic groups of the protected pentaglycine was performed with EDC. The protected N-terminal amine groups of modified silica particles were finally deprotected in the presence of piperidine.
The successful functionalization and modification of silica particles was monitored by FT-IR spectroscopy and thermal gravimetric analysis. The FT-IR spectrum of all chemically modified samples shows 2800–3000 C–H band which is related to the stretching vibration band of –CH2 bonds in alkyl chains of newly introduced functional groups (Fig. 2).
 | ||
| Fig. 2 FT-IR analysis of the (a) derivatives of GO nanoparticles (b) derivatives of silica. | ||
The band at 462.8 cm−1 indicates Si–O–Si or O–Si–O bending modes and the bands at 802 and 1105 cm−1 are assigned to the Si–O–Si symmetric stretching mode. The band around 3420 cm−1 revealed the existence of hydroxyl groups (O–H) even after modification of the surface. The absorption band around 1637 cm−1 in all the spectra also reveals the presence of amino group (N–H) on the surface. The vibration band around 1402 cm−1 reveals the in-plane bending vibration of CH2 which arises from newly introduced moieties. Furthermore, amide I band at 1600–1700 cm−1 and amide II band at 1565–1525 cm−1 could be identified in all derivatives of silica particles.
The presence of functional groups on the surface of silica after modification was also investigated by TGA. Fig. 3 shows TGA diagram of aminated silica. The result exhibits that the amine-modified carrier is thermally stable up to 250 °C. The small weight loss at around 100 °C can be attributed to the removing of physically adsorbed water molecules. The large weight loss of around 9%; starting from 250 °C, is due to the decomposition of amine moieties from silica surface. The result of TG analysis of silica–(Gly)5 displays almost a similar pattern with that observed for silica–NH2.
 | ||
| Fig. 3 TG analysis of silica particles after modification. | ||
In TGA diagram of silica–(Gly)5 (Fig. 3), it can be observed a large weight loss of around 4% in the range of 300–600 °C associated with removing of the introduced linker from the surface. A small weight loss of 0.1% is also observed for this sample corresponding to water removal from the solid. The immobilized derivative of SpA on silica–(Gly)5 was also subjected to TG analysis. Lower thermal stability is observed for this derivative compared to both silica–NH2 and silica–(Gly)5. As Fig. 3 shows, a large weight loss of 7% is started from 150 °C which can be attributed to removing of the protein molecules and also introduced linker from the surface of carrier.
3.2. Preparation of modified GO nanoparticles
In order to have amine functional groups on the surface of graphene oxide and provide –CH2–NH2 domain for sortase-mediated immobilization of SpA, two different methodologies were applied (Fig. 4).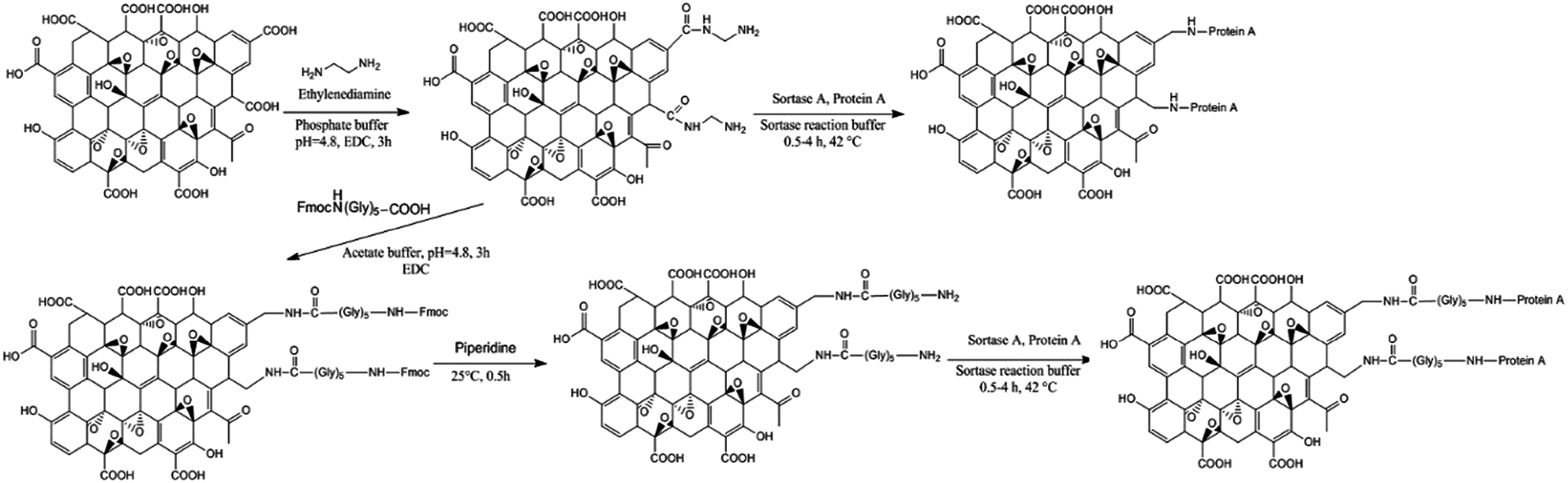 | ||
| Fig. 4 A schematic illustration of chemical modification of GO nanoparticles for oriented immobilization of protein A. | ||
Amidation of carboxylic acid groups of graphene oxide by using dimethylamine was the first approach. This linker as a non-amino acid nucleophile can provide the needed domain to increase the performance of SrtA. Further modification of the introduced amine groups by pentaglycine produced new –CH2–NH2 domain on the surface of graphene oxide. Similar to silica, the EDC based coupling chemistry was used in this approach. FT-IR spectroscopy and TGA/DTA were used to analyze the obtained modified supports in order to confirm the successful functionalization. Fig. 2 displays the FT-IR spectra of GO–NH2 and GO–(Gly)5 in which the samples represent almost a similar pattern. In both spectra, an intense peak is observed at 1755–1760 cm−1 which can be ascribed to the presence of carbonyl groups after chemical modification.39
The weak bands around 1585 cm−1 can be attributed to the bending vibration of remaining O–H and epoxide groups after modification. The weak vibration bands at around 1475 cm−1 may be attributed to the skeletal vibration of the graphene sheets. Moreover, an additional intense peak at around 1075 cm−1 corresponds to C–O band.
In addition, newly appeared peaks at 2850–2950 cm−1 belong to the asymmetric and symmetric stretching vibration of CH2 groups. The intense peaks at around 3400 cm−1 in the spectrum of both samples can be attributed to stretching vibration of O–H and amine groups. Compared with bare graphene nanoparticles, all these bands related with the containing functional groups and proving successful functionalization of the carrier.
Modification of graphene surface was also investigated by TGA/DTA. The thermogravimetric curves of graphene oxide and its functionalized forms were depicted in Fig. 5. The GO nanoparticles exhibit two steps of weight loss at 100 °C and 190 °C. An endothermal peak at around 100 °C with 11% weight loss should be attributed to the removing of physically adsorbed water. When the temperature reached 190 °C it shows an intense exothermic peak with a large weight loss of around 81% in the TGA curve, due to the irreversible removal of oxygen-containing groups and carbon oxidation.40 The amine-modified GO powders show almost similar TGA curve compared to the bare graphene oxide. Two degradation steps are observed for this derivative. Initially, a small endothermic peak with 11% weight loss starting at about 70 °C which belongs to the loss of adsorbed water, followed by another endothermic steep step (weight loss 27%) at 180–230 °C, which is assigned to the thermal elimination of functional groups. This curve does not show any significant weight loss up to 250 °C representing improved thermal stability of GO nanoparticles after modification with amine groups. Thermal analysis of GO nanoparticles modified with pentaglycine moieties has almost similar pattern compared to that observed for amine-modified GO. The GO–(Gly)5 is thermally more stable than the un-modified GO and losses weight in three stages. The first weight loss (8%) is observed at 80 °C with the corresponding endothermic peak in DTA curve due to evaporation of interstitial water molecules.
 | ||
| Fig. 5 TGA diagrams of derivatives of GO nanoparticles. | ||
The other two endothermic peaks with the large mass loss of 24% are observed at 140–250 °C and 300–400 °C which are coming from the decomposition of hydroxyl, carboxyl and pentaglycine moieties of the GO surface.
The GO particles before and after functionalization with amine and pentaglycine moieties were also characterized using XRD analysis. The XRD patterns of GO samples give a distinguishable peak centered at 2θ = 10.9°, corresponding to the (002) inter-planar spacing of 0.84 nm.41,42 As Fig. 6 shows, the XRD profile of GO nanoparticles after immobilization of protein A is not changed, suggesting that the crystalline structure of the support remained almost unchanged after protein attachment.
 | ||
| Fig. 6 XRD analysis of GO nanoparticles after modification and immobilization of SpA. | ||
3.3. Expression of sortase A Δ59 and protein A
To produce SrtAΔ59, the SrtA gene from the Staphylococcus Aureus (ATCC35556D) was cloned into the pQE-30 plasmids. Recombinant expression in Escherichia coli BL21 DE3 cells successfully produced the sortase A lacking 59 N-terminal amino acids which contain His-tag. SrtAΔ59 was purified by affinity chromatography. The expression and purification of SrtAΔ59 was confirmed by SDS-PAGE analysis (Fig. 7).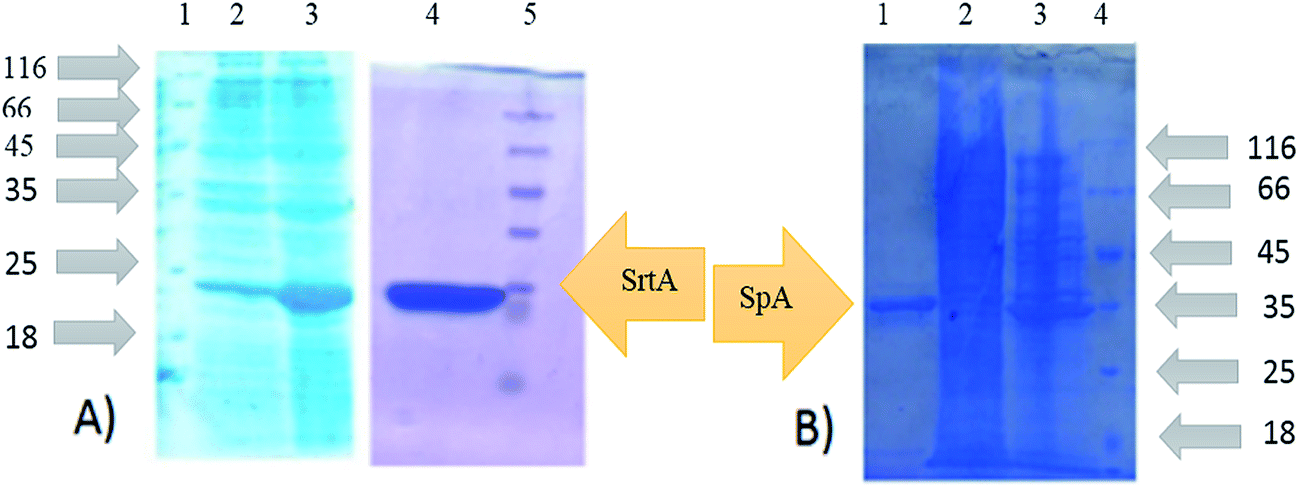 | ||
| Fig. 7 Expression and purification of recombinant SrtA and protein A, (A) lane 2: Srt AΔ59 negative control for expression, lane 3: Srt AΔ59 expression, lane 4: purified Srt AΔ59, lane 1 and 5: protein marker. (B) Lane 1: purified SpA, lane 2: SpA expression, lane 3: SpA control expression, lane 4: protein marker. | ||
Protein A as a surface protein is originally found in the cell wall of the bacterium S. aureus. SpA has about 42 KDa molecular weight and is encoded by the SpA gene and its regulation is controlled by DNA topology, cellular osmolality, and a two-component system called ArlS–ArlR. For the expression and purification of protein A, plasmid pET-26-SpA4 was transformed in E. coli BL21 DE3 followed by purification by affinity chromatography. Expression and purification of SpA was also analyzed by SDS-PAGE (Fig. 7).
3.4. Immobilization of protein A on modified silica and graphene supports
We selected two different compounds as carrier to investigate the performance of SrtA in immobilization process. For this, silica particles with a hydrophilic surface and GO nanoparticles with more hydrophobic nature were used as model carriers. Furthermore, for functionalization and introducing nucleophiles to the surface of these supports two distinct compounds were used both having amine functional group at the end point of their structures. On the other hand, SpA was used as a model protein to immobilize on the surface of the selected carriers. The used protein naturally has LPxTG domain on its structure and was used for immobilization without further modification. The target protein could be immobilized on the carrier directly after breaking bacterial cell and separation of cell debris of lysis cell solutions. In this way, the immobilization of target protein is performed from threonine of LPxTG motif which means that the target protein is immobilized from C-terminal.29 However, Williamson and co-workers have reported labeling of N-terminal of proteins that confirm the potential application of SrtA approach to covalent immobilization of proteins via N-terminal.43 A schematic illustration of C-terminal oriented-immobilization of protein A on modified silica particles using the sortase-mediated technique is shown in Fig. 1. Immobilization of the target protein on both derivatives of silica was performed at 42 °C and pH 8.2. This condition of reaction was applied according to the previous reports on optimum activity of SrtA.44 The results showed that in both carriers, SrtA is able to simultaneously purify and immobilize SpA from the crude extract after up to 4 h of incubation. The short immobilization time and using relatively mild condition for immobilization is the other advantage of enzymatic attachment of SpA on the solid carrier which diminishes possibility of protein denaturation during the process. The composition of crude protein before and after incubation also showed that almost all the SpA is captured from the crude extract by SrtA while non-target proteins are remained in the solution.In traditional covalent immobilization, to obtain higher protein loading and volumetric activity, the proteins need to be purified before immobilization. Recently with increasing the use of genetic engineering to produce recombinant proteins, a large number of studies have been reported to link protein purification with its immobilization process.45 Most of them are based on preparation of heterofunctional supports in which one functional group on the support provides initial adsorption of the protein followed by covalent attachment of the adsorbed protein by the other remaining functional groups. The other strategy to couple immobilization with purification is to immobilize proteins on a previously immobilized anti-target protein. Monoclonal or polyclonal antibodies are normally used in this procedure and permits an extremely selective protein immobilization.45 The results of our study confirm that sortase-mediated ligation as a highly selective immobilization process provides one-step purification and covalent immobilization of proteins for overcoming the problems derived from protein purification (time-consuming, high cost, inactivation, etc.). The used approach also provides site-specific immobilization of proteins on solid surfaces exclusively from the LPxTG domain while the nonspecific covalent attachment procedure targeting nucleophile groups of several amino acids. Nonspecific process, therefore enables immobilization of proteins from many sites of the protein surface in which immobilization reaction at or near the active site can damage the function of the immobilized protein. Up to now several methods and strategies have been reported for site-directed immobilization of proteins mainly based on introducing a sequence tag into the target protein. The use of sortase ligation, however, is recently reported to apply for oriented immobilization to improve functional properties of proteins.46
Another immobilization experiment was set up by the use of purified protein A for determination of immobilization yield on both derivatives. Silica–(Gly)5 showed higher capacity for protein immobilization by loading 7.8 mg of the purified protein on 1 g of its surface which is 2.7 times higher than the loading capacity of 1 g silica–NH2 (2.9 mg of protein A). This result is in good agreement with previously published results in sortase-mediated immobilization of proteins in which pentaglycine-conjugated supports has been explained as the most suitable domain to be recognized by SrtA.47 In fact, observation of maximal activity for pentaglycine is unsurprising since the Gram positive cell wall has naturally pentaglycine bridges for SrtA conjugation.
Immobilization of the crude extract of SpA and its purified form on amine and pentaglycine functionalized derivatives of graphene oxide was also carried out by sortase-mediated approach (Fig. 4). Both derivatives showed proper capacity in purification of SpA from the crude extract. The leaching experiment showed covalent attachment of the purified protein on the surface of GO–NH2 and GO–(Gly)5.
Our investigations also showed that opposed to the results obtained from silica, the GO–NH2 derivative is more efficient compared to GO–(Gly)5 in sortase-mediated immobilization process by loading 124 mg of SpA on 1 g of this carrier. This is almost 1.4 fold higher than the loading capacity of GO–(Gly)5 which could immobilize only 89.9 mg of protein A at the same condition. This may be related to more hydrophobic nature of GO–NH2 compared to GO–(Gly)5 which can create SrtA microenvironment more similar to the native environment of this enzyme. This observation is also against previous investigations in which pentaglycine has been introduced as the most effective nucleophile for sortase-mediated immobilization.28,47 The ability to reduce the nucleophile to a non-amino acid one (ethylenediamine) is of value due to elimination of pentaglycine preparation and reduction in the number of processes needed for functionalization of the support, thus decreasing the total cost of immobilization process.
3.5. Investigation of the functional properties of the immobilized derivatives
To address the efficiency of the used immobilization strategy and investigate the effect of nucleophile and carrier on the functional properties of the immobilized protein, biological activity test was conducted by the use of the immobilized preparations in Human Immunoglobulin G (IgG) purification followed by SDS-PAGE analysis. SpA reversibly binds to the Fc region of IgG through interaction with the heavy chain of this protein. It should be mentioned that SpA only removes the intact form of IgG from human serum through binding to its Fc region, without changing the structure of IgG. The structure alters when there is an S–S bond breaking agent, such as 2ME or DTT, present in the buffers as like as loading buffer, in which heavy chain and light chain of IgG are separated.Fig. 8 shows the results of SDS-PAGE analysis of IgG purification by the same amounts of silica–NH2 and silica–(Gly)5. The results showed that 30 mg of silica–(Gly)5–SpA was able to purify 0.9 mg of IgG, while 0.6 mg of IgG was purified by 30 mg of silica–NH2–SpA at the same condition. The higher efficiency of silica–(Gly)5 compared to silica–NH2 proves that altering the nucleophile on the surface can change the loading capacity of the support.
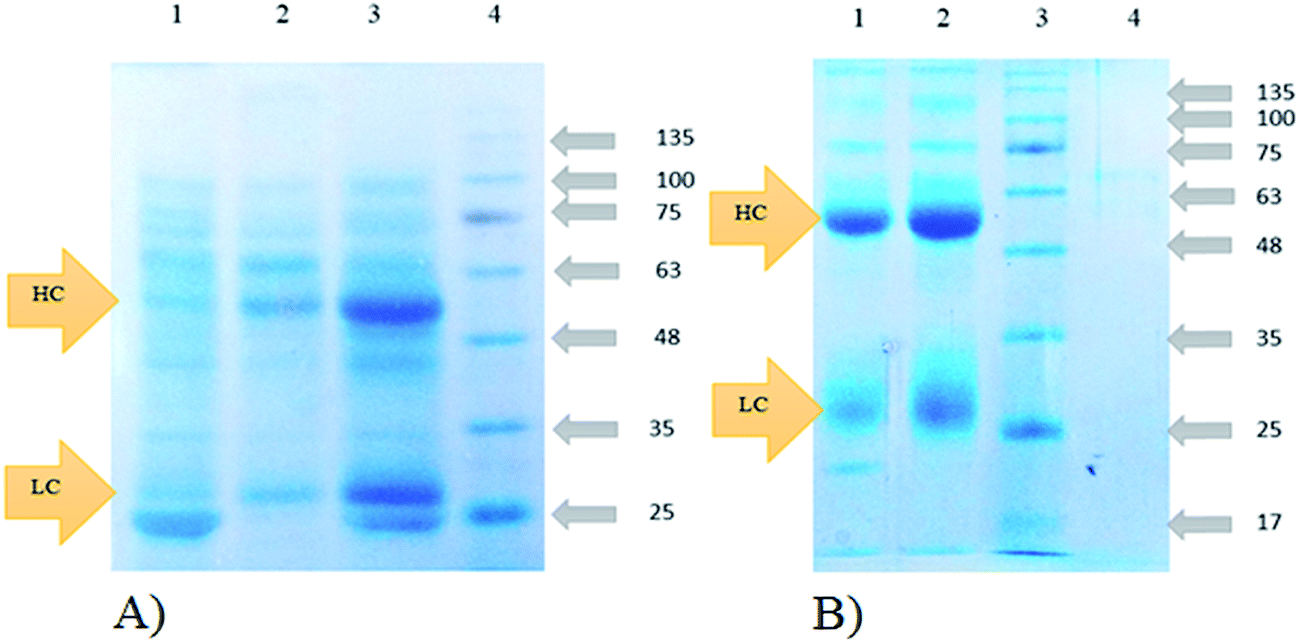 | ||
| Fig. 8 Functional assay of IgG purification from human serum (A) lane 1: un-modified silica as control, lane 2: silica–NH2–SpA, lane 3: silica–(Gly)5–SpA lane 4: protein marker. (B) Lane 1: GO–(Gly)5–SpA, lane 2: GO–NH2–SpA, lane 3: protein marker, lane 4: un-modified GO nanoparticles as control (HC: Heavy Chain, LC: Light Chain). | ||
The same purification experiment was performed by immobilized derivatives of GO nanoparticles.
As can be seen from Fig. 8, contrary to the results obtained from silica derivatives, Human IgG purification by immobilized GO preparations shows higher immobilization efficiency for GO–NH2 compared to GO–(Gly)5. While 10 mg of GO–NH2–SpA purifies 1.7 mg of IgG, GO–(Gly)5–SpA could purify 1.1 mg of IgG from the same amount of human serum. This means that the IgG concentration after purification by GO–NH2–SpA is almost 1.5 times more than the purified IgG by using the GO–(Gly)5–SpA at the same condition. It can be attributed to the higher loading capacity of GO–NH2 in immobilization process which increases the number of SpA molecules on its surface.
3.6. Reusability of the immobilized derivatives
For large-scale applications of an immobilized protein and ensuring the covalent linkage of protein on the solid support, reusability is crucial in terms of process economy. Therefore, the performance of the selected immobilized derivative of each carrier (GO–NH2–SpA and silica–(Gly)5–SpA) was examined by their repeatedly use in the purification of IgG from human serum (Fig. 9). For this, GO–NH2–SpA and silica–(Gly)5–SpA were used for six consecutive cycles, with sequential reactions separated by centrifugation and re-suspension. | ||
| Fig. 9 Reusability of immobilized derivative of silica–(Gly)5–SpA after 6 cycles in IgG purification. | ||
As can be seen from Fig. 9 for silica-no obvious loss of purification efficiency was observed after six cycles, further proving covalent binding of SpA on the support. Almost the same results were obtained for GO–NH2–SpA in terms of reusability and confirming the covalent attachment of SpA on this support.
4. Conclusion
We here present the C-terminal oriented attachment of protein A on amine and pentaglycine functionalized silica and GO nanoparticles by using sortase-mediated immobilization strategy. Simple producing SrtA in large amounts makes this method very cost effective. Furthermore, the site-directed coupling allows proteins to be orientated uniformly on carrier surface in such a way that the immobilized proteins retain their functional properties compared to proteins immobilized by random immobilization. The covalent coupling of proteins in this strategy permits for more stringent washing steps compared to proteins adsorbed on a surface with physical adsorption. This method also has the advantage of one step purification and immobilization of the target protein in a mild condition allowing short cuts in protein purification protocols. Our investigations showed that functionalization of silica particles with pentaglycine and subsequent immobilization of SpA on its surface produces immobilized derivative with higher functional properties compared to the derivative obtained from immobilization of SpA on amine-functionalized silica. On the other hand, amine-functionalized GO nanoparticles showed higher efficiency both in immobilization of SpA and purification of IgG from human serum compared to GO–(Gly)5. Comparison of the results obtained from silica derivatives with those obtained from GO nanoparticles clearly confirmed that not only the kind of amine nucleophile on the support but also the type of used carrier can affect the immobilization process thus altering the functional properties of the final product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for the project was provided by the National Institute of Genetic Engineering and Biotechnology (NIGEB) of Iran (Project no. 940901-I-537).References
- Z. Chen and A.-P. Zeng, Curr. Opin. Biotechnol., 2016, 42, 198–205 CrossRef CAS PubMed.
- M. Babaki, M. Yousefi, Z. Habibi, J. Brask and M. Mohammadi, Biochem. Eng. J., 2015, 101, 23–31 CrossRef CAS.
- M. Garmroodi, M. Mohammadi, A. Ramazani, M. Ashjari, J. Mohammadi, B. Sabour and M. Yousefi, Int. J. Biol. Macromol., 2016, 86, 208–215 CrossRef CAS PubMed.
- M. Mohammadi, M. Ashjari, S. Dezvarei, M. Yousefi, M. Babaki and J. Mohammadi, RSC Adv., 2015, 5, 32698–32705 RSC.
- R. I. Corona-González, R. Miramontes-Murillo, E. Arriola-Guevara, G. Guatemala-Morales, G. Toriz and C. Pelayo-Ortiz, Bioresour. Technol., 2014, 164, 113–118 CrossRef PubMed.
- A. Alghunaim, E. T. Brink and B.-m. Z. Newby, Polymer, 2016, 101, 139–150 CrossRef CAS.
- I. E. Moreno-Cortez, J. Romero-García, V. González-González, D. I. García-Gutierrez, M. A. Garza-Navarro and R. Cruz-Silva, Mater. Sci. Eng., C, 2015, 52, 306–314 CrossRef CAS PubMed.
- M. R. Mehrasbi, J. Mohammadi, M. Peyda and M. Mohammadi, Renewable Energy, 2017, 101, 593–602 CrossRef CAS.
- M. Mohammadi, S. Gandomkar, Z. Habibi and M. Yousefi, RSC Adv., 2016, 6, 52838–52849 RSC.
- Y. Liu and J. Yu, Microchim. Acta, 2016, 183, 1–19 CrossRef CAS.
- M. Mohammadi, Z. Habibi, S. Dezvarei, M. Yousefi, S. Samadi and M. Ashjari, Process Biochem., 2014, 49, 1314–1323 CrossRef CAS.
- Y. Ding, R. Cui, M. Hu, S. Li, Q. Zhai and Y. Jiang, J. Mater. Sci., 2017, 52, 10001–10012 CrossRef CAS.
- J. C. Y. Wu, C. H. Hutchings, M. J. Lindsay, C. J. Werner and B. C. Bundy, J. Biotechnol., 2015, 193, 83–90 CrossRef CAS PubMed.
- Y. Kwon, M. A. Coleman and J. A. Camarero, Angew. Chem., Int. Ed., 2006, 45, 1726–1729 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- B. T. Houseman, J. H. Huh, S. J. Kron and M. Mrksich, Nat. Biotechnol., 2002, 20, 270–274 CrossRef CAS PubMed.
- S. Dedola, S. A. Nepogodiev and R. A. Field, Org. Biomol. Chem., 2007, 5, 1006–1017 CAS.
- P. Jonkheijm, D. Weinrich, M. Köhn, H. Engelkamp, P. Christianen, J. Kuhlmann, J. C. Maan, D. Nüsse, H. Schroeder and R. Wacker, Angew. Chem., 2008, 120, 4493–4496 CrossRef.
- E. Steen Redeker, D. T. Ta, D. Cortens, B. Billen, W. Guedens and P. Adriaensens, Bioconjugate Chem., 2013, 24, 1761–1777 CrossRef CAS PubMed.
- Q. Shi, X. Chen, T. Lu and X. Jing, Biomaterials, 2008, 29, 1118–1126 CrossRef CAS PubMed.
- M. B. Soellner, K. A. Dickson, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 11790–11791 CrossRef CAS PubMed.
- D. Guan, Y. Kurra, W. Liu and Z. Chen, Chem. Commun., 2015, 51, 2522–2525 RSC.
- L. A. Marraffini, A. C. DeDent and O. Schneewind, Microbiol. Mol. Biol. Rev., 2006, 70, 192–221 CrossRef CAS PubMed.
- M. Buhl, B. Vonhören and B. J. Ravoo, Bioconjugate Chem., 2015, 26, 1017–1020 CrossRef CAS PubMed.
- A. D. de Araújo, J. M. Palomo, J. Cramer, O. Seitz, K. Alexandrov and H. Waldmann, Chem.–Eur. J., 2006, 12, 6095–6109 CrossRef PubMed.
- S. Tsukiji and T. Nagamune, ChemBioChem, 2009, 10, 787–798 CrossRef CAS PubMed.
- T. Ito, R. Sadamoto, K. Naruchi, H. Togame, H. Takemoto, H. Kondo and S.-I. Nishimura, Biochemistry, 2010, 49, 2604–2614 CrossRef CAS PubMed.
- S. Samantaray, U. Marathe, S. Dasgupta, V. K. Nandicoori and R. P. Roy, J. Am. Chem. Soc., 2008, 130, 2132–2133 CrossRef CAS PubMed.
- H. Ton-That, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424–12429 CrossRef CAS.
- R. G. Kruger, B. Otvos, B. A. Frankel, M. Bentley, P. Dostal and D. G. McCafferty, Biochemistry, 2004, 43, 1541–1551 CrossRef CAS PubMed.
- T. Matsumoto, T. Tanaka and A. Kondo, Langmuir, 2012, 28, 3553–3557 CrossRef CAS PubMed.
- M. Raeeszadeh-Sarmazdeh, R. Parthasarathy and E. T. Boder, Colloids Surf., B, 2015, 128, 457–463 CrossRef CAS PubMed.
- B. Kuropka, N. Royla, C. Freund and E. Krause, Proteomics, 2015, 15, 1230–1234 CrossRef CAS PubMed.
- S. Ghaedmohammadi, G. Rigi, R. Zadmard, E. Ricca and G. Ahmadian, Mol. Biotechnol., 2015, 57, 756–766 CrossRef CAS PubMed.
- J. Sambrook and D. W. S. Russell, The condensed protocols from molecular cloning: a laboratory manual, 2006 Search PubMed.
- C. Huang, R. Du, P. Zhang, H. Meng, H. Jia, Y. Song, M. Li, Y. Zhang and S. Sun, Acta Biochim. Biophys. Sin., 2011, 43, 110–117 CrossRef CAS PubMed.
- S. Sandhu, J. A. Schouten, J. Thompson, M. Davis and T. D. Bugg, Analyst, 2012, 137, 1130–1136 RSC.
- K. Ljungberg, A. Söderbärg and U. Jansson, Appl. Phys. Lett., 1995, 67, 650–652 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- S. S. Maktedar, G. Avashthi and M. Singh, RSC Adv., 2016, 6, 114264–114275 RSC.
- M. Mohammadi, M. Ashjari, M. Garmroodi, M. Yousefi and A. A. Karkhane, RSC Adv., 2016, 6, 72275–72285 RSC.
- G. Xie, P. Xi, H. Liu, F. Chen, L. Huang, Y. Shi, F. Hou, Z. Zeng, C. Shao and J. Wang, J. Mater. Chem., 2012, 22, 1033–1039 RSC.
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Angew. Chem., Int. Ed., 2012, 51, 9377–9380 CrossRef CAS PubMed.
- D. A. Levary, R. Parthasarathy, E. T. Boder and M. E. Ackerman, PLoS One, 2011, 6, e18342 CAS.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435–456 CrossRef CAS PubMed.
- M. Raeeszadeh-Sarmazdeh, R. Parthasarathy and E. T. Boder, Biotechnol. Prog., 2017, 33, 824–831 CrossRef CAS PubMed.
- S. Baer, J. Nigro, M. P. Madej, R. M. Nisbet, R. Suryadinata, G. Coia, L. P. Hong, T. E. Adams, C. C. Williams and S. D. Nuttall, Org. Biomol. Chem., 2014, 12, 2675–2685 CAS.
| This journal is © The Royal Society of Chemistry 2017 |