
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial alkaloids produced by the mangrove endophyte Penicillium brocae MA-231 using the OSMAC approach†
Ling-Hong Meng,
Xiao-Ming Li,
Yang Liu,
Gang-Ming Xu and
Bin-Gui Wang *
*
Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Laboratory of Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Nanhai Road 7, Qingdao 266071, P. R. China. E-mail: wangbg@ms.qdio.ac.cn
First published on 4th December 2017
Abstract
The OSMAC (one strain-many compounds) protocol was used in this study to obtain various metabolites from the mangrove-derived endophytic fungus Penicillium brocae MA-231. As a result, four new thiodiketopiperazine alkaloids, penicibrocazines F–I (1–4), together with two new N-containing p-hydroxyphenopyrrozin derivatives brocapyrrozins A and B (7 and 8) as well as six known alkaloids (5, 6, and 9–12), were identified from the extract of the Czapek culture medium of the fungus. Their structures and absolute configurations were determined through interpretation of their spectroscopic data and by X-ray crystallographic analysis. Compounds 7 and 9 exhibited potent activity against human pathogenic bacterium Staphylococcus aureus with MIC values of 0.125 and 0.5 μg mL−1, respectively.
Introduction
Microbial secondary metabolites are evidenced as a major source of leading compounds for drug research.1–3 However, mining of microbial genomes revealed the presence of numerous silent metabolite gene clusters in a single microbe, displaying underestimated metabolic potentials, which are not always expressed under common laboratory culture conditions.4 The “one strain-many compounds (OSMAC)” approach, introduced by Zeeck and co-workers, has been successfully applied to increase the chemical diversity and the yield of new secondary metabolites from a single microbial strain, in which the promising strain is cultured in a variety of media and/or under different culture conditions.5 In our previous reports, a series of epithiodioxopiperazines (ETPs) including brocazines A–F6 and penicibrocazines A–E,7 have been obtained from the PDB culture of the mangrove-derived endophytic fungus Penicillium brocae MA-231. To dig the metabolic potential of this fungus, Czapek medium culture was used during the fermentation optimization and it seemed that the potentially silent gene clusters were activated, which resulted in the identification of three new ETPs with a rare spirocyclic skeleton.8 Further work on the remaining fractions of the fungus resulted in the isolation of four new highly unsaturated thiodiketopiperazine derivatives, namely, penicibrocazines F–I (1–4), along with two new p-hydroxyphenopyrrozin derivatives brocapyrrozins A and B (7 and 8), as well as six known alkaloid analogues including epicoccin A (5),9 phomazine A (6),10 4-hydroxy-3-phenyl-1H-pyrrol-2(5H)-one (9),11 hexahydro-2-hydroxy-1-phenyl-1H-pyrrolizin-3-one (10) (CAS registry number 172548-51-5), phenopyrrozine (11),12 and p-hydroxyphenopyrrozin (12).12 It should be noted that although compound 10 is listed in the CAS registry file, there is no published NMR data for this compound. The fully assigned 1H and 13C NMR data of 10 were reported for the first time in the present report. Herein, the isolation, structure elucidation, and biological evaluation of compounds 1–12 are presented.Results and discussion
The Czapek culture extract of P. brocae MA-231 was purified by a combination of column chromatography with silica gel, Sephadex LH-20, Lobar LiChroprep RP-18, and semi-preparative HPLC, to yield compounds 1–12 (Fig. 1).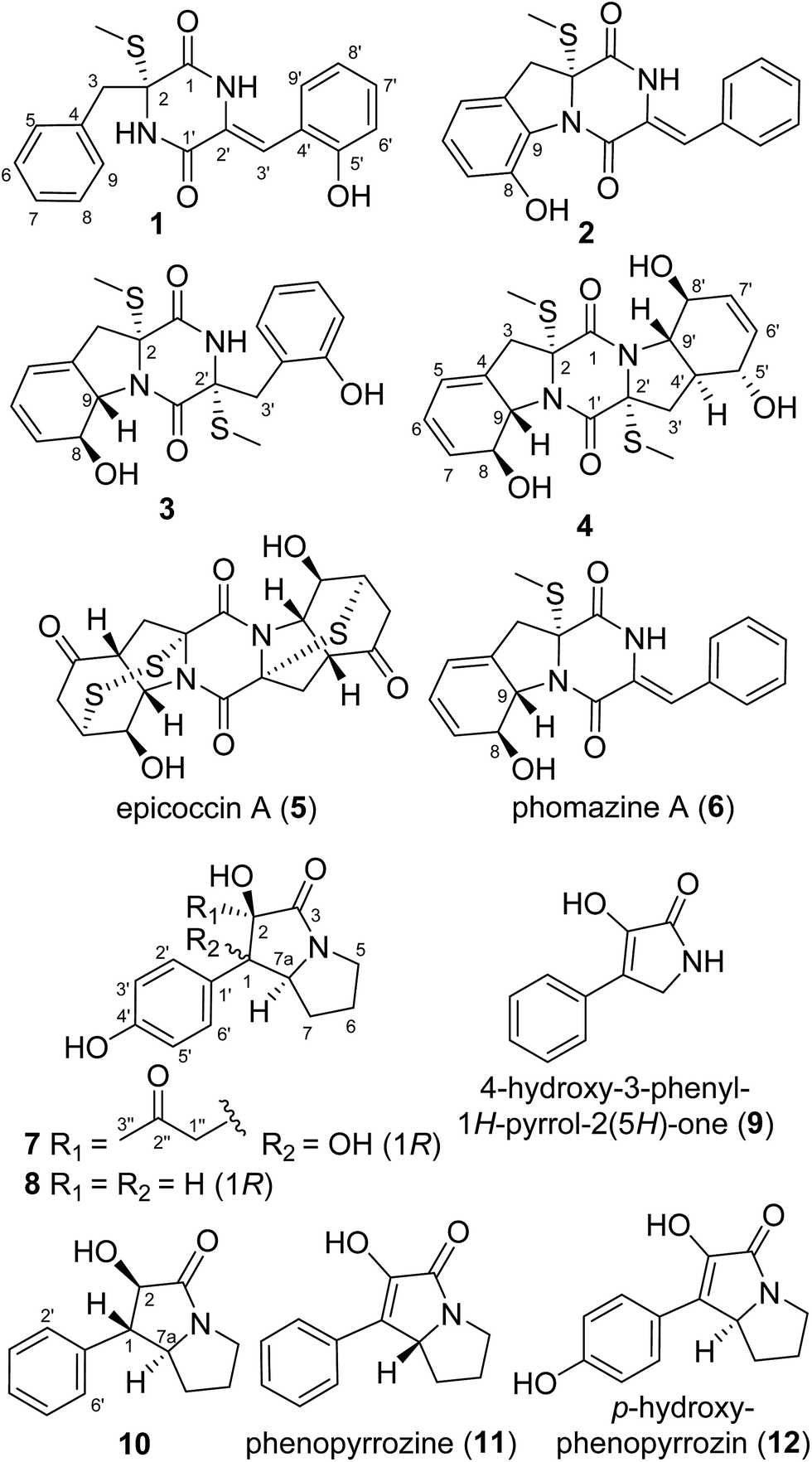 | ||
| Fig. 1 Structures of the isolated compounds 1–12. | ||
Compound 1 was obtained as colorless crystal and its molecular formula was determined as C19H18N2O3S on the basis of positive HR-ESI-MS data, indicating 12 degrees of unsaturation. The 1H NMR spectrum of 1 showed signals characteristic for a mono-substituted (H-5 to H-9) and an ortho-substituted (H-6′ to H-9′) phenyl units (Fig. 1 and Table 1), which were supported by the corresponding COSY and HMBC correlations as shown in Fig. 2. A total of 19 carbon atoms including one methyl, one methylene, ten aromatic/olefinic methines, and seven quaternary (two amide) carbon signals were observed in the 13C NMR and DEPT spectra (Table 1). Complete NMR assignments and connectivity of 1 were further determined by analysis of the 2D NMR spectroscopic data. HMBC correlations from H-3 to C-1 and C-2, from H-3′ to C-1′ and C-2′, and from NH-2′ to C-1 and C-3′ (Fig. 2) indicated the presence of a 2,5-diketopiperazine ring in the structure of 1. In addition, the clear HMBC cross-peaks from H-3 to C-4 and C-5 and from H-3′ to C-5′ verified the linkage of the 2,5-diketopiperazine to the above two benzene rings via C-3 and C-3′, respectively. Finally, the HMBC correlation from S-methyl to C-2 established a S-methyl moiety at C-2. The planar structure of 1 was thus established as shown in Fig. 1. An X-ray crystallographic experiment (Fig. 3) confirmed the structure of 1 and established the Z-geometry of the Δ2′(3′)-double bond. The Cu/Kα radiation experiments gave a refined Flack parameter 0.02(7) which allowed assignment of the absolute configuration of the stereogenic center in 1 as 2R. Based on the above data, the structure of 1 was determined and it was named as penicibrocazine F.
| Position | 1 (acquired in DMSO-d6) | 2 (acquired in CDCl3) | 3 (acquired in DMSO-d6) | 4 (acquired in DMSO-d6) | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 162.5, C | 163.8, C | 164.6, C | 167.2, C | ||||
| 2 | 67.5, C | 73.2, C | 73.4, C | 73.8, C | ||||
| 3 | α 3.55, d (13.4) β 3.01, d (13.4) | 42.1, CH2 | α 3.75, d (17.0) β 3.31, d (17.0) | 39.6, CH2 | α 2.82, d (15.5) β 2.12, d (15.5) | 37.8, CH2 | α 2.99, d (15.9) β 3.04, d (15.9) | 37.7, CH2 |
| 4 | 135.3, C | 127.0, C | 133.0, C | 133.6, C | ||||
| 5 | 7.33, d (7.4) | 130.6, CH | 6.80, d (7.3) | 116.3, CH | 5.90, brs | 119.1, CH | 5.98, brs | 118.9, CH |
| 6 | 7.26, t (7.4) | 127.9, CH | 7.14, t (7.3) | 129.7, CH | 5.61, d (9.6) | 130.5, CH | 5.89, d (9.8) | 123.4, CH |
| 7 | 6.77, t (7.4) | 119.0, CH | 6.91, d (7.3) | 118.5, CH | 5.86, d (9.6) | 123.3, CH | 5.63, d (9.8) | 130.7, CH |
| 8 | 7.26, t (7.4) | 127.9, CH | 147.4, C | 4.67, d (13.7) | 72.0, CH | 4.63, d (14.0) | 73.2, CH | |
| 9 | 7.33, d (7.4) | 130.6, CH | 131.0, C | 4.53, d (13.7) | 68.7, CH | 4.74, d (14.0) | 68.0, CH | |
| 1′ | 159.8, C | 158.5, C | 167.6, C | 168.3, C | ||||
| 2′ | 124.7, C | 125.8, C | 67.3, C | 72.4, C | ||||
| 3′ | 6.56, s | 112.4, CH | 7.18, s | 119.8, CH | α 3.46, d (14.0) β 3.20, d (14.0) | 40.2, CH2 | α 2.48, m overlap β 2.10, t (12.6) | 34.9, CH2 |
| 4′ | 120.5, C | 132.8, C | 121.1, C | 2.30, m | 43.2, CH | |||
| 5′ | 154.9, C | 7.43, d (7.3) | 129.8, CH | 156.0, C | 4.08, m | 68.8, CH | ||
| 6′ | 6.83, d (8.1) | 126.8, CH | 7.47, t (7.3) | 128.9, CH | 6.77, d (7.5) | 115.2, CH | 5.55, d (10.1) | 133.3, CH |
| 7′ | 7.13, dd (7.6, 8.1) | 129.9, CH | 7.39, t (7.3) | 129.4, CH | 7.03, t (7.5) | 128.2, CH | 5.69, d (10.1) | 129.9, CH |
| 8′ | 7.20, dd (7.6, 8.1) | 116.4, CH | 7.47, t (7.3) | 128.9, CH | 6.68, t (7.5) | 118.5, CH | 4.16, d (8.2) | 71.2, CH |
| 9′ | 7.19, d (7.6) | 131.7, CH | 7.43, d (7.3) | 129.8, CH | 6.96, d (7.5) | 131.2, CH | 3.44, dd (11.9, 8.2) | 67.5, CH |
| 2′-SMe | 2.27, s | 13.5, CH3 | 2.15, s | 14.1, CH3 | ||||
| 2-SMe | 2.18, s | 12.8, CH3 | 2.26, s | 13.9, CH3 | 2.14, s | 14.2, CH3 | 2.16, s | 14.3, CH3 |
| 8-OH | 7.83, s | 5.57, brs | 5.21, s | |||||
| 5′-OH | 9.33, brs | 5.30, d (5.8) | ||||||
| 8′-OH | 5.90, brs | |||||||
| 2′-NH | 9.15, s | 11.1, s | ||||||
| 2-NH | 10.65, brs | 9.33, brs | ||||||
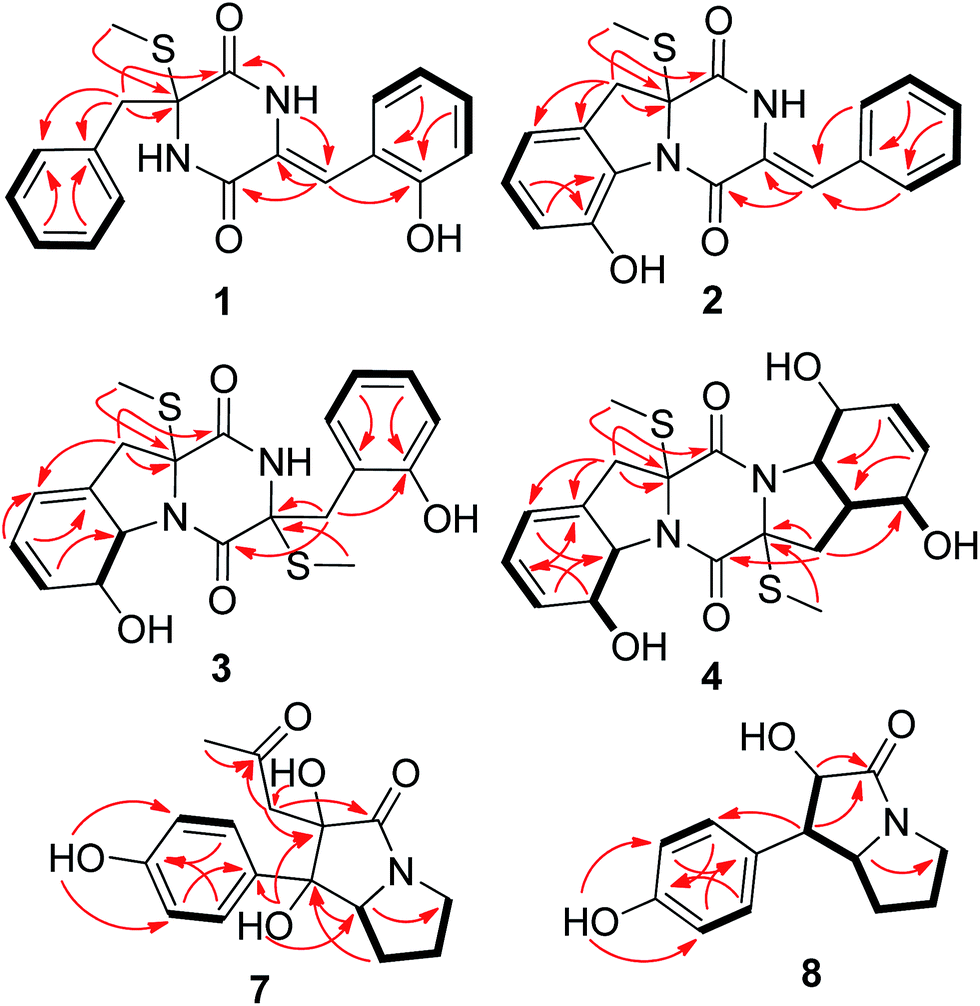 | ||
| Fig. 2 Key HMBC (arrows) and COSY (bold lines) correlations of compounds 1–4, 7, and 8. | ||
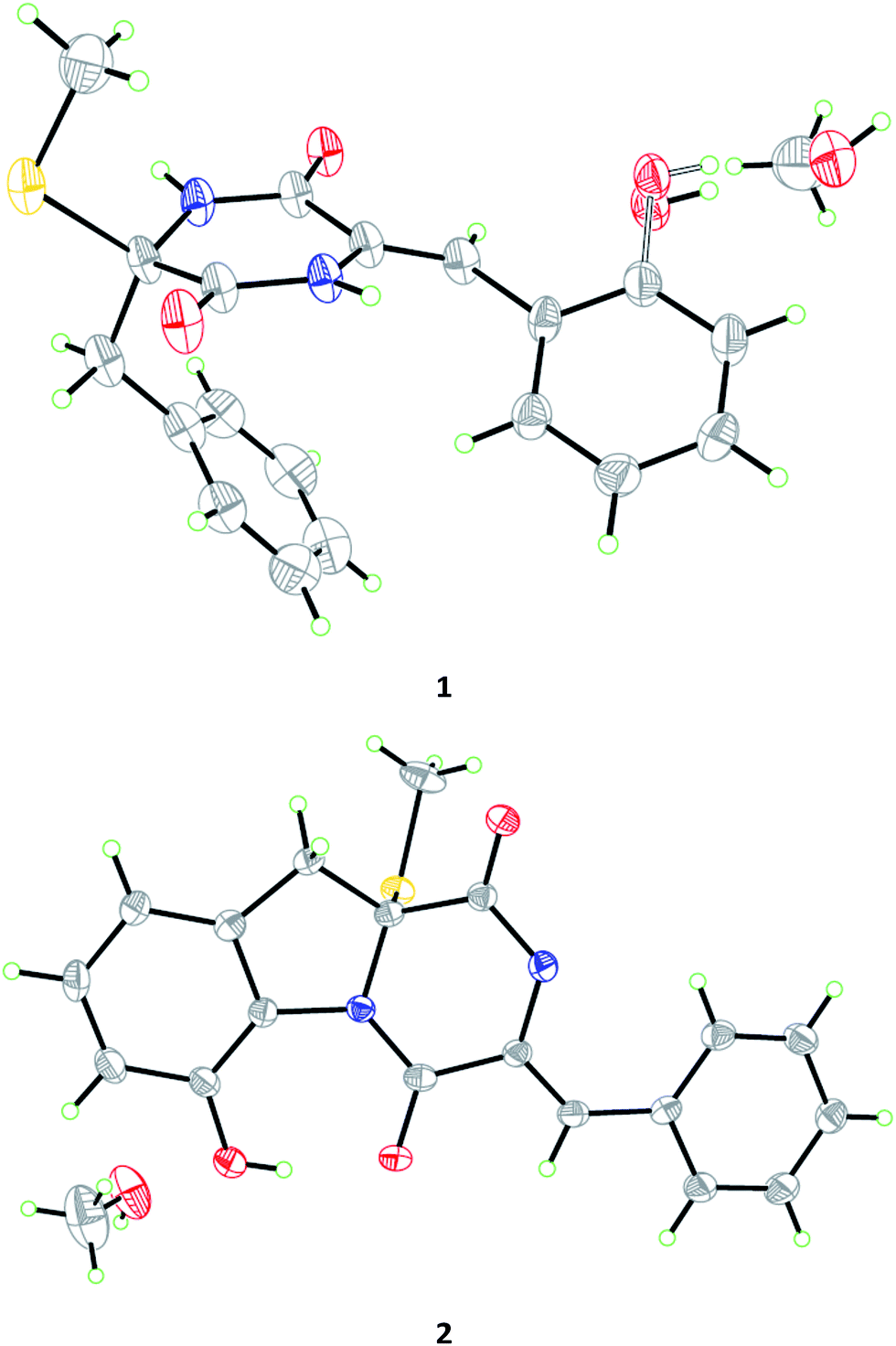 | ||
| Fig. 3 X-ray crystallographic structures of compounds 1 and 2 (inclusive with a MeOH molecule in each of the crystals). | ||
Compound 2, initially obtained as colorless amorphous powder, was determined to have a molecular formula C19H16N2O3S as established by the HR-ESI-MS analysis. The 1H and 13C NMR data (Table 1) revealed some structural similarities to phomazine A (6), a thiodiketopiperazine isolated from the marine-derived fungus Phoma sp. OUCMDZ-1847.10 However, signals for O- or N-substituted methines resonating at δC/H 74.3/4.64 (CH-8) and δC/H 69.6/4.87 (CH-9) in phomazine A were absent in the 1H and 13C NMR spectra of 2. Instead, resonances for two quaternary aromatic carbons at δC 147.4 (C-8) and δC 131.0 (C-9) were observed in the NMR spectra of 2 (Table 1). These observations were supported by the relevant COSY and HMBC correlations (Fig. 2). Upon slow evaporation of the solvent (MeOH) by storing the sample in a refrigerator, single crystals of 2 were cultivated, making feasible an X-ray diffraction analysis that unequivocally confirmed the structure (Fig. 3). The final refinement on the Cu/Kα data resulted in a Flack parameter of 0.00(2), allowing an assignment of the absolute configuration as 2R. Based on the above discussion, the structure of compound 2 was elucidated and the trivial name penicibrocazine G was assigned.
Compound 3 was obtained as yellow amorphous powder, with the molecular formula C20H22N2O4S2, as determined by the HR-ESI-MS data. The 1H NMR spectrum of 3 showed signals for an ortho-substituted phenyl unit, two thiomethyls, five methines, and two methylenes. The 13C NMR spectrum exhibited signals for 20 carbon atoms, corresponding to two methyl groups, two aliphatic methylenes, nine methines (seven aromatic/olefinic and two heteroatom-bonded), and seven non-protonated carbons (two amide carbonyls, two heteroatom-bonded, and three olefinic) (Table 1). The 1H and 13C NMR spectroscopic data of 3 were similar to those of 1 and phomazine A (6).10 The NMR chemical shifts for the left portion of 3 were nearly identical to that of 6, whereas the right portion showed some similarity to that of 1, except that a thiomethyl signal (δC/H 13.5/2.27, 2′-SMe), a methylene signal (δC/H 40.2/3.46, 3.20, CH-3′), and a heteroatom-bonded quaternary carbon signal (δC 67.3, C-2′) in 3 replaced the corresponding trisubstituted ethylene signals (δC/H 124.7, C-2′; 112.4/6.56, CH-3′) in the NMR spectra of 1 (Table 1). These observations were confirmed by relevant COSY and HMBC correlations (Fig. 2).
The relative configuration of 3 was determined by analysis of J-values and NOESY correlations (Fig. 4). The large coupling constant between H-8 and H-9 (J = 13.7 Hz) revealed the trans–orientation of the proton pair, while the key NOE correlations from 2-SMe to H-8 and 2′-SMe suggested the cofacial orientation of these groups. As might be expected, the CD spectrum of 3 showed similar profile as that of phomazine A (6),10 which exhibited a negative CE around 224 nm and a positive CE near 283 nm. There are three chromophores in 3: a skewed diene with an allylic alcohol moiety, a thiodiketopiperazin unit, and a substituted phenolic moiety. The latter is expected to contribute primarily to the 270 nm ECD transition.13 Therefore, the absolute configuration of compound 3 was assigned as 2R, 8S, 9S, and 2′R. Based on the above evidence, the structure of compound 3 was elucidated and the trivial name penicibrocazine H was assigned to this compound.
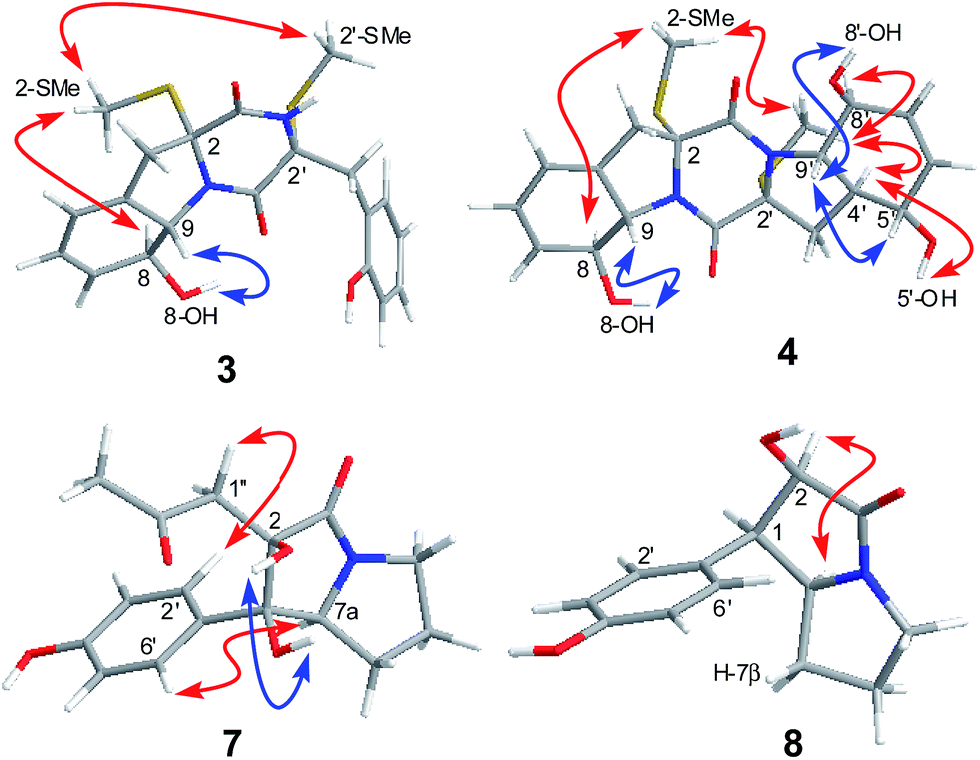 | ||
| Fig. 4 Key NOESY correlations for compounds 3, 4, 7, and 8. | ||
Penicibrocazine I (4) was assigned the molecular formula C20H24N2O5S2 on the basis of positive HR-ESI-MS data. Detailed analysis of the NMR data disclosed the structure of 4 to possess a disulfide diketopiperazine skeleton. Specifically, the 1H and 13C NMR chemical shifts for the right portion of 4 were nearly identical to those of penicibrocazine C, a symmetrical disulfide diketopiperazine identified from the PDB culture medium of P. brocae MA-231,7 whereas the left portion closely matched to that of 3. These observations were supported by the relevant COSY and HMBC correlations (Fig. 2). The relative configuration of 4 was deduced to be the same as that of 3 and penicibrocazine C, according to the NOESY experiment (Fig. 4) as well as the coupling patterns of the relevant protons (Table 1). The CD spectrum exhibited a negative CE at 218 nm and a positive CE at 276 nm, similar to those of 3 and penicibrocazine C. Therefore, the absolute configuration of compound 4 was assigned as 2R, 8S, 9S, 2′R, 4′S, 5′S, 8′S, and 9′S.
Brocapyrrozin A (7) was obtained as colorless crystals (DMSO) with a molecular formula of C16H19NO5 as established by the HR-ESI-MS analysis. The 13C and DEPT NMR spectra showed the presence of 16 carbon resonances (Table 2), including one methyl, four methylenes, four aromatic methines, one aliphatic methine, and six quaternary (with two carbonyl, two aromatic, and two oxygenated) carbons. The 1H NMR spectrum showed signals at δH 7.34 (2H, d, J = 8.3 Hz, H-2′/H-6′) and 6.75 (2H, d, J = 8.3 Hz, H-3′/H-5′), characteristic of a para-substituted phenyl group. In addition, signals corresponding to four methylene groups at δH 2.96/3.46, (H2-5), 1.92/1.87 (H2-6), 1.90/1.84 (H2-7), and 2.29/1.98 (H2-1′′), and one nitrogenated methine group at δH 4.46 (H-7a) as well as one methyl group at δH 2.09 (H-3′′), were also observed (Table 2). The COSY cross peaks H2-5/H2-6/H2-7/H-7a as well as HMBC correlations from H-7a to C-1, C-2, C-3, and C-5 and from H2-5 to C-3 established a bicyclic system in the molecular structure. The HMBC correlations from OH-1 to C-1′ connected the p-hydroxyphenyl group to the bicyclic system. In addition, the HMBC correlations from H3-3′′ to C-2′′ and from H2-1′′ to C-2′′, C-1, C-2, and C-3 confirmed the presence of the acetonyl unit and attached it to C-2.
| Position | 7 | 8 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 81.9, C | 3.67, dd (6.3, 6.8) | 50.9, CH | |
| 2 | 82.8, C | 4.69, d (6.8) | 73.8, CH | |
| 3 | 174.2, C | 174.1, C | ||
| 5 | α 2.96, m β 3.46, m | 42.5, CH2 | α 3.27, m β 2.95, m | 41.7, CH2 |
| 6 | α 1.92, m β 1.87, m | 23.5, CH2 | α 1.76, m β 1.51, m | 25.5, CH2 |
| 7 | α 1.90, m β 1.84, m | 25.3, CH2 | α 1.51, m β 0.83, m | 26.0, CH2 |
| 7a | 4.46, t (6.1) | 61.6, CH | 3.94, dd (6.3, 13.6) | 60.3, CH |
| 1′ | 129.7, C | 127.0, C | ||
| 2'/6′ | 7.34, d (8.3) | 128.3, CH | 6.74, d (8.3) | 130.8, CH |
| 3'/5′ | 6.75, d (8.3) | 114.7, CH | 6.61, d (8.3) | 115.1, CH |
| 4′ | 157.0, C | 156.4, C | ||
| 1′′ | α 2.29, d (13.0) β 1.98, d (13.0) | 48.7, CH2 | ||
| 2′′ | 206.4, C | |||
| 3′′ | 2.09, s | 31.4, CH3 | ||
| 1-OH | 5.53, s | |||
| 2-OH | 5.31, brs | 5.31, brs | ||
| 4′-OH | 9.45, s | 9.18, s | ||
The relative configuration of 7 was deduced from analysis of NOESY data (Fig. 4). NOE correlations from H-7a to H2-1′′ and H-2′/6′ revealed the cofacial orientation of these groups, while an NOE cross-peak from OH-1 to OH-2 placed these groups on the other face of the molecule. An X-ray crystallographic experiment confirmed the structure and relative configuration of 7 as depicted (Fig. 5). The Cu/Kα radiation for the X-ray diffraction with the refined Flack parameter of 0.02(7) allowed the assignment of absolute configuration of stereogenic centers in 7 as 1R, 2R, and 7a S.
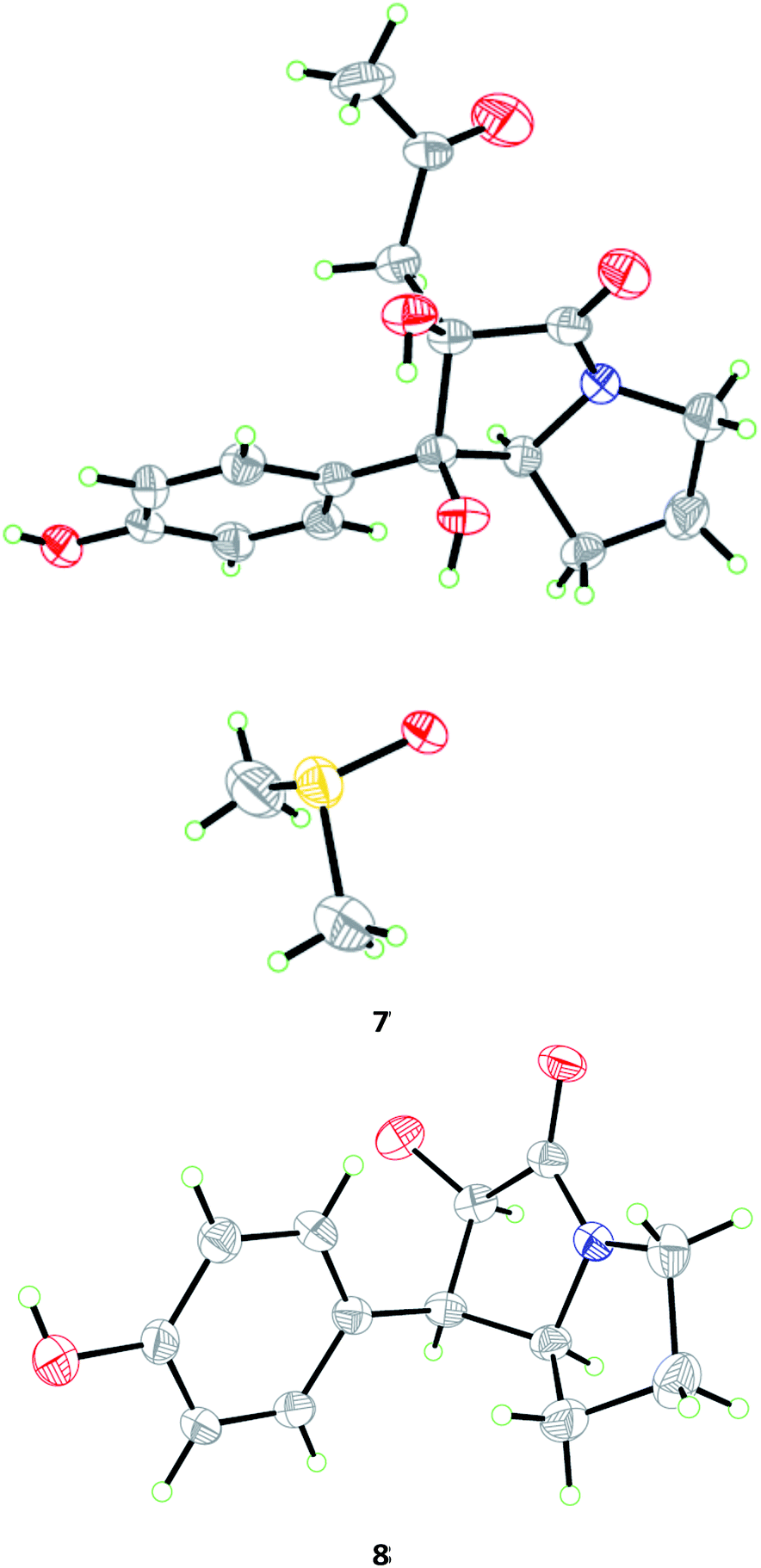 | ||
| Fig. 5 X-ray crystallographic structures of compounds 7 (inclusive with a DMSO molecule in the crystal) and 8. | ||
The molecular formula of brocapyrrozin B (8) was determined to be C13H15NO3 by HR-ESI-MS, having two protons more than that of p-hydroxyphenopyrrozin (12), which was previously isolated from a marine-derived fungus Chromocleista sp.12 Its NMR spectroscopic data were very similar to those of 12, except for the resonances of the olefinic quaternary carbon signals at δC 127.4 (C-1) and 141.9 (C-2) in the 13C NMR spectrum of 12 being replaced by signals of two aliphatic methine groups at δC/H 50.9/3.67 (CH-1) and δC/H 73.8/4.69 (CH-2) in the NMR spectra of 8 (Table 2). These data revealed that 8 is the 1,2-dihydrogenated derivative of 12. This deduction was further supported by the COSY and HMBC correlations (Fig. 2). The relative configuration of 8 was determined by analysis of NOESY correlations (Fig. 4). The NOE correlation from H-2 to H-7a indicated the cofacial orientation of these groups, while NOE cross-peak from H-2′/6′ to H-7β placed these groups on the opposite face. The absolute configurations for the stereogenic centers of 8 were also determined as 1R, 2R, and 7a S by the X-ray crystallographic analysis (Fig. 5).
The molecular formula of 10 was determined to be C13H15NO2 by HR-ESI-MS, having one oxygen atom less than that of 8. Its NMR spectroscopic data were very similar to 8. However, the signals of the quaternary carbon resonating at δC 156.4 (C-4′) and the corresponding exchangeable OH group at δH 9.18 (OH-4′) in the NMR spectra of 8 were replaced by a methine signal at δC 127.6/δH 7.30 (CH-4′) in 10 (Experimental section). This observation was further supported by the COSY and HMBC correlations (Fig. S41 and 43 in the ESI†). Its relative configuration was assigned by NOESY experiment (Fig. S44†). NOE correlations from H-2 to H-7a and H-2′/6′ revealed the cofacial orientation of these groups. The structure of 10 was thus assigned as hexahydro-2-hydroxy-1-phenyl-1H-pyrrolizin-3-one. This compound is listed in the CAS registry file (registry number 172548-51-5), but its NMR data were not reported, and the fully assigned data are now provided for the first time (Experimental Section).
Compounds 1–4 and 7–10 were screened for their antibacterial activity against several human and aquatic pathogenic bacteria (Escherichia coli, Micrococcus luteus, Staphylococcus aureus, Aeromonas hydrophilia, Edwardsiella tarda, Vibrio anguillarum, V. harveyi, and V. parahaemolyticus). Compounds 7 and 9 showed significant activity against S. aureus with MIC values of 0.125 and 0.5 μg mL−1, respectively, which are stronger than (7) or comparable to (9) that of the positive control, chloromycetin (MIC 0.5 μg mL−1). Moreover, compounds 3 and 4 exhibited activity against V. harveyi with MIC values of 16.0 and 32.0 μg mL−1, respectively, while the positive control chloromycetin has MIC value of 4 μg mL−1. Compound 3 also showed activity against E. coli, A. hydrophilia and V. parahaemolyticus with MIC values of 16.0, 32.0, and 16.0 μg mL−1, respectively, which are comparable to that of the positive control, chloromycetin (MIC = 2.0, 4.0, 2.0 μg mL−1). These data indicated that more S-methyl groups of the thiodiketopiperazine derivatives likely increased their antibacterial activity (3 and 4 vs. 1, 2, 5, and 6).
The antifungal activity of all the isolated compounds 1–12 was assessed using the microtiter broth dilution method against pathogenic fungi Alternaria brassicae, Colletotrichum gloeosprioides, Fusarium graminearum, and F. oxysporum. Compounds 7 and 9 showed potent activity against F. oxysporum with minimal inhibitory concentration (MIC) values of 0.25 and 0.125 μg mL−1, respectively, which are stronger than that of the positive control zeocin (with MIC value of 0.5 μg mL−1). Compound 8 also exhibited activity against F. oxysporum with MIC 64.0 μg mL−1. The result indicated that having the acetonyl group at C-2 (7 vs. 8) or lack of pyrrole unit (9 vs. 11) likely strengthened the activity against F. oxysporum.
Conclusions
In conclusion, four new unsaturated thiodiketopiperazine derivatives (penicibrocazines F–I, 1–4) and two new p-hydroxyphenopyrrozin alkaloids (brocapyrrozins A and B, 7 and 8) as well as six known alkaloid analogues (5, 6, 9–12) were obtained from the mangrove-derived endophytic fungus P. brocae MA-231 by culturing with Czapek medium, demonstrating that the OSMAC approach is a simple and useful method to dig various metabolites from a single fungal strain. Compounds 7 and 9 exhibited potent antimicrobial activities against the pathogenic bacterium Staphylococcus aureus or pathogenic fungus Fusarium oxysporum.Experimental section
General experimental procedures
Melting points were determined with an SGW X-4 micro-melting-point apparatus. Optical rotations were determined on an Optical Activity AA-55 polarimeter. Ultraviolet (UV) spectra were recorded on a Lengguang Gold Spectrumlab-54 spectrophotometer, λmax in nm. ECD spectra were acquired on a J-810 spectropolarimeter. The 1H-, 13C-, and 2D NMR spectra were obtained on a Bruker Avance 500 spectrometer (500 MHz for 1H and 125 MHz for 13C), δ in ppm with TMS as internal standard, J in Hz. Low and high ESIMS were obtained on a VG Auto spec 3000 spectrometer, in m/z. HPLC were performed on a Dionex HPLC system with a P680 pump, an ASI-100 automated sample injector, a TCC-100 column oven, a UVD 340U multiple wavelength detector, and a Dionex Acclaim ODS column (4.6 × 250 mm, 5 μm). Silica gel (SiO2; 100–200 mesh, 200–300 mesh, and GF254, Qingdao Haiyang Chemical Group Corporation, China), RP-18 reverse-phase silica gel (40–63 μm, Merck, Darmstadt), and Sephadex LH-20 (Merck, Darmstadt) were used for open column chromatography.Fungal cultivation
The endophytic fungal strain P. brocae MA-231 was isolated from the fresh tissue of the marine mangrove plant Avicennia marina that was collected at Hainan Island, P. R. China, in August 2012. The fungus was identified based on its ITS region of the rDNA as described previously6 and the sequence was deposited at GenBank, with accession number KM191342. A BLAST search result showed that the sequence was the most similar (99%) to the sequence of Penicillium brocae (compared to no. AF484394). The strain is preserved at the Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences.For chemical investigations, the fresh mycelium was inoculated into 1 L flask preloaded with 400 mL of Czapek medium (consisting of 14 g glucose, 9.6 g mannitol, 1.4 g NaNO3, 0.6 g KH2PO4, 0.3 g MgSO4, 1.2 g sodium glutamate, 0.6 g sodium tartrate, and 6 g sea salt) followed by a four-day culture at 200 rpm min−1 and 28 °C. The whole liquid (10 × 400 mL) collected from the flask was inoculated into a 50 L seed tank containing 38 L of sterilized Czapek medium (antifoam 0.5‰, aeration rate 2000 NL h−1, 150 rpm min−1). After a subsequent fermentation at 28 °C for 24 h, the afforded liquid was transferred as seed into a 500 L fermentor preloaded with 300 L of sterilized Czapek medium and cultivated at 28 °C for 4 days (antifoam 0.5‰, aeration rate 6000 NL h−1, 150 rpm min−1).
Extraction and isolation
The filtrate of the culture broth was extracted exhaustively with ethyl acetate, and concentrated under reduced pressure to give an extract (100 g), which was fractionated by silica gel vacuum liquid chromatography (VLC) using different solvents of increasing polarity from petroleum ether (PE) to MeOH to yield 9 fractions (Frs. 1–9) based on HPLC and TLC analysis. Fr.4 (6.9 g) was further purified by reversed-phase column chromatography (CC) over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 to 100:0) to afford five subfractions Fr.4–1 ∼ Fr.4–5. Fr.4–3 was further purified by CC on silica gel eluting with a CH2Cl2–MeOH gradient (from 100:1 to 40:1) and then by Sephadex LH-20 (MeOH) to afford compounds 1 (6.9 mg) and 2 (3.5 mg). Purification of Fr.4–4 by CC on silica gel with a CH2Cl2–MeOH gradient (from 100:1 to 30:1) and then by Sephadex LH-20 (MeOH) to afford compounds 3 (5.8 mg) and 5 (7.2 mg). Fr.4–5 was subjected to Sephadex LH-20 (MeOH) and preparative-TLC to afford compounds 4 (7.6 mg) and 6 (8.5 mg). Fr.6 (6.5 g) was also purified by CC over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 20:80 to 100:0) to afford six subfractions (Fr.6–1 ∼ Fr.6–6). Fr.6–2 was further purified by CC on Sephadex LH-20 (MeOH) and then by preparative TLC (plate: 20 × 20 cm, developing solvents: CH2Cl2–MeOH, 10:1) to yield compounds 8 (15.0 mg) and 12 (20.5 mg). Further purification of Fr.6–3 by CC on silica gel eluting with a CH2Cl2–MeOH gradient (from 50:1 to 5:1) yielded compounds 7 (21.0 mg) and 10 (25.8 mg). Fr.6–5 was further purified by CC over silica gel with a CH2Cl2–MeOH gradient (from 100:1 to 20:1) and then by Sephadex LH-20 (MeOH) to yield compounds 9 (29.4 mg) and 11 (28.1 mg).
:80 to 100:0) to afford five subfractions Fr.4–1 ∼ Fr.4–5. Fr.4–3 was further purified by CC on silica gel eluting with a CH2Cl2–MeOH gradient (from 100:1 to 40:1) and then by Sephadex LH-20 (MeOH) to afford compounds 1 (6.9 mg) and 2 (3.5 mg). Purification of Fr.4–4 by CC on silica gel with a CH2Cl2–MeOH gradient (from 100:1 to 30:1) and then by Sephadex LH-20 (MeOH) to afford compounds 3 (5.8 mg) and 5 (7.2 mg). Fr.4–5 was subjected to Sephadex LH-20 (MeOH) and preparative-TLC to afford compounds 4 (7.6 mg) and 6 (8.5 mg). Fr.6 (6.5 g) was also purified by CC over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 20:80 to 100:0) to afford six subfractions (Fr.6–1 ∼ Fr.6–6). Fr.6–2 was further purified by CC on Sephadex LH-20 (MeOH) and then by preparative TLC (plate: 20 × 20 cm, developing solvents: CH2Cl2–MeOH, 10:1) to yield compounds 8 (15.0 mg) and 12 (20.5 mg). Further purification of Fr.6–3 by CC on silica gel eluting with a CH2Cl2–MeOH gradient (from 50:1 to 5:1) yielded compounds 7 (21.0 mg) and 10 (25.8 mg). Fr.6–5 was further purified by CC over silica gel with a CH2Cl2–MeOH gradient (from 100:1 to 20:1) and then by Sephadex LH-20 (MeOH) to yield compounds 9 (29.4 mg) and 11 (28.1 mg).
X-Ray crystallographic analysis of compounds 1, 2, 7, and 8 (ref. 14)
All crystallographic data were collected on a Bruker Smart-1000 CCD diffractometer equipped with a graphite-monochromatic Cu-Kα radiation (λ = 1.54178) Å at 293(2) K. The data were corrected for absorption by using the program SADABS.15 The structures were solved by direct methods with the SHELXTL software package.16 All non-hydrogen atoms were refined anisotropically. The H atoms were located by geometrical calculations, and their positions and thermal parameters were fixed during the structure refinement. The structures were refined by full-matrix least-squares techniques.17Antimicrobial assays
Antimicrobial assays against pathogenic bacteria Escherichia coli, Micrococcus luteus, and Staphylococcus aureus and against aquatic pathogenic bacteria Aeromonas hydrophilia, Edwardsiella tarda, Vibrio anguillarum, V. harveyi, and V. parahaemolyticus as well as against pathogenic fungi Alternaria brassicae, Colletotrichum gloeosprioides, Fusarium graminearum, and F. oxysporum, were carried out using the well diffusion method.18 Chloromycetin was used as a positive control for the antibacterial assay, while zeocin was used as a positive control for the antifungal assay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (81673351 and 31600267) and by the Key Laboratory of Marine Bioactive Substance and Modern Analytical Technique, SOA (MBSMAT-2015-05). L.-H. M. thanks the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2017250) and the China Postdoctoral Science Foundation for project funding (2016M590667 and 2017T100521). B.-G. W. acknowledges the support of Taishan Scholar project from Shandong province.Notes and references
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2016, 33, 382–431 RSC.
- F. Cardoso-Martínez, J. M. de la Rosa, A. R. Díaz-Marrero, J. Darias, C. Cerella, M. Diederich and M. Cueto, Org. Biomol. Chem., 2015, 13, 7248–7256 Search PubMed.
- N. Gerhards and S. M. Li, Org. Biomol. Chem., 2017, 15, 8059–8071 CAS.
- A. A. Brakhage, Nat. Rev. Microbiol., 2013, 11, 21–32 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. HÖfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- L. H. Meng, X. M. Li, C. T. Lv, C. G. Huang and B. G. Wang, J. Nat. Prod., 2014, 77, 1921–1927 CrossRef CAS PubMed.
- L. H. Meng, P. Zhang, X. M. Li and B. G. Wang, Mar. Drugs, 2015, 13, 276–287 CrossRef CAS PubMed.
- L. H. Meng, C. Y. Wang, A. Mándi, X. M. Li, X. Y. Hu, M. U. Kassack, T. Kurtán and B. G. Wang, Org. Lett., 2016, 18, 5304–5307 CrossRef CAS PubMed.
- Y. G. Zhang, S. C. Liu, Y. S. Che and X. Z. Liu, J. Nat. Prod., 2007, 70, 1522–1525 CrossRef CAS PubMed.
- F. D. Kong, Y. Wang, P. P. Liu, T. H. Dong and W. M. Zhu, J. Nat. Prod., 2014, 77, 132–137 CrossRef CAS PubMed.
- A. Mallinger, B. Nadal, N. Chopin and T. L. Gall, Eur. J. Org. Chem., 2010, 1142–1148 CrossRef CAS.
- Y. C. Park, S. P. Gunasekera, J. V. Lopez, P. J. McCarthy and A. E. Wright, J. Nat. Prod., 2006, 69, 580–584 CrossRef CAS PubMed.
- J. M. Wang, N. Jiang, J. Ma, S. S. Yu, R. X. Tan, J. G. Dai, Y. K. Si, G. Z. Ding, S. G. Ma, J. Qu, L. Fang and D. Du, Tetrahedron, 2013, 69, 1195–1201 CrossRef CAS.
- Crystallographic data of compounds 1, 2, 7, and 8 have been deposited in the Cambridge Crystallographic Data Centre as CCDC 1541032 (for 1), 1541033 (for 2), 1541034 (for 7), and 1541035 (for 8).†.
- G. M. Sheldrick, SADABDS, Software for Empirical Absorption Correction, University of Gottingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL, Bruker Analytical X-ray System Inc., Structure Determination Software ProgramMadison, WI, 1997 Search PubMed.
- G. M. Sheldrick, SHELXTL-97 and SHELXTL-97, Program for X-ray Crystal Structure Solution and Refinement, University of Gottingen, Germany, 1997 Search PubMed.
- S. K. S. Al-Burtamani, M. O. Fatope, R. G. Marwah, A. K. Onifade and S. H. Al-Saidi, J. Ethnopharmacol., 2005, 96, 107–112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR (1–4, 7, 8, and 10) and ECD (1–4) spectra (PDF). X-ray crystallographic files (CIF) and CCDC numbers 1541032–1541035 for compounds 1, 2, 7, and 8, respectively. CCDC 1541032–1541035. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12081h |
| This journal is © The Royal Society of Chemistry 2017 |