
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of pH-responsive nanocomposites of gold nanoparticles and graphene oxide and their applications in SERS and catalysis
Aihua Yao†
 *ab,
Qingge Fu†c,
Ling Xub,
Yan Xub,
Wenqi Jiangb and
Deping Wangab
*ab,
Qingge Fu†c,
Ling Xub,
Yan Xub,
Wenqi Jiangb and
Deping Wangab
aKey Laboratory of Advanced Civil Engineering Materials, Ministry of Education, Tongji University, Shanghai 200092, China. E-mail: aihyao@126.com
bSchool of Materials Science and Engineering, Tongji University, Shanghai 200092, China
cDepartment of Emergency, Changhai Hospital, Second Military Medical University, Shanghai 200433, China
First published on 15th December 2017
Abstract
Poly(4-vinylpyridine) (P4VP), a pH-sensitive polymer, was employed to non-covalently functionalize graphene oxides (GOs) through π–π stacking interactions. Citrate-capped Au nanoparticles (Au NPs) were then immobilized onto the P4VP-grafted GO, yielding pH-responsive Au NPs–GO nanocomposites. Such Au NPs–GO nanocomposites displayed pH-responsive localized surface plasmon resonance (LSPR) changes and assembly behavior due to protonation and deprotonation of the pyridine units of P4VP molecules. The catalytic activity of the Au NPs–GO nanocomposites was evaluated using the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) as a model reaction, and demonstrated significantly high catalytic activity and stability at low pH (2.0), which was 4-fold higher than that at high pH (5.0). Furthermore, our results indicated that the Au NPs–GO nanocomposites induced active adsorption and selective SERS detection of negatively charged analytes under low pH conditions, and are therefore highly attractive for real-world chemical detection.
Introduction
Nanosized particles of noble metals, especially gold nanoparticles (Au NPs), have attracted increasing attention over the past few decades, due to their attractive electric, optical, and catalytic properties as well as potential applications in wide variety areas including physics, chemistry, biology, material science and their different interdisciplinary fields.1–3 However, Au NPs in solution tend to aggregate due to their high surface energy, which leads to graduate reduction in stability and catalytic activity.4 The limitation can be overcome by immobilizing Au NPs on diverse supports, such as SiO2, carbon, and polymer microspheres, etc.5–7 Among these supports, graphene and its derivatives (graphene oxide, GO, and reduced graphene oxide, RGO) have been studied extensively due to their planar structure, high surface area and excellent physicochemical properties.8 Compared to graphene, GO can be produced in a low-cost and high-yield manner, and is therefore more suitable for practical applications. Furthermore, the presence of abundant oxygen-containing functional groups on both basal planes and edges not only makes GO highly hydrophilic, but also provides a large number of active sites for surface functionalization and nanoparticle immobilization. So far, a considerable amount of research has been focused on the development of GO or RGO supported Au NPs for various applications involving catalysts, sensors, SERS, and medicine, etc.8–11 Au NPs–GO nanocomposites have proven to exhibit novel and enhanced physicochemical properties, which is considered to be beneficial from a strong synergistic interaction between these two components.12The most popular and effective strategies for synthesizing Au NPs–GO nanocomposites can be classified into two categories, namely, ex situ assembly and in situ growth approaches.13 In situ growth provides better control of the size, distribution and loading density of the nanoparticles on GO sheets when compared to ex situ methods. In ex situ approaches, the modification of either GO sheets or Au NPs (or both) with suitable functional groups is a critical step for efficient assembly of Au NPs onto GO surfaces and improving the interfacial interaction between them. A wide range of functional groups, such as amino, pyridyl, thiol, and carboxy, etc., have been utilized to immobilize Au NPs onto GO sheets, to yield Au NPs–GO nanostructured assemblies with desired properties.8,14–16
Poly(4-vinylpyridine) (P4VP) is an attractive polymer and has been widely used as a surface modifier because of the pyridyl group's strong affinity to transition metals.17,18 Additionally, P4VP is well-known as a pH-responsive polymer that undergoes molecular conformational changes at a critical transition pH value (∼4.5).19 At low pH (<4.5), the pyridyl segment of P4VP is protonated by reactions with acids and forms positively charged pyridinium groups, which results in the chain stretching under the electrostatic repulsion. Conversely, the pyridine exists in a deprotonated form at higher pH ranges (>4.5), and the polymer chains adopted a coil conformation. Such a pH effect of P4VP has been used to functionalize GO and Au NPs for various applications. For example, pH responsive Au@P4VP nanocomposites were developed utilizing Au NPs or nanorods decorated with P4VP molecules. It was found that the resulting nanocomposites exhibited pH-dependent localized surface plasmon resonance (LSPR) due to the conformational transition of P4VP in different pH environments, and therefore LSPR-based sensing or catalytic applications were explored.20–22 P4VP-grafted GO displayed reversible electrochemical response to pH changes. The protonation/deprotonation of pyridine units induced significant changes in the microenvironment of GO/P4VP assembles, and allowed for applications in pH-induced switchable bioelectronics, intelligent nanodevices and pH-triggered drug delivery system.11,23,24 Recently, Li et al.6 synthesized core–shell C@P4VP microspheres by coating P4VP shells on the surfaces of carbon spheres, and then the microspheres were used to support Au NPs for catalytic reduction of 4-nitrophenol. Their results indicated that the polymeric P4VP shell not only endowed the catalyst with smart pH-responsibility but also enhanced its catalytic activity and cycle stability.
Inspired by the studies above, we developed Au NPs–GO nanocomposites based on P4VP-grafted GO nanosheets decorated with Au NPs. It was expected that the conformational transition of P4VP chains would result in a tunable interfacial interaction between GO and Au NPs, and allowed for controlled performance. Our results indicated that the presence of the P4VP not only functionalized the GO surfaces, enabling the immobilization of Au NPs with high and uniform distribution on GO sheets, but also endowed the nanocomposites with pH-responsive behavior. We therefore explored their potential applications in SERS and catalysis, and found that the Au NPs–GO nanocomposites displayed pH-dependent catalytic activity towards the reduction of 4-NP, and selective SERS detection of negatively-charged dye molecules.
Experimental
Chemicals
Graphite flakes (nature, −10 mesh) were purchased from Alfa Aesar (Tianjin, China). HAuCl4·4H2O and sodium citrate (C6H5Na3O7·2H2O) were purchased from Shanghai Aladdin Chemical Reagent Company. Dimethyl sulfoxide (DMSO), and poly(4-vinylpyridine) (P4VP, Mw = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sigma-Aldrich. All reagents were analytical grade and used without further purification. All aqueous solutions were prepared with twice-distilled water (18.2 MΩ cm).
000) were purchased from Sigma-Aldrich. All reagents were analytical grade and used without further purification. All aqueous solutions were prepared with twice-distilled water (18.2 MΩ cm).
Synthesis of Au NPs–GO nanocomposites
000 rpm and washed with diluted HCl solution (pH 2.0) atleast three times to remove excess P4VP. The final P4VP-modified GO was redispersed in DMSO and stored; the final concentration of GO was about 2.5 mg mL−1.000 rpm and washed with deionized water three times and dried under vacuum at 60 °C.Results and discussion
Synthesis and characterization of Au NPs–GO nanocomposites
Initial GO suspensions were prepared according to the modified Hummers method from commercial graphite. Surface grafting was carried out using P4VP molecules with the aid of sonication. Further interactions between P4VP–GO conjugates and citrate-capped Au NPs resulted in Au NPs–GO nanocomposites. A successful decoration of Au NPs on GO sheets was verified by TEM observation. As shown in Fig. 1(a), the GO sample is composed of few-layer sheets and exhibits typical wrinkled morphology. After the assembly process, the GO sheets were decorated with well-dispersed spherical Au NPs (Fig. 1(b) and (c)). The mean size of Au NPs anchored on the GO was about 20 nm as determined by measuring diameters of total 200 particles from 5 TEM images (the inset of (c)). It is noteworthy that there are almost no free Au NPs outside the domain of the GO sheets, indicating a strong interaction between the Au NPs and P4VP-grafted GO. The high-resolution TEM image in Fig. 1(d) reveals that the Au NPs have a highly crystalline structure with an interplane distance of ∼0.235 nm that corresponds to the spacing of (111) plane of a face-centered cubic (fcc) Au crystal. | ||
| Fig. 1 (a–c) TEM images of GO sheets and Au NPs–GO nanocomposites with different magnifications; (c) HRTEM image of a representative Au NPs–GO nanoparticle. | ||
XRD pattern in Fig. 2(a) further verifies the fcc structure of Au NPs (JCPDS no. 04-0784). The average size of Au NPs was determined to be 21.2 nm according to the Scherrer formula, consistent with the size observed by TEM images. Moreover, a weak diffraction peak around 2θ = 25° corresponding to the (002) plane of GO is also detected. The elemental composition and interfacial interaction between GO and Au NPs were checked by XPS technique. The survey scan profile in Fig. 2(b) confirms that the Au NPs–GO nanocomposites contain C, O, Au, and N elements. The weak N signal originates from the pyridine moieties of P4VP chain, indicating the existence of P4VP molecules on GO surfaces. The deconvolution of the C 1s spectrum resulted in four different components. As shown in Fig. 2(c), the predominant peak at 284.6 eV corresponds to non-oxygenated carbon bonds in both GO and P4VP. The peak at 286.4 eV is attributed to the C–O bonds in GO and C–N bonds in P4VP. The broad peak at 287.8 eV corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in GO. The C 1s spectrum further shows a small signal at 291.7 eV, which is due to a π–π* shake-up signal characteristic of aromatic π electrons. This signal has been observed for similar polymer–GO composites.26,27 These results indicated that the GO sheets have been non-covalently functionalized with P4VP molecules through π–π interactions between the delocalized sp2 carbon structure of GO and the pyridine moieties of P4VP chains. The Au 4f core-level spectrum in Fig. 2(d) reveals Au 4f doublet peaks at 83.8 and 87.5 eV. These peaks are shifted to lower binding energies when compared to the standard XPS spectrum of metallic Au0 (84.0 and 87.7 eV), indicating the electron transfer from GO to Au NPs.28
O bonds in GO. The C 1s spectrum further shows a small signal at 291.7 eV, which is due to a π–π* shake-up signal characteristic of aromatic π electrons. This signal has been observed for similar polymer–GO composites.26,27 These results indicated that the GO sheets have been non-covalently functionalized with P4VP molecules through π–π interactions between the delocalized sp2 carbon structure of GO and the pyridine moieties of P4VP chains. The Au 4f core-level spectrum in Fig. 2(d) reveals Au 4f doublet peaks at 83.8 and 87.5 eV. These peaks are shifted to lower binding energies when compared to the standard XPS spectrum of metallic Au0 (84.0 and 87.7 eV), indicating the electron transfer from GO to Au NPs.28
 | ||
| Fig. 2 (a) XRD pattern of Au NPs–GO nanocomposites; (b–d) XPS survey scan, C 1s deconvolution and Au 4f analysis of Au NPs–GO nanocomposites. | ||
Fig. 3(a) shows UV-vis absorption spectra of P4VP-grafted GO (P4VP/GO) and Au NPs–GO nanocomposites dispersed in DMSO and are compared with neat GO and P4VP. The spectrum of GO (curve II) exhibits an absorption peak at 234 nm and a shoulder peak at 300 nm, which can be assigned to the π–π* transition of CC bonds and n–π* transition of CO bonds, respectively.29 A strong absorption peak at 287 nm arising from P4VP polymer is observed in the spectrum of P4VP/GO (curve III), which provides further evidence for the successful surface grafting of GO by the P4VP molecules. Further decoration of Au NPs on P4VP/GO results in a new absorption peak at ∼530 nm that corresponds to the LSPR of Au NPs (curve IV).
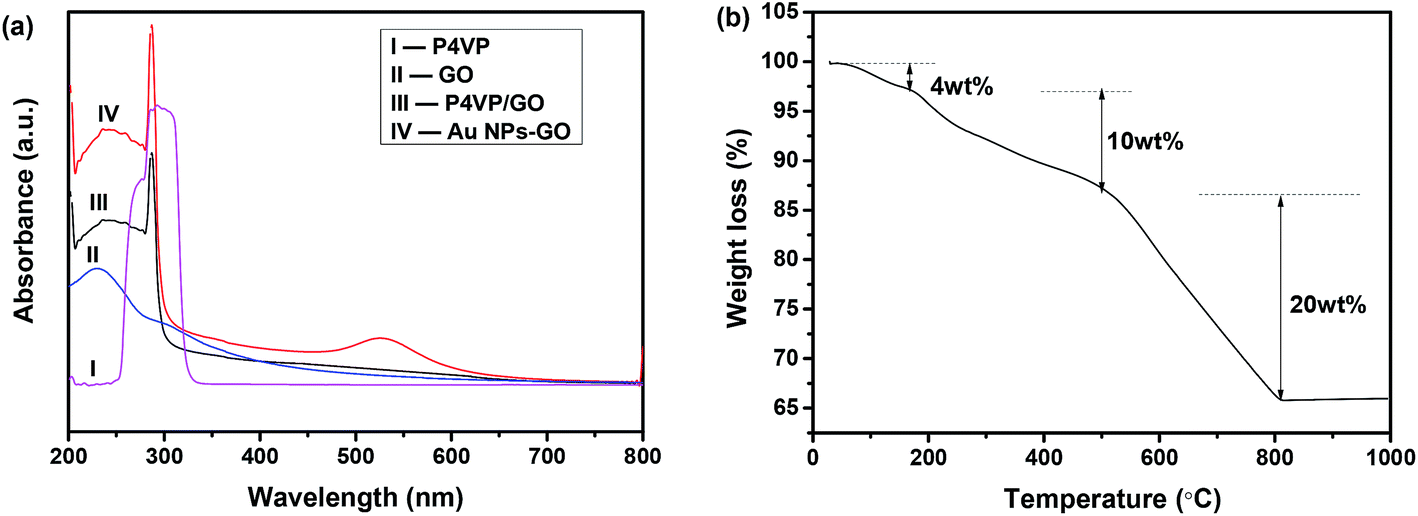 | ||
| Fig. 3 (a) UV-vis absorption spectra of P4VP-grafted GO and Au NPs–GO nanocomposites dispersed in DMSO, GO and P4VP; (b) TGA curve of Au NPs–GO nanocomposites. | ||
The mass percentage of Au in the Au NPs–GO nanocomposites was analysed via TGA. In Fig. 3(b), the small weight loss below 150 °C is attributed to loss of adsorbed water and residual DMSO. The weight loss of 10% between 200 and 500 °C is associated with the thermal degradation of P4VP molecules, citrate capping on the Au NPs, and some oxygen-containing functional groups of GO. Upon increasing the temperature up to 800 °C, a 50% weight loss is observed and is attributed to decomposition of the carbon skeleton of GO. Accordingly, the mass percentage of Au NPs is determined to be around 36% of the total nanocomposites based on the remaining weight at the end of the experiment.
Pyridine, with an aromatic structure, has a strong tendency to interact with GO sheets via π–π stacking. Simultaneously pyridine groups have good binding affinity to Au NPs via the formation of hydrogen bonding with the citrate capping on the surfaces of Au NPs. Therefore, after the formation of π–π stacking interactions with GO, P4VP still contains a large number of unbound pyridyl groups that are capable of binding to Au NPs. As a result of the free pyridines, the Au NPs are well anchored to the GO sheets with high coverage density, and therefore providing sufficient amount of catalytic active sites and highly efficient electron transfer between the Au NPs and GO, which may subsequently enhance the catalytic activity and SERS effect of the nanocomposites.
pH response of Au NPs–GO nanocomposites
To evaluate the pH response of Au NPs–GO nanocomposites, the samples were dispersed in dilute HCl solution of pH 2.0–5.0 and deionized water (pH 6.5), respectively. After equilibrium adsorption, the UV-vis absorption spectra of all samples were recorded from 400 to 700 nm. Fig. 4(a) clearly displays that the LSPR peaks of Au NPs change from 524 nm at pH 2.0 to 532 nm at pH 6.5, with around 8 nm red-shift. Simultaneously, broadening of the absorption peak occurs with an increase in pH. The LSPR peak positions of Au NPs were plotted vs. pH values and presented in Fig. 4(b). With increasing pH, the LSPR peak shows small changes at pH < 4.0, but a dramatic increase as pH changed from 4.0 to 5.0, and eventually reaches a nearly constant wavelength of ∼532 nm. Such pH-dependent LSPR absorption of the Au NPs–GO is similar to those observed in P4VP-grafted Au nanoparticles and nanorods.20,21 | ||
| Fig. 4 (a) UV-vis spectra of the Au NPs–GO nanocomposites dispersed in dilute HCl solution of pH 2.0–5.0 and DI water (pH 6.5); (b) LSPR peak positions changes of the Au NPs–GO nanocomposites at various pH; the inset of (b) shows scheme of pH-responsive morphology change of the Au NPs–GO nanocomposites. (c and d) TEM images of the Au NPs–GO nanocomposites at pH 2.0 and pH 5.0; the insets of (c) and (d) show photographs of the Au NPs–GO nanocomposites at pH 2.0 and pH 5.0. | ||
The pH-responsive behavior of the Au NPs–GO nanocomposites was further evaluated through zeta potential analysis and TEM observation. The insets in Fig. 4(c) and (d) show the photographs of the nanocomposites dispersed in dilute HCl solutions at pH 2.0 and 5.0. It was found that the dispersion of Au NPs–GO nanocomposites was dependent on the pH value of the medium. At pH = 2.0, the nanocomposites were well dispersed in aqueous solution, and formed a reddish black homogeneous dispersion. However, obvious aggregation occurred and resulted in black precipitates at the bottom of the vessel when the pH was changed to 5.0. If the medium pH was adjusted back to 2.0, the precipitates were able to be redispersed into the solution again, confirming a reversible pH-responsive behavior. The zeta potential measurements revealed that the particle zeta potential maintained a high value of ∼32.5 mV at pH 2.0, and such a high surface charge prevented the aggregation of the nanocomposites, thus allowing good dispersion. Conversely, at a higher pH (5.0), the zeta potential drastically decreased to 5.7 mV, and nearly went to zero under neutral conditions. Upon further increasing pH to 10.0, the zeta potential value was −7.2 eV. The negative zeta potential of the nanocomposites originated from the deprotonation of citrate anions capping on Au NPs as well as the carboxylic acid and phenolic hydroxyl groups on GO sheets. Nevertheless, the zeta potential values of the nanocomposites at pH > 5.0 are not enough to achieve good particle suspension stability. TEM images in Fig. 4(c) and (d) directly reveal the pH-response of the Au NPs–GO nanocomposites. Large agglomerates of Au NPs are obviously formed on the GO sheets as the pH value of the media is changed from 2.0 to 5.0.
Such a pH-responsive behavior of the Au NPs–GO nanocomposites is ascribed to the protonation/deprotonation of pyridine groups on the grafted P4VP. At low pH (<4.0), the pyridyl groups are protonated to form pyridinium ions, and renders the Au NPs–GO nanocomposites with positive charges, as evidenced by the high zeta potential. Meanwhile the positively charged polymer chains repulse each other, and result in an increased distance between the Au NPs. The increased distance between Au NPs is verified by the observed good dispersion of Au NPs on GO sheets at pH 2.0 versus poor dispersion at pH 5.0 due to a lack of electrostatic repulsion. Thus, large Au NPs agglomerations are observed on GO sheets at pH 5.0 as shown in TEM, accompanied by the redshift and broadening of UV-vis absorption peaks. However, the electrostatic repulsion between negatively charged citrate capped-Au NPs limited agglomeration to a certain degree, so that the SPR peaks shifts to a constant wavelength of about 532 nm. On the other hand, the P4VP chains collapse onto the GO surfaces at high pH, and increase the refractive index around Au NPs, which can also result in a red shift of Au LSPR peaks.30
pH-responsive catalysis activity of the Au NPs–GO nanocomposites
The catalytic activity of the Au NPs–GO nanocomposites was evaluated using the reduction of 4-NP to 4-AP as a model reaction. It has been reported that Au NPs act as an effective catalyst for the reduction of 4-NP in the presence of NaBH4 by facilitating electron transfer from BH4− to 4-NP.31,32 To compare the catalytic performance of the nanocomposites at various pH levels, the Au NPs–GO nanocomposites were initially dispersed in dilute HCl solution of pH 2.0 and 5.0. These solutions were subsequently injected into a mixture of 4-NP and NaBH4. It was found that the dark yellow color of mixture solution was quickly quenched. The fast reaction rate is evidenced by the sharp decrease of the absorption peaks at 400 nm in UV-vis spectra (Fig. 5(a)). Meanwhile, new peaks corresponding to 4-AP appeared at 300 nm appear and increase over time. As shown in Fig. 5(a), the reduction of 4-NP to 4-AP is finished within 100 s using Au NPs–GO nanocomposite catalysts at pH 2.0. Pseudo-first order kinetics was utilized to determine the reaction rate constant (k), which can be described by the equation:where C0 and Ct represent the 4-NP concentrations at time 0 and time t, respectively. Fig. 5(b) shows a well-defined linear relationship between ln(Ct/C0) and time, and the corresponding rate constants are determined from the slopes of the plotted lines. As observed, the reaction rate constant (k) is raised significantly from 0.223 to 0.954 min−1 when the pH values of the reaction change from 5.0 to 2.0. For comparison purpose, the same amount of citrate-capped Au NPs as those anchored on the GO sheets were also evaluated under the same conditions and the k value was calculated to be 0.308 min−1. These comparative results demonstrate the superior catalytic capability of the Au NPs–GO catalyst at pH = 2.0. Furthermore, it is noteworthy that the present Au NPs–GO nanocomposites at low pH value exhibit a higher catalytic activity than recently reported Au-based catalysts,26,33 and are even comparable to Pt-based catalysts for the reduction of 4-NP.26

 | ||
| Fig. 5 UV-vis spectra of the reduction reaction of 4-NP to 4-AP with Au NPs–GO nanocomposites at different time intervals; (b) the ln(Ct/C0) vs. t plots for the reduction reaction of 4-NP in the presence of Au NPs–GO nanocomposites at pH 2.0 and 5.0 and Au NPs. Conditions: [4-NP] = 10 mM; [Au NPs–GO catalysts] = 0.2 mg mL−1; [Au NPs catalysts] = 0.08 mg mL−1; [NaBH4] = 0.2 M; (c) reusability test of Au NPs–GO nanocomposites as catalysts for the reduction of 4-NP. | ||
For the Au NPs–GO nancomposites incubated in the HCl solution at pH 2, the protonated polymer chains of P4VP tend to extend maximally under the electrostatic repulsion. In this case, the Au NPs bound to the P4VP can easily access the reagents (4-NP and HB4−), which increase the number of accessible catalytic active sites. Further, the extended chain morphology facilitate the electron transfer between the GO and Au NPs, increasing local electron concentration. The higher electron concentration favors the uptake of electrons by 4-NP molecules.34 Meanwhile, BH4− ions are readily to absorbed on the positively charged Au NPs–GO nanocomposites and relayed electrons to the active sizes for reduction of 4-NP.35 However in the case of pH 5.0, the P4VP chains adopted a coiled conformation, resulting in the inclusion of Au NPs within the polymer chains and thus partial catalytic active sites were shielded. The reduction of catalytic active sites resulted in relatively low catalytic activity.
Since catalytic stability is highly desirable for catalyst in practical applications, the reusability of the Au NPs–GO nanocomposites was evaluated by successively testing their catalytic performance. For this purpose, the catalyst was collected, washed and redispersed in dilute HCl solution of pH 2.0 for the next cycle. The conversion of 4-NP to 4-AP was calculated as 1 − At/A0, where At represents the absorption of 4-NP at 400 nm at 100 s, and A0 is the absorption of 4-NP in original reactant. From Fig. 5(c), no measured decease in the catalytic activity was observed after running for 5 successive cycles, demonstrating the excellent catalytic stability of the Au NPs–GO nanocomposites.
SERS activity of the Au NPs–GO nancomposites
Inspired by the high zeta potential of the Au NPs–GO nanocomposites at pH < 4.0, we hypothesized that the negatively charged analyte molecules would preferentially adsorb onto sample surfaces, and subsequently the anchored Au NPs enhanced their Raman signals; whereas the adsorption of positively charged analytes would be unfavorable due to electrostatic repulsion, and therefore enabled the charge-selective SERS analysis of dye molecules. Herein two positively charged analytes, rhodamine 6G (R6G) and methylene blue (MB), and two negatively charged analytes, trypan blue (TB) and ponceau S (PS), were employed as representative Raman probes to demonstrate the hypothesis. The Au NPs–GO nanocomposites that were well-dispersed in the HCl solution of pH 2.0 were mixed absolutely with the aqueous solution of each analyte, and then the mixture solution was dropped onto glass slides for SERS measurement.Fig. 6(a) compares the SERS spectra of 1 mM aqueous solution of R6G, MB, TB and PS recorded under the same conditions. Distinct Raman bands corresponding to TB and PS molecules were observed, suggesting a strong SERS enhancement for the negatively charged analyte molecules. Conversely, the SERS spectra collected from R6G and MB molecules are similar to that of the Au NPs–GO nanocomposites. Only typical D and G bands of the GO component at 1345 and 1580 cm−1 were observed, but no characteristic Raman bands of the analyte molecules were detected. The charge selectivity was further tested using a mixed solution of TB and R6G (concentration of 1 mM each). The SERS spectrum in Fig. 6(b) demonstrates a decrease in intensity of characteristic TB Raman signals compared with single TB solution, yet is still clearly distinguishable. These results clearly indicate the high sensitivity and selectivity of SERS detection for negatively charged analytes with the Au NPs–GO nanocomposites under low pH conditions.
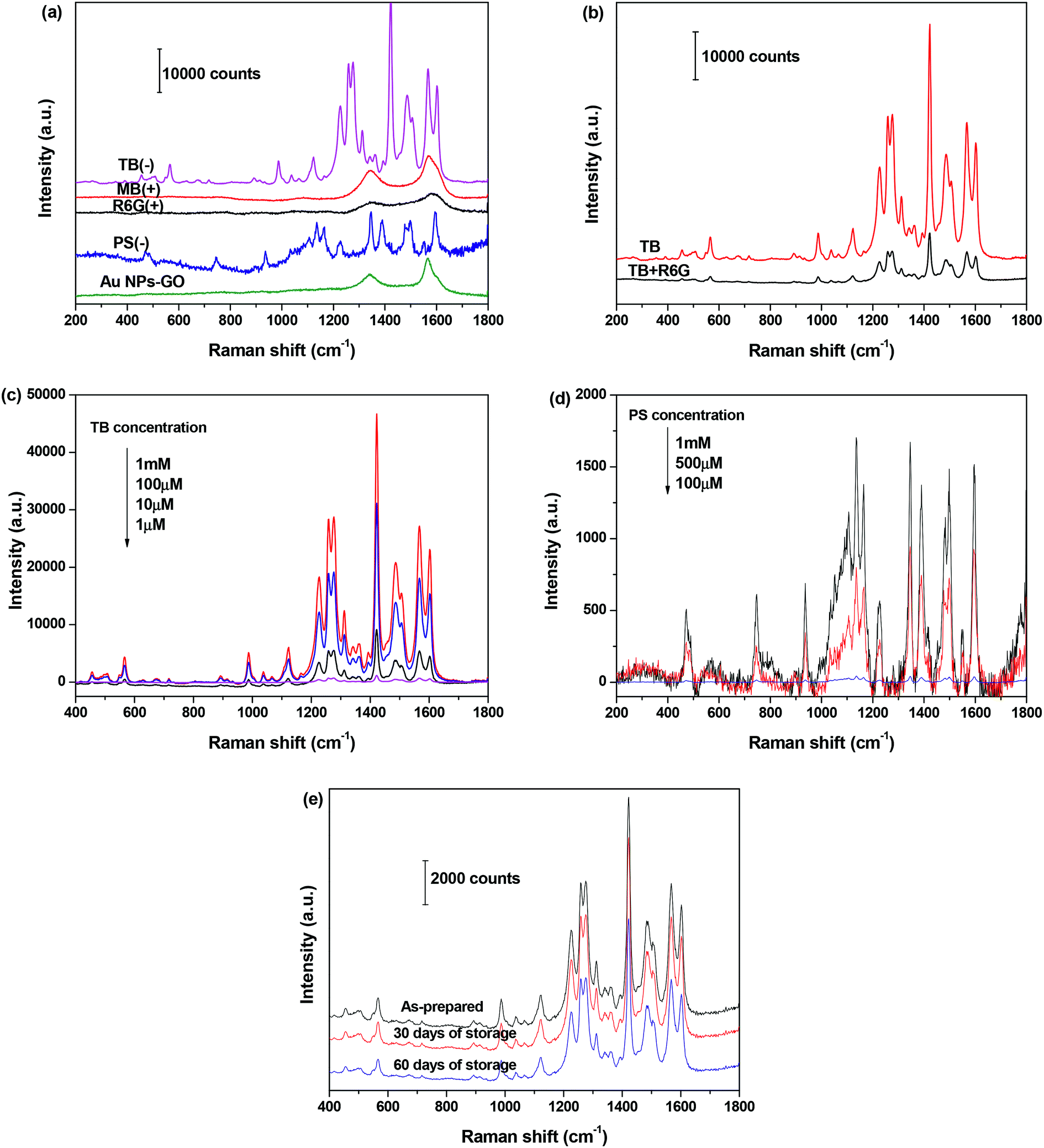 | ||
| Fig. 6 SERS spectra of 1 mM aqueous solutions of R6G (+), MB (+), TB (−) and PS (−) adsorbed on the Au NPs–GO nanocomposites at pH 2.0; (b) SERS spectra of 1 mM TB and dye mixture containing R6G and TB (1 mM each) on the Au NPs–GO nanocomposites; (c and d) SERS spectra of different concentrations of TB and PS adsorbed on Au NPs–GO nanocomposites; (e) SERS spectra of TB (10 μM) absorbed on Au NPs–GO after storage in the refrigerator for 0, 30, or 60 days. | ||
We further measured the SERS spectra as a function of the dye concentration and determined the detection sensitivity. As expected, the Raman signal intensities of TB and PS decreases with decreasing concentrations (Fig. 6(c) and (d)). For TB, the Raman band is clearly distinguishable down to 1 μM concentration, which was 100 times lower than that of PS. The difference in detection sensitivity between the two analysts is attributed to a stronger affinity between TB and substrate than PS and the substrate.
The stability of Au NPs–GO nanocomposites was assessed by determining the Raman peak intensity of adsorbed TB (10 μM) after storage in the refrigerator for 0, 30, or 60 days. The spectra in Fig. 6(e) presents that the Raman signal intensity of TB decreases to approximately 8% and 30% of its initial value after 30 and 60 days of storage, respectively. These results indicate that the Au NPs–GO nanocomposites are stable as a substrate and produce reliable SERS measurements.
Conclusions
The Au NPs–GO nanocomposites were successfully synthesized by decorating Au NPs onto P4VP-grafted GO sheets. The presence of P4VP polymer not only enabled the immobilization of Au NPs with high and uniform distribution on GO sheets, but also endowed the nanocomposites with pH-sensitive characteristics. We demonstrated that the as-prepared Au NPs–GO nanocomposites displayed pH-responsive LSPR changes and assembly behavior. The Au NPs–GO nanocomposites were employed as efficient catalysts for the reduction of 4-NP, and demonstrated pH-dependent catalytic activity. Further investigation indicated that the nanocomposites exhibited selective adsorption and high-efficient SERS detection of negatively-charged dye molecules. Therefore, the Au NPs–GO nanocomposites provide a new opportunity for real-world chemical detection and catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 50702037), Natural Science Foundation of Shanghai Municipality (No. 16ZR1400700), Shanghai Health and Family Planning Commission Project (No. 2012y193) and Research Funds for the Central Universities.References
- E. Priyadarshini and N. Pradhan, Sens. Actuators, B, 2017, 238, 888–902 CrossRef CAS.
- W. Zhou, X. Gao and D. B. Liu, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS PubMed.
- L. Wang, H. L. Li, J. Q. Tian and X. P. Sun, ACS Appl. Mater. Interfaces, 2010, 11, 2987–2991 Search PubMed.
- W. N. Zhang, G. Lu, C. L. Cui, Y. Y. Liu, S. Z. Li, W. J. Yan, C. Xing, Y. G. Robin Chi, Y. H. Yang and F. W. Huo, Adv. Mater., 2014, 26, 4056–4060 CrossRef CAS PubMed.
- J. E. Lee, C. S. Bera, Y. S. Choi and W. I. Lee, Appl. Catal., B, 2017, 214, 15–22 CrossRef CAS.
- J. Li, Z. T. Huang, M. Yang, L. Yan, X. W. Zhang, H. Y. Gao, Y. H. Tang, Q. Q. Ma and G. Wang, New J. Chem., 2015, 39, 2949–2955 RSC.
- J. Wang, X. L. Lu, N. Huang, H. F. Zhang, R. Z. Li and W. K. Li, Mater. Sci. Eng., B, 2017, 224, 1–8 CrossRef CAS.
- S. Bai and X. P. Shen, RSC Adv., 2012, 2, 64–98 RSC.
- Y. W. Zhang, S. Liu, W. B. Lu, L. Wang, J. Q. Tian and X. P. Sun, Catal. Sci. Technol., 2011, 1, 1142–1144 CAS.
- T. Fujigaya, C. Kim, Y. Hamasaki and N. Nakashima, Sci. Rep., 2016, 6, 21314 CrossRef CAS PubMed.
- O. Parlak, A. P. F. Turner and A. Tiwari, J. Mater. Chem. B, 2015, 3, 7434–7439 RSC.
- P. T. Yin, S. Shah, M. Chhowalla and K. B. Lee, Chem. Rev., 2015, 115, 2483–2531 CrossRef CAS PubMed.
- W. G. Ma, X. Y. Lv, D. X. Han, F. H. Li, X. D. Dong and L. Niu, J. Electroanal. Chem., 2013, 690, 111–116 CrossRef CAS.
- J. Lan, H. Y. Zou, Q. Wang, P. Zeng, Y. F. Li and C. Z. Huang, Talanta, 2016, 161, 482–488 CrossRef CAS PubMed.
- Y. Y. Liu, H. C. Li, B. Guo, L. J. Wei, B. Chen and Y. Y. Zhang, Biosens. Bioelectron., 2017, 91, 734–740 CrossRef CAS PubMed.
- Y. Shen, D. J. Rao, Q. L. Sheng and J. B. Zheng, Microchim. Acta, 2017, 184, 3591–3601 CrossRef CAS.
- S. M. Kim and Y. T. Jeong, Trans. Electr. Electron. Mater., 2009, 11, 85–88 CrossRef.
- K. Kim, H. Ryoo, Y. M. Lee and K. S. Shin, J. Colloid Interface Sci., 2010, 342, 479–484 CrossRef CAS PubMed.
- S. Malynych, I. Luzinov and G. Chumanov, J. Phys. Chem. B, 2002, 106, 1280–1285 CrossRef CAS.
- D. X. Li, A. He, Y. Cui and J. B. Li, Chem. Mater., 2007, 19, 412–417 CrossRef CAS.
- D. X. Li, Y. J. Jang, J. U. Lee, J. E. Lee, S. T. Kochuveedu and D. H. Kim, J. Mater. Chem., 2011, 21, 16453–16460 RSC.
- J. E. Lee, K. C. Chung, Y. H. Jang, Y. J. Jang, S. T. Kochuveedu, D. X. Li and D. H. Kim, Anal. Chem., 2012, 84, 6494–6500 CrossRef CAS PubMed.
- H. B. Wang, B. Y. Xia, Y. Yan, N. Li, J. Y. Wang and X. Wang, J. Phys. Chem. B, 2013, 117, 5606–5613 CrossRef CAS PubMed.
- T. Kavitha, I. K. Kang and S. Y. Park, Polym. Int., 2015, 64, 1660–1666 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Y. Cho, H. Y. Seo, Y. S. Yeom, P. Kumar, A. S. Lee, K. Y. Baek and H. G. Yoon, Carbon, 2016, 105, 340–352 CrossRef.
- H. Nandivada, H. Y. Chen, L. Bondarenko and J. Lahann, Angew. Chem., Int. Ed., 2006, 45, 3360–3363 CrossRef CAS PubMed.
- Y. C. Wan, H. F. Teoh, E. S. Tok and C. H. Sow, J. Appl. Phys., 2015, 117, 054304–054312 CrossRef.
- F. Zhang, H. Q. Cao, D. M. Yue and Z. F. Zhou, J. Phys. Chem. C, 2013, 117, 3513–3519 CAS.
- D. X. Li, Q. He, Y. Yang, H. Möhward and J. B. Li, Macromolecules, 2008, 41, 7254–7256 CrossRef CAS.
- T. Ma, W. S. Yang, S. M. Liu, H. J. Zhang and F. Liang, Catalysts, 2017, 7, 38–47 CrossRef.
- G. H. Chang, Y. L. Luo, W. B. Lu, X. Y. Qin, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Catal. Sci. Technol., 2012, 2, 800–806 CAS.
- H. P. Li, L. L. N. Han, J. J. Cooper-White and H. Kim, Nanoscale, 2012, 4, 1355–1361 RSC.
- Y. J. Song, J. H. Lü, B. X. Liu and C. L. Lü, RSC Adv., 2016, 6, 64937–64945 RSC.
- J. S. Fang, Y. W. Zhang, Y. M. Zhou, C. Zhang, S. Zhao, H. X. Zhang and X. L. Sheng, Appl. Surf. Sci., 2017, 392, 36–45 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |