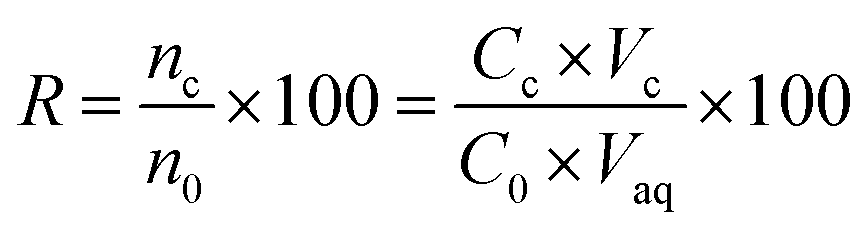DOI:
10.1039/C7RA11030H
(Paper)
RSC Adv., 2017,
7, 56528-56536
Freezing temperature controlled deep eutectic solvent dispersive liquid–liquid microextraction based on solidification of floating organic droplets for rapid determination of benzoylureas residual in water samples with assistance of metallic salt†
Received
7th October 2017
, Accepted 10th December 2017
First published on 15th December 2017
Abstract
An efficient and novel salt-assisted deep eutectic solvent (DES) dispersive liquid–liquid microextraction based on solidification of floating organic droplets combined with high performance liquid chromatography was developed for extraction and determination of four benzoylureas (BUs) in water samples. In this method, a hydrophobic deep eutectic solvent (DES) used as extractant was dissolved in a dispersive-demulsified solvent containing FeCl3. The influence of main factors on the efficiency of this procedure was investigated by one-factor experimental design and central composite design (CCD) successively. Under optimal conditions, the proposed method manifest good recoveries in the range of 82.36–93.82% and high precision (relative standard deviations below 5%). The enrichment factors for the analytes changed from 91 to 97. The limits of detection varied from 0.11 to 0.35 μg L−1 with the linear coefficients greater than 0.999. The method is successfully applied to determine the BUs in different water samples, which proved the potential use of this method in real samples.
1. Introduction
Water is extremely important to living beings, but this natural resource has been continually polluted.1 Pollution sources including industrial manufacture, agricultural production and residents living, among which pesticides used for agricultural might be one contributing factor.2,3 Benzoylureas (BUs) are widely used insect growth regulators, which blocking the molting process of target insects through the inhibition of insect cuticle chitin synthesis. This mechanism will make insects ruptured for malformed cuticle or dead for starvation.4,5
As for the absence of chitin in plants and vertebrates, BUs own low mammalian toxicity.6 While, due to the vast consumption of BUs in farming, their residues may enter to waters by surface runoff, rainfall and subsoil water. Then, human beings may be harmed via the food chain causing chronic exposure and long-term toxicity effects. For above reasons, maximal residue limits (MRLs) of pesticides in water were established by the European Union to evaluate the security of different waters.7 The concentration of pesticide residue is generally lower than the detection limit of the analytical instruments. The matrix effects from other compounds may interfere the determination results. Thus, a preliminary sample preparation before the instrument detection is necessary. This process should be capable to promote the extraction recovery and enrichment of the analytes and remove possible interferences.
Several sample preparation methods have been developed for the extraction of BUs residues from different matrixes. Solid-phase microextraction (SPME) was used to detect BUs in water and juice,8 and dispersive micro-solid-phase (D-μSPE) was used to determine insecticides in honey samples.9 Besides above, some microextraction methods based on liquid extractant were employed for residual detection, like dispersive liquid–liquid microextraction (DLLME), hollow fiber liquid-phase microextraction (HF-LPME) and floated organic drop microextraction (FODME).10–12 Liquid-phase microextraction (LPME) is an important and frequently used mode to analyze trace-level residues of metal and organic compounds.13,14 LPME is on the principle of analytes partitioning as that of liquid–liquid extraction (LLE) but which requires minimal amounts of organic solvent. The advantages of this sample preparation technology are low cost, efficient, and environmentally friendly.15
DLLME is one microextraction form based on LPME with a ternary component solvent system. This sample preparation method was first introduced by Rezaee et al. at 2006 (ref. 16) and has been widely used in the research of pesticides residue.17–19 Three solvents in the conventional DLLME system are extractant, dispersive solvent and sample solution. The dispersive solvent must be miscible with the other two solvents and the extractant must be immiscible with the aqueous phase. After the quick injection of the mixture of extractant and dispersive solvent, the aqueous system turns emulsified immediately. Thanks to the finely dispersive droplets, the extraction equilibrium attained rapidly and efficiently.20
In the conventional DLLME, non-polar, high density and toxic chlorinated solvents are usually used as extraction solvent.21 Hence, centrifugation and micro-syringe were applied to separate the phases and collect the organic extraction solvents settled at the bottom of the centrifugation tube. On the other hand, the number of available extraction solvents is very small which limits the use of the DLLME. As an attempt to overcome these limitations, Leong and Huang introduced a novel sample preparation method named as dispersive liquid–liquid microextraction based on solidification of floating organic droplet (DLLME-SFOD).22 DLLME-SFOD uses extractant with a lower density, a low toxicity and a melting point near room temperature (10–30 °C), such as 1-undecanol, 1-dodecanol, 2-dodecanol and hexadecane. In the DLLME-SFOD, the sample solution in the tube was transferred into an ice bath where the floating organic droplet was allowed to solidify before direct analysis. This method has proved to be efficiently to extract organic and inorganic compounds in environmental samples.23,24 However, the experimental temperature was greatly restricted by the melting point of the available organic extractant. Therefore, we need to search some new extractant to enlarge the application range of the DLLME-SFOD.
Ionic liquids (ILs) are salts completely composed of ions, both inorganic or organic anions which includes asymmetric organic cations involving nitrogen or phosphorus atoms. IL is a class of non-molecular solvents possess a wide variety of physicochemical properties including low melting points, negligible vapor pressure at room temperature and high chemical and electrochemical stability.25 The melting points of the ILs are mostly below ambient temperature, so this sort of solvent was commonly referred to as room temperature ionic liquids (RTILs).26 Due to their special structure, the ILs could be designed by the assembly of the appropriate anion, cation, and substituent alkyl chain in different length. Because of the unique properties of ILs, they have played an important role in sample preparation technique.27–29 And more researchers pay attention to the modification of IL and their usage in the pesticides residue analysis.
From another point of view, the DLLME-SFOD considered as a procedure to make the sample emulsified. Followed the injection of the mixture of dispersive solvent and extraction solvent into the aqueous solution, an oil/water (O/W) emulsion formed. To break up the dispersed system, some chemicals were introduced as demulsifiers, such as acetone and high concentration of salts including NaCl and AlCl3.30–32 These demulsifiers were commonly added into the samples after extraction, which increased the experimental steps and cost.33
Freezing point is the most important factor affecting the choice of extractants in SFOD-DLLME. 1-Dodecanol is a general used extractant, but its melting point is around 25 °C which is not suitable for the microextraction under room temperature due to its easy solidification. Deep eutectic solvents (DESs) are usually made by mixing two different chemicals, a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA). Due to hydrogen bond interactions between them, DESs have lower freezing points than either HBD or HBA.34 Tricaprylmethylammonium chloride ([N8,8,8,1]Cl) is a water-insoluble quaternary ammonium salt, which can relatively uniformly distribute in the solution. In this work, hydrophobic [N8,8,8,1]Cl mixed with 1-dodecanol to form a novel kind of DES. The freezing point of this solvent changed with the mole ratio of the [N8,8,8,1]Cl. After the addition of DES, the liquid–liquid emulsive system is stable which cannot be destroyed even via centrifugation. It has known that the vital step to break down the emulsion is to break the interfacial film.35 Hence, the demulsifier was needed to facilitate the phase separation. The demulsifiers usually used are water miscible organic solvents, which may decrease partition coefficients of analytes into the extraction solvent.36,37 Electrolyte should be another choice. Electrolytes worked as demulsifiers will reduce the zeta potential and the stability of the interfacial film, resulting in the agglomeration of fine droplets of extraction solvent and easy phase separation.38 Metal chlorine salt, like FeCl3, is an appropriate candidate for the demulsifier. Meanwhile, FeCl3 is easily soluble in organic solvents, which makes it possible to prepare an organic solution containing Fe3+ as dispersive solvent and demulsifier.
In this study, we successfully fabricated a freezing temperature controlled DES and proposed using Fe3+ contained organic solution as dispersive-demulsified solvent. Combine the advantages of DES and the dispersive-demulsified solvent, we developed a novel extraction method named as salt-assisted DES dispersive liquid–liquid microextraction based on solidification of floating organic droplets (SA-DES-DLLME-SFOD) to enrich BUs from water samples. In the SA-DES-DLLME-SFOD, the extractant can be designed according to the experiment demands and the salts contained solvent can make the extractant dispersive and sample aqueous demulsified. Above improvements prompted this method to be more efficient and green. Experimental parameters that affected the extraction efficiency were assessed by one-factor experimental design and then optimized by central composite design (CCD). In final, the novel proposed method was evaluated by real water samples with satisfactory results.
2. Experimental
2.1. Reagents and materials
Triflumuron, hexaflumuron, flufenoxuron and lufenuron with 99% purity were bought from Aladdin Industrial Corporation (Shanghai, China). Tricaprylmethylammonium chloride ([N8,8,8,1]Cl), 1-dodecanol, dodecanoic acid and decanoic acid were purchased from Shanghai Macklin Biochemcial Co., LTD (Shanghai, China). HPLC-grade methanol and acetonitrile were obtained from Fisher chemical (Thermo Fisher Scientific, Shanghai, China). Analytical-grade sodium chloride (NaCl) were purchased from Beijing Chemical Factory (Beijing, China). Ferric chloride and aluminium chloride were acquired from Sinopharm Chemical Reagent Company (Beijing, China). Deuterium dimethyl sulfoxide was bought from Sigma-Aldrich (USA). Deionized water was purified using a Milli-Q SP Reagent Water System (Millipore, Bedford, MA, USA). Stock standard solutions were prepared in acetonitrile at the concentration of 100 mg L−1 and stored in the dark at 4 °C. The working standard solutions were prepared by diluting the stock standard solutions to various concentrations in acetonitrile. The calibration curve for each insecticide was obtained by fitting different concentration of pesticides against its HPLC peak area.
2.2. Instrumental and analytical conditions
Quantitative HPLC analysis was performed on an Agilent 1200 series HPLC system (CT, USA) equipped with a binary high-pressure pump, a column oven, an auto-sampler, and a variable-wavelength detector (VWD). The chromatographic separation was performed by a Spursil C18 column (5 μm, 4.6 × 250 mm, Dikma Limited) with a Spursil C18 guard cartridge (5 μm, 2.1 × 10 mm, Dikma Limited) at 25 °C. The mobile phase was composed of acetonitrile and water at the volume ratio of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25. The flow rate was set as 1 mL min−1 with absorb wavelength at 254 nm. The sample injection volume was 10 μL. The analytes were weighed on a Mettler-Toledo AL104 electronic balance (Shanghai, China). A low-speed refrigerated centrifuge (Anting TDL-40B, Shanghai, China) was used for sample treatment, and 0.45 μm micropore membranes (Jingteng, Tianjing, China) were used for sample filtering. 10 mL and 2.5 mL Teflon centrifugal tubes and a 500 μL microsyringe (Anting, Shanghai, China) were used during the microextraction procedure. Fourier transform infrared spectra (FTIR) were recorded by a Spectrum 100 (PerkinElmer, USA), using KBr pellet technique. Freezing point was recorded through a cryogenic thermometer.
:25. The flow rate was set as 1 mL min−1 with absorb wavelength at 254 nm. The sample injection volume was 10 μL. The analytes were weighed on a Mettler-Toledo AL104 electronic balance (Shanghai, China). A low-speed refrigerated centrifuge (Anting TDL-40B, Shanghai, China) was used for sample treatment, and 0.45 μm micropore membranes (Jingteng, Tianjing, China) were used for sample filtering. 10 mL and 2.5 mL Teflon centrifugal tubes and a 500 μL microsyringe (Anting, Shanghai, China) were used during the microextraction procedure. Fourier transform infrared spectra (FTIR) were recorded by a Spectrum 100 (PerkinElmer, USA), using KBr pellet technique. Freezing point was recorded through a cryogenic thermometer.
2.3. Preparation of the real samples
Four water samples from different sources were used as representative samples. These water samples were collected from Yongding River (Fengtai District, Being), Xiaoyue River (Haidian District, Beijing), a well in Liangxiang (Fangshan District, Beijing) and a swimming pool (Haidian District, Being). The water samples were stored in a refrigerator at 4 °C before analysis.
2.4. Preparation of DESs
DESs with different ratio of IL were synthesized as follows: mixing [N8,8,8,1]Cl with 1-dodecanol at the required molar ratio in a beaker at 40 °C with 45 min constant stirring until the liquid turn transparent. Then, the mixture was cooled to room temperature and applied for the microextraction procedure.
2.5. Preparation of dispersive-demulsified solvent
This solvent was prepared by dissolving different amount of metal chloride in organic solvents like methanol, acetonitrile and ethanol.
2.6. Microextraction procedure
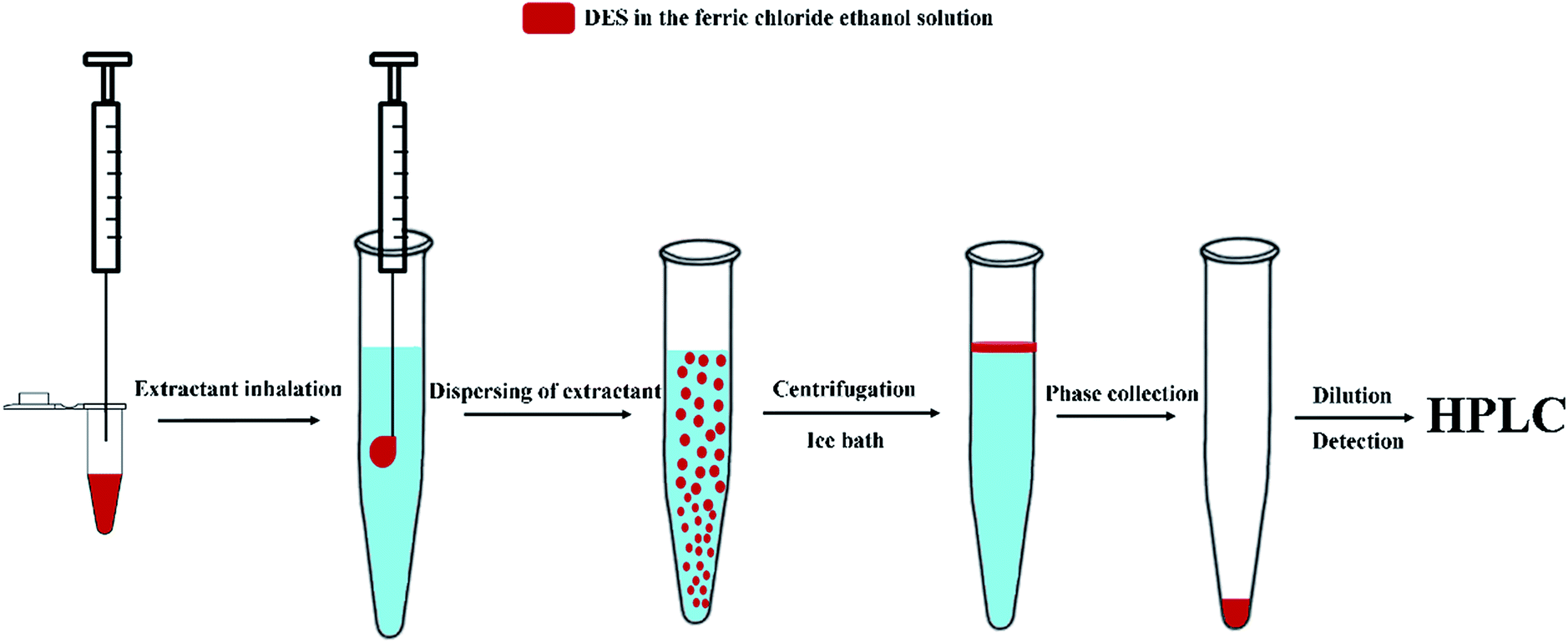
8 mL aqueous solution containing target analytes at different concentrations was introduced into the 10 mL polytetrafluoroethylene tube. Then, 250 μL of 1.2 mol L−1 ferric chloride ethanol solution contained 50 μL DES as dispersive-demulsified solvent was rapidly injected into the solution using a syringe. Immediately, a cloudy solution was formed in the extraction vessel. The tube was closed and centrifuged for 2 min at 2000 rpm and then placed into ice bath for 10 min until the upper phase turn solidified. The bottom water phase was quickly poured out. Finally, the collected DES was diluted by 30 μL ethanol before final HPLC analysis. The microextraction procedure is schematically illustrated in Fig. 1.
 |
| | Fig. 1 The scheme of the microextraction procedure. | |
2.7. Data handling and processing
The design and analysis of the experiments were performed using the MINITAB® Release 16 Statistical Software (State College, PA, USA).
2.8. Calculation of enrichment factor and extraction recovery
The extraction recovery (ER) was defined as the ratio between the amount of the analytes in the organic phase (n0) and the initial amount of the analytes (no) within the sample. The recoveries (R%) and enrichment factors (EFs) of this method were obtained from the following equations:| |
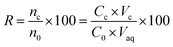 | (1) |
| |
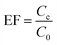 | (2) |
where Cc is defined as the analyte concentration in the organic phase; C0 is the initial concentration of the analytes in water sample; and Vc and Vaq are the volumes of the collected DES phase and sample solution, respectively.
3. Results and discussion
3.1. Freezing points of DES
A eutectic mixture was composed of two or three components and formed through hydrogen bond. Due to the hydrogen bond interaction, the new product will obtain a lower freezing point than all or either of the individual constituents. On the other hand, the freezing point of the extractant will also affect the extraction efficiency in the SFOD. The freezing points of DESs with different mole ratio of [N8,8,8,1]Cl were showed in Fig. 2. It is clear to see that the DES exhibited the lowest melt point when the mole ratio of IL is 50% and this might ascribe to the relatively stronger hydrogen-bond interaction. The freezing points of these DESs were all lower than that of 1-dodecanol. And the freezing points can be fitted to two different linear equations as shown in Table 1.
 |
| | Fig. 2 Freezing points of DESs at different ratio of IL. | |
Table 1 Equations for the freezing pointsa
| Mole ratio of IL |
Linear equation |
R2 |
| Y stands for the freezing point, X represents the mole ratio of IL. |
| <50% |
Y = −72.00X + 23.11 |
0.9946 |
| >50% |
Y = −18.00X − 1.81 |
0.9861 |
3.2. FTIR of DES
FTIR spectra is a useful tool to illustrate the structure of a compound. The FTIR spectra of 1-dodecanol, [N8,8,8,1]Cl and DES(1:2.5) with a mole ratio between IL and 1-dodecanol at 1:2.5 were shown in Fig. 3. In the spectrum of 1-dodecanol, peaks located at 3670 cm−1, 2930 cm −1, and 1060 cm−1 ascribed to the stretching vibrations of the O–H, C–H, and C–O groups, respectively. As for the spectrum of [N8,8,8,1]Cl, the absorption at around 3400 cm−1 should belong to the stretching vibration of O–H. The peak at 1058 cm−1 is ascribed to the C–N vibration. The absorption at 2930 and 2855 cm−1 belong to the asymmetric and symmetric stretching of C–H, respectively. These characteristic absorption of [N8,8,8,1]Cl and 1-dodecanol were presented in the DES(1:2.5) spectrum. The strong band near 1750 cm−1 of DES(1:2.5) can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration. This absorption enhancement should due to the formation of intermolecular hydrogen bond between [N8,8,8,1]Cl and 1-dodecanol.
O vibration. This absorption enhancement should due to the formation of intermolecular hydrogen bond between [N8,8,8,1]Cl and 1-dodecanol.
 |
| | Fig. 3 FTIR spectra of [N8,8,8,1]Cl, 1-dodecanol and DES(1:2.5). | |
3.3. Optimization of SA-DES-DLLME conditions
3.3.1 Effect of the type of microextraction solvent and mode. The type of extractant was an extremely important factor for the extraction process. In this study, four kinds of solvents were used as potential extractant according to the principle of SFOD. These solvents were 1-dodecanol, the mixture of 1-dodecanol and dodecanoic acid at the mole ration of 1:1, the mixture of 1-dodecanol and decylic acid at the mole ration of 1:1 and the mixture of 1-dodecanol and [N8,8,8,1]Cl at the mole ration of 1:1 [DES(1:1)]. We found that if use DES(1:1) as extractant and organic solution as dispersive solvent, the sample solution was still turbid even after centrifugation. However, when we use DES(1:1) and ferric chloride ethanol solution in the DLLME process, the DES(1:1) can be easily obtained. Different phenomena of microextraction processes using DES(1:1) after centrifugation with different concentration of FeCl3 showed in Fig. S1.†Fig. 4 shows the recoveries of target analytes by different extractants. A, B and C represent the methods using 1-dodecanol, the mixture of 1-dodecanol and dodecanoic and the mixture of 1-dodecanol and decylic acid, respectively. D stands for the extraction process which uses DES(1:1) as extractant and 500 mg NaCl as extra demulsifying agent. While, E shows recoveries of the microextraction that applies DES(1:1) as extractant and 1.06 mol L−1 ferric chloride ethanol solution as dispersive-demulsified solvent. Method C demonstrates excellent performance in extracting triflumuron, but poor extraction capacity of other three pesticides. Method E exhibited comparatively better extraction efficiency than others, especially for flufenoxuron and lufenuron. These results indicate that the method, under the assistance of Fe3+, employed [N8,8,8,1]Cl and 1-dodecanol consisted DES as extractant might be an ideal way to determination BUs in water samples.
 |
| | Fig. 4 Effect of the type of microextraction solvent and mode on the extraction of BUs. Extraction conditions: analytes concentration, 50 μg L−1; volume of extractant, 50 μL; dispersive solvent (methanol), 200 μL. Error bars correspond to the relative standard deviation of the mean recovery for n = 5 replicates. | |
3.3.2 Effect of the composition of DES. On the basis of preceding experiments, we found that this IL composed DES was an outstanding extractant. In general, viscosities of eutectic mixtures are mainly affected by the molar ratio between HBD and a HBA. The viscosity of the DES directly affects its application and we found the DES with a lower proportion of 1-dodecanol was not convenient to manipulate in the extraction. In order to get a suitable proportion of the extractant, we explored DESs with different molar ratios (1:1, 1:1.5, 1:2, 1:2.5 and 1:3). Recoveries for different DESs are summarized in Fig. 5. From the results, we can see that DES(1:2.5) has the best extraction ability to retrieve BUs from water samples. Therefore, DES(1:2.5) was selected for further experiments.
 |
| | Fig. 5 Effect of the composition of DES on the extraction of BUs. Extraction conditions: analytes concentration, 50 μg L−1; DES volume, 50 μL; dispersive solvent (methanol), 200 μL; concentration of FeCl3: 1.10 mol L−1. Error bars correspond to the relative standard deviation of the mean recovery for n = 5 replicates. | |
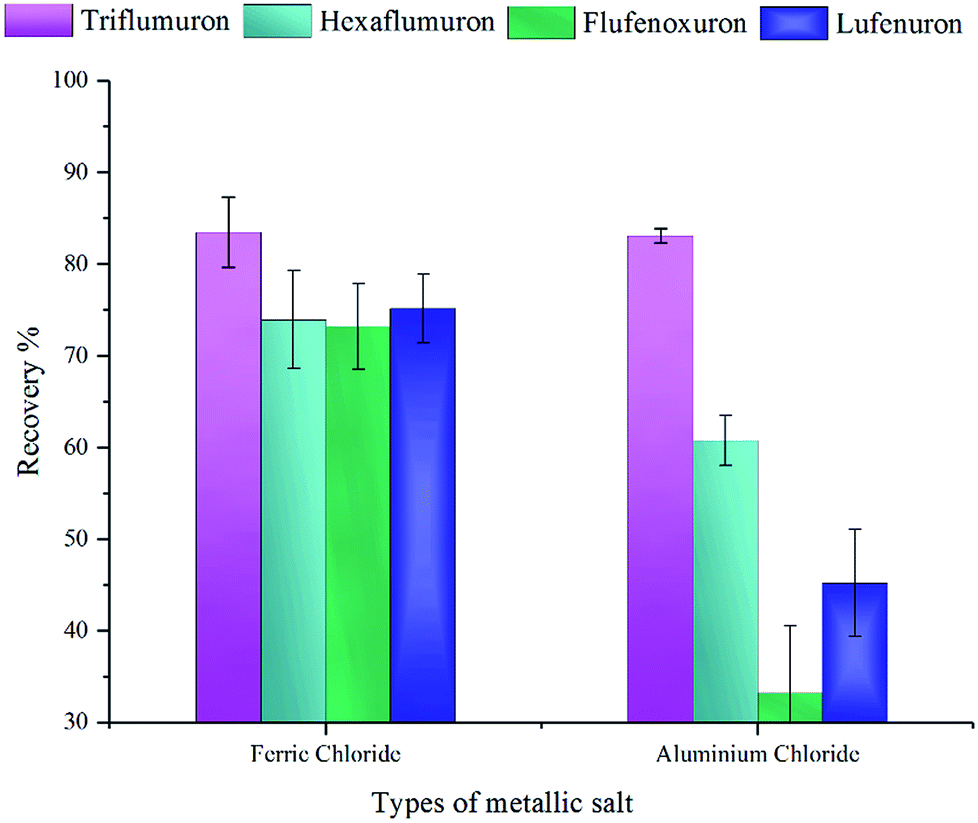
3.3.3 Selection of the types of metallic salts. Salts in this method worked as a demulsifier to decrease the stability of the emulsion solution generated by the adding of DES. The addition of the salts will increase the ionic strength of the emulsion, leading to the flocculation and coalescence of the extractant. And ion in salt with high valence can reduce the zeta potential effectively. Thus, we compared the usage of FeCl3 and AlCl3 ethanol solution. The concentration and volume for these two potential dispersive-demulsified solvents are same. The results in Fig. 6 showed FeCl3 has better extraction ability, especially in the recovery of flufenoxuron. This may due to the stronger interaction between the salt and the emulsion solution. FeCl3 was chosen as the demulsifier in the further study.
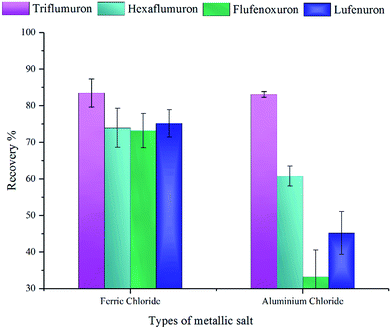 |
| | Fig. 6 Effect of the kind of salts on the extraction of BUs. Extraction conditions: analytes concentration, 50 μg L−1; DES(1:2.5) volume, 50 μL; dispersive solvent (methanol), 200 μL. Error bars correspond to the relative standard deviation of the mean recovery for n = 5 replicates. | |
3.3.4 Selection of the types of solution medium of the dispersive-demulsified solvent. In this method, the dispersive-demulsified solvent not only act as the dispersive solvent to dissolve and disperse the DES, but also served as a solution medium to solve the melt salt. Hence, this solution medium should be miscible with the salt, DES and water sample. Due to this character, methanol, acetonitrile and ethanol were investigated. Fig. 7 showed their effect on extraction according to the value of recovery. It is clearly seen that ethanol can extract four BUs from water samples with best recovery. In addition, ethanol is comparatively more environmentally friendly. Based on these results, ethanol was chosen as the solution medium of the dispersive-demulsified solvent.
 |
| | Fig. 7 Effect of the solution medium of the dispersive-demulsified solvent on the extraction of BUs. Extraction conditions: analytes concentration, 50 μg L−1; DES(1:2.5) volume, 50 μL; dispersive solvent, 200 μL; concentration of FeCl3: 1.10 mol L−1. Error bars correspond to the relative standard deviation of the mean recovery for n = 5 replicates. | |
3.3.5 Central composite design. There are several other significant parameters that affect the recovery of BUs, including volume of dispersive-demulsified solvent (VD), volume of extractant (VE) and concentration of Fe3+ (C). Therefore, an optimization design was required after the one-factor-one-step methodology. In this particular case, a second-order model central composite design (CCD) was used. This optimization involves estimating the coefficients by fitting the response functions to the average recoveries of four BUs, predicting the response of the fitted model and checking its adequacy.39 This statistics-based optimizing strategy makes it possible to evaluate the interacting effects between variables and variable optimization procedure.In this study, a circumscribed CCD with five levels (−α, −1, 0, 1, α) was employed. These three quantity related variables were studied in a multivariate study with 20 random experimental trails. The design with different levels of factors were showed in Table S1.†
The coefficient of determination (R2 and adjusted-R2) can represent the quality of fit of the model equation in some degree. The R2 and adjusted-R2 acquired by this approach were 0.971 and 0.944, respectively. These values showed a good relationship between experimental data and fitted model, as well as the high potential of model in prediction of response. Data analysis gave a semi-empirical expression of the recovery of BUs in the following equation. In this study, the response Y was the average recovery of four BUs (%), VE, VD and C represent the related variable factors.
| Y = −21.325 − 0.062VE + 125.058C + 0.191VD − 0.006VE2 − 68.009C2 − 0.001VD2 − 0.001VD2 + 0.785VEC − 0.001VEVD + 0.024VDC |
The significance of these factors in this model and their interaction were evaluated by the analysis of variance (ANOVA) test. The results were showed in Table 2. The mean squares were determined by dividing the sum of squares for each variation source by their degrees of freedom. The F-value was acquired via dividing the model mean square by the residual mean square. The P-value of factor C and factor VD are lower than 0.05, implying that variations that occur in the recovery should be closely related to the model, rather than with the experimental error. Whereas, 0.05 < P value <0.10 indicates a relative effect on the extraction. As can be seen, factor VE also affect the extraction recovery of BUs.
Table 2 ANOVA for central composite design
| Source of variation |
Sum of square |
Dfa |
Mean square |
F-Value b |
P-Value |
| Df: Degrees of freedom. Test for comparing model variance with residual (error) variance. |
| VE |
978.48 |
1 |
0.307 |
0.05 |
0.832 |
| C |
137.68 |
1 |
135.241 |
20.95 |
0.001 |
| VD |
224.75 |
1 |
79.514 |
12.32 |
0.006 |
| VE × VE |
30.26 |
1 |
77.143 |
11.95 |
0.006 |
| C × C |
200.11 |
1 |
260.376 |
40.33 |
0.000 |
| VD × VD |
430.39 |
1 |
430.389 |
66.67 |
0.000 |
| VE × C |
123.31 |
1 |
13.308 |
19.10 |
0.001 |
| VE × VD |
20.57 |
1 |
20.57 |
3.19 |
0.105 |
| C × VD |
2.88 |
1 |
2.885 |
0.45 |
0.519 |
| Lack of fit |
64.56 |
5 |
12.911 |
|
|
| Pure error |
0.00 |
5 |
0.00 |
|
|
| Total SS |
2213.00 |
19 |
|
|
|
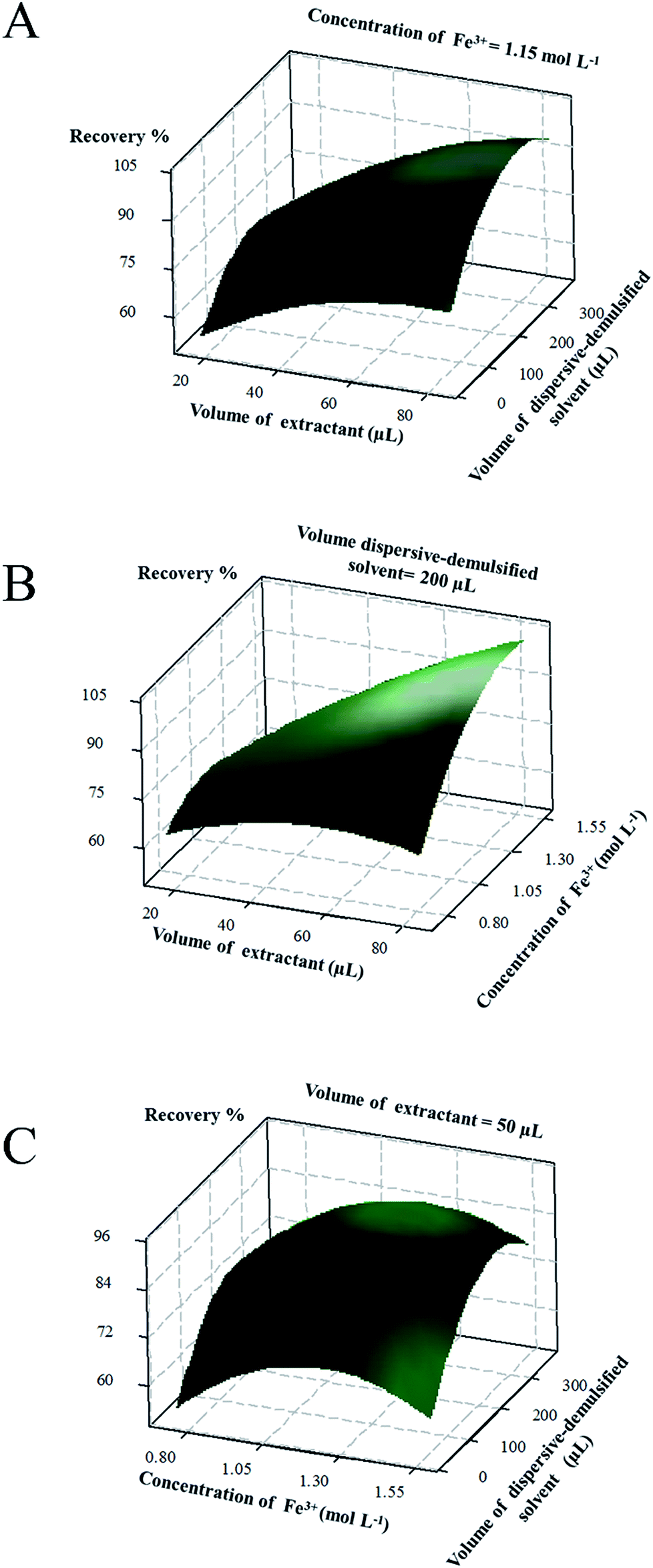
Three-dimensional response surface plot for the average recovery (%) versus variables were represented in Fig. 8. Fig. 8a depicts the response surface obtained by plotting DES volume versus dispersive-demulsified solvent volume with the salt concentration fixed at 1.15 mol L−1. From the results, we can see that the maximum recovery for the BUs was achieved with DES volume between 40 and 60 μL and dispersive-demulsified solvent volume between 150 and 250 μL. Higher DES amount tended to result in a decrease of the recovery of the target compounds. This can be explained as that the increasing of the amount of DES increased the partitioning of the analytes into extractant, but excess amount of DES did little to enhance the recovery. Simultaneously, a small volume of dispersive-demulsified solvent cannot effectively disperse the DES, whereas, a large volume of solvent increases the solubility of the analytes in water leading to a low recovery. Fig. 8b demonstrated the effects of ferric chloride concentration and DES volume. It showed that concurrently decreasing the concentration of ferric chloride enhances the performance of the microextraction process. This situation might be explained by the salting-out effect and emulsion break-up ability derived from strong electrolyte salts. The increasing of the salt concentration will decrease the solubility of the target analytes in the aqueous phase and enhance their transference to the organic phase consequently. The concentration of FeCl3 and dispersive-demulsified solvent volume in the model equation had greater relevance as seen in Fig. 8c. Their interaction was significant and simultaneously changing of these two factors (increasing the salt concentration to 1.2 mol L−1 and the volume to 250 μL) makes the extraction performance enhanced remarkably.
 |
| | Fig. 8 Response surface model for average recovery of four BUs. (A) Volume of extractant (μL) vs. volume of dispersive-demulsified solvent (μL), (B) volume of extractant (μL) vs. concentration Fe3+ (mol L−1); (C) concentration Fe3+ (mol L−1) vs. volume of dispersive-demulsified solvent (μL). | |
According to the model equation and the response surface plots, the final optimal extraction conditions for the SA-DES-DLLME-SFOD method were chosen as 50 μL of DES, 1.2 mol L−1 of ferric chloride and 250 μL of dispersive-demulsified solvent.
3.4. Analytical features of the method
After the optimization of relevant parameters, quantitative characteristics of this proposed method were studied and the analytical features were summarized in Table 3. The linearity range of calibration curve for triflumuron, hexaflumuron, flufenoxuron and lufenuron was 2.5–500, 2.5–500, 5–500 and 5–500 μg L−1, respectively. The correlation coefficients were changed from 0.9991 to 0.9994. The limit of detection (LOD) was defined as a signal-to-noise ratio (S/N) of 3, and the values in this proposed method for the target BUs were between 0.11 to 0.35 μg L−1. Moreover, the reproducibility and precision in terms of intra- and inter-day precision were evaluated by carrying out independent measurements of the BUs at 50 μg L−1 each day with five replications and on five consecutive days with three replications, respectively. The precision, expressed as relative standard deviations (RSDs) of recovery, was lower than 5% for all the BUs. The EFs were in the range of 90–97 and the extraction recoveries changed from 82.36 to 93.82%. These analytical merits indicated the high sensitivity and acceptable repeatability of the SA-DES-DLLME-SFOD.
Table 3 The analytical performance characteristics of the SA-DES-DLLME-SFOD combined with HPLC-DADa
| Fungicides |
Linear equation |
R2 |
Linearity (μg L−1) |
Intra-day precisionb (RSD, %) |
Inter-day precisionc (RSD, %) |
LOD (μg L−1) |
EF |
Recovery (%) |
| RSD: relative standard deviation; LOD: limits of detection (S/N = 3). Method precision within a day (for every concentration, n = 5). Method precision among three days (for every concentration, n = 3). |
| Triflumuron |
Y = 2.174X + 4.516 |
0.9993 |
2.5–500 |
3.26 |
3.32 |
0.11 |
97 |
93.82 |
| Hexaflumuron |
Y = 2.590X + 1.985 |
0.9991 |
2.5–500 |
2.05 |
2.74 |
0.11 |
93 |
91.19 |
| Flufenoxuron |
Y = 1.393X − 0.912 |
0.9991 |
5–500 |
2.25 |
4.63 |
0.21 |
91 |
82.36 |
| Lufenuron |
Y = 1.885X − 1.185 |
0.9994 |
5–500 |
2.27 |
3.12 |
0.35 |
90 |
82.86 |
3.5. Application of real samples
Four water samples (Yongding River water, Xiaoyue River water, well water and swimming pool water) treated with the target pesticides were sampled and analyzed following SA-DES-DLLME-SFOD procedure to clarify its applicability and reliability. No target compounds were detected in blank water samples showed these water samples were free of BUs contamination. For the sake of assessing the matrix effect of this proposed method, the water samples were fortified at 200 and 50 μg L−1 of four BUs. The recoveries of the target chemicals in spiking water samples were summarized in Table 4. As can be seen, the extraction recoveries were changed from 75.7 to 105.2% with RSDs in the range of 1.0–6.9%. These results demonstrated that the SA-DES-DLLME-SFOD method could be used in trace analysis of BUs in real water samples with good relative recoveries and no obvious interferences. Fig. 9 shows the chromatograms attained from the Yongding River based on the optimum SA-DES-DLLME-SFOD method.
Table 4 Analytical performance of the method for environmental samples (n = 5)a
| Sample |
Analytes |
Spiked level (μg L−1) |
Recovery ± RSD (%) |
Sample |
Analytes |
Spiked level (μg L−1) |
Recovery ± RSD (%) |
| A, Yongding River water; B, Xiaoyue River water; C, well water; D, swimming pool water. |
| A |
Triflumuron |
200 |
106.28 ± 1.9 |
B |
Triflumuron |
200 |
105.20 ± 1.5 |
| 50 |
89.20 ± 3.7 |
50 |
85.27 ± 5.4 |
| Hexaflumuron |
200 |
82.22 ± 4.9 |
Hexaflumuron |
200 |
77.57 ± 4.2 |
| 50 |
87.06 ± 5.0 |
50 |
78.56 ± 6.9 |
| Flufenoxuron |
200 |
75.94 ± 2.9 |
Flufenoxuron |
200 |
81.84 ± 2.0 |
| 50 |
84.28 ± 2.6 |
50 |
79.6 ± 6.5 |
| Lufenuron |
200 |
96.31 ± 5.1 |
Lufenuron |
200 |
93.38 ± 6.0 |
| 50 |
88.75 ± 4.3 |
50 |
78.82 ± 3.9 |
| C |
Triflumuron |
200 |
100.10 ± 3.4 |
D |
Triflumuron |
200 |
97.34 ± 2.2 |
| 50 |
88.67 ± 6.0 |
50 |
86.50 ± 2.2 |
| Hexaflumuron |
200 |
75.66 ± 5.7 |
Hexaflumuron |
200 |
84.69 ± 3.2 |
| 50 |
85.32 ± 3.7 |
50 |
84.91 ± 5.9 |
| Flufenoxuron |
200 |
81.42 ± 3.6 |
Flufenoxuron |
200 |
88.78 ± 2.9 |
| 50 |
77.94 ± 3.0 |
50 |
97.45 ± 2.3 |
| Lufenuron |
200 |
85.54 ± 2.6 |
Lufenuron |
200 |
82.65 ± 5.6 |
| 50 |
76.07 ± 1.0 |
50 |
79.66 ± 1.8 |
 |
| | Fig. 9 The typical chromatograms of benzoylureas in Yongding River water. (1) Triflumuron, (2) hexaflumuron, (3) flufenoxuron, (4) lufenuron. | |
4. Conclusions
In this work, a novel SA-DES-DLLME-SFOD was developed for trace analysis of BUs in water samples. Frequently-used highly toxic organic extractant in DLLME was substituted by a kind of hydrophobic DES whose freezing point can be controlled. The hydrophobic DES, synthesized from [N8,8,8,1]Cl and 1-dodecanol, with lower density than water was suitable to use in the DLLME-SFOD. During the extraction procedure, ferric chloride ethanol solution was applied as the dispersive-demulsified solvent to disperse the extractant and break up the emulsion system in sequence. Owing to the introduction of DES and dispersive-demulsified solvent, we can employ the SFOD in relatively low temperature avoid using extra heating equipment and demulsifier. Hence, the extraction efficiency was significantly increased with extraction time reducing. The parameters that affected the extraction performance were optimized by CCD. And the performance of this proposed method was satisfactory for the determination of BUs in different environmental water samples. The SA-DES-DLLME-SFOD was proved to be a potential sample preparation technique to detect BUs in real water samples.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support of the Fundamental Research Funds for the Central public welfare research institutes (Project No. ZKKT17007).
References
- J. L. de O. Arias, C. Rombaldi, S. S. Caldas and E. G. Primel, J. Chromatogr. A, 2014, 1360, 66–75 CrossRef CAS PubMed.
- S. S. Caldas, C. M. Bolzan, M. B. Cerqueira, D. Tomasini, E. B. Furlong, C. Fagundes and E. G. Primel, J. Agric. Food Chem., 2011, 59, 11918–11926 CrossRef CAS PubMed.
- O. Novillo, J. F. Pertusa and J. Tomas, Sci. Total Environ., 2017, 598, 1130–1139 CrossRef CAS PubMed.
- Q. Zhou, G. Wang and G. Xie, J. Sep. Sci., 2013, 36, 2323–2329 CrossRef CAS PubMed.
- M. M. Galera, M. D. Garcia and R. S. Valverde, Talanta, 2008, 76, 815–823 CrossRef CAS PubMed.
- L. Chen, J. Chen, Y. Guo, J. Li, Y. Yang, L. Xu and F. Fu, Food Chem., 2014, 143, 405–410 CrossRef CAS PubMed.
- X. Yang, M. Yang, B. Hou, S. Li, Y. Zhang, R. Lu and S. Zhang, J. Sep. Sci., 2014, 37, 1996–2001 CrossRef CAS PubMed.
- M. Mei, X. Huang, K. Liao and D. Yuan, Anal. Chim. Acta, 2015, 860, 29–36 CrossRef CAS PubMed.
- P. Zhang, X. Cui, X. Yang, S. Zhang, W. Zhou, H. Gao and R. Lu, J. Sep. Sci., 2016, 39, 412–418 CrossRef CAS PubMed.
- C. Ruan, X. Zhao and C. Liu, J. Sep. Sci., 2015, 38, 2931–2937 CrossRef CAS PubMed.
- J. Zhou, R. Liu, G. Song and M. Zhang, Anal. Lett., 2009, 42, 1805–1819 CrossRef CAS.
- C.-H. Wang, X.-X. Ma, C. Wang, Q.-H. Wu and Z. Wang, Chin. Chem. Lett., 2014, 25, 1625–1629 CrossRef CAS.
- S. Dadfarnia and A. M. Haji Shabani, Anal. Chim. Acta, 2010, 658, 107–119 CrossRef CAS PubMed.
- M. Andraščíková, E. Matisová and S. Hrouzková, Sep. Purif. Rev., 2014, 44, 1–18 CrossRef.
- L. Guo, S. Tan, X. Li and H. K. Lee, J. Chromatogr. A, 2016, 1438, 1–9 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- B. Jin, L. Xie, Y. Guo and G. Pang, Food Res. Int., 2012, 46, 399–409 CrossRef CAS.
- P. Jovanov, V. Guzsvány, M. Franko, S. Lazić, M. Sakač, I. Milovanović and N. Nedeljković, Food Res. Int., 2014, 55, 11–19 CrossRef CAS.
- E. G. Primel, S. S. Caldas, L. C. Marube and A. L. V. Escarrone, Trends Environ. Anal. Chem., 2017, 14, 1–18 CrossRef CAS.
- W. Ahmad, A. A. Al-Sibaai, A. S. Bashammakh, H. Alwael and M. S. El-Shahawi, TrAC, Trends Anal. Chem., 2015, 72, 181–192 CrossRef CAS.
- L. Kocúrová, I. S. Balogh, J. Šandrejová and V. Andruch, Microchem. J., 2012, 102, 11–17 CrossRef.
- M. I. Leong and S. D. Huang, J. Chromatogr. A, 2008, 1211, 8–12 CrossRef CAS PubMed.
- X. You, Z. Xing, F. Liu and X. Zhang, Anal. Chim. Acta, 2015, 875, 54–60 CrossRef CAS PubMed.
- X. C. Wang, B. Shu, S. Li, Z. G. Yang and B. Qiu, Talanta, 2017, 162, 90–97 CrossRef CAS PubMed.
- J. An, M. J. Trujillo-Rodriguez, V. Pino and J. L. Anderson, J. Chromatogr. A, 2017, 1500, 1–23 CrossRef CAS PubMed.
- H. N. Abdelhamid, TrAC, Trends Anal. Chem., 2016, 77, 122–138 CrossRef CAS.
- M. Vian, C. Breil, L. Vernes, E. Chaabani and F. Chemat, Current Opinion in Green and Sustainable Chemistry, 2017, 5, 44–48 CrossRef.
- O. Filippou, D. Bitas and V. Samanidou, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1043, 44–62 CrossRef CAS PubMed.
- L. Ruiz-Aceituno, M. L. Sanz and L. Ramos, TrAC, Trends Anal. Chem., 2013, 43, 121–145 CrossRef CAS.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Talanta, 2015, 132, 769–774 CrossRef CAS PubMed.
- P. Sricharoen, N. Limchoowong, S. Techawongstien and S. Chanthai, Food Chem., 2016, 203, 386–393 CrossRef CAS PubMed.
- P. Sricharoen, N. Limchoowong, S. Techawongstien and S. Chanthai, Arabian J. Chem., 2017 DOI:10.1016/j.arabjc.2016.12.020.
- Y. Wang, M. Wang, H. Wang, W. Wang, J. Wu and X. Wang, Food Chem., 2015, 173, 1213–1219 CrossRef CAS PubMed.
- M. B. Taysun, E. Sert and F. S. Atalay, J. Chem. Eng. Data, 2017, 62, 1173–1181 CrossRef CAS.
- E. Del Carpio, S. Rodríguez, M. Rondón and B. Borges, J. Pet. Sci. Eng., 2014, 122, 542–550 CrossRef CAS.
- Y. Xia, M. Cheng, F. Guo, X. Wang and J. Cheng, Anal. Chim. Acta, 2012, 724, 47–53 CrossRef CAS PubMed.
- H. Chen, R. Chen and S. Li, J. Chromatogr. A, 2010, 1217, 1244–1248 CrossRef CAS PubMed.
- G. M. Fair, J. C. Geyer and D. A. Okun, Water and Wastewater Engineering, John Wiley & Sons, Inc., New York, 1968 Search PubMed.
- R. Sousa, V. Homem, J. L. Moreira, L. M. Madeira and A. Alves, Anal. Methods, 2013, 5, 2736 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11030h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*