DOI:
10.1039/C7RA11860K
(Paper)
RSC Adv., 2017,
7, 54881-54891
Late stage iodination of biologically active agents using a one-pot process from aryl amines†
Received
27th October 2017
, Accepted 26th November 2017
First published on 1st December 2017
Abstract
A simple and effective one-pot tandem procedure that generates aryl iodides from readily available aryl amines via stable diazonium salts has been developed. The operationally simple procedure and mild conditions allow late-stage iodination of a wide range of aryl compounds bearing various functional groups and substitution patterns. A novel synthetic strategy involving the preparation of nitroaryl compounds followed by a chemoselective tin(II) dichloride reduction and the use of the one-pot diazotisation–iodination transformation was also developed. The general applicability of this approach was demonstrated with the preparation of a number of medicinally important compounds including CNS1261, a SPECT imaging agent of the N-methyl-D-aspartate (NMDA) receptor and IBOX, a compound used to detect amyloid plaques in the brain.
Introduction
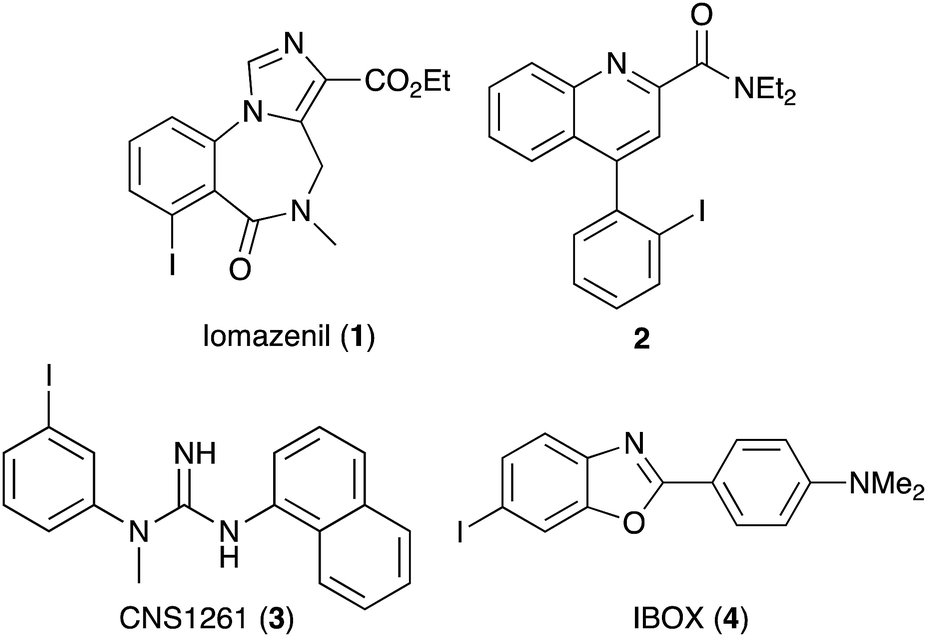
Aryl iodides are ubiquitous synthetic building blocks, used routinely in organic chemistry. The reactivity of the carbon–iodine bond allows facile oxidative addition and subsequent application as organometallic reagents in cross-coupling reactions.1 Aryl iodides are also found as structural components in a wide-range of medicinally important compounds, such as iomazenil (1, Fig. 1),2 a specific ligand for the central-type benzodiazepine receptor and quinoline-2-carboxamide 2,3 a high affinity agent for the translocator protein (TSPO). Aryl and heteroaryl iodides in radioiodinated form are routinely used in single photon emission computed tomography (SPECT) imaging for the clinical diagnosis of disease and drug development.4 For example, [123I]CNS1261 (3) is a SPECT imaging agent for the N-methyl-D-aspartate (NMDA) receptor,5 while a radioiodine version of IBOX (4) is used to detect amyloid plaques in the brain.6
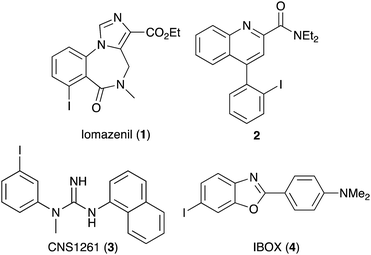 |
| | Fig. 1 Structures of biologically active aryl iodides. | |
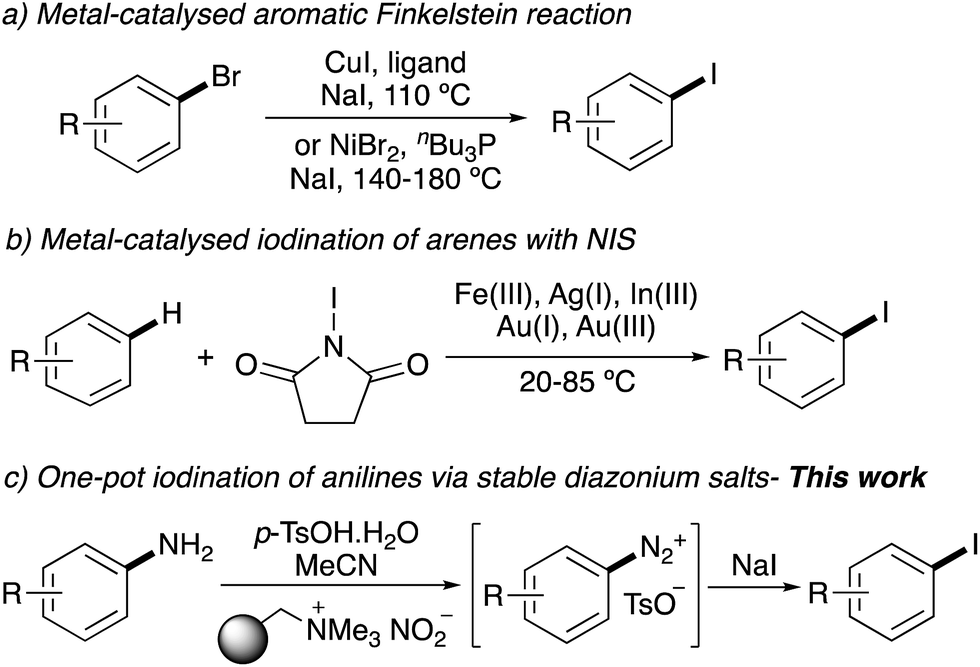
The importance of aryl iodides has led to a wide range of general synthetic approaches. Traditional methods include electrophilic aromatic substitution7 and directed ortho-lithiation–iodination sequences.8 Current developments and understanding of transition metal catalysis has allowed the discovery of many aryl iodination reactions.9 These include the copper- or nickel-catalysed Finkelstein reaction of aryl bromides (Scheme 1a).10,11 Copper-catalysed iododeboronation of aryl boronic acids has also been used for the preparation of aryl iodides,12 and recently, this transformation has been extended for the incorporation of radioiodine.13 Another approach that has received much attention is the Lewis acid activation of N-iodosuccinimide (NIS) and the subsequent iodination of activated arenes (Scheme 1b).14,15 Lewis acids such as silver(I) triflimide have been used to tune the NIS activation for clean mono-iodination of highly activated arenes.14f Despite these advances, there are still limitations with many of these transformations. The metal-catalysed iodination of aryl bromides requires elevated temperatures, while the Lewis acid-NIS method is restricted to activated arenes.
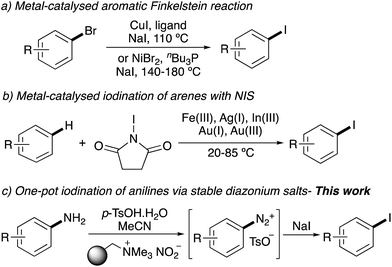 |
| | Scheme 1 Recently developed approaches for iodination of aryl compounds. | |
The other traditional approach for the preparation of aryl iodides involves the conversion of aryl amines to diazonium salts, followed by a Sandmeyer-type reaction with iodide nucleophiles.16 However, the standard conditions of diazonium salt formation involves strong acids (e.g. HCl or H2SO4) and the use of sodium nitrite, which can lead to release of nitrogen oxides. In addition, various diazonium salts are unstable and potentially explosive. These issues have restricted the large-scale application of this approach, particularly for complex biologically active targets. A number of groups have overcome the limitations associated with the standard conditions of diazonium salt formation, by using polymer-supported nitrite reagents.17 It has been shown that these can be used in combination with mild acidic conditions (e.g. p-TsOH) for the preparation of thermally stable tosylate diazonium salts.17c,18
Using these advances as a starting point, we were interested in developing a general one-pot, two-step tandem iodination of readily available aryl amines via stable diazonium salts. In addition, we wanted to develop a novel synthetic strategy that would allow the assembly of biologically relevant compounds, that could then be used with this mild one-pot procedure for late-stage iodination.19 Here we report an operationally simple, one-pot tandem iodination of aryl amines using a polymer supported nitrite reagent and p-toluenesulfonic acid as the proton source (Scheme 1c). A general strategy for the preparation of iodinated medicinal agents using a chemoselective tin(II) chloride reduction of nitroaryl precursors followed by the one-pot tandem diazotisation–iodination procedure is also described.
Results and discussion
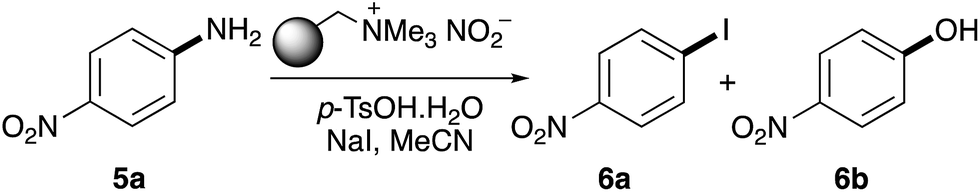
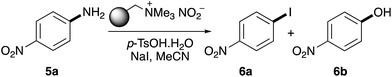
Previous studies have shown that polymer-supported nitrite reagents can be easily formed by the ion exchange of tetraalkylammonium functionalised resins with an aqueous solution of sodium nitrite.17 In this current study, nitrite supported by the tetraalkylammonium resin Amberlyst A-26 was prepared and used to investigate the optimisation of the one-pot diazotisation–iodination of 4-nitroaniline (5a). Initial trials at room temperature (20 °C) and using acetonitrile as the solvent, showed that while diazonium salt formation was fast, the subsequent substitution reaction formed phenol 6b as the major product (Table 1, entry 1). However, phenol formation could be completely suppressed by cooling the reaction to 10 °C. After a one-hour reaction time, this gave 53% conversion to iodide 6a (entry 2). Analysis of each step of the process at this temperature showed that, while diazonium salt formation was still fast (<15 minutes), iodination required a longer reaction time. Increasing the overall reaction time up to two hours showed an increase in conversion to 85% (entry 4). This was further optimised by initially conducting the reaction at 10 °C and then warming the mixture to room temperature after 1 hour (entry 5). This led to the generation of iodide 6a as the sole product in quantitative conversion.
Table 1 Optimisation of the one-pot diazotisation–iodination of 4-nitroaniline (5a)
|
| Entry |
Temp. (°C) |
Time (h) |
6aa (%) |
6ba (%) |
| Conversion measured by 1H NMR spectroscopy. |
| 1 |
20 |
1 |
15 |
60 |
| 2 |
10 |
1 |
53 |
— |
| 3 |
10 |
1.5 |
70 |
— |
| 4 |
10 |
2 |
85 |
— |
| 5 |
10–20 |
2 |
>95 |
— |
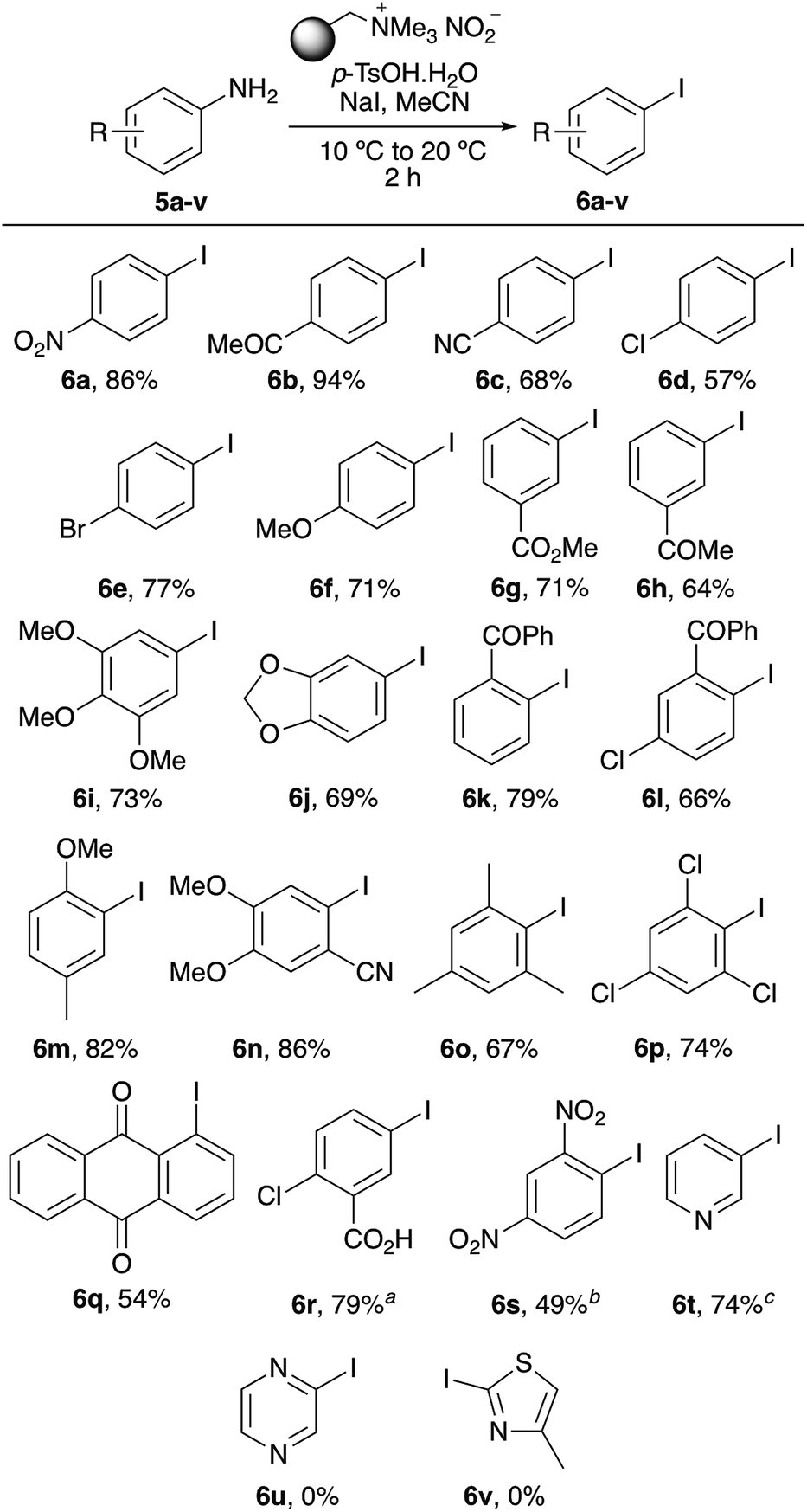
Using the optimised protocol, the scope of the one-pot tandem diazotisation–iodination process was explored as a general method for the iodination of aryl amines (Scheme 2). The process was found to be compatible with a wide range of substitution patterns and functional groups and could tolerate electron-rich or electron-deficient substituents. In general, the majority of substrates investigated all gave the iodides as sole products, typically in good to high yields. Sterically encumbered 2,6-disubstituted anilines such as 5o and 5p also yielded the corresponding iodides (6o and 6p), in 67% and 74%, respectively, after a two-hour reaction time. Only very electron-deficient substrates required further optimisation. Anilines bearing two strongly electron-deficient groups [e.g. 5-amino-2-chlorobenzoic acid (5r) or 2,4-dinitroaniline (5s)] required longer reaction times, while 3-aminopyridine (5t) could be converted to the corresponding iodide 6t in 74% yield but required more forcing conditions (60 °C). The only amino-substituted aryl compounds that were found not to react during this process were heterocyclic ring systems bearing multiple heteroatoms (e.g. 5u and 5v). The presence of multiple heteroatoms deactivates the amino group such that diazonium salt formation under these mild conditions is not possible.
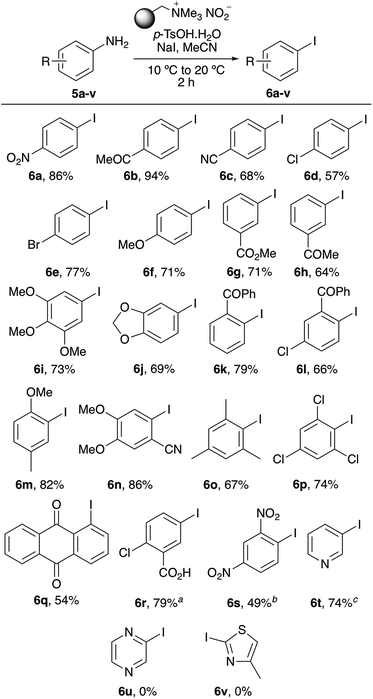 |
| | Scheme 2 Scope of one-pot diazotisation–iodination process. For each reaction 3 eq. of polymer-supported nitrite/p-TsOH and 2 eq. of NaI were used. aReaction required 24 h. bReaction required 16 h. cReaction was performed at 60 °C for 16 h. | |
As well as a broad substrate scope, avoiding toxic reagents and harsh reaction conditions, the other key advantage of this one-pot tandem approach is a very mild work-up procedure, involving simple filtration of the polymer-supported nitrite reagent and an aqueous wash of the organic layer. The Amberlyst A-26 resin can also be re-used by further ion exchange reactions with sodium nitrite.
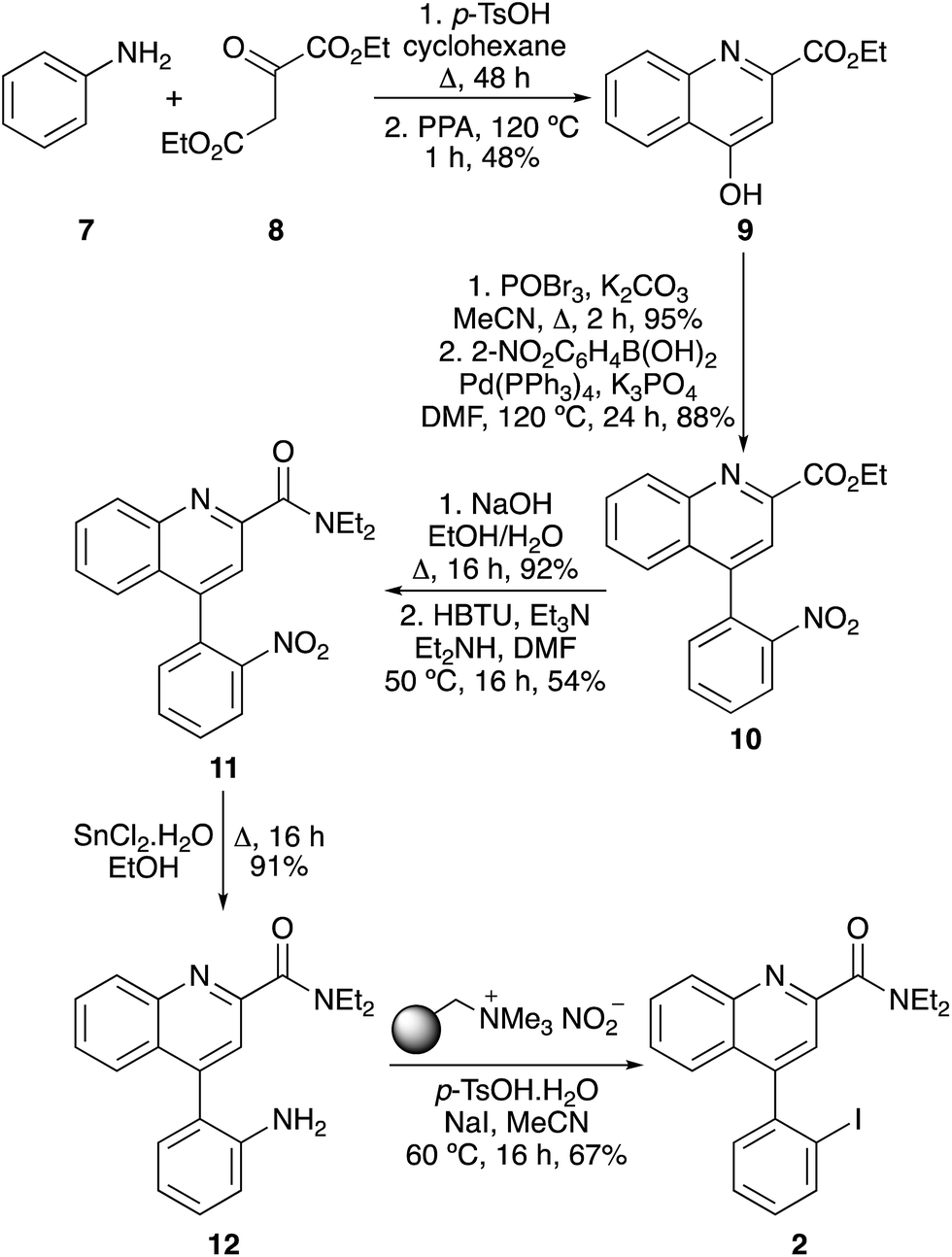
The next stage of this work was the application of this one-pot, two step method for the synthesis of more complex biologically active aryl iodides. This required the development of a synthetic strategy where the key amino group was masked during the early stages of each synthetic route, allowing preparation of the target scaffold, before being released at the penultimate stage for the final step iodination. For each target investigated in this study, it was found that the use of a nitro group permitted facile synthesis of each biologically active precursor, which could then be reduced to give the required amino group for the one-pot process. The first target was quinoline-2-carboxamide 2, a high affinity agent (Ki 5 nM) for the translocator protein (TSPO), an important biological target for the study of focal neuroinflammation in diseases such as brain cancer, stroke and neurodegeneration.3,20 The quinoline-2-carboxamide core was rapidly assembled by condensation of aniline (7) with diethyl oxalacetate (8), followed by ring closure of the resulting imine under acidic conditions (Scheme 3).3 Bromination of 9, followed by a Suzuki–Miyaura reaction with 2-nitrophenylboronic acid completed the preparation of the 4-phenylquinoline scaffold 10, with introduction of the key nitro group. Hydrolysis of the ethyl ester and coupling with diethylamine in the presence of HBTU gave the corresponding diethylamide 11. Following the preparation of 2′-nitrophenyl analogue 11, it was found that chemoselective reduction to the corresponding aniline could be achieved efficiently under mild conditions using tin(II) chloride.21 The one-pot tandem diazotisation–iodination process was then optimised for the final stage conversion to TSPO affinity agent 2. Due to the bulky ortho-substituent, the reaction required a temperature of 60 °C for 16 h to achieve completion. This gave quinoline-2-carboxamide 2 as the sole product in 67% yield.
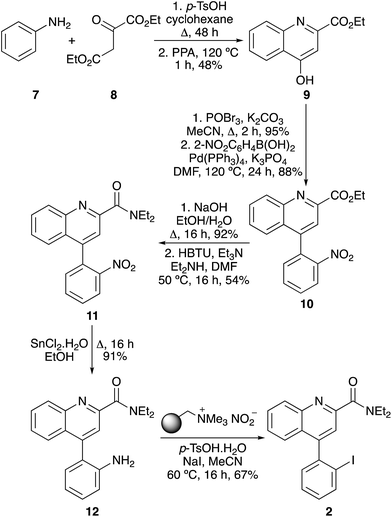 |
| | Scheme 3 Synthesis of TSPO targeting agent 2. | |
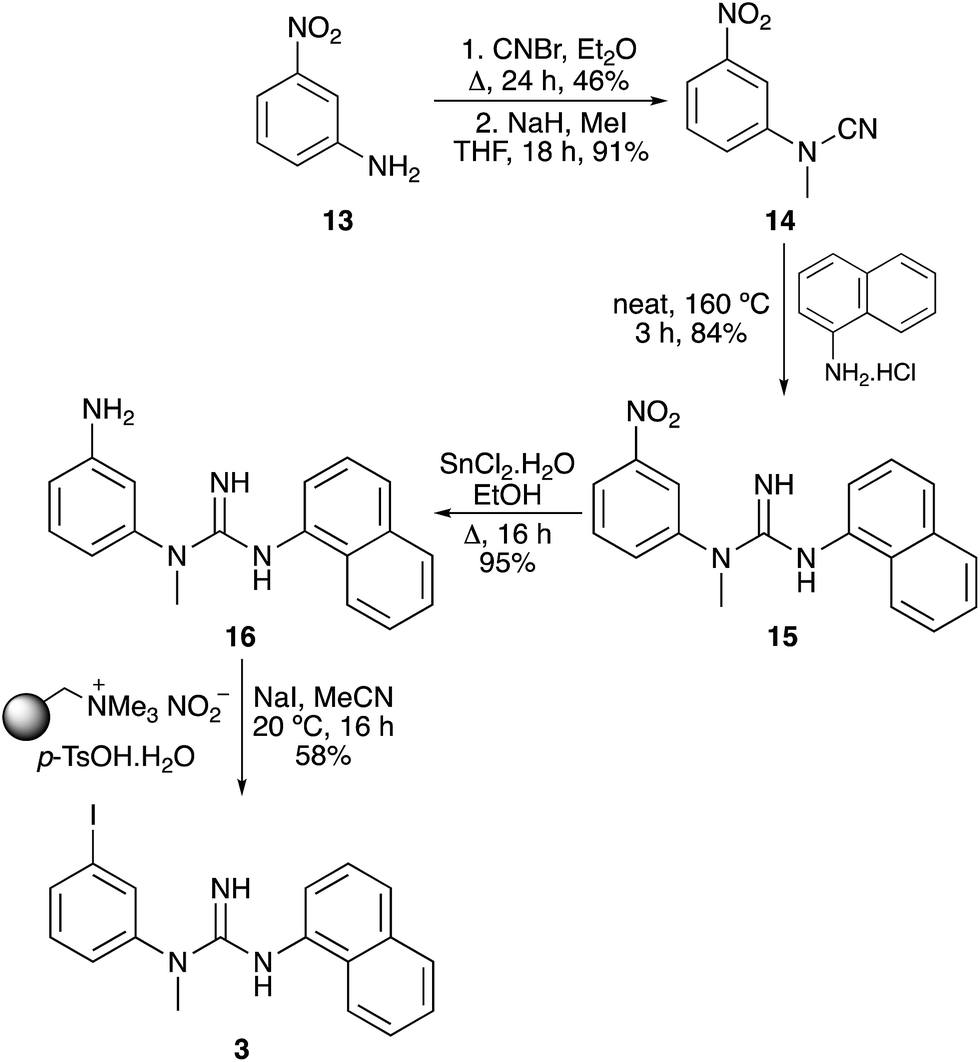
The next target was CNS1261 (3), a SPECT imaging agent of N-methyl D-aspartate (NMDA) receptor activation.5 Again, a synthetic route to a nitroaryl precursor was developed, before implementation of the tin(II) chloride reduction and the one-pot tandem iodination process (Scheme 4). Cyanation and methylation of 3-nitroaniline (13) under standard conditions, followed by thermal coupling of the resulting cyanamide 14 with 1-naphthylamine hydrochloride, gave biaryl guanidine 15. After a highly efficient tin(II) chloride reduction of the nitroaryl group, the one-pot tandem iodination process of meta-substituted aniline 16 was found to proceed at room temperature resulting in a 58% yield of CNS1261 (3).
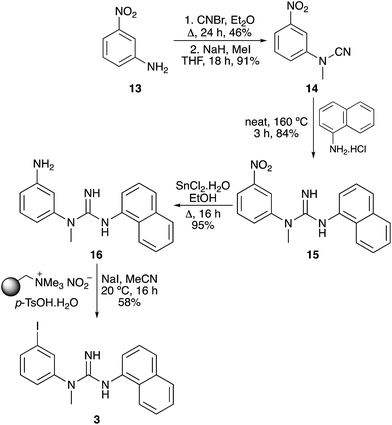 |
| | Scheme 4 Synthesis of CNS1261 (3). | |
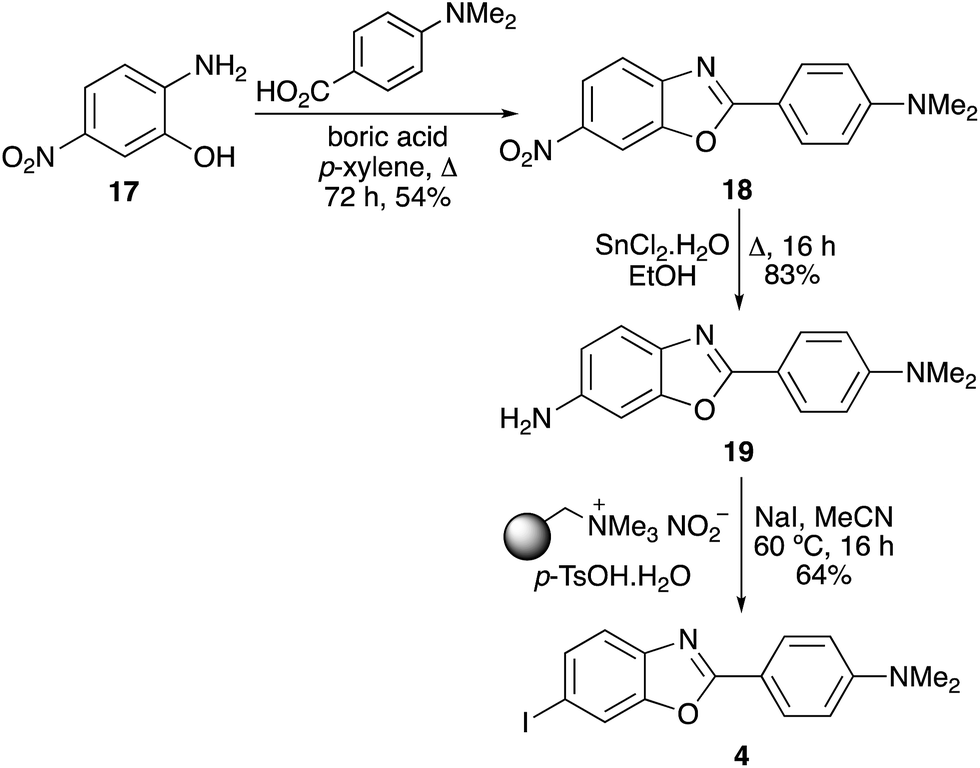
The final target in this study was IBOX (4), an imaging agent of amyloid plaques in the brain.6 The same strategy of nitroaryl precursor formation, followed by reduction and application of the one-pot process was used for the rapid synthesis of IBOX (Scheme 5). The benzoxazole ring system of IBOX bearing a nitro group was formed by condensation of 2-hydroxy-4-nitroaniline (17) with 4-dimethylaminobenzoic acid in the presence of boric acid. Efficient tin dichloride reduction of the nitro group, followed by the one-pot tandem diazotisation–iodination process completed the three-pot synthesis of IBOX (4). The 53% overall yield achieved for IBOX (4) from 18 using the tin(II) chloride reduction and the one-pot tandem iodination process was a significant improvement on the literature synthesis.6 This involved reductive hydrogenolysis of 18, followed by stepwise diazotisation and iodination of amine 19 under traditional conditions (NaNO2/HCl, then KI), giving 4 in 13% yield.6
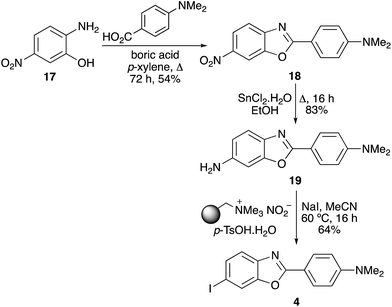 |
| | Scheme 5 Synthesis of IBOX (4). | |
Conclusions
In conclusion, a rapid and efficient tandem process has been developed for the general preparation of aryl iodide compounds bearing a range of functional groups and substitution patterns. The mild conditions for diazonium salt formation, the facile removal of the polymer-supported nitrite reagent and simple work up and purification allowed the clean and efficient synthesis of a small library of iodinated compounds. The only amino-substituted aryl compounds found not to undergo this transformation were deactivated amino-substituted heterocycles bearing multiple heteroatoms. The process was exemplified with the preparation of a range of important biologically active agents, including CNS1261 and IBOX, compounds that are used to study various neurological diseases. In this part of the study, a novel strategy was developed to allow the preparation of biologically relevant structures with the amine group masked, before release of the amine and the late-stage incorporation of the iodine atom. Nitroaryl precursors were used to prepare the core structures, before a combination of a highly efficient chemoselective tin(II) chloride reduction, followed by the one-pot tandem iodination process allowed rapid access to these targets. We expect this method to be of general use for the iodination of a wide range of aryl compounds and are currently investigating further applications.
Experimental
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a PureSolv 500 MD solvent purification system. All reactions were performed under an atmosphere of argon unless otherwise mentioned. Brine refers to a saturated solution of sodium chloride. Flash column chromatography was performed using Fisher matrix silica 60. Macherey-Nagel aluminium-backed plates pre-coated with silica gel 60 (UV254) were used for thin layer chromatography and were visualised by staining with KMnO4. 1H and 13C NMR spectra were recorded on a Bruker DPX 400 (1H: 400 MHz; 13C: 101 MHz) spectrometer or a Bruker 500 (1H: 500 MHz; 13C: 126 MHz) spectrometer with chemical shift values reported in ppm relative to a residual solvent peak and in the solvent stated. Assignment of 1H NMR signals is based on COSY experiments. Assignment of 13C NMR signals is based on HSQC and/or DEPT experiments. All coupling constants, J, are quoted in Hz. Mass spectra were obtained using a JEOL JMS-700 spectrometer for EI and CI or a Bruker Microtof-q for ESI. Infrared spectra were obtained neat using a Shimadzu IRPrestige-21 spectrometer. Melting points were determined on a Reichert platform melting point apparatus.
General procedure for preparation of polymer-supported nitrite17
The polymer-supported nitrite reagent was prepared by the addition of Amberlyst® A26 hydroxide form resin (1.00 g, 4.00 mmol) to a solution of sodium nitrite (0.55 g, 8.00 mmol) in water (20 mL). The mixture was stirred at room temperature for 0.5 h. The polymer-supported nitrite was filtered and washed with water until the pH of the filtrate became neutral. The content of the polymer-supported nitrite was 3.5 mmol of NO2 per g.17
General procedure for one-pot diazotisation and iodination
4-Iodonitrobenzene (6a)22. To a solution of p-toluenesulfonic acid monohydrate (0.27 g, 1.1 mmol) in acetonitrile (2 mL) was added 4-nitroaniline (5a) (0.050 g, 0.36 mmol). The solution was cooled in a water bath to 10–15 °C. Polymer-supported nitrite (0.31 g, containing 1.1 mmol of NO2) was added, followed by sodium iodide (0.11 g, 0.54 mmol) in water (0.2 mL). The reaction mixture was stirred for 1 h then warmed to room temperature and stirred for 2 h in total. The mixture was filtered from the resin and the resin was washed with diethyl ether (50 mL). The reaction mixture and diethyl ether washings were combined and washed with water (50 mL). The aqueous layer was then extracted with diethyl ether (3 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) to give 4-iodonitrobenzene (6a) as a pale yellow solid (0.080 g, 86%). Mp 170–172 °C (lit.22 169–171 °C); δH (400 MHz, CDCl3) 7.91 (2H, d, J 8.0 Hz, 3-H and 5-H), 7.94 (2H, d, J 8.0 Hz, 2-H and 6-H); δC (101 MHz, CDCl3) 102.8 (C), 125.0 (2× CH), 138.8 (2× CH), 148.0 (C); m/z (CI) 250 (MH+. 60%), 209 (12), 193 (15), 124 (30), 113 (22), 85 (78), 69 (100).
4-Iodoacetophenone (6b)23. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-aminoacetophenone (5b) (0.050 g, 0.39 mmol), p-toluenesulfonic acid monohydrate (0.24 g, 1.1 mmol), polymer-supported nitrite (0.32 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.74 mmol). The crude material was purified using flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) to give 4-iodoacetophenone (5b) as a white solid (0.090 g, 94%). Mp 84–85 °C (lit.23 82–84 °C); δH (400 MHz, CDCl3) 2.57 (3H, s, COCH3), 7.66 (2H, d, J 8.6 Hz, 3-H and 5-H), 7.83 (2H, d, J 8.6 Hz, 2-H and 6-H); δC (101 MHz, CDCl3) 26.6 (CH3), 101.2 (C), 129.9 (2× CH), 136.5 (C), 138.1 (2× CH), 197.5 (C); m/z (ESI) 269 (MNa+. 80%).
4-Iodobenzonitrile (6c)24. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-aminobenzonitrile (5c) (0.050 g, 0.38 mmol), p-toluenesulfonic acid monohydrate (0.24 g, 1.1 mmol), polymer-supported nitrite (0.32 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.74 mmol). Purification by flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) gave 4-iodobenzonitrile (6c) as a white solid (0.080 g, 68%). Spectroscopic data were consistent with the literature.24 Mp 122–124 °C; δH (400 MHz, CDCl3) 7.36 (2H, d, J 8.6 Hz, 3-H and 5-H), 7.84 (2H, d, J 8.6 Hz, 2-H and 6-H); δC (101 MHz, CDCl3) 100.4 (C), 111.9 (C), 118.3 (C), 133.3 (2× CH), 138.6 (2× CH); m/z (EI) 229 (M+. 80%), 102 (100), 84 (95), 69 (31), 49 (38).
4-Iodochlorobenzene (6d)25. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-chloroaniline (5d) (0.050 g, 0.41 mmol), p-toluenesulfonic acid monohydrate (0.22 g, 1.2 mmol), polymer-supported nitrite (0.34 g, containing 1.2 mmol of NO2) and sodium iodide (0.12 g, 0.79 mmol). The crude material was purified using flash column chromatography eluting with 5% ethyl acetate in petroleum ether (40–60) to give 4-iodochlorobenzene (6d) as a white solid (0.058 g, 57%). Mp 55–56 °C (lit.25 54–55 °C); δH (400 MHz, CDCl3) 7.09 (2H, d, J 8.7 Hz, 3-H and 5-H), 7.61 (2H, d, J 8.7 Hz, 2-H and 6-H); δC (101 MHz, CDCl3) 91.3 (C), 130.7 (2× CH), 134.4 (C), 138.9 (2× CH); m/z (EI) 238 (M+. 52%), 111 (50), 84 (100), 49 (40).
4-Iodobromobenzene (6e)26. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-bromoaniline (5e) (0.050 g, 0.28 mmol), p-toluenesulfonic acid monohydrate (0.17 g, 0.87 mmol), polymer-supported nitrite (0.25 g, containing 0.87 mmol of NO2) and sodium iodide (0.090 g, 0.58 mmol). Purification by flash column chromatography eluting with 5% diethyl ether in petroleum ether (40–60) gave 4-iodobromobenzene (6e) as a white solid (0.063 g, 77%). Mp 91–92 °C (lit.26 90 °C); δH (400 MHz, CDCl3) 7.23 (2H, d, J 8.6 Hz, 3-H and 5-H), 7.54 (2H, d, J 8.6 Hz, 2-H and 6-H); δC (101 MHz, CDCl3) 92.2 (C), 122.4 (C), 133.6 (2× CH), 139.2 (2× CH); m/z (EI) 282 (M+. 48%), 155 (32), 84 (100), 49 (48).
4-Iodoanisole (6f)27. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-methoxyaniline (5f) (0.050 g, 0.40 mmol), p-toluenesulfonic acid monohydrate (0.23 g, 1.2 mmol), polymer-supported nitrite (0.34 g, containing 1.2 mmol of NO2) and sodium iodide (0.12 g, 0.80 mmol). The crude material was purified using flash column chromatography eluting with 5% ethyl acetate in petroleum ether (40–60) to give 4-iodoanisole (6f) as a white solid (0.070 g, 71%). Mp 47–48 °C (lit.27 49–50 °C); δH (400 MHz, CDCl3) 3.78 (1H, s, OCH3), 6.68 (2H, d, J 9.0 Hz, 2-H and 6-H), 7.56 (2H, d, J 9.0 Hz, 3-H and 5-H); δC (101 MHz, CDCl3) 55.5 (CH3), 82.9 (C), 116.5 (2× CH), 138.4 (2× CH), 159.6 (C); m/z (EI) 234 (M+. 100%), 219 (22), 191 (11), 92 (14).
Methyl 3-iodobenzoate (6g)28. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using methyl 3-aminobenzoate (5g) (0.050 g, 0.33 mmol), p-toluenesulfonic acid monohydrate (0.19 g, 0.99 mmol), polymer-supported nitrite (0.28 g, containing 0.99 mmol of NO2) and sodium iodide (0.10 g, 0.66 mmol). Purification by flash column chromatography eluting with 20% diethyl ether in petroleum ether (40–60) gave methyl 3-iodobenzoate (6g) as a yellow solid (0.060 g, 71%). Mp 52–53 °C (lit.28 54–56 °C); δH (400 MHz, CDCl3) 3.91 (3H, s, OCH3), 7.17 (1H, t, J 7.9 Hz, 5-H), 7.87 (1H, ddd, J 7.9, 1.7, 1.2 Hz, 4-H), 7.99 (1H, ddd, J 7.9, 1.7, 1.2 Hz, 6-H), 8.37 (1H, t, J 1.7 Hz, 2-H); δC (101 MHz, CDCl3) 52.5 (CH3), 93.9 (C), 128.9 (CH), 130.2 (CH), 132.2 (C), 138.6 (CH), 141.9 (CH), 165.7 (C); m/z (EI) 262 (M+. 28%), 231 (30), 203 (9), 84 (100), 49 (40).
3-Iodoacetophenone (6h)29. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 3-aminoacetophenone (5h) (0.050 g, 0.40 mmol), p-toluenesulfonic acid monohydrate (0.21 g, 1.1 mmol), polymer-supported nitrite (0.32 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.74 mmol). Purification by flash column chromatography eluting with 20% diethyl ether in petroleum ether (40–60) gave 3-iodoacetophenone (6h) as a colorless oil (0.060 g, 64%). Spectroscopic data were consistent with the literature.29 δH (500 MHz, CDCl3) 2.58 (3H, s, COCH3), 7.21 (1H, t, J 7.9 Hz, 5-H), 7.86–7.93 (2H, m, 4-H and 6-H), 8.28 (1H, t, J 1.6 Hz, 2-H); δC (126 MHz, CDCl3) 26.7 (CH3), 94.6 (C), 127.6 (CH), 130.5 (CH), 137.5 (CH), 139.0 (C), 142.0 (CH), 196.7 (C); m/z (EI) 246 (M+. 78%), 231 (100), 203 (48), 181 (20), 84 (80), 76 (56).
3,4,5-Trimethoxyiodobenzene (6i)30. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 3,4,5-trimethoxyaniline (5i) (0.050 g, 0.29 mmol), p-toluenesulfonic acid monohydrate (0.16 g, 0.82 mmol), polymer-supported nitrite (0.23 g, containing 0.82 mmol of NO2) and sodium iodide (0.080 g, 0.55 mmol). The crude material was purified using flash column chromatography eluting with 20% diethyl ether in petroleum ether (40–60) to give 3,4,5-trimethoxyiodobenzene (6i) as a yellow solid (0.060 g, 73%). Mp 84–86 °C (lit.30 83–85 °C); δH (400 MHz, CDCl3) 3.80 (3H, s, OCH3), 3.82 (6H, s, 2× OCH3), 6.87 (2H, s, 2× ArH); δC (101 MHz, CDCl3) 56.5 (2× CH3), 61.0 (CH3), 86.2 (C), 115.1 (2× CH), 138.4 (C), 154.1 (2× C); m/z (EI) 294 (M+. 100%), 279 (58), 251 (19), 236 (18), 124 (21), 109 (12), 84 (70), 49 (25).
1-Iodo-3,4-methylenedioxybenzene (6j)31. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 3,4-(methylenedioxy)aniline (5j) (0.050 g, 0.36 mmol), p-toluenesulfonic acid monohydrate (0.21 g, 1.1 mmol), polymer-supported nitrite (0.31 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.73 mmol). Purification by flash column chromatography eluting with 5% ethyl acetate in petroleum ether (40–60) gave 1-iodo-3,4-methylenedioxybenzene (6j) as a pale yellow oil (0.06 g, 69%). Spectroscopic data were consistent with the literature.31 δH (400 MHz, CDCl3) 5.96 (2H, s, CH2), 6.59 (1H, d, J 8.1 Hz, 5-H), 7.12 (1H, d, J 1.6 Hz, 2-H), 7.14 (1H, dd, J 8.1, 1.6 Hz, 6-H); δC (101 MHz, CDCl3) 82.3 (C), 101.5 (CH2), 110.6 (CH), 117.8 (CH), 130.7 (CH), 148.0 (C), 148.8 (C); m/z (EI) 248 (M+. 100%), 247 (68), 183 (10), 121 (42), 63 (34).
2-Iodobenzophenone (6k)32. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2-aminobenzophenone (5k) (0.050 g, 0.25 mmol), p-toluenesulfonic acid monohydrate (0.14 g, 0.76 mmol), polymer-supported nitrite (0.22 g, containing 0.76 mmol of NO2) and sodium iodide (0.080 g, 0.51 mmol). The crude material was purified using flash column chromatography eluting with 5% diethyl ether in petroleum ether (40–60) to give 2-iodobenzophenone (6k) as an orange solid (0.060 g, 79%). Mp 30–31 °C (lit.32 31–32 °C); δH (400 MHz, CDCl3) 7.15–7.21 (1H, m, ArH), 7.30 (1H, dd, J 7.6, 1.5 Hz, ArH), 7.42–7.49 (3H, m, 3× ArH), 7.58–7.63 (1H, m, ArH), 7.79–7.83 (2H, m, 2× ArH), 7.93 (1H, dd, J 8.0, 0.8 Hz, ArH); δC (101 MHz, CDCl3) 92.3 (C), 127.9 (CH), 128.6 (CH), 128.8 (CH), 130.6 (2× CH), 131.2 (2× CH), 133.8 (CH), 135.7 (C), 139.8 (CH), 144.5 (C), 197.3 (C); m/z (ESI) 331 (MNa+. 100%).
5-Chloro-2-iodobenzophenone (6l)33. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2-amino-5-chlorobenzophenone (5l) (0.050 g, 0.22 mmol), p-toluenesulfonic acid monohydrate (0.13 g, 0.66 mmol), polymer-supported nitrite (0.19 g, containing 0.66 mmol of NO2) and sodium iodide (0.070 g, 0.44 mmol). Purification by flash column chromatography eluting with 10% diethyl ether in petroleum ether (40–60) gave 5-chloro-2-iodobenzophenone (6l) as a yellow solid (0.050 g, 66%). Mp 72–74 °C (lit.33 80–82 °C); δH (400 MHz, CDCl3) 7.18 (1H, dd, J 8.4, 2.5 Hz, 4-H), 7.28 (1H, d, J 2.5 Hz, 6-H), 7.46–7.52 (2H, m, 2× ArH), 7.61–7.67 (1H, m, ArH), 7.78–7.82 (2H, m, 2× ArH), 7.84 (1H, d, J 8.4 Hz, 3-H); δC (101 MHz, CDCl3) 89.4 (C), 128.6 (CH), 129.0 (2× CH), 130.6 (2× CH), 131.4 (CH), 134.2 (CH), 134.8 (C), 135.2 (C), 141.0 (CH), 146.0 (C), 195.9 (C); m/z (EI) 342 (M+. 100%), 265 (20), 215 (20), 152 (14), 105 (59), 77 (26), 51 (11).
2-Iodo-4-methylanisole (6m)34. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2-methoxy-5-methylaniline (5m) (0.050 g, 0.36 mmol), p-toluenesulfonic acid monohydrate (0.21 g, 1.1 mmol), polymer-supported nitrite (0.31 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.73 mmol). The crude material was purified using flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) to give 2-iodo-4-methylanisole (6m) as a white solid (0.070 g, 82%). Spectroscopic data were consistent with the literature.34 Mp 28–29 °C; δH (500 MHz, CDCl3) 2.26 (3H, s, 4-CH3), 3.85 (3H, s, OCH3), 6.72 (1H, d, J 8.3 Hz, 6-H), 7.10 (1H, dd, J 8.3, 1.6 Hz, 5-H), 7.60 (1H, d, J 1.6 Hz, 3-H); δC (126 MHz, CDCl3) 20.1 (CH3), 56.6 (CH3), 85.9 (C), 110.9 (CH), 130.1 (CH), 132.2 (C), 140.0 (CH), 156.2 (C); m/z (EI) 248 (M+. 100%), 233 (28), 121 (11), 84 (28), 78 (17).
4,5-Dimethoxy-2-iodobenzonitrile (6n)35. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2-amino-4,5-dimethoxybenzonitrile (5n) (0.050 g, 0.28 mmol), p-toluenesulfonic acid monohydrate (0.16 g, 0.84 mmol), polymer-supported nitrite (0.24 g, containing 0.84 mmol of NO2) and sodium iodide (0.080 g, 0.56 mmol). Purification by flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) gave 4,5-dimethoxy-2-iodobenzonitrile (6n) as an orange solid (0.070 g, 86%). Mp 98–99 °C (lit.35 103–104 °C); δH (400 MHz, CDCl3) 3.87 (3H, s, OCH3), 3.91 (3H, s, OCH3), 7.02 (1H, s, 3-H), 7.24 (1H, s, 6-H); δC (101 MHz, CDCl3) 56.4 (CH3), 56.6 (CH3), 88.8 (C), 112.3 (C), 115.9 (CH), 119.9 (C), 121.7 (CH), 149.5 (C), 153.0 (C); m/z (ESI) 312 (MNa+. 100%).
2,4,6-Trimethyliodobenzene (6o)36. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2,4,6-trimethylaniline (5o) (0.050 mL, 0.37 mmol), p-toluenesulfonic acid monohydrate (0.21 g, 1.1 mmol), polymer-supported nitrite (0.32 g, containing 1.1 mmol of NO2) and sodium iodide (0.11 g, 0.74 mmol). Purification by flash column chromatography eluting with 20% diethyl ether in petroleum ether (40–60) gave 2,4,6-trimethyliodobenzene (6o) as a yellow solid (0.061 g, 67%). Mp 29–30 °C (lit.36 29–30 °C); δH (400 MHz, CDCl3) 2.25 (3H, s, CH3), 2.44 (6H, s, 2× CH3), 6.90 (2H, s, 2× ArH); δC (101 MHz, CDCl3) 20.8 (CH3), 29.7 (2× CH3), 104.4 (C), 128.1 (2× CH), 137.5 (C), 141.9 (2× C); m/z (EI) 246 (M+. 100%), 119 (90), 91 (68), 84 (82), 77 (20).
1-Iodo-2,4,6-trichlorobenzene (6p)37. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 1-amino-2,4,6-trichlorobenzene (5p) (0.050 g, 0.26 mmol), p-toluenesulfonic acid monohydrate (0.15 g, 0.77 mmol), polymer-supported nitrite (0.22 g, containing 0.77 mmol of NO2) and sodium iodide (0.080 g, 0.51 mmol). Purification by flash column chromatography eluting with 5% diethyl ether in petroleum ether (40–60) gave 1-iodo-2,4,6-trichlorobenzene (6p) as a white solid (0.060 g, 74%). Mp 53–54 °C (lit.37 53 °C); δH (400 MHz, CDCl3) 7.37 (2H, s, 2× ArH); δC (101 MHz, CDCl3) 101.7 (C), 127.4 (2× CH), 135.3 (C), 141.4 (2× C); m/z (EI) 308 (M+. 83%), 181 (30), 143 (12), 84 (100).
1-Iodoanthraquinone (6q)38. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 1-aminoanthraquinone (5q) (0.050 g, 0.22 mmol), p-toluenesulfonic acid monohydrate (0.13 g, 0.67 mmol), polymer-supported nitrite (0.19 g, containing 0.67 mmol of NO2) and sodium iodide (0.070 g, 0.45 mmol). The crude material was purified using flash column chromatography eluting with 10% diethyl ether in petroleum ether (40–60) to give 1-iodoanthraquinone (6q) as a yellow solid (0.040 g, 54%). Mp 201–202 °C (lit.38 204–205 °C); δH (400 MHz, CDCl3) 7.40 (1H, t, J 7.8 Hz, 3-H), 7.77–7.85 (2H, m, 2× ArH), 8.25–8.30 (1H, m, ArH), 8.33–8.38 (1H, m, ArH), 8.41 (1H, dd, J 7.8, 1.2 Hz, ArH), 8.45 (1H, dd, J 7.8, 1.2 Hz, ArH); δC (101 MHz, CDCl3) 93.4 (C), 127.0 (CH), 127.9 (CH), 128.5 (CH), 132.5 (C), 132.8 (C), 134.0 (CH), 134.0 (C), 134.2 (CH), 134.7 (CH), 136.0 (C), 148.8 (CH), 181.7 (C), 182.0 (C); m/z (ESI) 357 (MNa+. 100%).
2-Chloro-5-iodobenzoic acid (6r)39. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 5-amino-2-chlorobenzoic acid (5r) (0.050 g, 0.29 mmol), p-toluenesulfonic acid monohydrate (0.17 g, 0.87 mmol), polymer-supported nitrite (0.25 g, containing 0.87 mmol of NO2) and sodium iodide (0.090 g, 0.58 mmol). The reaction mixture was stirred at room temperature for 24 h. This gave 2-chloro-5-iodobenzoic acid (6r) as a white solid (0.070 g, 79%). Mp 155–156 °C (lit.39 155.5–156 °C); δH (400 MHz, CDCl3) 7.22 (1H, d, J 8.4 Hz, 3-H), 7.78 (1H, dd, J 8.4, 2.2 Hz, 4-H), 8.32 (1H, d, J 2.2 Hz, 6-H); δC (101 MHz, CDCl3) 90.9 (C), 130.2 (C), 133.2 (CH), 134.9 (C), 141.1 (CH), 142.5 (CH), 168.8 (C); m/z (EI) 282 (M+. 100%), 265 (30), 149 (8), 84 (59), 49 (50).
2,4-Dinitroiodobenzene (6s)23. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2,4-dinitroaniline (5s) (0.050 g, 0.27 mmol), p-toluenesulfonic acid monohydrate (0.16 g, 0.82 mmol), polymer-supported nitrite (0.23 g, containing 0.82 mmol of NO2) and sodium iodide (0.080 g, 0.55 mmol). The reaction mixture was stirred at room temperature for 16 h. The crude material was purified using flash column chromatography eluting with 40% ethyl acetate in petroleum ether (40–60) to give 2,4-dinitroiodobenzene (6s) as a pale yellow solid (0.040 g, 49%). Mp 88–89 °C (lit.23 88–89 °C); δH (500 MHz, CDCl3) 8.11 (1H, dd, J 8.6, 2.5 Hz, 5-H), 8.31 (1H, d, J 8.6 Hz, 6-H), 8.68 (1H, d, J 2.5 Hz, 3-H); δC (126 MHz, CDCl3) 94.9 (C), 120.6 (CH), 127.1 (CH), 143.6 (CH), 148.2 (C), 153.4 (C); m/z (EI) 294 (M+. 53%), 201 (10), 84 (100), 49 (58).
3-Iodopyridine (6t)40. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 3-aminopyridine (5t) (0.030 g, 0.32 mmol), p-toluenesulfonic acid monohydrate (0.18 g, 0.96 mmol), polymer-supported nitrite (0.27 g, containing 0.96 mmol of NO2) and sodium iodide (0.10 g, 0.64 mmol). The reaction mixture was heated to 60 °C and stirred for 16 h. The crude material was purified using flash column chromatography eluting with 60% ethyl acetate in petroleum ether (40–60) to give 3-iodopyridine (6t) as a white solid (0.050 g, 74%). Mp 50–51 °C (lit.40 52–53 °C); δH (400 MHz, CDCl3) 7.11 (1H, dd, J 6.4, 4.6 Hz, 5-H), 8.02 (1H, dt, J 6.4, 1.4 Hz, 4-H), 8.56 (1H, dd, J 4.6, 1.4 Hz, 6-H), 8.84 (1H, d, J 1.4, Hz, 2-H); δC (101 MHz, CDCl3) 93.8 (C), 125.5 (CH), 144.6 (CH), 148.2 (CH), 155.9 (CH); m/z (EI) 205 (M+. 80%), 139 (44), 84 (78), 44 (100).
Ethyl 4-hydroxyquinoline-2-carboxylate (9)41. Aniline (7) (1.11 g, 11.9 mmol), diethyl oxaloacetate (8) (2.50 g, 11.9 mmol) and p-toluenesulfonic acid (2.26 g, 11.9 mmol) in cyclohexane (50 mL) were stirred vigorously under Dean–Stark conditions for 48 h. After cooling to room temperature, the reaction mixture was filtered and washed with cyclohexane before concentrating in vacuo to give the imine as a yellow oil. Neat polyphosphoric acid (∼5 g) was added and the reaction mixture was stirred vigorously at 120 °C for 1 h. After cooling to room temperature, a saturated solution of aqueous sodium hydrogen carbonate (75 mL) was slowly added. Chloroform (100 mL) was added and the two layers separated. The aqueous layer was extracted with chloroform (3 × 100 mL). The organic layers were dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 10% methanol in dichloromethane to give ethyl 4-hydroxyquinoline-2-carboxylate (9) as a yellow solid (1.24 g, 48%). Mp 214–215 °C (lit.41 213 °C); δH (400 MHz, CDCl3) 1.44 (3H, t, J 7.1 Hz, OCH2CH3), 4.48 (2H, q, J 7.1 Hz, OCH2CH3), 7.00 (1H, s, 3-H), 7.38 (1H, t, J 7.6 Hz, ArH), 7.45 (1H, d, J 8.2, Hz, ArH), 7.65–7.68 (1H, m, ArH), 8.35 (1H, dd, J 8.2, 0.9 Hz, ArH), 8.98 (1H, br s, OH); δC (101 MHz, CDCl3) 14.1 (CH3), 63.4 (CH2), 111.6 (CH), 118.0 (CH), 124.5 (CH), 126.3 (CH), 126.4 (C), 133.1 (CH), 136.5 (C), 139.0 (C), 163.0 (C), 179.7 (C); m/z (ESI) 240 (MNa+. 100%).
Ethyl 4-bromoquinoline-2-carboxylate42. Ethyl 4-hydroxyquinoline-2-carboxylate (9) (0.840 g, 3.87 mmol) was dissolved in acetonitrile (50 mL) before addition of phosphorus oxybromide (3.33 g, 11.6 mmol) and potassium carbonate (1.60 g, 11.6 mmol). The mixture was heated under reflux for 2 h. After cooling to room temperature, the solution was concentrated in vacuo and water added. The product was extracted with ethyl acetate (3 × 100 mL) and the organic layers dried (MgSO4), filtered and concentrated in vacuo to give ethyl 4-bromoquinoline-2-carboxylate as a brown solid (1.03 g, 95%). Mp 88–90 °C (lit.42 91–92 °C); δH (500 MHz, CDCl3) 1.49 (3H, t, J 7.1 Hz, OCH2CH3), 4.56 (2H, q, J 7.1 Hz, OCH2CH3), 7.74 (1H, ddd, J 8.4, 6.9, 1.2 Hz, 6-H), 7.83 (1H, ddd, J 8.4, 6.9, 1.2 Hz, 7-H), 8.23 (1H, dd, J 8.4, 1.2 Hz, 5-H), 8.31 (1H, dd, J 8.4, 1.2 Hz, 8-H), 8.46 (1H, s, 3-H); δC (126 MHz, CDCl3) 14.5 (CH3), 62.7 (CH2), 125.2 (CH), 126.8 (CH), 129.0 (C), 130.0 (CH), 131.2 (CH), 131.4 (CH), 135.3 (C), 148.1 (C), 148.2 (C), 164.5 (C); m/z (CI) 280 (MH+. 100%), 202 (20), 81 (17), 69 (24).
Ethyl 4-(2′-nitrophenyl)quinoline-2-carboxylate (10)3. Ethyl 4-bromoquinoline-2-carboxylate (0.500 g, 1.79 mmol) was dissolved in N,N′-dimethylformamide (25 mL) before addition of 2-nitrophenylboronic acid (0.360 g, 2.14 mmol), tetrakis(triphenylphosphine)palladium(0) (0.210 g, 0.180 mmol) and potassium phosphate (0.450 g, 2.14 mmol). The reaction mixture was stirred at 120 °C for 24 h, before addition of 2-nitrophenylboronic acid (0.360 g, 2.14 mmol) and potassium phosphate (0.450 g, 2.14 mmol) and further stirring at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted in ethyl acetate (50 mL) and washed with a 5% aqueous solution of lithium chloride (3 × 50 mL) followed by brine (50 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 40% ethyl acetate in petroleum ether (40–60) to give ethyl 4-(2′-nitrophenyl)quinoline-2-carboxylate (10) as a yellow solid (0.510 g, 88%). Mp 149–150 °C (lit.3 156–157 °C); δH (500 MHz, CDCl3) 1.49 (3H, t, J 7.1 Hz, OCH2CH3), 4.51–4.63 (2H, m, OCH2CH3), 7.44–7.48 (2H, m, 2× ArH), 7.57 (1H, ddd, J 8.4, 7.0, 1.6 Hz, ArH), 7.72 (1H, ddd, J 8.6, 8.0, 1.6 Hz, ArH), 7.77–7.82 (2H, m, 2× ArH), 8.07 (1H, s, 3-H), 8.26 (1H, dd, J 8.0, 1.0 Hz, ArH), 8.40 (1H, d, J 8.6 Hz, ArH); δC (126 MHz, CDCl3) 14.5 (CH3), 62.6 (CH2), 120.5 (CH), 124.6 (CH), 125.1 (CH), 127.7 (C), 129.3 (CH), 130.1 (CH), 130.5 (CH), 131.6 (CH), 132.5 (CH), 132.8 (C), 133.6 (CH), 146.5 (C), 147.8 (C), 148.0 (C), 148.7 (C), 165.3 (C); m/z (EI) 322 (M+. 25%), 278 (52), 250 (100), 205 (41), 190 (31), 130 (30), 84 (39).
4-(2′-Nitrophenyl)-N,N-diethylquinoline-2-carboxamide (11). Ethyl 4-(2′-nitrophenyl)quinoline-2-carboxylate (10) (0.380 g, 1.18 mmol) was dissolved in 50% aqueous ethanol (20 mL) before addition of ground sodium hydroxide (0.190 g, 4.72 mmol). The mixture was stirred under reflux for 16 h. After cooling to room temperature, the ethanol was removed in vacuo and the water layer acidified (pH 4) with 1 M hydrochloric acid solution. The product was extracted with dichloromethane (3 × 50 mL), washed with water (2 × 50 mL), dried (MgSO4), filtered and concentrated in vacuo to give 4-(2′-nitrophenyl)quinoline-2-carboxylic acid as a brown solid (0.320 g, 92%), which was used without further purification. 4-(2′-Nitrophenyl)quinoline-2-carboxylic acid (0.0900 g, 0.300 mmol) was dissolved in N,N′-dimethylformamide (10 mL), before addition of triethylamine (61.0 μL, 0.440 mmol) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (0.120 g, 0.320 mmol). The mixture was stirred at room temperature for 1 h and then heated to 50 °C, with the addition of diethylamine (30.0 μL, 0.440 mmol). The reaction mixture was stirred at 50 °C for 16 h. Water was added and the mixture stirred for an additional 1 h. After cooling to room temperature, the reaction mixture was diluted in ethyl acetate (20 mL) and washed with a 5% aqueous solution of lithium chloride (3 × 20 mL), followed by brine (20 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 60% ethyl acetate in petroleum ether (40–60) to give 4-(2′-nitrophenyl)-N,N-diethylquinoline-2-carboxamide (11) as a colorless oil (0.055 g, 54%). νmax/cm−1 (neat) 2974 (CH), 1630 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1528, 1346, 1275, 1098, 766; δH (400 MHz, CDCl3) 1.27 (3H, t, J 7.2 Hz, NCH2CH3), 1.32 (3H, t, J 7.2 Hz, NCH2CH3), 3.40–3.72 (4H, m, 2× NCH2CH3), 7.42 (1H, dd, J 8.4, 1.2 Hz, ArH), 7.47–7.53 (2H, m, 2× ArH), 7.55 (1H, s, 3-H), 7.66–7.80 (3H, m, 3× ArH), 8.16–8.24 (2H, m, 2× ArH); δC (101 MHz, CDCl3) 13.1 (CH3), 14.6 (CH3), 40.7 (CH2), 43.7 (CH2), 119.9 (CH), 124.6 (CH), 124.9 (CH), 126.4 (C), 128.1 (CH), 129.9 (CH), 130.2 (CH), 130.5 (CH), 132.5 (CH), 132.9 (C), 133.5 (CH), 145.9 (C), 146.9 (C), 148.8 (C), 154.4 (C), 168.5 (C); HRMS (ESI) m/z 372.1303 (MNa+. C20H19N3O3Na requires 372.1319).
O), 1528, 1346, 1275, 1098, 766; δH (400 MHz, CDCl3) 1.27 (3H, t, J 7.2 Hz, NCH2CH3), 1.32 (3H, t, J 7.2 Hz, NCH2CH3), 3.40–3.72 (4H, m, 2× NCH2CH3), 7.42 (1H, dd, J 8.4, 1.2 Hz, ArH), 7.47–7.53 (2H, m, 2× ArH), 7.55 (1H, s, 3-H), 7.66–7.80 (3H, m, 3× ArH), 8.16–8.24 (2H, m, 2× ArH); δC (101 MHz, CDCl3) 13.1 (CH3), 14.6 (CH3), 40.7 (CH2), 43.7 (CH2), 119.9 (CH), 124.6 (CH), 124.9 (CH), 126.4 (C), 128.1 (CH), 129.9 (CH), 130.2 (CH), 130.5 (CH), 132.5 (CH), 132.9 (C), 133.5 (CH), 145.9 (C), 146.9 (C), 148.8 (C), 154.4 (C), 168.5 (C); HRMS (ESI) m/z 372.1303 (MNa+. C20H19N3O3Na requires 372.1319).
4-(2′-Aminophenyl)-N,N-diethylquinoline-2-carboxamide (12). 4-(2′-Nitrophenyl)-N,N-diethylquinoline-2-carboxamide (11) (0.050 g, 0.15 mmol) was dissolved in ethanol (2 mL) before addition of tin(II) chloride dihydrate (0.18 g, 0.77 mmol). The solution was stirred under reflux for 16 h. After cooling to room temperature, the reaction was quenched by addition of a saturated aqueous solution of sodium hydrogen carbonate and the product was extracted with ethyl acetate (3 × 5 mL). The organic layers were dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 1% methanol in dichloromethane to give 4-(2′-aminophenyl)-N,N-diethylquinoline-2-carboxyamide (12) as a colorless oil (0.045 g, 91%). νmax/cm−1 (neat) 3348 (NH), 2973 (CH), 1620 (CO), 1486, 1451, 1275, 1099, 750; δH (500 MHz, CDCl3) 1.27 (3H, t, J 7.2 Hz, NCH2CH3), 1.32 (3H, t, J 7.2 Hz, NCH2CH3), 3.42–3.67 (6H, m, 2× NCH2CH3 and NH2), 6.83 (1H, dd, J 8.2, 1.2 Hz, ArH), 6.88 (1H, td, J 8.2, 1.2 Hz, ArH), 7.14 (1H, dd, J 8.2, 1.2 Hz, ArH), 7.28 (1H, td, J 8.2, 1.2 Hz, ArH), 7.53 (1H, ddd, J 8.2, 7.2, 1.2 Hz, ArH), 7.64 (1H, s, 3-H), 7.71–7.78 (2H, m, 2× ArH), 8.17 (1H, d, J 8.2 Hz, ArH); δC (126 MHz, CDCl3) 13.1 (CH3), 14.6 (CH3), 40.5 (CH2), 43.6 (CH2), 115.9 (CH), 118.6 (CH), 121.4 (CH), 122.7 (C), 126.2 (CH), 126.7 (C), 127.6 (CH), 129.9 (CH), 130.2 (CH), 130.4 (CH), 130.8 (CH), 143.9 (C), 147.3 (C), 147.4 (C), 155.0 (C), 168.8 (C); HRMS (ESI) m/z 320.1743 (MH+. C20H22N3O requires 320.1757).
4-(2-Iodophenyl)-N,N-diethylquinoline-2-carboxamide (2)3. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 4-(2-aminophenyl)-N,N-diethylquinoline-2-carboxamide (12) (0.040 g, 0.12 mmol), p-toluenesulfonic acid monohydrate (0.070 g, 0.38 mmol), polymer-supported nitrite (0.11 g, containing 0.38 mmol of NO2) and sodium iodide (0.038 g, 0.25 mmol). The reaction mixture was heated to 60 °C and stirred for 16 h. The crude material was purified using flash column chromatography eluting with 40% ethyl acetate in petroleum ether (40–60) to give 4-(2-iodophenyl)-N,N-diethylquinoline-2-carboxamide (2) as a pale yellow solid (0.037 g, 67%). Mp 58–59 °C (lit.3 64–66 °C); δH (500 MHz, CDCl3) 1.25 (3H, t, J 7.1 Hz, NCH2CH3), 1.33 (3H, t, J 7.1 Hz, NCH2CH3), 3.38–3.60 (2H, m, NCH2CH3), 3.63 (2H, q, J 7.1 Hz, NCH2CH3), 7.18 (1H, td, J 7.8, 1.6 Hz, 4′-H), 7.33 (1H, dd, J 7.8, 1.6 Hz, 5-H), 7.44–7.55 (4H, m, 3-H, 6-H, 5′-H and 6′-H), 7.75 (1H, ddd, J 8.4, 7.8, 1.6 Hz, 7-H), 8.01 (1H, dd, J 7.8, 1.6 Hz, 3′-H), 8.19 (1H, d, J 8.4 Hz, 8-H); δC (126 MHz, CDCl3) 13.1 (CH3), 14.7 (CH3), 40.6 (CH2), 43.6 (CH2), 98.5 (C), 120.8 (CH), 125.9 (CH), 126.5 (C), 127.6 (CH), 128.3 (CH), 130.1 (CH), 130.1 (CH), 130.2 (CH), 130.3 (CH), 139.5 (CH), 142.6 (C), 147.2 (C), 151.3 (C), 154.5 (C), 168.7 (C); m/z (EI) 430 (M+. 25%), 359 (84), 331 (91), 294 (23), 203 (100), 176 (26), 149 (26), 72 (78), 69 (31).
N-(3-Nitrophenyl)cyanamide43. Cyanogen bromide (0.12 g, 1.08 mmol) in diethyl ether (5 mL) was added dropwise to a stirred solution of 3-nitroaniline (13) (0.10 g, 0.72 mmol) in diethyl ether (5 mL) at 0 °C. The reaction mixture was heated under reflux for 24 h. After cooling to room temperature, the reaction mixture was filtered and washed with ethyl acetate. The ethyl acetate washings were washed with 10% hydrochloric acid (2 × 10 mL), water (10 mL) and brine (10 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give (3-nitrophenyl)cyanamide as a yellow solid (0.056 g, 46%). Mp 133–135 °C (lit.43 133–135 °C); δH (400 MHz, CD3OD) 7.38 (1H, ddd, J 8.1, 2.2, 0.8 Hz, 6-H), 7.59 (1H, t, J 8.1 Hz, 5-H), 7.81 (1H, t, J 2.2 Hz, 2-H), 7.91 (1H, ddd, J 8.1, 2.2, 0.8 Hz, 4-H); δC (126 MHz, CD3OD) 110.8 (CH), 112.1 (C), 118.5 (CH), 122.1 (CH), 132.1 (CH), 141.7 (C), 150.6 (C); m/z (EI) 163 (M+. 100%), 117 (32), 90 (61), 57 (27).
N-Methyl-N-(3-nitrophenyl)cyanamide (14)44. Sodium hydride in 60% mineral oil (0.930 g, 2.33 mmol) was washed with hexane (3 × 10 mL) and dried. This was then added to (3-nitrophenyl)cyanamide (0.190 g, 1.17 mmol) in THF (3 mL) and heated under reflux for 2 h. The reaction mixture was cooled to 0 °C and methyl iodide (0.186 μL, 2.91 mmol) was added dropwise. The reaction mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was then diluted with methanol (5 mL) and water (10 mL) and extracted with chloroform (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography eluting with 1% methanol in dichloromethane gave N-methyl-N-(3-nitrophenyl)cyanamide (14) as a white solid (0.190 g, 91%). Spectroscopic data were consistent with the literature.43 Mp 106–107 °C; δH (400 MHz, CDCl3) 3.44 (3H, s), 7.48 (1H, ddd, J 8.2, 2.3, 0.9 Hz, 6-H), 7.57 (1H, t, J 8.2 Hz, 5-H), 7.86 (1H, t, J 2.3 Hz, 2-H), 7.95 (1H, ddd, J 8.2, 2.3, 0.9 Hz, 4-H); δC (126 MHz, CDCl3) 37.2 (CH3), 109.4 (CH), 112.7 (C), 118.2 (CH), 121.0 (CH), 130.8 (CH), 142.0 (C), 149.3 (C); m/z (EI) 177 (M+. 100%), 152 (67), 131 (74), 104 (61), 90 (26), 77 (60), 63 (25), 50 (18).
N-(1-Naphthyl)-N′-(3-nitrophenyl)-N′-methylguanidine (15). 1-Naphthylamine (0.13 g, 0.75 mmol) was dissolved in diethyl ether (1 mL) and converted to its hydrochloride salt by dropwise addition of aqueous hydrochloric acid (1 M) in diethyl ether (1 mL) to give a pink precipitate. This was filtered and dried. This was added to N-methyl-N-(3-nitrophenyl)cyanamide (14) (0.13 g, 0.75 mmol) under argon and the neat reaction mixture was stirred at 160 °C for 3 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (10 mL) and washed with 0.1 M sodium hydroxide (10 mL). The aqueous layer was extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 1% methanol and 1% triethylamine in dichloromethane to give N-(1-naphthyl)-N′-(3-nitrophenyl)-N′-methylguanidine (15) as a viscous oil (0.20 g, 84%). νmax/cm−1 (neat) 3485 (NH), 3386 (NH), 2924 (CH), 1631, 1567, 1522 (NO), 1482, 1379, 1345 (NO), 1234, 960, 854, 783, 771, 732, 684; δH (400 MHz, CDCl3) 3.60 (3H, s, NCH3), 7.02 (1H, dd, J 7.2, 1.0 Hz, 2′-H), 7.39–7.58 (5H, m, 5× ArH), 7.72 (1H, ddd, J 8.1, 2.1, 1.0 Hz, 6-H), 7.79–7.86 (1H, m, ArH), 8.03 (1H, ddd, J 8.1, 2.1, 1.0 Hz, 4-H), 8.07–8.13 (1H, m, ArH), 8.23 (1H, t, J 2.1 Hz, 2-H); δC (101 MHz, CDCl3) 39.0 (CH3), 117.5 (CH), 120.1 (CH), 120.6 (CH), 122.8 (CH), 124.0 (CH), 125.4 (CH), 126.2 (CH), 126.5 (CH), 128.1 (CH), 128.6 (C), 130.2 (CH), 131.9 (CH), 134.9 (C), 145.9 (C), 146.5 (C), 149.1 (C), 150.1 (C); HRMS (ESI) m/z 321.1333 (MH+. C18H17N4O2 requires 321.1346).
N-(1-Naphthyl)-N′-(3-aminophenyl)-N′-methylguanidine (16). The reaction was carried out according to the procedure for 4-(2′-aminophenyl)-N,N-diethylquinoline-2-carboxyamide (12) using N-(1-naphthyl)-N′-(3-nitrophenyl)-N′-methylguanidine (15) (0.23 g, 0.73 mmol) and tin(II) chloride dihydrate (0.82 g, 3.6 mmol) in ethanol (10 mL). The reaction mixture was stirred under reflux for 16 h to give N-(1-naphthyl)-N′-(3-aminophenyl)-N′-methylguanidine (16) as a brown solid (0.12 g, 95%). Mp 96–98 °C; νmax/cm−1 (neat) 3322 (NH), 3200 (NH), 3051, 1618, 1599, 1580, 1560, 1491, 1383, 1275, 1227, 970, 781, 750; δH (400 MHz, CDCl3) 3.50 (3H, s, NCH3), 6.53 (1H, ddd, J 7.9, 2.2, 0.9 Hz, 4-H), 6.63 (1H, t, J 2.2 Hz, 2-H), 6.70 (1H, ddd, J 7.9, 2.2, 0.9 Hz, 6-H), 7.05 (1H, dd, J 7.4, 1.1 Hz, 2′-H), 7.15 (1H, t, J 7.9 Hz, 5-H), 7.40 (1H, dd, J 8.4, 7.4 Hz, 3′-H), 7.43–7.49 (2H, m, 2× ArH), 7.52 (1H, br d, J 8.4 Hz, 4′-H), 7.78–7.84 (1H, m, ArH), 8.13–8.19 (1H, m, ArH); δC (101 MHz, CDCl3) 39.0 (CH3), 113.4 (CH), 113.5 (CH), 116.7 (CH), 118.4 (CH), 122.5 (CH), 124.1 (CH), 125.2 (CH), 126.0 (CH), 126.4 (CH), 128.0 (CH), 129.2 (C), 130.6 (CH), 134.9 (C), 145.5 (C), 145.7 (C), 148.0 (C), 151.2 (C); HRMS (EI) m/z 290.1528 (M+. C18H18N4 requires 290.1531).
N-(1-Naphthyl)-N′-(3-iodophenyl)-N′-methylguanidine (3). The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using N-(1-naphthyl)-N′-(3-aminophenyl)-N′-methylguanidine (16) (0.061 g, 0.21 mmol), p-toluenesulfonic acid monohydrate (0.12 g, 0.62 mmol), polymer-supported nitrite (0.18 g, containing 0.62 mmol of NO2) and sodium iodide (0.062 g, 0.41 mmol). The reaction mixture was stirred at room temperature for 16 h. Purification by flash column chromatography eluting with 60–100% ethyl acetate in petroleum ether gave N-(1-naphthyl)-N′-(3-iodophenyl)-N′-methylguanidine (3) as a viscous oil (0.049 g, 58%). νmax/cm−1 (neat) 3482 (NH), 3385 (NH), 3053, 2924 (CH), 1624, 1557, 1474, 1376, 1266, 1233, 943, 782, 693; δH (400 MHz, CDCl3) 3.53 (3H, s, NCH3), 7.03 (1H, dd, J 7.2, 1.1 Hz, 2′-H), 7.13 (1H, t, J 8.0 Hz, 5-H), 7.35 (1H, ddd, J 8.0, 2.0, 0.9 Hz, 6-H), 7.41 (1H, dd, J 8.1, 7.2, Hz, 3′-H), 7.44–7.49 (2H, m, 2× ArH), 7.52 (1H, br d, J 8.1 Hz, 4′-H), 7.58 (1H, ddd, J 8.0, 2.0, 0.9 Hz, 4-H), 7.73 (1H, t, J 2.0 Hz, 2-H), 7.79–7.85 (1H, m, ArH), 8.09–8.16 (1H, m, ArH); δC (101 MHz, CDCl3) 39.2 (CH3), 94.6 (C), 117.9 (CH), 122.6 (CH), 124.1 (CH), 125.3 (CH), 126.1 (CH), 126.2 (CH), 126.5 (CH), 128.1 (CH), 129.0 (C), 131.1 (CH), 134.9 (C), 135.5 (CH), 135.9 (CH), 145.9 (C), 146.2 (C), 150.5 (C); HRMS (ESI) m/z 402.0445 (MH+. C18H17IN3 requires 402.0462).
2-(4′-Dimethylaminophenyl)-6-nitrobenzoxazole (18). 4-(Dimethylamino)benzoic acid (0.530 g, 3.25 mmol), 5-nitro-2-aminophenol (17) (0.500 g, 3.25 mmol) and boric acid (0.200 g, 3.25 mmol) in p-xylene (50 mL) were stirred under Dean–Stark conditions for 72 h. After cooling to room temperature, the reaction mixture was filtered and washed with p-xylene (100 mL) before concentrating in vacuo. Water (20 mL) was added to the residue and the mixture made basic (pH 9–10) with 1 M sodium hydroxide solution. The aqueous layer was extracted with dichloromethane (3 × 20 mL) and the organic layers were combined, dried (MgSO4), filtered and concentrated in vacuo. The crude material was purified using flash column chromatography eluting with 40% ethyl acetate in petroleum ether (40–60) to give 2-(4′-dimethylaminophenyl)-6-nitrobenzoxazole (18) as an orange solid (0.485 g, 54%). Mp 215–217 °C; νmax/cm−1 (neat) 3422, 3315, 2924 (CH), 1615 (CC), 1510, 1491, 1357, 822; δH (500 MHz, CDCl3) 3.10 (6H, s, 2× NCH3), 6.75 (2H, d, J 9.1 Hz, 3′-H and 5′-H), 7.69 (1H, d, J 8.7 Hz, 4-H), 8.10 (2H, d, J 9.1 Hz, 2′-H and 6′-H), 8.26 (1H, dd, J 8.7, 2.1 Hz, 5-H), 8.37 (1H, d, J 2.1 Hz, 7-H); δC (126 MHz, CDCl3) 40.2 (2× CH3), 106.7 (CH), 111.7 (2× CH), 112.5 (C), 118.5 (CH), 120.9 (CH), 130.1 (2× CH), 144.2 (C), 148.6 (C), 149.9 (C), 153.3 (C), 168.9 (C); HRMS (EI) m/z 283.0956 (M+. C15H13N3O3 requires 283.0957).
2-(4′-Dimethylaminophenyl)-6-aminobenzoxazole (19). The reaction was carried out according to the procedure for 4-(2′-aminophenyl)-N,N-diethylquinoline-2-carboxyamide (12) using 2-(4′-dimethylaminophenyl)-6-nitrobenzoxazole (18) (0.12 g, 0.42 mmol) and tin(II) chloride dihydrate (0.48 g, 2.1 mmol) in ethanol (10 mL). The reaction mixture was stirred under reflux for 16 h. The crude material was purified using flash column chromatography eluting with 1–5% methanol in dichloromethane to give 2-(4′-dimethylaminophenyl)-6-aminobenzoxazole (19) as an orange solid (0.089 g, 83%). Mp 186–187 °C; νmax/cm−1 (neat) 3430 (NH), 3314 (NH), 2916 (CH), 1613 (CC), 1508, 1489, 1356, 1126, 949, 820; δH (500 MHz, CDCl3) 3.05 (6H, s, 2× NCH3), 6.65 (1H, dd, J 8.4, 2.1 Hz, 5-H), 6.75 (2H, d, J 9.0 Hz, 3′-H and 5′-H), 6.84 (1H, d, J 2.1 Hz, 7-H), 7.45 (1H, d, J 8.4 Hz, 4-H), 8.03 (2H, d, J 9.0 Hz, 2′-H and 6′-H); δC (126 MHz, CDCl3) 40.3 (2× CH3), 96.8 (CH), 111.8 (2× CH), 112.8 (CH), 115.0 (C), 119.4 (CH), 128.6 (2× CH), 135.3 (C), 144.1 (C), 151.9 (C), 152.1 (C), 162.6 (C); HRMS (CI) m/z 254.1289 (MH+. C15H16N3O requires 254.1293).
2-(4′-Dimethylaminophenyl)-6-iodobenzoxazole (4)6. The reaction was carried out according to the general procedure for 4-iodonitrobenzene (6a) using 2-(4′-dimethylaminophenyl)-6-aminobenzoxazole (19) (0.050 g, 0.20 mmol), p-toluenesulfonic acid monohydrate (0.11 g, 0.60 mmol), polymer-supported nitrite (0.17 g, containing 0.60 mmol of NO2) and sodium iodide (0.060 g, 0.40 mmol). The reaction mixture was heated to 60 °C and stirred for 16 h. Purification by flash column chromatography eluting with 20% ethyl acetate in petroleum ether (40–60) gave 2-(4′-dimethylaminophenyl)-6-iodobenzoxazole (19) as a yellow solid (0.043 g, 64%). Spectroscopic data were consistent with the literature.6 Mp 183–184 °C; δH (400 MHz, CDCl3) 3.08 (6H, s, 2× NCH3), 6.77 (2H, d, J 9.0 Hz, 3′-H and 5′-H), 7.43 (1H, d, J 8.4 Hz, 4-H), 7.60 (1H, dd, J 8.4, 1.4 Hz, 5-H), 7.86 (1H, d, J 1.4 Hz, 7-H), 8.08 (2H, d, J 9.0 Hz, 2′-H and 6′-H); δC (101 MHz, CDCl3) 40.3 (2× CH3), 86.5 (C), 111.9 (2× CH), 113.8 (C), 119.5 (CH), 120.7 (CH), 129.5 (2× CH), 133.5 (CH), 142.7 (C), 151.5 (C), 152.7 (C), 164.6 (C); m/z (ESI) 387 (MNa+. 100%).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from Medical Research Scotland (studentship to N. L. S.), G.E. Healthcare Ltd and the University of Glasgow is gratefully acknowledged.
Notes and references
-
(a) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed;
(b) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC;
(c) Organic Bromine and Iodine Compounds, in Handbook of Environmental Chemistry, ed. A. H. Neilson, Springer, Heidelberg, Berlin, 2003 Search PubMed.
-
(a) S. S. Zoghbi, R. M. Baldwin, J. P. Seibyl, M. S. Al-Tikriti, Y. Zea-Ponce, M. Laruelle, E. H. Sybirska, S. W. Woods, A. W. Goddard, R. T. Malison, R. Zimmerman, D. S. Charney, E. O. Smith, P. B. Hoffer and R. B. Innis, Nucl. Med. Biol., 1992, 19, 881 CAS;
(b) L. H. Pinborg, C. Videbæk, S. G. Hasselbalch, S. A. Sørensen, A. Wagner, O. B. Paulson and G. M. Knudsen, J. Neurol., Neurosurg. Psychiatry, 2001, 70, 657 CrossRef CAS;
(c) K. Mori, T. Mori, Y. Toda, E. Fujii, M. Miyazaki, M. Harada and S. Kagami, Brain Dev., 2012, 34, 478 CrossRef PubMed.
- A. Blair, L. Stevenson, D. Dewar, S. L. Pimlott and A. Sutherland, MedChemComm, 2013, 4, 1461 RSC.
-
(a) M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153 RSC;
(b) S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149 RSC.
-
(a) J. Owens, A. A. Tebbutt, A. L. McGregor, K. Kodama, S. S. Magar, M. E. Perlman, D. J. Robins, G. J. Durant and J. McCulloch, Nucl. Med. Biol., 2000, 27, 557 CrossRef CAS PubMed;
(b) R. J. J. Knol, K. De Bruin, B. L. F. Van Eck-Smit, S. Pimlott, D. J. Wyper and J. Booij, Synapses, 2009, 67, 557 CrossRef PubMed.
- Z.-P. Zhuang, M.-P. Kung, C. Hou, K. Plössl, D. Skovronsky, T. L. Gur, J. Q. Trojanowski, V. M.-Y. Lee and H. F. Kung, Nucl. Med. Biol., 2001, 28, 887 CrossRef CAS PubMed.
- R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1999, pp. 619–626 Search PubMed.
- V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS.
- For reviews, see:
(a) T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043 RSC;
(b) N. L. Sloan and A. Sutherland, Synthesis, 2016, 48, 2969 CrossRef.
-
(a) A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844 CrossRef CAS PubMed;
(b) M. Chen, S. Ichikawa and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 263 CrossRef CAS PubMed.
-
(a) A. A. Cant, R. Bhalla, S. L. Pimlott and A. Sutherland, Chem. Commun., 2012, 48, 3993 RSC;
(b) A. A. Cant, S. Champion, R. Bhalla, S. L. Pimlott and A. Sutherland, Angew. Chem., Int. Ed., 2013, 52, 7829 CrossRef CAS PubMed.
-
(a) H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5652 CrossRef CAS PubMed;
(b) R. H. Tale, G. Toradmal and V. B. Gopula, RSC Adv., 2015, 5, 84910 RSC;
(c) B. M. Partridge and J. F. Hartwig, Org. Lett., 2013, 15, 140 CrossRef CAS PubMed.
-
(a) T. C. Wilson, G. McSweeney, S. Preshlock, S. Verhoog, M. Tredwell, T. Cailly and V. Gouverneur, Chem. Commun., 2016, 52, 13277 RSC;
(b) P. Zhang, R. Zhuang, Z. Guo, X. Su, X. Chen and X. Zhang, Chem.–Eur. J., 2016, 22, 16783 CrossRef CAS PubMed.
-
(a) F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028 CrossRef CAS PubMed;
(b) C.-Y. Zhou, J. Li, S. Peddibhotla and D. Roma, Org. Lett., 2010, 12, 2104 CrossRef CAS PubMed;
(c) D. Qiu, F. Mo, Z. Zheng, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 5474 CrossRef CAS PubMed;
(d) D. Leboeuf, J. Ciesielski and A. J. Frontier, Synlett, 2014, 25, 399 CAS;
(e) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782 CrossRef CAS PubMed;
(f) D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, J. Org. Chem., 2016, 81, 772 CrossRef CAS PubMed.
- Brønsted acid catalysed activation of NIS for aryl iodide formation has also been developed: G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770 CrossRef CAS PubMed.
- H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS PubMed.
-
(a) M. Caldarelli, I. R. Baxendale and S. V. Ley, Green Chem., 2000, 2, 43 RSC;
(b) J. Merrington, M. James and M. Bradley, Chem. Commun., 2002, 140 RSC;
(c) V. D. Filimonov, M. Trusova, P. Postnikov, E. A. Krasnokutskaya, Y. M. Lee, H. Y. Hwang, H. Kim and K.-W. Chi, Org. Lett., 2008, 10, 3961 CrossRef CAS PubMed.
-
(a) E. A. Krasnokutskaya, N. I. Semenischeva, V. D. Filimonov and P. Knochel, Synthesis, 2007, 81 CrossRef CAS;
(b) M. E. Trusova, E. A. Krasnokutskaya, P. S. Postnikov, Y. Choi, K.-W. Chi and V. D. Filimonov, Synthesis, 2011, 2154 CAS.
- We have recently reported a one-pot iodination of anilines using diazonium salts specifically for radioiodination: N. L. Sloan, S. K. Luthra, G. McRobbie, S. L. Pimlott and A. Sutherland, Chem. Commun., 2017, 53, 11008 RSC.
-
(a) A. M. Scarf, L. M. Ittner and M. Kassiou, J. Med. Chem., 2009, 52, 581 CrossRef CAS PubMed;
(b) A. Blair, F. Zmuda, G. Malviya, A. A. S. Tavares, G. D. Tamagnan, A. J. Chalmers, D. Dewar, S. L. Pimlott and A. Sutherland, Chem. Sci., 2015, 6, 4772 RSC.
- L. Gilfillan, A. Blair, B. J. Morris, J. A. Pratt, L. Schweiger, S. Pimlott and A. Sutherland, MedChemComm, 2013, 4, 1118 RSC.
- S. H. Korzeniowski and G. W. Gokel, Tetrahedron Lett., 1977, 18, 3519 CrossRef.
- A. Zarei, A. R. Hajipour and L. Khazdooz, Synthesis, 2009, 941 CrossRef CAS.
- L. Malet-Sanz, J. Madrzak, R. S. Holvey and T. Underwood, Tetrahedron Lett., 2009, 50, 7263 CrossRef CAS.
- Y. Suzuki, Y. Ishiwata, K. Moriyama and H. Togo, Tetrahedron Lett., 2010, 51, 5950 CrossRef CAS.
- M. Banerjee, R. Shukla and R. Rathore, J. Am. Chem. Soc., 2009, 131, 1780 CrossRef CAS PubMed.
- G. Jakab, A. Hosseini, H. Hausmann and P. R. Schreiner, Synthesis, 2013, 45, 1635 CrossRef CAS.
- D. A. Offermann, J. E. McKendrick, J. J. P. Sejberg, B. Mo, M. D. Holdom, B. A. Helm, R. J. Leatherbarrow, A. J. Beavil, B. J. Sutton and A. C. Spivey, J. Org. Chem., 2012, 77, 3197 CrossRef CAS PubMed.
- C.-Y. Liu, H. Ren and P. Knochel, Org. Lett., 2006, 8, 617 CrossRef CAS PubMed.
- M. G. Banwell, E. Hamel, D. C. R. Hockless, P. Verdier-Pinard, A. C. Willis and D. J. Wong, Bioorg. Med. Chem., 2006, 14, 4627 CrossRef CAS PubMed.
- D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 7908 CrossRef CAS PubMed.
- W. A. Lees and A. Burawoy, Tetrahedron, 1964, 20, 1533 CrossRef CAS.
- E. J. Trybulski, E. Reeder, J. F. Blount, A. Walser and R. I. Fryer, J. Org. Chem., 1982, 47, 2441 CrossRef CAS.
- S. Wan, S. R. Wang and W. Lu, J. Org. Chem., 2006, 71, 4349 CrossRef CAS PubMed.
- B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786 CrossRef CAS PubMed.
- A. Bachki, F. Foubelo and M. Yus, Tetrahedron, 1994, 50, 5139 CrossRef CAS.
- R. Bolton and J. P. B. Sandall, J. Chem. Soc., Perkin Trans. 2, 1977, 278 RSC.
- H. O. House, D. G. Koepsell and W. J. Campbell, J. Org. Chem., 1972, 37, 1003 CrossRef CAS.
- C. W. Huffman, E. M. Godar and D. C. Torgeson, J. Agric. Food Chem., 1967, 15, 976 CrossRef CAS.
- M. A. Bartucci and J. W. Ciszek, J. Org. Chem., 2014, 79, 5586 CrossRef CAS PubMed.
- B. Riegel, C. J. Albisetti Jr, G. R. Lappin and R. H. Baker, J. Am. Chem. Soc., 1946, 68, 2685 CrossRef CAS.
- C. E. Kaslow and M. M. Marsh, J. Org. Chem., 1947, 12, 456 CrossRef CAS PubMed.
- H. Ghosh, R. Yella, A. R. Ali, S. K. Sahoo and B. K. Patel, Tetrahedron Lett., 2009, 50, 2407 CrossRef CAS.
- P. J. Klein, M. Chomet, A. Metaxas, J. A. M. Christiaans, E. Kooijman, R. C. Schuit, A. A. Lammertsma, B. N. M. Van Berckel and A. D. Windhorst, Eur. J. Med. Chem., 2016, 118, 143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c7ra11860k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a