
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of substituted pyrazines from N-allyl malonamides†
Frederic Ballaschk,
Hellmuth Erhardt and
Stefan F. Kirsch *
*
Organic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: sfkirsch@uni-wuppertal.de
First published on 7th December 2017
Abstract
The synthesis of pyrazines is described using a sequence that begins with the diazidation of N-allyl malonamides followed by thermal or copper-mediated cyclization. The pyrazine products possess an ester and a hydroxy group at 2- and 3-positions of the heterocyclic core, while alkyl- and aryl groups may be introduced at the other positions. We also show how to modify the pyrazines obtained with our method; examples regarding alkylations, side-chain brominations, hydrogenations and cross-couplings are presented.
Amongst the countless classes of nitrogen-containing heterocycles, pyrazines1 are distinguished from their kin by the ease of how to access the symmetrical members. For example, the selfcondensation of α-amino ketones easily provides symmetrical pyrazines in an efficient way.2 However, this approach provides mixtures of pyrazines when two different α-amino ketones are used.3 The condensation of 1,2-diamines with 1,2-diketones (followed by dehydrogenation) is another typical method for the preparation of the pyrazine core that has severe limitations with respect to the regiospecifity.4 In addition to these classical methods, a broad range of alternative methods were developed with the goal of addressing as many substituents as possible.5 Pyrazines were synthesized starting from α-hydroxy ketones,6 α-halo ketones,7 nitro epoxides,8 and 2H-azirines.9 The pyrazine core was modified through lithiation;10 halogenated pyrazines were submitted to palladium-catalyzed cross-coupling reactions.11 A surprisingly small number of reports describe the synthesis of the pyrazine core through cyclization strategies, including electrocyclizations and cyclocondensations.12
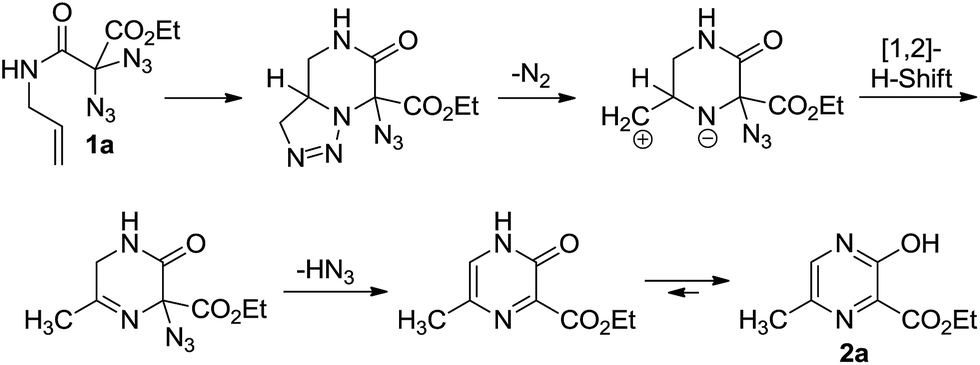
As part of our ongoing studies to unveil the reactivity of geminal diazido compounds,13 we recently showed that 2,2-diazido-3-oxohept-6-enoates gave 3-hydroxypyridines upon thermolysis (Scheme 1).14 We then questioned whether, starting with diazido compounds 1 derived from N-allyl malonamides, pyrazines 2 may become accessible in a fully analogous way. The challenging and densely substituted pyrazine core of 2 shows potential to produce several variants through further heterocycle modifying endeavors.
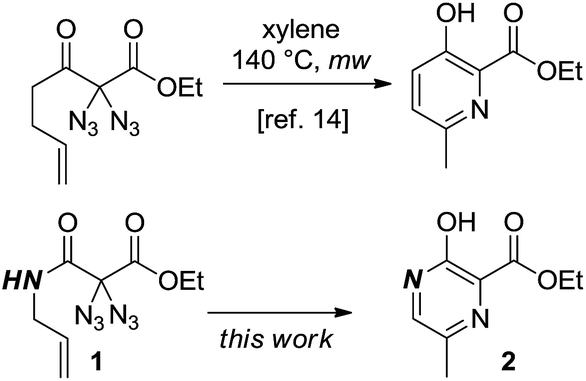 | ||
| Scheme 1 Formation of heterocycles through the thermolysis of geminal diazides. | ||
Earlier studies demonstrated how nitrogen-containing heterocycles can be synthesized from precursor compounds possessing the geminal diazido moiety.15 To our knowledge, this is the first study to successfully convert diazidated N-allyl malonamides into pyrazines through simple thermolysis. We began this study with the synthesis of the diazides 1 as summarized in Scheme 2. To this end, the malonic acid monoethylester 3 was submitted to standard amide-forming conditions using the respective allylic amine 4, HOBT and EDC at room temperature in dichloromethane. The subsequent diazidation of the N-allyl malonamides 5 was achieved with iodine and sodium azide in aqueous DMSO at room temperature, a method we recently developed as a general entry toward diazides derived from 1,3-dicarbonyl compounds.16 This two-step sequence provided an easy and rapid access to a range of diazidated N-allyl malonamides (1a–1e). Caution! Diazidated compounds are potentially hazardous and should be handled with care. Although we never encountered any problems, we advise the use of protective gear, in particular for scales > 1 mmol.
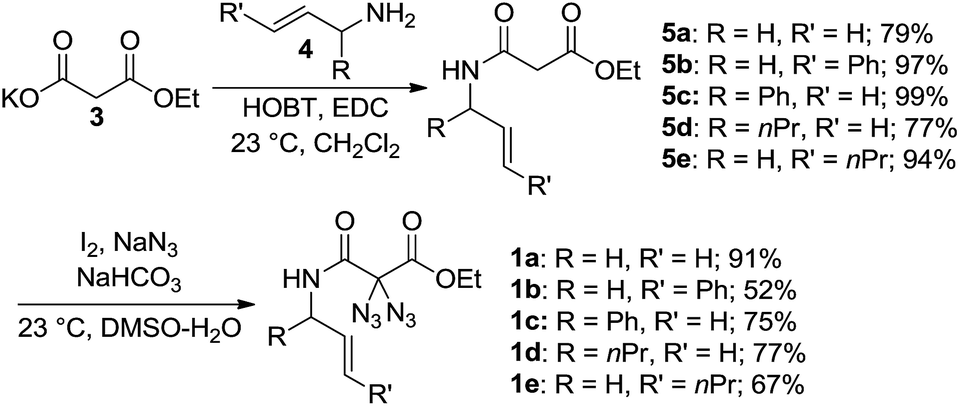 | ||
| Scheme 2 Synthesis of gem-diazido N-allyl malonamides. | ||
To our delight, conventional heating of geminal diazide 1a (R = H, R′ = H) in xylene gave the desired pyrazine core. Moderate yields were obtained when the reaction was carried out in xylene at 140 °C with a concentration of 0.05 mol L−1 affording the product 2a in 43%. Unfortunately, our search for better reaction conditions was mainly fruitless: other solvents and temperatures led to markedly reduced yields. A range of transition metal complex additives had simply no effect on the pyrazine formation; in a few cases (e.g., with iron and palladium salts) pyrazine products were not even detected, despite complete consumption of the diazides. The use of stoichiometric amounts of copper(I)iodide in acetic acid at 120 °C, on the other hand, led to a substantial increase in yield, and the pyrazine 2a was obtained in 68% yield in a fully reproducible manner. Although these conditions appeared somewhat unusual in terms of stochiometry and solvent, they typically gave better yields for pyrazine formation, in direct comparison to the simple heating in xylene, while the number of unidentified by-products was reduced.
As summarized in Scheme 3, several diazidated amides 1 were successfully converted into the pyrazines 2 by relying on either the pure heating in xylene (A) or the copper-mediated version in acetic acid (B). Besides the parent structure 2a with a methyl group at 6-position (R = H, R′ = H), the method also allowed for the construction of pyrazines having other substituents at 6-position (e.g., 2b with R′ = Ph). It was also possible to introduce additional substituents at 5-position as exemplified through the formation of 2c (R = Ph) and 2d (R = nPr).
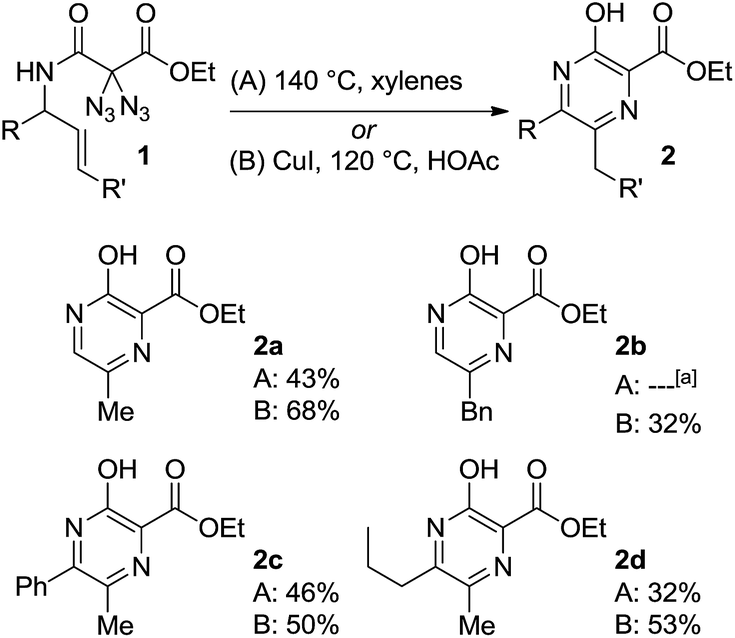 | ||
| Scheme 3 Synthesis of pyrazines 2. [a] = mixture of inseperable compounds. | ||
We point out that 3-hydroxy-2-pyrazinecarboxylic acid derivatives 2 are, in priciple, capable of prototropic tautomerism. The IR spectra of the neat compounds measured with ATR probes actually suggest the existence of the tautomeric 3-oxo-3,4-dihydropyrazine form since the spectra typically show IR signals for two independent carbonyl groups. On the other hand, NMR data in CDCl3-solution appear to provide evidence for the aromatic pyrazine system: as exemplified with compound 2a, the chemical shifts of −42.0 ppm for N-1 and −85.4 ppm for N-4 (approximated from 1H-15N-HMBC correlations) indicate the pyrazine core. This view is further supported by the downfield shift for H-5 (8.29 ppm, 1H NMR spectrum). Although NMR data on derivatives of pyrazinecarboxylic acid are rare,17 a recent study by Holzer, Eller and co-workers18 on 3-amino-2-pyrazinecarboxylic acid and 3,4-dihydro-3-oxo-2-pyrazinecarboxylic acid confirms our structural assignment and suggests that we have an unusual case where the 3-hydroxy-2-pyrazinecarboxylic acid core is existent in solution, rather than its more common 3-oxo-3,4-dihydropyrazine tautomer.
Our rationale for the reaction is analogous to what we proposed for the formation of 3-hydroxypyridines through the thermolysis of diazides:14 the reaction starts with an intramolecular 1,3-dipolar cycloaddition between one azide moiety and the olefin to provide a triazoline intermediate. After loss of elemental nitrogen and a 1,2-hydride shift, elimination of hydrazoic acid may lead to the heterocyclic product (Scheme 4). As shown in eqn (1), the reaction of diazide 6 having an alkyne moiety led to the bicyclic compound 7 where the triazole substructure remained stable, and the hydrazoic acid was still attached. This structural assignment was supported by NMR, HRMS and IR data, including the characteristic IR band at 2133 cm−1.
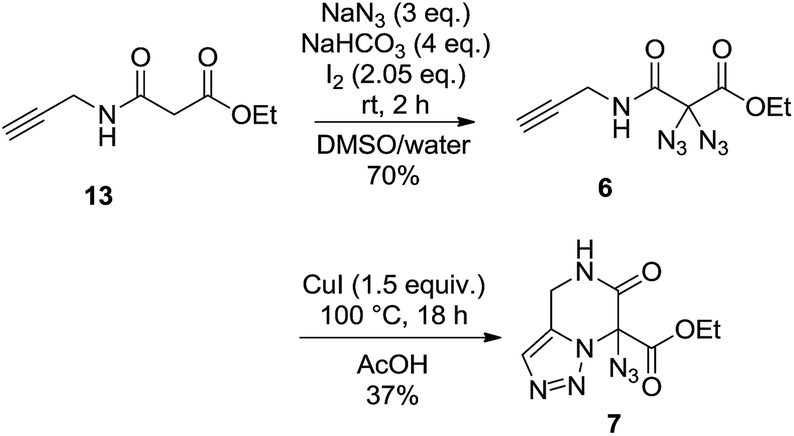 | (1) |
 | ||
| Scheme 4 Possible mechanism for the formation of pyrazine 2a. | ||
Due to the great importance of flexibility and diversity in the application of this method, the direct modification of the 3-hydroxypyrazine 2a was then investigated as summarized in Scheme 5. We showed that it is easily possible to modify the parent structure 2a in various ways: for example, the nitrogen was selectively methylated with methyl iodide and cesium carbonate in acetonitrile at room temperature, leading to the N-methylated dihydro pyrazinone 8 (path a). On the other hand, the alkylation of the hydroxy oxygen was achieved through the use of silver carbonate giving the ethers 9a (with X = Me) and 9b (with X = iPr) (path b). High yields were also obtained for the silylation (e.g., formation of 9d and 9e), and the triflate 9c was formed with triflic anhydride under standard conditions (path b). The latter compound became an ideal starting point for further diversifications with classical palladium-catalyzed cross coupling methodologies. For example, Suzuki coupling of 9c with various boronic acids provided a range of interesting structures 10a–e featuring the densely functionalized pyrazine core (path c). With pyrazines not possessing a free hydroxyl, a further functionalization of the methyl group at 6-position was possible through radical bromination under Wohl–Ziegler conditions; for example, the brominated pyrazines 11a and 11b were formed in moderate yields. The hydrogenation of the aromatic core was achieved using rhodium on alumina in ethanol at 30 bar of hydrogen. The oxo-piperazine 12 was isolated as a mixture of diastereomers in 87% yield. Unfortunately, we were not able to install halogen atoms at the 5-position of the existing pyrazine core 9a; all attempts using metalation or electrophilic substitution protocols failed, at least in our hands.
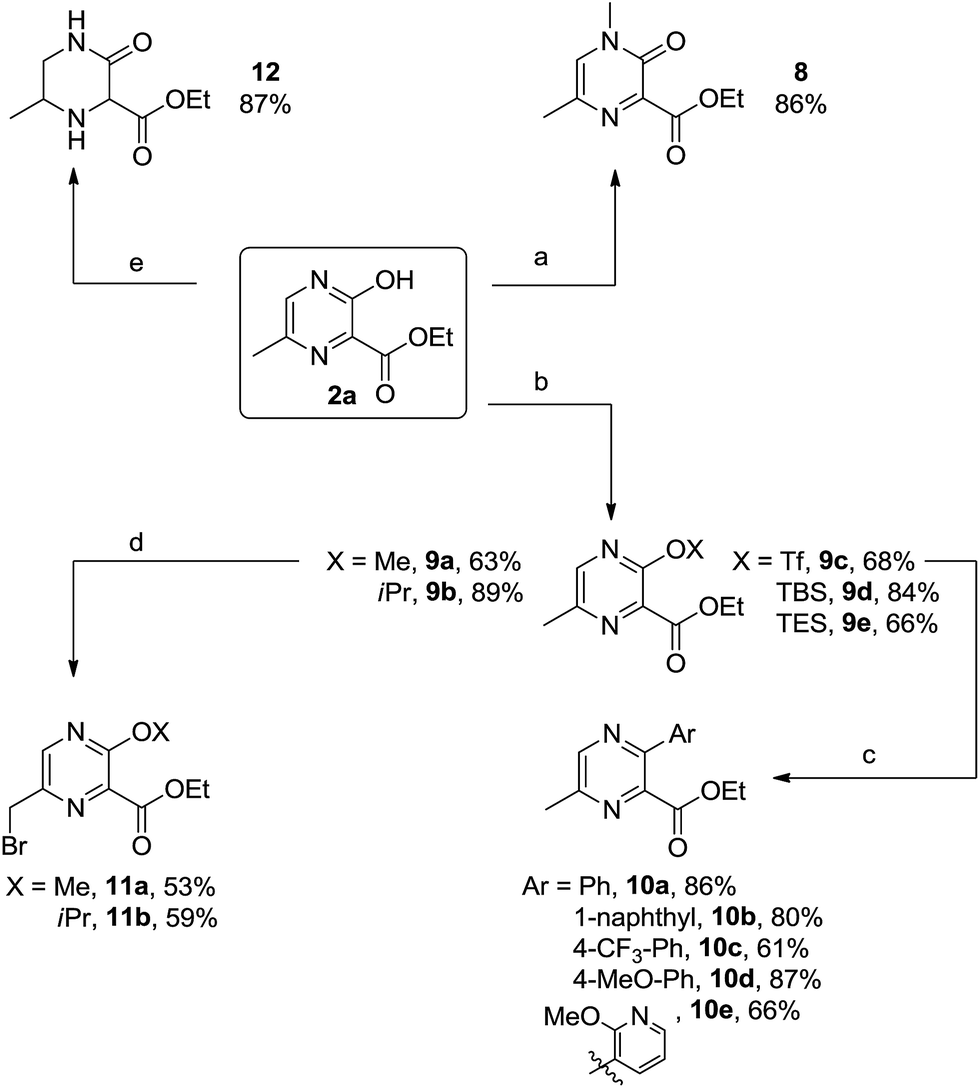 | ||
| Scheme 5 Derivatization of the pyrazine 2a. Reaction conditions: (a) MeI (1.2 eq.), Cs2CO3 (1.2 eq.), MeCN, 25 °C; (b) 9a, 9b: MeI (2 eq.) or iPrI (2 eq.), Ag2CO3 (1.2 eq.), n-hexane/benzene, 90 °C; 9c: Tf2O (1.3 eq.), NEt3 (3 eq.), DCM; −20 °C to 0 °C, 100 min; 9d: TBSCl (3 eq.), imidazole (3 eq.), pyridine, 25 °C; 9e: TESOTf (3 eq.), 2,6-lutidine (3 eq.), DCM, 25 °C; (c) Ar–B(OH)2 (1.5 eq.), Cs2CO3 (3 eq.), Pd(PPh3)4 (5 mol%), dioxane/water, 85 °C; (d) AIBN (10 mol%), NBS (1 eq.), CCl4, 75 °C; (e) H2 (30 bar), Rh/Al2O3 (10 mol%), EtOH, 25 °C. | ||
Conclusions
In summary, we have introduced a new and simple method for the synthesis of densely substituted pyrazines 2. Starting from N-allyl malonamides, the title compounds are accessible through a two-step sequence consisting of diazidation and subsequent thermolysis. We have also shown how the parent pyrazine structure 2 can be further modified at almost every position, in a simple and straightforward manner, thus rendering the pyrazines 2 highly valuable as building blocks for the synthesis of naturally occurring and pharmaceutically active compounds.Experimental
General procedure for the synthesis of gem-diazido N-allyl malonamides 1
N-Allyl malonamides 5 (1 eq.) were dissolved in DMSO/water (2/1) (0.1 M), and NaN3 (4 eq.) and NaHCO3 (3 eq.) were added under heavy stirring. The suspension was cooled to 0 °C, and iodine (2.05 eq.) was slowly added. The reaction maintained at this temperature for 10 min before warming to room temperature. The mixture was stirred at room temperature until complete consumption of the starting material (monitored via TLC) (∼2 h). The reaction mixture was quenched with aqueous saturated solution of Na2S2O3 until complete discoloration appeared. The mixture was extracted with EtOAc, and the combined organic phases were washed with brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. Purification by flash-chromatography on silica gel furnished the corresponding gem-diazido N-allyl malonamides 1.Caution! Diazidated compounds are potentially hazardous. We advise the use of protective gear, in particular for scales > 1 mmol.
General procedure for the synthesis of pyrazines 2
A: The reaction was carried out under argon atmosphere. Gem-diazido N-allyl malonamide 1 (1 eq.) was dissolved in xylenes (0.05 M) and stirred at 140 °C for 24 h. The reaction mixture was cooled to room temperature and the solvent was evaporated under reduced pressure. Purification by flash-chromatography on silica gel furnished the corresponding pyrazines 2.B: The reaction was carried out under argon atmosphere. Gem-diazido N-allyl malonamide 1 (1 eq.) was dissolved in glacial acetic acid (0.05 M). Copper(I)iodide (1.5 eq.) was added, and the reaction was stirred at 120 °C for 24 h. The reaction mixture was cooled to room temperature and filtered through a short pad of celite. The filtrate was evaporated under reduced pressure. Purification by flash-chromatography on silica gel furnished the corresponding pyrazines 2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For general reviews, see: (a) N. J. Race, I. R. Hazelden, A. Faulkner and J. F. Bower, Chem. Sci., 2017, 57, 10257 Search PubMed; (b) E. Marcantoni and M. Petrini, Adv. Synth. Catal., 2016, 358, 3657 CrossRef CAS; (c) W. Unsworth and R. Taylor, Synlett, 2016, 2051 CrossRef CAS; for selected applications of pyrazines, see: (d) D. Garella, E. Borretto, A. Di Stilo, K. Martina, G. Cravotto and P. Cintas, MedChemComm, 2013, 4, 1323 RSC; (e) G. Roymahapatra, S. M. Mandal, W. F. Porto, T. Samanta, S. Giri, J. Dinda, O. L. Franco and P. K. Chattaraj, Curr. Med. Chem., 2012, 19, 4184 CrossRef CAS PubMed; (f) R. Mondal, S. Ko and Z. J. Bao, Mater. Chem., 2010, 20, 10568 RSC; (g) A. Adams and N. de Kimpe, Food Chem., 2009, 115, 1417 CrossRef CAS; (h) D. P. De Schutter, D. Saison, F. Delvaux, G. Derdelinckx, J.-M. Rock, H. Neven and F. R. Delvaux, J. Agric. Food Chem., 2008, 56, 246 CrossRef CAS PubMed.
- (a) V. Chandrashaker, M. Taniguchi, M. Ptaszek and J. S. Lindsey, Tetrahedron, 2012, 68, 6957 CrossRef CAS; (b) N. Rojas, Y. Grillasca, A. Acosta, I. Audelo and G. G. J. de la Mora, J. Heterocycl. Chem., 2013, 50, 982 CrossRef CAS; (c) S. Albrecht, M. Al-Lakkis-Wehbe, A. Orsini, A. Defoin, P. Pale, E. Salomon, C. Tarnus and J.-M. Weibel, Bioorg. Med. Chem., 2011, 19, 1434 CrossRef CAS PubMed; (d) M. Peña-López, M. M. Martínez, L. A. Sarandeses and J. P. Sestelo, Org. Lett., 2010, 12, 852 CrossRef PubMed; (e) T. Focken and A. B. Charette, Org. Lett., 2006, 8, 2985 CrossRef CAS PubMed; (f) T. Chiba, H. Sakagami, M. Murata and M. J. Okimoto, J. Org. Chem., 1995, 60, 6764 CrossRef CAS.
- J. S. Dickschat, H. Reichenbach, I. Wagner-Döbler and S. Schulz, Eur. J. Org. Chem., 2005, 4141 CrossRef CAS.
- (a) P. Ganji and P. W. N. M. van Leeuwen, J. Org. Chem., 2017, 82, 1768 CrossRef CAS PubMed; (b) K. K. D. R. Viswanadham, M. Prathap Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517 RSC; (c) P. Ghosh, A. Mandal and R. Subba, Catal. Commun., 2013, 41, 146 CrossRef CAS; (d) P. Hazarika, P. Gogoi and D. Konwar, Synth. Commun., 2007, 37, 3447 CrossRef CAS; (e) A. Kleineweischede and J. Mattay, Eur. J. Org. Chem., 2006, 947 CrossRef CAS; (f) A. Ohta, S. Masano, S. Iwakura, A. Tamura, H. Watahiki, M. Tsutsui, Y. Akita, T. Watanabe and T. J. Kurihara, J. Heterocycl. Chem., 1982, 19, 465 CrossRef CAS.
- (a) N. S. Y. Loy, S. Kim and C.-M. Park, Org. Lett., 2015, 17, 395 CrossRef CAS PubMed; (b) M. Ganesh Kumar, V. J. Thombare, R. D. Bhaisare, A. Adak and H. N. Gopi, Eur. J. Org. Chem., 2015, 135 CrossRef CAS; (c) Z. Chen, D. Ye, G. Xu, M. Ye and L. Liu, Org. Biomol. Chem., 2013, 11, 6699 RSC; (d) D. F. Taber, P. W. DeMatteo and K. V. Taluskie, J. Org. Chem., 2007, 72, 1492 CrossRef CAS PubMed; (e) H. Matsushita, S.-H. Lee, K. Yoshida, B. Clapham, G. Koch, J. Zimmermann and K. D. Janda, Org. Lett., 2004, 6, 4627 CrossRef CAS PubMed; (f) A. Hossain, S. K. Pagire and O. Reiser, Synlett, 2017, 28, 1707 CrossRef CAS.
- (a) F. Tamaddon, A. D. Tafti and F. Pooramini, Synthesis, 2016, 48, 4295 CrossRef CAS; (b) L. Fan, W. Chen and C. Qian, Tetrahedron Lett., 2013, 54, 231 CrossRef CAS; (c) A. L. Sauers, D. M. Ho and S. J. Bernhard, J. Org. Chem., 2004, 69, 8910 CrossRef CAS PubMed.
- (a) J. G. Barlind, U. A. Bauer, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, R. D. M. Davies, J. W. Eriksson, C. D. Hammond and R. J. Hovland, Med. Chem., 2012, 55, 10610 CrossRef CAS PubMed; (b) T. Utsukihara, H. Nakamura, M. Watanabe and C. A. Horiuchi, Tetrahedron Lett., 2006, 47, 9359 CrossRef CAS.
- (a) A. Vidal-Albalat, S. Rodríguez and F. V. González, Org. Lett., 2014, 16, 1752 CrossRef CAS PubMed; (b) R. H. Fischer and H. M. Weitz, Synthesis, 1976, 53 CrossRef CAS.
- (a) S. Choi, S. Ha and C.-S. Park, Chem. Commun., 2017, 53, 6054 RSC; (b) F. Palacios, A. M. Ochoa de Retana, J. I. Gil and R. López de Munain, Org. Lett., 2002, 4, 2405 CrossRef CAS PubMed; (c) R. Flammang, S. Lacombe, A. Laurent, A. Maquestiau, B. Marquet and S. Novkova, Tetrahedron, 1986, 42, 315 CrossRef CAS.
- (a) L. Decrane, N. Plé and A. J. Turck, J. Heterocycl. Chem., 2005, 42, 509 CrossRef CAS; (b) W. Liu, D. S. Wise and L. B. J. Townsend, J. Org. Chem., 2001, 66, 4783 CrossRef CAS PubMed; (c) C. Y. Zhang and J. M. Tour, J. Am. Chem. Soc., 1999, 121, 8783 CrossRef CAS; (d) N. Plé, A. Turck, A. Heynderickx and G. Quéguiner, Tetrahedron, 1998, 54, 4899 CrossRef.
- (a) T. Hosoya, R. Iimori, S. Yoshida, Y. Sumida, Y. Sahara-Miura, J.-I. Sato and S. Inouye, Org. Lett., 2015, 17, 3888 CrossRef CAS PubMed; (b) A. Petrosyan, P. Ehlers, S. Reimann, T. V. Ghochikyan, A. S. Saghyan, A. Spannenberg, S. Lochbrunner and P. Langer, Tetrahedron, 2015, 71, 6803 CrossRef CAS; (c) C.-G. Yang, G. Liu and B. J. Jiang, J. Org. Chem., 2002, 67, 9392 CrossRef CAS PubMed; (d) W. J. Thompson, J. H. Jones, P. A. Lyle and J. E. Thies, J. Org. Chem., 1988, 53, 2052 CrossRef CAS.
- (a) N. V. Rostovskii, J. O. Ruvinskaya, M. S. Novikov, A. F. Khlebnikov, I. A. Smetanin and A. V. J. Agafonova, J. Org. Chem., 2017, 82, 256 CrossRef CAS PubMed; (b) Y. Okada, H. Taguchi, Y. Nishiyama and T. Yokoi, Tetrahedron Lett., 1994, 35, 1231 CrossRef CAS; (c) G. Büchi and J. J. Galindo, J. Org. Chem., 1991, 56, 2605 CrossRef.
- (a) P. Biallas, A. P. Häring and S. F. Kirsch, Org. Biomol. Chem., 2017, 15, 3184 RSC; (b) A. P. Häring, P. Biallas and S. F. Kirsch, Eur. J. Org. Chem., 2017, 1526 CrossRef; (c) H. Erhardt, F. Mohr and S. F. Kirsch, Eur. J. Org. Chem., 2016, 5629 CrossRef CAS; (d) H. Erhardt, F. Mohr and S. F. Kirsch, Chem. Commun., 2016, 52, 545 RSC.
- H. Erhardt, K. A. Kunz and S. F. Kirsch, Org. Lett., 2017, 19, 178 CrossRef CAS PubMed.
- (a) C. O. Kappe and G. J. Färber, J. Chem. Soc., Perkin Trans. 1, 1991, 5, 1342 RSC; (b) W. Ogilvie and W. Rank, Can. J. Chem., 1987, 65, 166 CrossRef CAS; (c) T. Kappe, G. Lang and E. Pongratz, J. Chem. Soc., Chem. Commun., 1984, 338 RSC; (d) R. M. Moriarty and P. Serridge, J. Am. Chem. Soc., 1971, 93, 1534 CrossRef CAS.
- (a) H. Erhardt, A. P. Häring, A. Kotthaus, M. Roggel, M. L. Tong, P. Biallas, M. Jübermann, F. Mohr and S. F. Kirsch, J. Org. Chem., 2015, 80, 12460 CrossRef CAS PubMed; (b) T. Harschneck, S. Hummel, S. F. Kirsch and P. Klahn, Chem.–Eur. J., 2012, 18, 1187 CrossRef CAS PubMed.
- S. Tobias and H. Günther, Tetrahedron Lett., 1982, 23, 4785 CrossRef CAS.
- W. Holzer, G. A. Eller, B. Datterl and D. Habicht, Magn. Reson. Chem., 2009, 47, 617 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data and copies of 1H and 13C NMR-spectra. See DOI: 10.1039/c7ra11529f |
| This journal is © The Royal Society of Chemistry 2017 |