
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelating agents as coating molecules for iron oxide nanoparticles†
Debora Bonvin a,
Jessica A. M. Bastiaansenbc,
Matthias Stuberbc,
Heinrich Hofmanna and
Marijana Mionić Ebersold*abc
a,
Jessica A. M. Bastiaansenbc,
Matthias Stuberbc,
Heinrich Hofmanna and
Marijana Mionić Ebersold*abc
aPowder Technology Laboratory, Institute of Materials, Ecole Polytechnique Fédérale de Lausanne, Switzerland
bDepartment of Radiology, University Hospital (CHUV), University of Lausanne (UNIL), Switzerland. E-mail: marijanamionic@gmail.com
cCenter of Biomedical Imaging (CIBM), Lausanne, Switzerland
First published on 8th December 2017
Abstract
Due to their high chemical affinity towards metal ions, chelating agents (CAs) have been used for decades for water purification, but also for protection against metal intoxication and in nanomedicine as linking molecules at the surface of nanoparticles. However, this strong chemical activity could also impact their colloidal behavior, which is essential for biomedical applications. Therefore, we coated iron oxide nanoparticles (IONPs) with four CAs, differing in their number of active chemical groups, with variations from 2 to 5 dents containing carboxylic groups: iminodiacetic acid (IDA), nitrilotriacetic acid (NTA), ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA). We found large differences between the CA-coated IONPs depending on the CA nature, especially regarding their agglomeration state and colloidal behavior, but also consequently their cellular uptake. Surprisingly, although CAs have been widely used for biomedical applications, CA-coated IONPs, especially IDA- and EDTA-coated IONPs, showed non-negligible toxicity. Moreover, for their application as contrast agents for MRI, we found that CA-coated IONPs displayed high r2 relaxivities, which differed according to their agglomeration state. Overall, our study suggests that CAs, depending on their chemical nature, can induce agglomeration and toxicity, which could be harmful in a clinical setting.
Introduction
Chelating agents (CAs) are organic molecules with two or more electron donor groups (e.g. nitrogen, oxygen, sulphur), which coordinatively bind to polyvalent metal ions, forming a cage. Thanks to this affinity for metal ions, CAs have been historically used as agents protecting against metal intoxication.1 In other words, they form cages around metal ions to protect the body from the toxic effects of the metal. This allows elimination of the metal ions from the body, mainly via the urinary excretion tract, which is possible for molecules smaller than 30 nm.2 The first CA that has been used in medicine was dimercaprol (British Anti Lewisite, BAL) to treat acute poisoning by arsenic,3 and later by mercury, gold and lead. In addition, gadolinium chelates are among the mostly used contrast agents for magnetic resonance imaging (MRI) in medicine.4The interest in using CAs in such applications essentially originates from the presence of multiple dents, each containing specific chemical groups (e.g. carboxylic or nitrogen groups), which form multiple bonds with the target metal ion. These dents can be flexible, giving to CAs highly reactive and versatile binding possibilities not only to form complexes with metal ions, but also to bind to metal oxide surfaces.5–7 A typical CA molecules have a minimum of two dents and can contain another active chemical group, such as one or more nitrogen groups. The versatility of these chemical groups can also cause the binding by bridging between two molecules/surfaces. Thus, the reactivity of CAs' chemical groups has been previously widely studied, especially the interaction of CAs with iron oxide surfaces,8 such as α-Fe2O3.5–7 Today, the versatility of CAs is also exploited in nanotechnology, where CAs were bound on one side to metal oxide nanoparticles (NPs) and on the other side to various chemical compounds. Mostly, CAs (e.g. nitrilotriacetic acid, NTA, or iminodiacetic acid, IDA) were employed around magnetic NPs for the separation of histidine tags9–12 or metal-containing/metal binding proteins13–15 in immobilized metal affinity chromatography. For clinical applications, Au-NPs16 and superparamagnetic IONPs17 were coupled to diethylenetriaminepentaacetic acid (DTPA) to chelate Gd3+ inside the coating. Also, CAs were used as linkers between NPs and fluorescent molecules, chemotherapeutic agents or even targeting peptides.18–20
CAs have therefore been essentially used for metal detoxification, protein isolation and metal sensing, but also in nanomedicine as linkers between multifunctional molecules and NPs. Therefore, for medical applications, NPs coated with CAs without using linking molecules have to be colloidally stable and biocompatible. Indeed, CAs occur even in the human body (e.g. chlorophyll, hemoglobin) and could therefore be considered as biocompatible per se. However, for example, lanthanide-based luminescent NPs coated with ethylenediaminetetraacetic acid (EDTA) induced necrosis in different cell lines, which was explained to be caused by the EDTA coating itself.21 Unfortunately, the agglomeration state of these NPs was not studied, but could be the origin of the observed toxicity. Therefore, studying both the ability of CAs to stabilize NPs and their toxicity, which are two indispensable attributes to use them in medicine, would open novel insights into the utility and potential of CAs for clinical applications.
Here, we coated iron oxide nanoparticles (IONPs), which are one of the mostly studied metal oxide NPs for medical applications,22–25 with the following CAs: IDA, NTA, EDTA and DTPA. These four CAs essentially differ in the number of dents containing carboxylic groups (2, 3, 4 and 5 respectively) and in the number of nitrogen groups (1, 1, 2 and 3 respectively). Firstly, the surface of CA-coated IONPs was characterized by Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and UV-visible spectroscopy. In order to evaluate the potential use of IDA, NTA, EDTA and DTPA as coating molecules, we studied CA-coated IONPs by transmission electron microscopy (TEM) and by dynamic light scattering (DLS), which was used to measure their zeta potentials and hydrodynamic diameters in water and biological medium and to evaluate their agglomeration state. Also, we evaluated the potential of CA-coated IONPs for MRI by measuring their MRI relaxivity in a clinical 3 T MRI scanner. Importantly, we performed a preliminary assessment of the toxicity of CA-coated IONPs with the MTS test and we studied their interaction with cells via their cellular uptake.
Materials and methods
IONPs
IONPs were synthesized following a protocol modified from Bonvin et al. described previously.26 Briefly, IONPs were synthesized by co-precipitation in combination with a hydrothermal treatment performed at 120 °C for 15 h.Coating of IONPs with CAs
IONPs' suspension in 10 mM HNO3 containing 50 mg IONPs (35 mg Fe) were mixed with water and the volume was adjusted to 10 ml. 68 mg of the following coating molecules were dissolved in 15 ml water: (i) iminodiacetic acid (IDA, Merck KGaA); (ii) nitrilotriacetic acid (NTA, AppliChem); (iii) ethylenediaminetetraacetic acid (EDTA, Carl Roth); (iv) diethylenetriaminepentaacetic acid (DTPA, AppliChem). 5 ml of the dissolved coating molecules were added to the IONPs' suspensions, which were rotated for 5 min with a rotator (tube rotator from VWR) placed on a shaker at 500 rpm. This procedure was repeated until the whole solution of coating molecules was added to the IONPs' suspension. The volume was adjusted to 35 ml with water to approximately reach a final concentration of 1 mgFe ml−1 and the IONPs' suspensions were rotated for 30 min with the rotator placed on the shaker at 500 rpm. After 30 min, the IONPs' suspensions were dialyzed (Spectra/Por®; 12–14 kDa) against water for 72 h by changing the dialysis solution every 10–12 h, and finally, the obtained suspensions were stored at 4 °C.IONPs' characterization
For transmission electron microscopy (TEM), 5 μl drops of IONPs' suspensions were deposited on holey carbon grids (Plano GmbH) placed onto absorbing filter paper and left to dry at room temperature. TEM micrographs were taken with a Talos F200X FEI electron microscope operated at an acceleration voltage of 200 kV and coupled to a CMOS-based FEI CETA 4k × 4k camera. The primary particle size of uncoated IONPs (diameter of IONPs, in the text referred as TEM diameter) of 1000 IONPs was measured manually from randomly taken TEM micrographs using the ImageJ software (the National Institutes of Health, U.S.).The hydrodynamic diameters (dh) and the zeta potentials of 1 ml IONPs' suspensions at concentration of 100 μgFe ml−1 in water (pH ∼ 6.1) and in RPMI medium (pH ∼ 7.6) were measured at room temperature in acrylic cuvettes (Sarstedt) with a Zetasizer Nano ZS (Malvern Instruments). The reported values of dh were obtained from the average of 3 × 12 measurements. The refractive index of γ-Fe2O3 and absorbance were set to 2.95 and 0.1, respectively. The dh of IONPs' suspensions were also measured by laser diffraction with the Mastersizer 3000 (Malvern) using the small volume sample dispersion unit. The refractive index and density of γ-Fe2O3-IONPs were set to 2.95 and 4.92 g ml−1, respectively, while the absorbance was set to 0.1. The dh of 50 μl of IONPs' suspensions at 200 μgFe ml−1 were measured at room temperature by centrifugal force with a disc centrifuge (CPS Instruments, Inc.) at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The refractive index and density of γ-Fe2O3-IONPs were set to 2.95 and 4.92 g ml−1, respectively, while the absorbance was set to 0.1. A sucrose gradient from 8% up to 24% sucrose was used. For all samples the iron concentration was determined by ICP-EOS. For this purpose, the volume of 80 μl of IONPs stock suspension of as-synthesized-IONPs was mixed with 920 μl of 6 M HCl. After three days, IONPs were fully dissolved and 500 μl of the obtained solution containing the dissolved Fe-ions was diluted in 2.5 ml of water. ICP-EOS was performed with ICP-EOS 9000 from Shimadzu.
000 rpm. The refractive index and density of γ-Fe2O3-IONPs were set to 2.95 and 4.92 g ml−1, respectively, while the absorbance was set to 0.1. A sucrose gradient from 8% up to 24% sucrose was used. For all samples the iron concentration was determined by ICP-EOS. For this purpose, the volume of 80 μl of IONPs stock suspension of as-synthesized-IONPs was mixed with 920 μl of 6 M HCl. After three days, IONPs were fully dissolved and 500 μl of the obtained solution containing the dissolved Fe-ions was diluted in 2.5 ml of water. ICP-EOS was performed with ICP-EOS 9000 from Shimadzu.
IONPs' suspensions were lyophilized for 4 days with an alpha 1–2 LD plus freeze dryer. Fourier transform IR (FTIR) spectra of IONPs' powder pellets were obtained with the Perkin Elmer Spectrum One spectrometer (series: 69288). Transmittance from 4600 cm−1 to 400 cm−1 were given as the average of measured 64 scans for each curve with a resolution of 4.00 cm−1. UV-visible spectra of IONPs' suspensions in water and in RPMI medium were measured at concentrations of 100 μgFe ml−1 in Brand® UV-cuvettes with a Cary 100 Bio spectrometer between 190 and 900 nm. The average time was set to 0.1 s, the data interval to 1 nm and the scan rate to 600 nm min−1. X-Ray Photoelectron Spectroscopy (XPS) measurements were carried out using a PHI VersaProbe II scanning XPS microprobe (Physical Instruments AG, Germany). Analysis was performed using a monochromatic Al Kα X-ray source of 24.8 W power with a beam size of 100 μm. The spherical capacitor analyser was set at 45° take-off angle with respect to the sample surface. The pass energy was 46.95 eV yielding a full width at half maximum of 0.91 eV for the Ag 3d5/2 peak. Curve fitting was performed using the PHI Multipak software.
Agglomerate density measurement
1 ml of IONPs at 100 μgFe ml−1 were dispensed into TPP PCV tubes (Techno Plastic Products, Trasadingen, Switzerland) and centrifuged at 2000 rpm for 1 h (Eppendorf centrifuge 5702 R, A-4-38 rotor). Agglomerate pellet volumes were measured using a TPP “easy read” measuring device and the agglomerate density of nine samples per condition were calculated as previously described:27
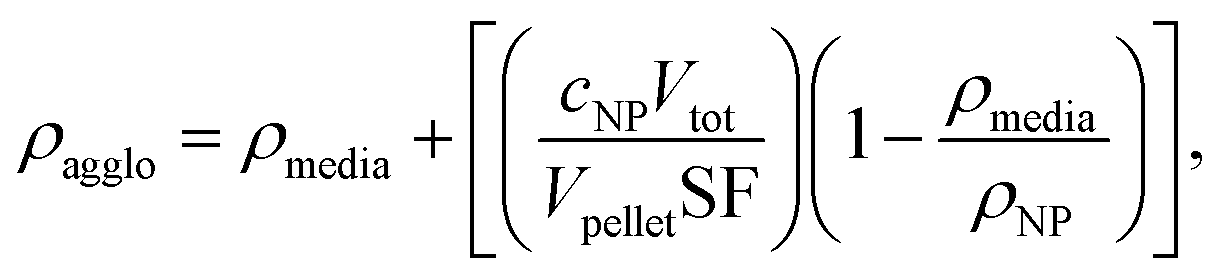 | (1) |
MRI
IONPs were suspended in a 2% agarose gel in 0.5 ml Eppendorf cups to obtain final concentrations ranging from 0 to 30 μgFe ml−1. All cups were placed in a water-containing phantom for subsequent T1 and T2 measurements, which were performed at room temperature on a 3.0 T clinical MRI scanner (MAGNETOM Prisma, Siemens AG, Erlangen, Germany), and data was acquired using a body array coil. The measurement of longitudinal relaxation time T1 was performed using a 2D spin echo sequence preceded by a 180° inversion pulse with different inversion times TI. The transversal relaxation times T2 measurements were performed using a 2D spin echo sequence with varying echo times TE. Imaging parameters for longitudinal relaxation times T1: TR 15 s, TE 7.1 ms, slice thickness 5 mm, FOV 250 × 150 mm2, matrix 384 × 310, RF excitation angle 90°, receiver bandwidth of 651 Hz per pixel, TI 23, 50, 100, 20, 500, 10000, 5000, 10000 ms. Imaging parameters for transversal relaxation times T2: TR 15 s, TE 7.1, 15, 25, 35, 65, 120, 240 ms, slice thickness 5 mm, FOV 250 × 150 mm2, matrix 384 × 310, RF excitation angle 90°, receiver bandwidth of 651 Hz per pixel. The signal evolution S as function of TI and TE was fitted to derive the T1 and T2 of each γ-Fe2O3 NPs suspension respectively, and is described as follows:
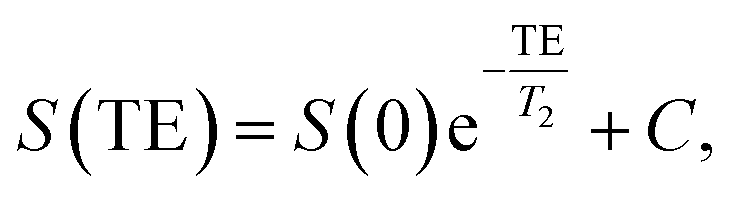 | (2) |
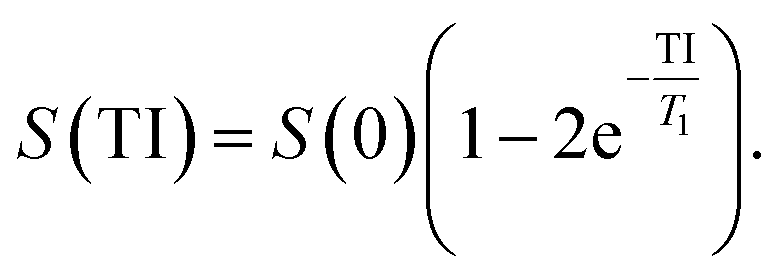 | (3) |
The T1 and T2 values as function of their γ-Fe2O3 NPs concentration were subsequently fitted to obtain the relaxivities r1 and r2 described as:
 | (4) |
All signal evolutions were analysed and fitted with Matlab (The MathWorks, Natick, MA, USA).
In vitro toxicity study
To determine the cell viability of LnCaP cells from ATCC (ATCC CRL-1740), the MTS test was used. 40000 LnCaP cells per well were cultured in 96-well plates at 37 °C, and exposed to 100 μl of different concentrations of IONPs (0, 20, 40, 60, 80 and 100 μgFe ml−1) for 24 h. Cells treated only with medium served as negative controls. After 24 h incubation, the supernatant of each well was removed. 100 μl of MTS solution (CellTiter 96® AQueous One Solution Cell Proliferation Assay from Promega, diluted 6 times in medium) was added to the cells. After 2 h incubation, the absorbance of the formazan product was measured with a microplate reader (Tecan Infinite M200) at a wavelength of 490 nm. The obtained values were used for calculation of non-corrected cell viabilities. These values were also corrected (by subtraction) for the absorbance of the corresponding deposited dose of coated or uncoated IONPs, which was measured separately for 100 μl of different concentrations of IONPs (0, 20, 40, 60, 80 and 100 μgFe ml−1) at 490 nm with the same microplate reader. All experiments were performed minimum in triplicates. Results are given as means (with standard deviations) of the so-obtained values.
Cellular uptake study
LnCaP cells were cultured in RPMI 1640 medium (ATCC), supplemented with 10% fetal bovine serum (Life Technologies) and 1.5% 10000 U ml−1 Penicillin Streptomycin (Life Technologies). 40000 LnCaP cells per well were cultured on plastic 13 mm diameter sterile coverslips (Nunc™ Thermanox™) in 12 well plates at 37 °C. Cells were exposed to 1 ml of 100 μgFe ml−1 of IONPs for 24 h. Cells treated only with medium served as negative controls. After 24 h, cells were washed three times with PBS and fixed for 1 h at room temperature with 2% paraformaldehyde and 2.5% glutaraldehyde in PBS (pH 7.4). After 1 h, coverslips were washed three times with 0.1 M cacodylate buffer (pH 7.4), stained with 1% osmium tetroxide in 0.1 M cacodylate buffer at room temperature for 1 h and with 2% uranyl acetate in water for 40 min. After dehydrating the coverslips with increasing percentage of ethanol, up to 100%, they were then embedded in 50% durcupan (in ethanol) for 30 min and in 100% durcupan for 2 h. The resin was left to polymerize overnight at 60 °C. The next day, the resin embedded samples were separated from the glass slides by plunging them alternately into liquid nitrogen and hot water. The cells were then thin sectioned at a thickness of 50 nm with a diamond knife (Diatome) and ultramicrotome (Leica Microsystems) and collected on a formvar film on copper slot grids. Samples were imaged with transmission electron microscope operating at 80 kV (FEI Company, Tecnai Spirit).
Results and discussion
Coating of IONPs with CAs
IONPs were synthesized with a method modified from a previously developed protocol.26,28 Fig. 1a and S1† show the TEM micrographs and the size distribution of the obtained uncoated IONPs (mean TEM diameter was 16.7 ± 4.5 nm). These IONPs were then coated with four different CAs: (i) IDA, (ii) NTA, (iii) EDTA and (iv) DTPA. The chemical structures of these four CA molecules are shown in Fig. 1. The binding of CAs to the IONPs' surface was performed at pH 2.45 ± 0.01, because a previous study by Matijević's group reported a maximum and almost instantaneous adsorption of NTA, EDTA and DTPA at pH around 2.5 at 25 °C on α-Fe2O3 (ref. 6) and on β-FeOOH.29 Taking into consideration the pKa values of the studied CAs (see Table S1†), at pH 2.45 all four CAs are essentially present as monovalent anions (CA−). However, NTA, EDTA and DTPA also exist as divalent anions (CA2−), especially EDTA, which has the lowest pKa2 (2.64).30 DTPA at this interaction pH also exists as a trivalent anion (CA3−). Therefore, at pH 2.45, the anionic forms of these CAs electrostatically interact with the strongly positively charged IONPs' surface (ξ is about +50 mV).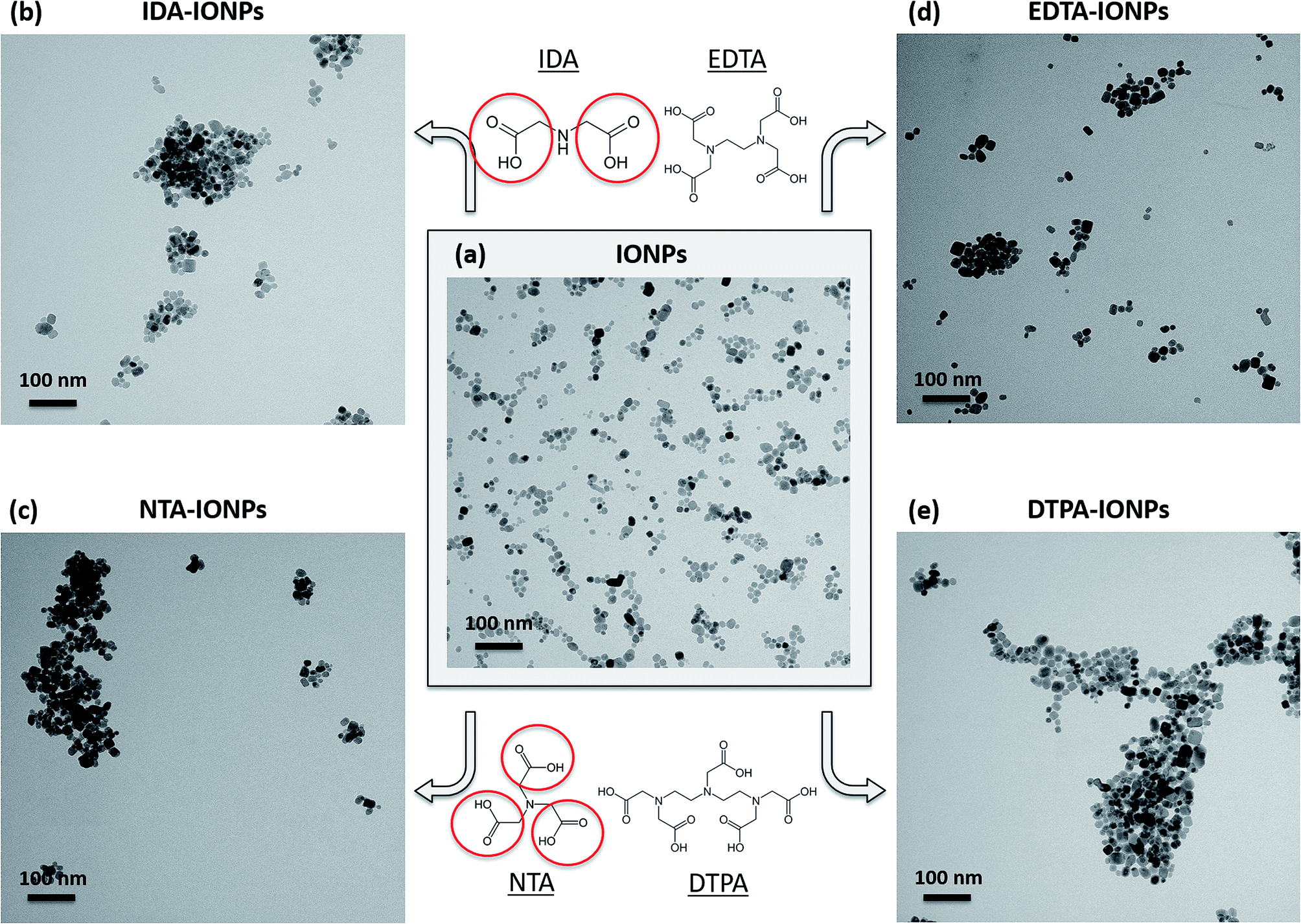 | ||
| Fig. 1 Representative TEM micrographs of uncoated IONPs (a), IDA-IONPs (b), NTA-IONPs (c), EDTA-IONPs (d) and DTPA-IONPs (e), all kept in water. The insert in (a) shows the distribution of the TEM diameter of uncoated IONPs. The chemical structure of the four CAs, which were used in this study, is given in the middle part above and below panel (a), and as an example dents containing carboxylic groups are marked by red circles in IDA and NTA molecules. | ||
Surface characterization of CA-coated IONPs
Firstly, the surface of uncoated and CA-coated IONPs was studied by XPS. Fig. S2† shows the overall XPS spectra. For all studied IONPs' samples, the Fe 2p3/2 was found at 710 eV (Fig. S2†). In addition, the presence of the CA molecules on the surface of IONPs in CA-coated IONPs is indicated by the detection of the N 1s peaks in CA-coated IONPs, which was not found in uncoated IONPs (Fig. S2†). The elemental compositions (Fe, O, C and N) determined from the XPS spectra are given in Table S2† as atomic percentages for uncoated and CA-coated IONPs. The presence of CAs in CA-coated IONPs was confirmed by the atomic percentages of N ranging from 1.1% to 3.9% obtained in CA-coated IONPs, which were 0% in uncoated IONPs. For CAs with larger number of nitrogen atoms (EDTA and DTPA), we observed a higher atomic percentage of N (EDTA-IONPs: 3.9%; DTPA-IONPs: 3.1%), while for CAs with lower number of nitrogen atoms (IDA and NTA), we found a lower atomic percentage of N (IDA-IONPs: 1.1%; NTA-IONPs: 1.7%).The attachment of CA molecules to the IONPs' surface was also investigated by FTIR (Fig. 2). We found that all CA-coated IONPs have characteristic peaks in the spectral region from 900 to 1800 cm−1 (see Fig. 2e). Typically the characteristic bands of the carboxylic group are in the 1570–1610 cm−1 region for its antisymmetrical vibrations, and in the 1350–1450 cm−1 region for its symmetrical vibrations.31–33 However, for all CA-coated IONPs the peak of antisymmetrical vibrations of the carboxylic group was found to be shifted towards ∼1620 cm−1. This was previously reported to be associated to the inductive effect of the protonated nitrogen atom on both the –COO− and –COOH groups.34 Besides this, the peak of symmetrical vibrations of the carboxylic group was found at the expected value of ∼1400 cm−1. The observed FTIR peaks in this region suggest that the COO− groups in CA-coated IONPs are interacting with other species (as also previously reported),31 such as with nitrogen dentates of other CA molecules or with IONPs. In addition to these characteristic peaks for the carboxylic group, we also observed in the FTIR spectra of CA-coated IONPs smaller peaks between 950 and 1250 cm−1, where typical stretching of C–O of carboxylic acid groups are visible. In this region, we identified the strongest peaks in DTPA-IONPs (which had a multivalent anionic state) and in EDTA-IONPs (which had a bivalent anionic state and thus an expected binding to the IONP's surface), while in NTA-IONPs and IDA-IONPs this peak was a low amplitude broad peak marginally above the background. These clear differences in this region of the spectrum could indicate different binding of carboxylic groups between the different CAs to IONPs or even to other chemical species (e.g. nitrogen groups of CA molecules).
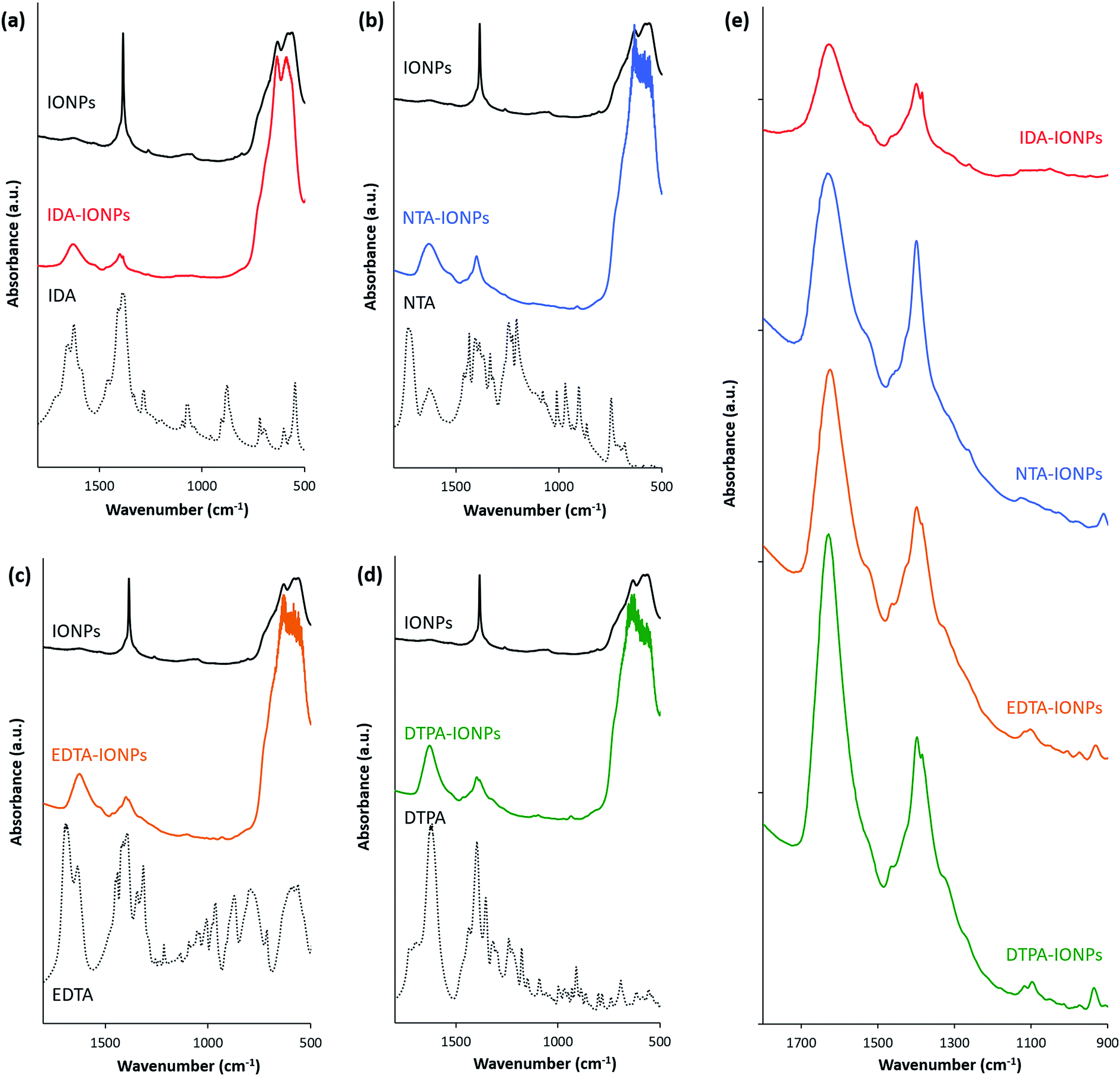 | ||
| Fig. 2 Absorbance of IDA-IONPs (a), NTA-IONPs (b), EDTA-IONPs (c) and DTPA-IONPs (d) as compared to uncoated IONPs (highlighted with black lines) and to each of the coating molecules (IDA, NTA, EDTA and DTPA, respectively; highlighted with black dashed lines) measured by FTIR. (e) Absorbance of CA-coated IONPs measured by FTIR for a narrow wavenumber range (900–1800 cm−1). | ||
In order to assess the influence of CAs on the surface properties of IONPs, we measured the zeta potential (ξ) of uncoated and CA-coated IONPs in water (Table 1). We found that the ξ changed between uncoated and CA-coated IONPs, suggesting that the IONPs' surfaces were coated with the CA molecules. In addition, the ξ was different between the four CA-coated IONPs. One would expect that the nitrogen dentate(s) would be oriented away from the surface at pH below the isoelectric point of IONPs (∼7), as at the coating pH of 2.45. Indeed, for IDA-IONPs, we found a positive ξ (26.2 ± 0.6). However, for IONPs coated with all other CAs, negative ξ were measured, suggesting that in these samples the carboxylic groups are exposed on the IONPs' surface. Especially NTA-IONPs have the strongest negative value of ξ (−17.6 ± 4.8), suggesting that this sample has the highest number of carboxylic groups exposed on the surface. This could be due to the possible conformation of the NTA molecule, in which not all carboxylic groups are able to turn and bind to the IONPs' surface. Interestingly, EDTA and DTPA contain more nitrogen groups, which could bind to the exposed carboxylic groups and therefore reduce the overall negative charges, which were found for EDTA-IONPs (−10.9 ± 1.0) and DTPA-IONPs (−6.0 ± 4.3).
| dh in H2O (nm) | PDI in H2O (—) | ρagglo in H2O (g cm−3) | ξ in H2O (mV) | dh in RPMI (nm) | PDI in RPMI (—) | ξ in RPMI (mV) | |
|---|---|---|---|---|---|---|---|
| IONPs | 25 ± 2 | 0.13 | 1.62 | 6.3 ± 0.8 | 115 ± 1 | 0.47 | −7.6 ± 0.6 |
| IDA-IONPs | 74 ± 27 | 0.31 | 1.41 | 26.2 ± 0.6 | 173 ± 180 | 0.25 | −7.8 ± 0.3 |
| NTA-IONPs | 67 ± 27 | 0.46 | 2.24 | −17.6 ± 4.8 | 364 ± 448 | 0.42 | −7.5 ± 0.7 |
| EDTA-IONPs | 35 ± 11 | 0.77 | 1.49 | −10.9 ± 1.0 | 429 ± 199 | 0.27 | −7.9 ± 0.1 |
| DTPA-IONPs | 132 ± 89 | 0.34 | 3.47 | −6.0 ± 4.3 | 1064 ± 683 | 0.41 | −7.5 ± 0.2 |
IONPs used for biomedical applications are in contact with biological medium, which contains biomolecules, especially proteins, which are known to change the surface properties of IONPs through the formation of a protein corona.35 Since proteins can modify the IONPs' surface, we also measured the ξ in RPMI medium containing 10% of protein-containing serum (Table 1). As expected, we measured different ξ values in RPMI medium as compared to water. Importantly, we measured the same ξ value of around −7.5 mV in RPMI medium for all studied IONPs, confirming that the surface of uncoated and CA-coated IONPs is covered with proteins, which modify their surface charge and hide their inherent surface properties.
The different binding of CAs to IONPs, and therefore their different surfaces observed by XPS and by FTIR, as well as their different ξ can affect their optical properties. We therefore measured the absorbance spectra of CA-coated IONPs by UV-visible spectroscopy in water, but also in RPMI medium, which can as well strongly impact the IONPs' absorbance (Fig. S3†). In water, the main difference between CA-coated IONPs was observed in the intensities of the main peak at around 388 nm, confirming the different surfaces that CA-coated IONPs have. In the RPMI medium, the main peak at 365 nm in the spectrum of uncoated IONPs shifted to 398 nm in the spectra of all CA-coated IONPs. This suggests that the attachment of proteins to the IONPs' surface is different between uncoated and CA-coated IONPs, as previously reported,36 which can be attributed to the presence of CAs.
Agglomeration state of CA-coated IONPs
TEM allowed us to do a preliminary study of the agglomeration states of uncoated and CA-coated IONPs, which can be seen in representative TEM micrographs shown in Fig. 1a–e, respectively. For uncoated IONPs, we found individual IONPs or agglomerates of only few IONPs. However, in the TEM micrographs of CA-coated IONPs, large agglomerates of ∼0.2–1 μm could be observed. Among the four studied CAs, the largest agglomerates were found in DTPA-IONPs. But, TEM characterization provides information about the agglomeration state of IONPs in a dried material. However, for biomedical application, the material resides in aqueous conditions and therefore the agglomeration state of IONPs in liquid needs to be investigated. We therefore measured the hydrodynamic diameter (dh) and the agglomerate densities (ρagglo) of uncoated and CA-coated IONPs in water (Table 1). The obtained dh values were also confirmed with another DLS instrument, as well as by centrifugal force (Fig. 3). We found that dh of DTPA-IONPs was the largest among the four CA-coated IONPs, which confirms the presence of agglomerates in this sample observed by TEM. The agglomeration found in all CA-IONPs is the first observation in this study that raises concerns regarding the use of nanomedicines coupled to CAs for bio-medical applications. Moreover, we also measured the highest ρagglo for this sample, suggesting that the IONPs' content in such agglomerates is high (Table 1). This is in agreement with the lowest ξ measured for this sample (−6.0 ± 4.3), which was insufficient for electrostatic stabilization. These results suggest strong aggregation in this sample. This could be due to the trivalent anionic state of DTPA, which could bind by bridging with other IONPs, but also with other nitrogen groups of the DTPA molecule on another IONP. In contrary, the smallest dh was found for EDTA-IONPs, which also had one of the lowest ρagglo among CA-coated IONPs, suggesting the presence of weak agglomerates rather than aggregates in this sample. This is also supported by the binding of the bivalent anionic state of EDTA, which could bind to the IONPs' surface via 1-2 carboxylic groups. As-such, the conformation of EDTA does not allow either the bridging between different IONPs (as possible in DTPA-IONPs) or the interactions of nitrogen dentate(s) of one bound EDTA molecule on one IONP with carboxylic moieties of other EDTA molecules on another IONP (as possible in all other CA). These measured dh and ρagglo in EDTA-IONPs were very similar to values obtained in uncoated IONPs, mostly comprising of well dispersed single IONPs. Also, IDA- and NTA-coated IONPs had the largest absolute values of ξ, and thus similar dh values.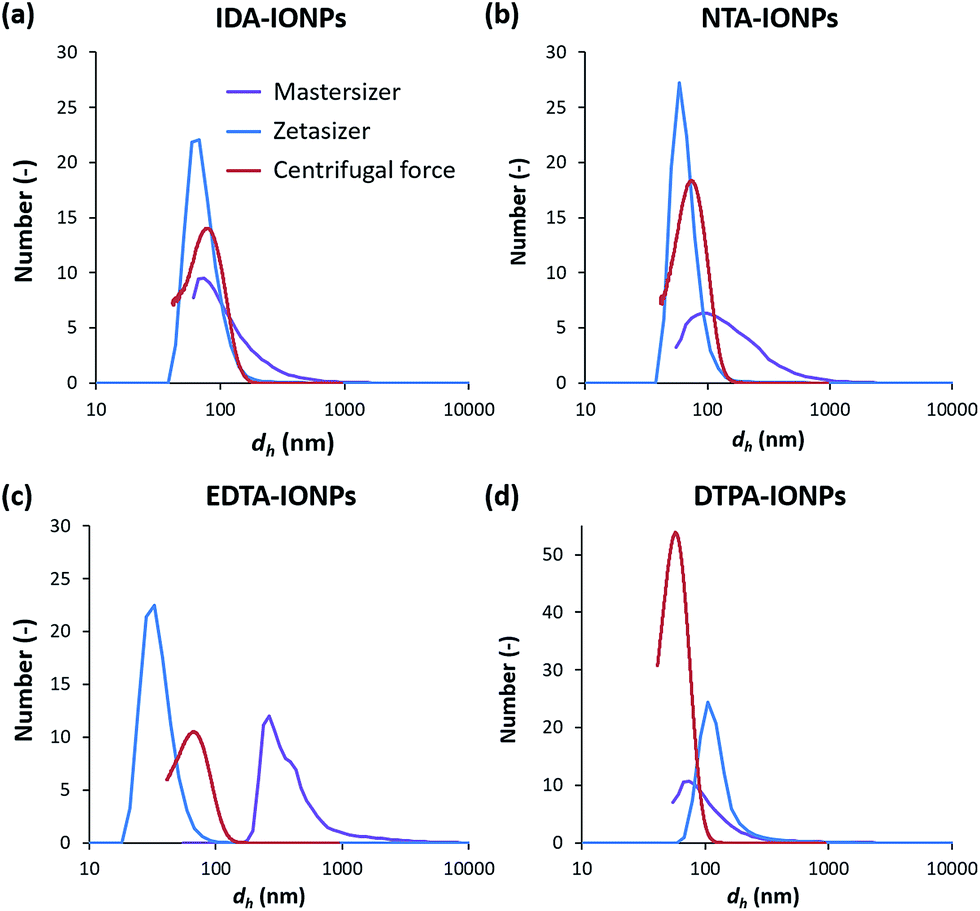 | ||
| Fig. 3 Number weighted distribution of the hydrodynamic diameter, dh, of IDA-IONPs (a), NTA-IONPs (b), EDTA-IONPs (c) and DTPA-IONPs (d) measured by dynamic light scattering (DLS) with two instruments (Mastersizer or Zetasizer), or by centrifugal force. | ||
A critical parameter for the use of IONPs in medicine is the colloidal behaviour of IONPs in a biological environment that contains biomolecules, especially proteins. In fact, proteins are known to modify the surface of NPs, forming the so-called protein corona.35 In other words, the medium, containing variable amounts of proteins, influences the protein corona, which can modify the agglomeration state of IONPs, previously shown to change their interaction with cells37–41 and their toxicity.42,43 In order to assess the agglomeration state of IONPs in the presence of proteins, we measured dh of uncoated and CA-coated IONPs in RPMI medium (Table 1). We found that dh of CA-coated IONPs in medium was larger than dh of CA-coated IONPs in water, indicating that proteins bind to the IONPs' surface and therefore modify their agglomeration state. Furthermore, even though similar surface charges were measured in RPMI medium between CA-coated IONPs, different dh and therefore agglomeration states were found between these samples. Interestingly, the dh measured in RPMI medium increased with increasing number of dentates in CAs. The highest dh was found for DTPA-IONPs (dh of ∼1064 nm), while the smallest dh was found for IDA-IONPs (dh or ∼173 nm). In fact, a higher number of active chemical groups increases the number of possibilities for binding. Therefore, DTPA, which has the largest number of carboxylic and nitrogen groups among the studied CAs, also has the largest possibilities to form multiple bonds not only with other DTPA-coated IONPs, but also with surrounding proteins, inducing agglomeration.
MRI relaxivity of coated IONPs
In order to evaluate the potential of CA-coated IONPs as MRI contrast agents, we measured their longitudinal (r1) and transverse (r2) relaxivities in a clinical 3 T MRI scanner, and determined their relaxivity ratio r2/r1 (Table 2). We found that the r1 values of CA-coated IONPs were all two times smaller than uncoated IONPs. In fact, previous studies also reported that the coating, and therefore the size of coated IONPs, significantly affects the r1.44 Besides the r1 value, the r2 relaxivity and the r2/r1 ratio are the most important parameters for characterizing IONPs for MRI applications. A high r2 constitutes an effective shortening of the transverse relaxation time T2 of neighbouring protons, being therefore classified as T2 contrast agents. The r2 values measured for NTA-IONPs and DTPA-IONPs were 20–25% lower than the r2 of uncoated IONPs, which is in agreement with previous studies, which reported that the surface coating can decrease the access of water protons to the IONPs' surface and consequently reduce the r2 relaxivity.44 These two samples also had the largest ρagglo, where IONPs are densely packed, therefore resulting in a lower r2. Interestingly, the r2 value of IDA-IONPs was similar to that of uncoated IONPs, while that of EDTA-IONPs was even 15% higher than for uncoated IONPs. These two samples had also the smallest ρagglo, where IONPs are more loosely packed, therefore, as expected, resulting in a higher contribution to r2. However, the increase of the r2 value of EDTA-IONPs as compared to uncoated IONPs was rarely observed after the addition of a surface coating and could be associated with an increase in available iron oxide surfaces in these two samples as compared to uncoated IONPs.45–47 Indeed, the value of dh of EDTA-IONPs measured in water was the lowest dh of the CA-coated IONPs (as seen in Table 1) and were similar to dh values of uncoated IONPs, comprising of mostly single well dispersed IONPs. This suggests that IONPs could be well separated by EDTA, even more than in uncoated IONPs, resulting in larger available surfaces to relax neighbouring protons and therefore to higher r2 values as compared to uncoated IONPs.| r1 (mM−1 s−1) | r2 (mM−1 s−1) | r2/r1 (mM−1 s−1) | |
|---|---|---|---|
| IONPs | 10.6 ± 0.2 | 1330.2 ± 33.5 | 125.4 |
| IDA-IONPs | 3.8 ± 0.1 | 1335.8 ± 64.0 | 353.9 |
| NTA-IONPs | 3.5 ± 0.1 | 1040.8 ± 27.3 | 293.2 |
| EDTA-IONPs | 5.5 ± 0.3 | 1543.6 ± 62.3 | 282.6 |
| DTPA-IONPs | 4.0 ± 0.1 | 1008.9 ± 24.4 | 249.7 |
Importantly, the r2 values found for all CA-coated IONPs (between 1009 mM−1 s−1 for DTPA-IONPs up to 1544 mM−1 s−1 for EDTA-IONPs) are up to 10 times higher than previously reported values also measured at 3 T (50–164 mM−1 s−1) obtained with IONPs coated with large coating molecules, such as polymers or sugars.48–50 It therefore seems that the use of small molecules, such as the CAs used here, significantly increases the r2 values, which can even be higher than uncoated IONPs.51 Given the high r2 and low r1 values measured for CA-coated IONPs, we consequently found large r2/r1 ratios for these samples (Table 2), which were at least two times larger than for uncoated IONPs.51 Since both r2 and r2/r1 ratios have to be high for a good T2 contrast agent, it seems that the coating of IONPs (which have high r2) by small molecules, such as folic acid,52 pyridoxal phosphate,53 ATP or NADPH,47 is a good strategy to obtain coated IONPs with high r2.
Toxicity evaluation
The in vitro toxicity of CA-coated IONPs was assessed with the MTS viability test performed on lymph node metastatic cells, LnCaP cells, which were also used for the uptake study shown below. Cells were incubated for 24 h with different concentrations of uncoated and CA-coated IONPs of up to 100 μgFe ml−1, which was the concentration used for the uptake study presented below. The obtained cell viabilities are shown in Fig. 4a. By comparing the obtained cell viabilities of all samples at concentrations between 1 and 10 μgFe ml−1, we found that more than 85% of the cells remained viable. At higher concentrations (50–100 μgFe ml−1), NTA- and DTPA-coated IONPs were non-toxic for the cells (NTA-IONPs: ξ of ∼−18 mV and dh of ∼67 nm; DTPA-IONPs: ξ of ∼−6 mV and dh of ∼132 nm), as expected for negatively charged IONPs.54 However, for uncoated IONPs and IDA-IONPs, the cell viability decreased already at 50 μgFe ml−1 (79.2% and 74.6% cell viability for uncoated and IDA-coated IONPs), but also at 100 μgFe ml−1 for EDTA-IONPs (81% cell viability). We have to note that coated IONPs have none-zero absorbance at 490 nm, where the cell viability was evaluated by the MTS test. In other words, the measured absorbance for higher studied concentrations of coated IONPs would contain both the absorbance originated from the cell viability and from the deposited dose of IONPs. Consequently, the cell viability for higher IONPs' doses is realistically lower than what we can see in Fig. 4a. We previously developed a method to correct the absorbance-based toxicity test, such as the MTS test. This method is based on the subtraction of the absorbance of the deposited dose of NPs calculated with the in vitro sedimentation diffusion and dosimetry (ISDD) model. In this case, the deposited dose is equal to the administrated dose, because 100% of all CA-coated IONPs sedimented after less than 24 h (the fraction of the deposited dose which interacts with cells could not be calculated). Thus, we measured the absorbance of the corresponding deposited doses of coated and uncoated IONPs and used the obtained values to correct the cell viabilities. The so-obtained corrected cell viabilities (Fig. 4b) were lower than the corresponding values without corrections in all samples for concentrations above 50 μgFe ml−1. As expected, the difference between the corresponding corrected and non-corrected cell viabilities were the most pronounced for a concentration of 100 μgFe ml−1 (up to ∼33% for NTA-IONPs), where all values of corrected cell viabilities were below 80%. This suggests a non-negligible toxicity of CA-IONPs. So far, the toxicity of IONPs has very often been reported to be caused by their physico-chemical properties.55 Even though IDA- and EDTA-coated IONPs showed similar small toxicities, we did not find any correlation with their ξ and dh. Indeed, they had opposite surface charges (ξ of ∼26 mV for IDA-IONPs and ∼−11 mV for EDTA-IONPs) and different dh (∼74 nm for IDA-IONPs and ∼35 nm for EDTA-IONPs). However, for both IDA-IONPs and EDTA-IONPs, small ρagglo were measured (1.41 g cm−3 for IDA-IONPs and 1.49 g cm−3 for EDTA-IONPs), suggesting that a higher surface of material was exposed to cells in these two samples, which can be at the origin of the higher toxicities measured for these two samples. Therefore, even though these CAs, especially EDTA,56–58 are widely used for biomedical applications, care has to be taken with the use of CA-coated IONPs for such applications, which seem to induce non-negligible toxicities. Further detailed studies should therefore deeper investigate the toxicity of CAs for their use in medicine.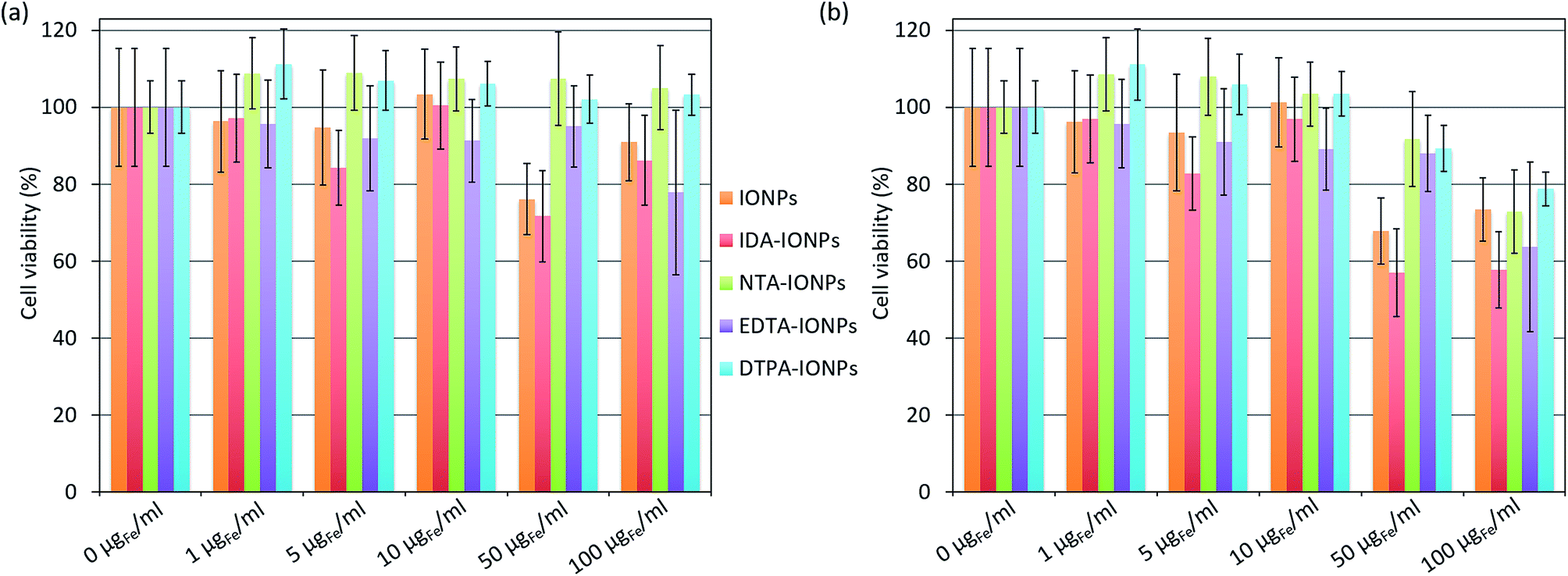 | ||
| Fig. 4 Viability of LnCaP cells incubated for 24 h with different concentrations (0, 1, 5, 10, 50 and 100 μgFe ml−1) of uncoated IONPs, IDA-IONPs, NTA-IONPs, EDTA-IONPs and DTPA-IONPs measured with the MTS test. The cell viabilities are the percentages obtained from the absorbance of cells treated with IONPs non-corrected (a) or corrected (b) for the absorbance of the administrated dose of the corresponding coated or non-coated IONPs and normalized with the absorbance of cells without IONPs (0 μgFe ml−1). All values are given as average ± standard deviation. | ||
Interaction of IONPs with cells
In order to evaluate how the surface properties and agglomeration states of the different CA-coated IONPs influence their biological behaviour, we studied their uptake in the same cell type used for the toxicity study: LnCaP cells. We fixed cells and sectioned them into 50 nm thick sections, which were stained with uranyl acetate to enhance the cellular contrast and were examined as such by TEM. Fig. 5 and S4† show representative TEM micrographs of such sections incubated for 24 h with 100 μgFe ml−1 of uncoated or CA-coated IONPs. We found that uncoated IONPs were taken up in large quantities by cells and were well dispersed inside cells (see red arrows in Fig. 5b), but also around cells (see green arrows in Fig. 5b). However, when incubating cells with CA-coated IONPs, we found much lower uptake of IDA-IONPs, NTA-IONPs and DTPA-IONPs as compared to uncoated IONPs (see red arrows in Fig. 5c, d and f), while the uptake of EDTA-IONPs was slightly lower or comparable to uncoated IONPs (see red arrows in Fig. 5e). Furthermore, the morphology of the vesicles, in which IONPs were located, was different between the uncoated and the CA-coated IONPs. While uncoated IONPs were mostly located inside large vesicles of 1–2 μm, CA-coated IONPs were found in smaller vesicles.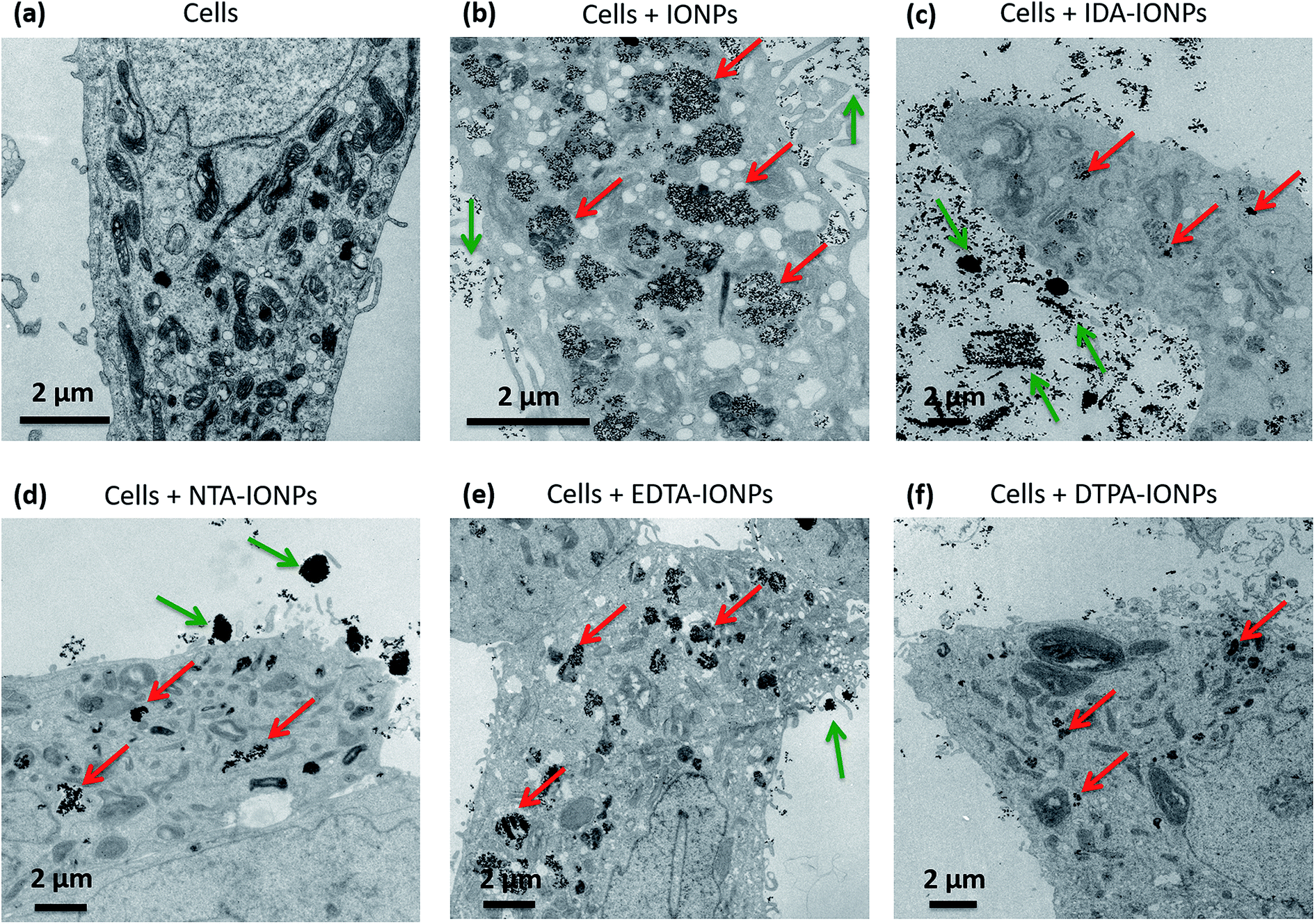 | ||
| Fig. 5 Representative TEM micrographs of 50 nm-thick sections of LnCaP cells without IONPs (a), and LnCaP cells incubated for 24 h with uncoated IONPs (b), IDA-IONPs (c), NTA-IONPs (d), EDTA-IONPs (e) and DTPA-IONPs (f). Green and red arrows show IONPs outside and inside cells, respectively. | ||
Differences in the cellular uptake of IONPs have been in the past often associated to surface charge and size effects.59–61 Here, CA-coated IONPs displayed similar ξ in RPMI medium, but they had very different surface charges in water. NTA-IONPs and EDTA-IONPs had the strongest negative surface charges and also showed the highest cellular uptake among CA-coated IONPs, which is consistent with previous reports.59,60 However, both uncoated and IDA-IONPs had positive ξ, but were taken up in large and small amounts, respectively. This is in agreement with previous reports, that the surface charge is not the only parameter influencing the uptake of CA-coated IONPs but also others, such as the size.59–61 Here, uncoated and CA-coated IONPs had very different dh in both water and RPMI medium. Interestingly, in the TEM micrographs of the cell sections, we observed large agglomerates of IONPs of up to 1–2 μm, especially for IDA-IONPs, NTA-IONPs and EDTA-IONPs. For example, we found elongated agglomerates of IDA-IONPs of 1–5 μm in length (see green arrows in Fig. 5c), as well as round agglomerates of NTA-IONPs (see green arrows in Fig. 5d) and EDTA-IONPs (see green arrows in Fig. 5e) of up to 5 μm. Interestingly, these agglomerates were found only outside cells and correlated with the large dh measured in RPMI medium (Table 1). It therefore seems that cells did not take up the μm-sized agglomerates of CA-coated IONPs. Indeed, uncoated IONPs were well dispersed in RPMI medium (dh = 115 ± 1 nm, see Table 1) and were taken up in large quantities by LnCaP cells, while CA-coated IONPs showed μm-sized agglomerates (dh ∼ 200–1100 nm) and were taken up in low amounts in cells. The lowest uptake among CA-coated IONPs was found for DTPA-IONPs, which was also the most agglomerated sample (dh = 1064 ± 683 nm in RPMI medium). Interestingly, the uptake behaviour of CA-coated IONPs was in accordance with their measured toxicities. While EDTA-IONPs had both the highest uptake and the highest toxicity among the four CAs, IDA-IONPs, NTA-IONPs and DTPA-IONPs showed much lower uptake and toxicities. Overall, it seems that the size of IONPs, i.e. their dh, strongly influenced the uptake of CA-coated IONPs, as well as their toxicities, as also observed in numerous studies of other coatings.61–63 It has to be highlighted that the charge and size of IONPs are among the most cited IONPs' properties influencing the cellular uptake. However, other factors could be at the origin of the differences observed in the cellular uptake between CA-coated IONPs and uncoated IONPs, as well as in between the CA-coated IONPs. Regarding this subject, there have been a lot of studies on the interaction of different types of NPs with cells with respect to different chemical groups present on the surface of NPs, different surface charges, etc.64,65 From that point of view, we also found different surface charges and chemical species exposed on the surface of CA-coated IONPs. Furthermore, ρagglo is also strongly influencing the sedimentation and diffusion behaviour of IONPs, and therefore their deposited dose66,67 and cellular uptake. Specifically, we found that 100% of all CA-coated IONPs have sedimented after less than 24 h by taking into consideration the values of ρagglo and dh (Table 1) and by using the In vitro Sedimentation, Diffusion and Dosimetry (ISDD) model.27,66 Therefore, the influence of ρagglo on the cellular uptake of IONPs could be important, but it has been unfortunately neglected so far.
Conclusions
We here report the coating of IONPs with four CAs (IDA, NTA, EDTA and DTPA), which differ in their number of carboxylic dents (2, 3, 4 and 5, respectively) and in their number of nitrogen groups. We found differences among the four CA-coated IONPs regarding their surface chemistry, surface charges and especially regarding their agglomeration state in water and in biological medium. In particular in water, the largest agglomerates were found for DTPA-IONPs (dh = 132 ± 89 nm), while we measured the smallest agglomerates for EDTA-IONPs (dh = 35 ± 11 nm). In addition, we measured both high r2 and r2/r1 ratios for all CA-coated IONPs, suggesting that all four types of IONPs have suitable relaxometric properties for their use as T2 MRI contrast agents. Since different CA-coated IONPs had different physico-chemical properties, we also found differences in their toxicity and uptake in LnCaP cells. The uptake of CA-coated IONPs, which formed agglomerates in RPMI medium (dh ∼ 200–1100 nm for CA-coated IONPs), was lower than the uptake of uncoated IONPs, which were well dispersed in medium (dh = 115 ± 1 nm for uncoated IONPs). Importantly, the toxicity of CA-coated IONPs was in accordance with the observed cellular uptake, especially for EDTA-IONPs, for which the uptake and the toxicity were the highest among the four CAs. Interestingly, we observed similar toxicity and uptake for CA-coated IONPs with similar ρagglo. Specifically, CA-coated IONPs with low ρagglo (IDA- and EDTA-coated IONPs), suggesting that they are weakly agglomerated and higher exposed surfaces, displayed higher toxicities and higher uptake as compared to CA-coated IONPs with high ρagglo (NTA- and DTPA-coated IONPs). Our study shows that care has to be taken when one intends to use any CA for biomedical applications. Although the strong chemical affinity of CA could be beneficial, as in the case of the chelation of toxic ions, this high chemical reactivity can simultaneously cause the aggregation of IONPs, as found in DTPA-IONPs. Also, this can induce uncontrolled interactions in a biological environment, where a lot of biomolecules are present, which also contain possible binding sites to bind with these CA molecules. Our study therefore suggests that CAs could be harmful in a clinical setting, something that should be further studied in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by: the Nanoscreen project funded by Competence Centre for Materials Science and Technology (CCMX), the Nanoneck2 project funded by Eurostars (Project E!9309 Nanoneck2) and the MagnetoTeranostics project that was scientifically evaluated by the Swiss National Science Foundation (SNSF), financed by the Swiss Confederation and funded by Nano-Tera.ch (Magnetotheranostics project No 530 627).References
- S. J. S. Flora and V. Pachauri, Int. J. Environ. Res. Public Health, 2010, 7, 2745–2788 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- R. A. Peters, L. A. Stocken and R. H. S. Thompson, Nature, 1945, 156, 616–619 CrossRef CAS PubMed.
- C. Olchowy, K. Cebulski, M. Łasecki, R. Chaber, A. Olchowy, K. Kałwak and U. Zaleska-Dorobisz, PLoS One, 2017, 12, e0171704 Search PubMed.
- H.-C. Chang and E. Matijević, J. Colloid Interface Sci., 1983, 92, 479–488 CrossRef CAS.
- H.-C. Chang, T. W. Healy and E. Matijević, J. Colloid Interface Sci., 1983, 92, 469–478 CrossRef CAS.
- J. Eisenlauer and E. Matijević, J. Colloid Interface Sci., 1980, 75, 199–211 CrossRef CAS.
- B. Nowack, Environ. Sci. Technol., 2002, 36, 4009–4016 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, X. Zhong, Z. Guo, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 3392–3393 CrossRef CAS PubMed.
- Y.-C. Li, Y.-S. Lin, P.-J. Tsai, C.-T. Chen, W.-Y. Chen and Y.-C. Chen, Anal. Chem., 2007, 79, 7519–7525 CrossRef CAS PubMed.
- M. Bloemen, L. Vanpraet, M. Ceulemans, T. N. Parac-Vogt, K. Clays, N. Geukens, A. Gils and T. Verbiest, RSC Adv., 2015, 5, 66549–66553 RSC.
- K. Salimi, D. D. Usta, I. Kocer, E. Celik and A. Tuncel, RSC Adv., 2017, 7, 8718–8726 RSC.
- C. Fang, L. Zhang, X. Zhang and H. Lu, Analyst, 2015, 140, 4197–4205 RSC.
- J. Li, M. Chen, Z. Gao, J. Du, W. Yang and M. Yin, Colloids Surf., B, 2016, 146, 468–474 CrossRef CAS PubMed.
- M. Zhang, X. He, L. Chen and Y. Zhang, Nanotechnology, 2011, 22, 065705 CrossRef PubMed.
- P.-J. Debouttière, S. Roux, F. Vocanson, C. Billotey, O. Beuf, A. Favre-Réguillon, Y. Lin, S. Pellet-Rostaing, R. Lamartine, P. Perriat and O. Tillement, Adv. Funct. Mater., 2006, 16, 2330–2339 CrossRef.
- W. Zhao, H. Huang, Y. Sun, X. Zhang, Y. Li and J. Wang, RSC Adv., 2015, 5, 97675–97680 RSC.
- M. Das, D. Mishra, P. Dhak, S. Gupta, T. K. Maiti, A. Basak and P. Pramanik, Small, 2009, 5, 2883–2893 CrossRef CAS PubMed.
- S. Chattopadhyay, S. K. Dash, T. Ghosh, D. Das, P. Pramanik and S. Roy, Cancer Nanotechnol., 2013, 4, 103–116 CrossRef PubMed.
- A. Narsireddy, K. Vijayashree, J. Irudayaraj, S. V. Manorama and N. M. Rao, Int. J. Pharm., 2014, 471, 421–429 CrossRef CAS PubMed.
- L. Galluzzi, L. Chiarantini, E. Pantucci, R. Curci, J. Merikhi, H. Hummel, P. K. Bachmann, E. Manuali, G. Pezzotti and M. Magnani, Nanomedicine, 2011, 7, 393–409 CrossRef PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. Rechenberg, J. Magn. Magn. Mater., 2005, 293(1), 483–496 CrossRef CAS.
- P.-E. Le Renard, R. Lortz, C. Senatore, J.-P. Rapin, F. Buchegger, A. Petri-Fink, H. Hofmann, E. Doelker and O. Jordan, J. Magn. Magn. Mater., 2011, 323, 1054–1063 CrossRef CAS.
- J. I. Schwerdt, G. F. Goya, M. Pilar Calatayud, C. B. Herenu, P. C. Reggiani and R. G. Goya, Curr. Gene Ther., 2012, 12, 116–126 CrossRef CAS PubMed.
- D. Bonvin, H. Hofmann and M. Mionic Ebersold, J. Nanopart. Res., 2016, 18, 376 CrossRef.
- G. DeLoid, J. M. Cohen, T. Darrah, R. Derk, L. Rojanasakul, G. Pyrgiotakis, W. Wohlleben and P. Demokritou, Nat. Commun., 2014, 5, 3514 Search PubMed.
- D. Bonvin, A. Arakcheeva, A. Millan, R. Pinol, H. Hofmann and M. Mionic Ebersold, RSC Adv., 2017, 7, 13159–13170 RSC.
- J. Rubio and E. Matijević, J. Colloid Interface Sci., 1979, 68, 408–421 CrossRef CAS.
- P. Thakur, J. N. Mathur, R. C. Moore and G. R. Choppin, Inorg. Chim. Acta, 2007, 360, 3671–3680 CrossRef CAS.
- D. T. Sawyer, Ann. N. Y. Acad. Sci., 1960, 88, 307–321 CrossRef CAS PubMed.
- S. Hou, S. Su, M. L. Kasner, P. Shah, K. Patel and C. J. Madarang, Chem. Phys. Lett., 2010, 501, 68–74 CrossRef CAS.
- S. Mohapatra, D. Pal, S. K. Ghosh and P. Pramanik, J. Nanosci. Nanotechnol., 2007, 7, 3193–3199 CrossRef CAS PubMed.
- H. G. Langer, Inorg. Chem., 1963, 2, 1080–1082 CrossRef CAS.
- D. Bonvin, U. Aschauer, D. T. L. Alexander, D. Chiappe, M. Moniatte, H. Hofmann and M. Mionić Ebersold, Small, 2017, 1700409 CrossRef PubMed.
- S. Yu, A. Peralvarez-Marin, C. Minelli, J. Faraudo, A. Roig and A. Laromaine, Nanoscale, 2016, 8, 14393–14405 RSC.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2012, 116, 8901–8907 CrossRef CAS PubMed.
- A. Bigdeli, S. Palchetti, D. Pozzi, M. R. Hormozi-Nezhad, F. Baldelli Bombelli, G. Caracciolo and M. Mahmoudi, ACS Nano, 2016, 10, 3723–3737 CrossRef CAS PubMed.
- S. Palchetti, L. Digiacomo, D. Pozzi, G. Peruzzi, E. Micarelli, M. Mahmoudi and G. Caracciolo, Nanoscale, 2016, 8, 12755–12763 RSC.
- Y. K. Lee, E.-J. Choi, T. J. Webster, S.-H. Kim and D. Khang, Int. J. Nanomed., 2014, 10, 97–113 Search PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- L. E. W. LaConte, N. Nitin, O. Zurkiya, D. Caruntu, C. J. O'Connor, X. Hu and G. Bao, J. Magn. Reson. Imag., 2007, 26, 1634–1641 CrossRef PubMed.
- T. Ahmad, H. Bae, I. Rhee, Y. Chang, J. Lee and S. Hong, Curr. Appl. Phys., 2012, 12, 969–974 CrossRef.
- G. Huang, H. Li, J. Chen, Z. Zhao, L. Yang, X. Chi, Z. Chen, X. Wang and J. Gao, Nanoscale, 2014, 6, 10404–10412 RSC.
- D. Bonvin, J. Bastiaansen, M. Stuber, H. Hofmann and M. M. Ebersold, J. Mater. Chem. B, 2017, 5, 8353–8365 RSC.
- S. Aryal, J. Key, C. Stigliano, J. S. Ananta, M. Zhong and P. Decuzzi, Biomaterials, 2013, 34, 7725–7732 CrossRef CAS PubMed.
- S. Srivastava, R. Awasthi, N. S. Gajbhiye, V. Agarwal, A. Singh, A. Yadav and R. K. Gupta, J. Colloid Interface Sci., 2011, 359, 104–111 CrossRef CAS PubMed.
- G. H. Simon, J. Bauer, O. Saborovski, Y. Fu, C. Corot, M. F. Wendland and H. E. Daldrup-Link, Eur. Radiol., 2006, 16, 738–745 CrossRef PubMed.
- D. Bonvin, D. T. L. Alexander, A. Millán, R. Piñol, B. Sanz, G. F. Goya, A. Martínez, J. A. M. Bastiaansen, M. Stuber, K. J. Schenk, H. Hofmann and M. Mionić Ebersold, Nanomaterials, 2017, 7, 225 CrossRef PubMed.
- D. Bonvin, J. A. M. Bastiaansen, M. Stuber, H. Hofmann and M. M. Ebersold, Dalton Trans., 2017, 46, 12692–12704 RSC.
- D. Bonvin, U. J. Aschauer, J. A. M. Bastiaansen, M. Stuber, H. Hofmann and M. Mionić Ebersold, Nanomaterials, 2017, 7, 202 CrossRef PubMed.
- K. R. Di Bona, Y. Xu, P. A. Ramirez, J. DeLaine, C. Parker, Y. Bao and J. F. Rasco, Reprod. Toxicol., 2014, 50, 36–42 CrossRef CAS PubMed.
- J. Rose, M. Auffan, O. Proux, V. Niviere and J.-Y. Bottero, in Encyclopedia of Nanotechnology, ed. B. Bhushan, Springer Netherlands, Dordrecht, 2012, p. 2085 Search PubMed.
- B. Bajaj, B. D. Malhotra and S. Choi, Biomol. Electron. Org. Nanotechnol. Environ. Preserv., 2010, vol. 519, pp. 1219–1223 Search PubMed.
- E. Ghasemi, A. Heydari and M. Sillanpää, Microchem. J., 2017, 131, 51–56 CrossRef CAS.
- Y. Yi, Y. Zhang, Y. Wang, L. Shen, M. Jia, Y. Huang, Z. Hou and G. Zhuang, Nanoscale Res. Lett., 2014, 9, 27 CrossRef PubMed.
- V. Ayala, A. P. Herrera, M. Latorre-Esteves, M. Torres-Lugo and C. Rinaldi, J. Nanoparticle Res., 2013, 15, 1874 CrossRef PubMed.
- F. Geinguenaud, C. Banissi, A. F. Carpentier and L. Motte, Nanomaterials, 2015, 5, 1588–1609 CrossRef CAS PubMed.
- Y. Ge, Y. Zhang, J. Xia, M. Ma, S. He, F. Nie and N. Gu, Colloids Surf., B, 2009, 73, 294–301 CrossRef CAS PubMed.
- K. H. Müller, M. Motskin, A. J. Philpott, A. F. Routh, C. M. Shanahan, M. J. Duer and J. N. Skepper, Biomaterials, 2014, 35, 1074–1088 CrossRef PubMed.
- O. T. Marisca, K. Kantner, C. Pfeiffer, Q. Zhang, B. Pelaz, N. Leopold, W. J. Parak and J. Rejman, Nanomaterials, 2015, 5, 1418–1430 CrossRef CAS PubMed.
- C. C. Hanot, Y. S. Choi, T. B. Anani, D. Soundarrajan and A. E. David, Int. J. Mol. Sci., 2016, 17, 54 CrossRef PubMed.
- S. Mondini, M. Leonzino, C. Drago, A. M. Ferretti, S. Usseglio, D. Maggioni, P. Tornese, B. Chini and A. Ponti, Langmuir, 2015, 31, 7381–7390 CrossRef CAS PubMed.
- P. M. Hinderliter, K. R. Minard, G. Orr, W. B. Chrisler, B. D. Thrall, J. G. Pounds and J. G. Teeguarden, Part. Fibre Toxicol., 2010, 7, 36 CrossRef CAS PubMed.
- D. Bonvin, H. Hofmann and M. M. Ebersold, Analyst, 2017, 142, 2338–2342 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08217g |
| This journal is © The Royal Society of Chemistry 2017 |