
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Achieving a high-efficiency dual-core chromophore for emission of blue light by testing different side groups and substitution positions†
Hwangyu Shin‡
b,
Beomjin Kim‡a,
Hyocheol Junga,
Jaehyun Leeb,
Hayoon Leea,
Seokwoo Kanga,
Jiwon Moonb,
Joonghan Kim b and
Jongwook Park*a
b and
Jongwook Park*a
aDepartment of Chemical Engineering, Kyung Hee University, Gyeonggi-do 17104, Republic of Korea. E-mail: jongpark@khu.ac.kr
bDepartment of Chemistry, The Catholic University of Korea, Bucheon, 420-743, Republic of Korea
First published on 7th December 2017
Abstract
Hetero dual-core derivatives that combine anthracene and pyrene were systematically studied for the purpose of producing highly efficient blue light-emitting materials applicable to organic light-emitting diode (OLED) lighting. Five compounds were designed in order to (1) determine which one of the two core chromophores in a hetero dual-core moiety, if any, acts as the main contributor to the optical and electronic properties of the final compounds, (2) control the electron-donating ability of the side group, and (3) change the substitution position. 1-[1,1′;3′,1′′]terphenyl-5′-yl-6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyrene (TP-AP-TP) was used as the reference material, and four other materials, including diphenyl-[10-(6-[1,1′;3′,1′′]terphenyl-5′-yl-pyren-1-yl)-anthracen-9-yl]-amine (DPA-AP-TP), diphenyl-[6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyren-1-yl]-amine (TP-AP-DPA), diphenyl-{4-[10-(6-[1,1′;3′,1′′]-terphenyl-5′-yl-pyren-1-yl)-anthracen-9-yl]-phenyl}-amine (TPA-AP-TP) and diphenyl-{4-[6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyren-1-yl]-phenyl}-amine (TP-AP-TPA), were synthesized as model compounds. The synthesized materials showed absorption wavelength peaks at 403–410 nm in the film state and exhibited PL emissions of 458–505 nm. Also, anthracene was shown to be the main core contributing to the optical and electronic properties. Among the synthesized molecules, the TPA-AP-TP molecule, in which triphenylamine, with its optimum electron-donating ability, was substituted into anthracene, showed excellent electroluminescence (EL) performance for OLED lighting with a current efficiency of 8.05 cd A−1, external quantum efficiency of 6.75%, and narrow EL FWHM of 53 nm.
1. Introduction
Organic light-emitting diodes (OLEDs) have attracted considerable attention in both academic and industrial circles since the pioneering work of Tang and Vanslyke in 1987. Because of advantageous properties such as self-emission, full-color emission, low driving voltage, flexibility and fast response time, OLEDs have recently been developed and applied extensively in the small and medium display market, and many studies are underway for their applications in television and lighting.1 Certain properties of OLEDs make them especially attractive in the lighting market, including area emission characteristics not found for other existing sources of light, environmentally friendly efficient use of energy, large area, ultra-light weight, and ultra-thin shape. It has thus become increasingly important to carry out investigations of the applications of OLEDs if they are to become the next-generation source of lighting and replace existing fluorescent and incandescent lamps.2 White OLEDs for lighting can be divided into those that produce white light by combining red-light, green-light, and blue-light (R, G, B) emitters3 and those that produce white light by combining emitters of sky-blue light and orange light or of sky-blue light and red light.4 Investigations have been more actively pursued for the latter two-color white OLEDs whose structures are relatively simple, because low cost is regarded as important in the lighting field, unlike in the display field. In addition, since the emission spectra of organic materials generally have peaks with broader wavelengths than do the emission spectra of inorganic materials, only two types of organic emitters, in particular the emitters of sky-blue and orange light or of sky-blue and red light, need to be combined to produce white light when using OLEDs. Many studies have achieved high efficiency and long lifetime for materials that emit orange or red light, but it has been a challenge to develop materials that emit pure blue light with high efficiency because of the large difference in the energy levels of adjacent hole- and electron-transporting layers caused by the wide band gap.5To overcome such issues and increase the efficiency of materials that emit blue light, most existing studies have used polycyclic aromatic hydrocarbon (PAH) moieties based on a single-core chromophore, such as anthracene,6 pyrene,7 and fluorene,8 which have excellent fluorescence characteristics. Studies have been actively conducted, especially with the anthracene core, to increase color purity and electroluminescence (EL) efficiency by adjusting dihedral angles of the core of the molecule or by introducing bulky groups or electron donor and acceptor groups as side groups.9 Furthermore, the most recent studies have focused on molecules consisting of multicore chromophore structures by combining two or more of the same PAH core moieties or by combining different such moieties.10 Our group has recently reported dual-core chromophores of the “AP-core type” where anthracene and pyrene cores were directly connected. These dual-core derivatives showed outstanding performances, with the EL efficiency and device lifetime increased by more than a factor of two compared to light-emitting materials that applied anthracene or pyrene as a single-core chromophore.11 Unlike the corresponding single-core materials, these dual-core structures displayed high efficiency because both anthracene and pyrene contributed to the absorption and emission processes.12 However, few studies on such hetero-type dual cores have been published, and there are also few published papers on the roles of core, types of side group and various substitution changes regarding on molecular structure. Accordingly, in order to develop blue light-emitting materials for highly efficient OLED lighting devices, this study examined hetero dual-core moieties in which anthracene and pyrene are connected. We specifically investigated not only which, if any, of the two core chromophores act as the main contributor to the absorption and emission processes, but also the effects of changing the side group on the performance properties of the final compounds. Therefore, (1) a hetero dual-core moiety consisting of anthracene and pyrene (AP-core) was selected as the standard core chromophore; (2) a nitrogen atom was introduced instead of a phenyl ring in order to increase electron donation to the dual core and hence increase device efficiency, (3) a bulky side group was introduced to suppress intermolecular interactions, and (4) the position at which the electron-donating side group is bonded to the dual core was changed to study the effects of changes in the substitution position on the characteristics of the material.
2. Results and discussion
2.1 Molecular design, synthesis, and optical properties
As previously reported by our group, 1-[1,1′;3′,1′′]terphenyl-5′-yl-6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyrene (TP-AP-TP), which includes anthracene and pyrene as a hetero dual core, yielded a better performance, with about two times higher EQE values and two times longer blue emission lifetimes than their single core counterparts.11Moreover, oscillator strength is closely associated with the electronic transition of the molecule, and an increase of the oscillator strength indicates a contribution of greater energy to the electronic transition at that absorption wavelength. For the AP core, the oscillator strength value of anthracene was calculated to be 2.5 times greater than the oscillator strength value of pyrene. Comparing oscillator strength values indicated anthracene to be the primary core and pyrene to be the secondary core in this dual-core structure.12 The primary and the secondary cores mean the main and the secondary contributors to absorbance and emission of light.
Since anthracene is the primary core of the dual core, substitution of an electron-donating side group into anthracene is expected to yield greater changes of the intrinsic properties of the final compound compared to a substitution into pyrene.
To determine the effects of changing the substitution position of the side group, four types of molecules that introduced m-terphenyl, diphenylamine, and triphenylamine moieties as side groups were synthesized: diphenyl-[10-(6-[1,1′;3′,1′′]terphenyl-5′-yl-pyren-1-yl)-anthracen-9-yl]-amine (DPA-AP-TP), diphenyl-[6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyren-1-yl]-amine (TP-AP-DPA), diphenyl-{4-[10-(6-[1,1′;3′,1′′]terphenyl-5′-yl-pyren-1-yl)-anthracen-9-yl]-phenyl}-amine (TPA-AP-TP) and diphenyl-{4-[6-(10-[1,1′;3′,1′′]terphenyl-5′-yl-anthracen-9-yl)-pyren-1-yl]-phenyl}-amine (TP-AP-TPA) (see Scheme 1). Diphenylamine, an electron-donating group, was substituted into the anthracene and pyrene cores to make DPA-AP-TP and TP-AP-DPA, respectively. These molecules were classified as types (A-1) and (A-2), respectively. TPA-AP-TP and TP-AP-TPA were formed by the substitution of triphenylamine, a side group in which an additional phenyl ring was added to the link between the core and diphenylamine side group in the type (A) form, and were classified as type (B-1) and (B-2), respectively. These synthesized molecules were compared with TP-AP-TP, which consists only of phenyl rings that do not include any hetero-atom. The five compounds were synthesized using boronylation and Suzuki aryl–aryl coupling reactions as illustrated in Fig. S1,† and the synthesized materials were characterized by nuclear magnetic resonance spectroscopy, elemental analysis, and fast atom bombardment mass analysis.
 | ||
| Scheme 1 The chemical structures of the synthesized light-emitting materials based on dual-core chromophores. | ||
To determine the thermal properties of the synthesized molecules, thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used, with the results summarized in Table 1. All of the synthesized molecules were quite thermally stable, with Tg values of at least 189 °C and decomposition temperature (Td) values of at least 488 °C. In particular, TPA-AP-TP (B-1) and TP-AP-TPA (B-2), perhaps due to their increased molecular weights, displayed excellent Td values of 516 and 530 °C, respectively. Molecules with high Td values have an advantage in that the morphology of the material is not easily changed by the heat generated during operation of the OLED device.13
| Compounds | Solutiona | Filmb | Tg | Tm | Td | HOMO (eV) | LUMO (eV) | Band gap (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UV (nm) | PL (nm) | FWHM (nm) | UV (nm) | PL (nm) | FWHM (nm) | |||||||
| a 1 × 10−5 M in chloroform.b Film thickness on the glass: 50 nm. | ||||||||||||
| TP-AP-TP | 379, 399 | 446 | 56 | 382, 403 | 458 | 53 | 243 | — | 480 | −6.01 | −3.05 | 2.96 |
| DPA-AP-TP | 357, 428 | 513 | 65 | 365, 437 | 505 | 59 | 194 | 317 | 497 | −5.55 | −3.04 | 2.51 |
| TP-AP-DPA | 383, 404 | 481 | 67 | 388, 410 | 487 | 106 | 199 | — | 488 | −5.44 | −2.76 | 2.68 |
| 502 | ||||||||||||
| TPA-AP-TP | 379, 401 | 479 | 63 | 382, 405 | 473 | 57 | 192 | 313 | 516 | −5.42 | −2.65 | 2.77 |
| TP-AP-TPA | 381, 400 | 467 | 67 | 384, 404 | 468 | 65 | 189 | 338 | 530 | −5.41 | −2.58 | 2.83 |
Ultraviolet-visible (UV-vis) absorption spectra and related data of the synthesized compounds are shown in Fig. 1 and Table 1. To understand the changes in optical properties, changes in the intramolecular electron density of the molecules were confirmed by density functional theory (DFT) calculations (CAM-B3LYP/6-31G(d) level). Frontier molecular orbitals of the optimized S0 state molecular structures are shown in Fig. 2. TP-AP-TP, with its bulky aromatic side group, showed an absorption wavelength peak of 399 nm in the solution state (Fig. 1a). On the other hand, DPA-AP-TP (A-1) and TP-AP-DPA (A-2), each containing the electron-donating diphenylamine group, showed absorption wavelength peaks of 428 nm and 404 nm. TPA-AP-TP (B-1) and TP-AP-TPA (B-2) substituted with a triphenylamine group showed absorption wavelength peaks of 401 nm and 400 nm. The bathochromic (red) shifts of the absorption wavelength peaks of these four compounds relative to that of TP-AP-TP may have been due to their having increased conjugation lengths in contrast to TP-AP-TP, which only consists of phenyl rings, the type (A) and type (B) molecules substituted with aromatic amine groups were calculated in the HOMO state to display a wide distribution of conjugated electrons, including to the side groups (Fig. 2). The especially large red shift of DPA-AP-TP (A-1) (by 29 nm compared to that of TP-AP-TP) may have been related to its side group substitution being in the anthracene core, whereas in TP-AP-DPA (A-2), with its smaller (5 nm) red shift, the substitution was done into the pyrene core. The particularly small magnitudes of the red shifts displayed by TPA-AP-TP (B-1) (by only 2 nm compared to that of TP-AP-TP) and TP-AP-TPA (B-2) (by only 1 nm), despite the change in their substitution positions, may have been due to the substituted triphenylamine group of these type (B) molecules, with its additional benzene ring, having a relatively reduced electron-donating ability, compared to the diphenylamine group of the type (A) molecules. The UV-vis absorption peak values in the film state (Fig. 1b) were similar to those in the solution state: in the film state, TP-AP-TP showed an absorption wavelength peak of 403 nm, DPA-AP-TP (A-1) and TP-AP-DPA (A-2) showed absorption wavelength peaks of 437 nm and 410 nm, and TPA-AP-TP (B-1) and TP-AP-TPA (B-2) showed absorption wavelength peak of 405 nm and 404 nm, which were shorter than those of the type (A) molecules.
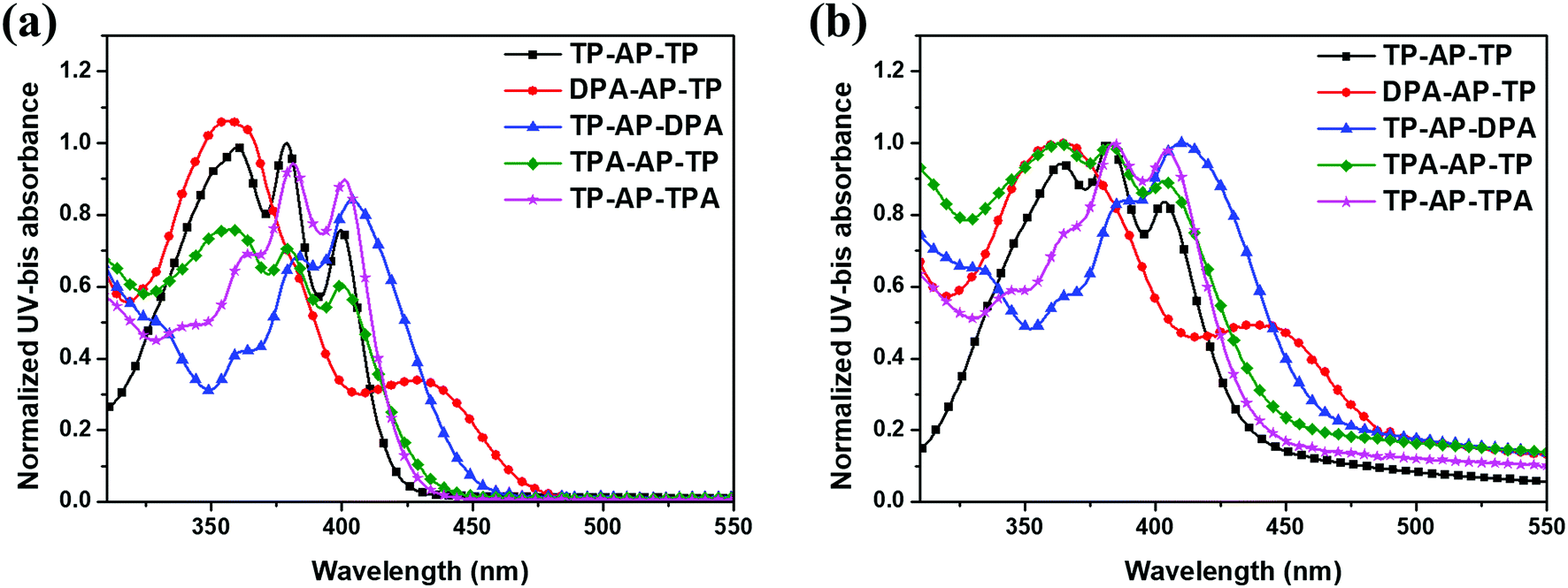 | ||
| Fig. 1 UV-vis absorption spectra of the synthesized compounds: (a) solution state, (b) film state. | ||
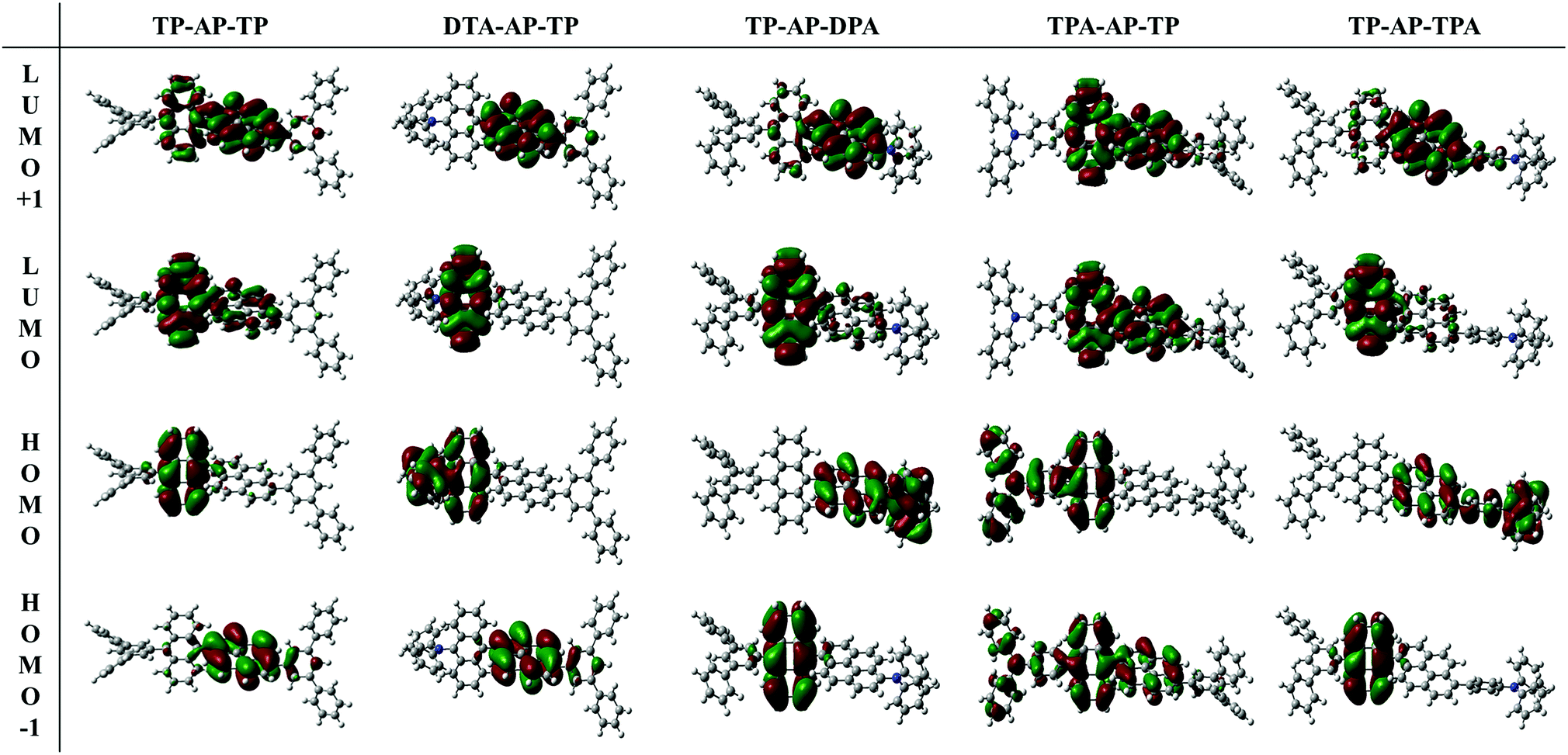 | ||
| Fig. 2 HOMO−1, HOMO, LUMO and LUMO+1 electronic density distributions of the S0 states of the compounds calculated at the CAM-B3LYP/6-31G(d) level. | ||
HOMO, LUMO and band gap levels of the molecules were measured and calculated using the UV-vis absorption data in the film state and ultraviolet photoelectron spectroscopy (Riken-Keiki, AC-2), as presented in Table 1. The HOMO and LUMO levels of TP-AP-TP were −6.01 eV and −3.05 eV, yielding a band gap of 2.96 eV. Since both type (A) and type (B) molecules have longer conjugation lengths compared to TP-AP-TP, their band gaps were decreased, by 0.26 eV on average: regarding the type (A) molecules, the band gap was 2.51 eV for DPA-AP-TP (A-1) (i.e., reduced by 0.45 eV compared to that of TP-AP-TP) and was 2.68 eV for TP-AP-DPA (A-2) (i.e., reduced by 0.28 eV); regarding the type (B) molecules, the band gap was 2.77 eV for TPA-AP-TP (B-1) (i.e., reduced by 0.19 eV compared to that of TP-AP-TP) and 2.83 eV for TP-AP-TPA (B-2) (i.e., reduced by 0.13 eV). The reduced band gaps of DPA-AP-TP (A-1) and of TPA-AP-TP (B-1), whose electron-donating groups were substituted into the anthracene core, compared, respectively, to the band gaps of TP-AP-DPA (A-2) and TP-AP-TPA (B-2), whose side groups were substituted in the pyrene core, occurred despite DPA-AP-TP (A-1) and TP-AP-DPA (A-2) having the same substituted aromatic amine side group (i.e., diphenylamine) and despite TPA-AP-TP (B-1) and TP-AP-TPA (B-2) having the same substituted aromatic amine side group (i.e., triphenylamine).
Time dependent (TD)-DFT calculations (TD-CAM-B3LYP/6-31G(d)) of the absorption wavelengths and oscillator strengths (f) of the molecules were carried out, and the results of the calculations are summarized in Table 2. The wavelength shift tendencies of the UV-vis absorption for the compounds obtained by calculations were similar to the experimental results.
| Compounds | λabs (nm) (oscillator strength (f)) | Characteristic transition | Distribution of charactera | Contributionb (%) |
|---|---|---|---|---|
| a A: anthracene, P: pyrene, DPA: diphenylamine, TPA: triphenylamine.b When the sum of contributions is less than 100%, the remaining contributions include various small portions of transitions. Only the main process was indicated. | ||||
| TP-AP-TP | 352 (0.642) | HOMO → LUMO | A → A + P | 75 |
| 319 (0.568) | HOMO−1 → LUMO+1 | P → A + P | 55 | |
| HOMO−1 → LUMO | P → A + P | 24 | ||
| DPA-AP-TP | 381 (0.362) | HOMO → LUMO | A + DPA → A | 95 |
| 331 (0.322) | HOMO−2 → LUMO | A + DPA → A | 81 | |
| TP-AP-DPA | 358 (0.910) | HOMO → LUMO+1 | P + DPA → P + A | 43 |
| HOMO−1 → LUMO | A → A + P | 40 | ||
| 344 (0.043) | HOMO−1 → LUMO | A → A + P | 45 | |
| HOMO → LUMO+1 | P + DPA → P + A | 37 | ||
| TPA-AP-TP | 355 (0.722) | HOMO → LUMO | A + TPA → A + P | 33 |
| HOMO−1 → LUMO | TPA + A + P → A + P | 23 | ||
| HOMO → LUMO+1 | A + TPA → A + P | 18 | ||
| 319 (0.579) | HOMO−2 → LUMO | TPA + A + P → A + P | 30 | |
| HOMO−1 → LUMO+1 | TPA + A + P → A + P | 27 | ||
| HOMO−2 → LUMO+1 | TPA + A + P → A + P | 21 | ||
| TP-AP-TPA | 353 (0.842) | HOMO−1 → LUMO | A → A | 81 |
| 328 (0.643) | HOMO → LUMO+1 | P + TPA → P | 59 | |
| HOMO−2 → LUMO+1 | P + TPA → P | 14 | ||
From the determined contribution of the electronic transition in the synthesized compound, TP-AP-TP showed an oscillator strength value of 0.642 at a wavelength of 352 nm, and HOMO → LUMO was shown to be the main process characterizing the transition here. The electron density distribution (Fig. 2) showed the anthracene core to be the primary source of this process. The oscillator strength value was 0.568 at 319 nm, and HOMO−1 → LUMO+1 and HOMO−1 → LUMO were found to be the main processes characterizing the electronic transition at this wavelength. The pyrene core was the main source of this process. Comparison of the oscillator strength values at these two wavelengths confirmed the electronic transition in which anthracene participated, rather than that in which the pyrene participated, to be the main factor in the overall process.12
Among the type (A) molecules, DPA-AP-TP (A-1), in which the electron-donating diphenylamine group was substituted into the anthracene core, showed oscillator strength values of 0.362 and 0.322 at 381 nm and 331 nm, respectively. In these two electronic transition processes, the contribution of the anthracene core was found to be the main factor. On the other hand, TP-AP-DPA (A-2), in which the electron-donating group was substituted into the pyrene core, showed an oscillator strength value of 0.910 at 358 nm. Here, the contribution of the electronic transition related to the pyrene core was clearly increased to show the transition process of pyrene.
Similar to type (A) molecules, type (B) molecules also showed an increased electronic transition contribution from each core to which an aromatic amine group was connected. TPA-AP-TP (B-1), in which the triphenylamine group was substituted into the anthracene core, showed oscillator strength values of 0.722 and 0.579 at 355 nm and 319 nm, respectively, and anthracene acted as the main core. TP-AP-TPA (B-2) showed a high oscillator strength value of 0.842 at 353 nm, with a large contribution of the anthracene core. An oscillator strength value of 0.643 was also shown at 328 nm, and the pyrene core provided the main source of this electronic transition.
While the contribution of the pyrene core to the electronic transition was increased in TP-AP-TPA (B-2) showed electronic transition characteristics similar to those of TP-AP-TP despite substitution of triphenylamine into the pyrene core. This similarity is due to the electron-donating ability of triphenylamine being smaller than that of diphenylamine and hence the triphenylamine group could not change the contribution of pyrene by much. The computational results are consistent with the experimental results, where, among the four type (A) and type (B) materials, TP-AP-TPA (B-2) showed the smallest difference in the band gap compared to that of TP-AP-TP.
The direct attachment of an aromatic amine group to the primary anthracene core results in electron donation to this core. This effect is larger for the connection to an anthracene core than that of the connection to a secondary pyrene core. Thus, as was shown from the results of the optical experiments (such as UV-vis absorption and band gaps), the properties of the DPA-AP-TP (A-1) and TPA-AP-TP (B-1) molecules differed from those of TP-AP-TP to a greater extent than did the properties of the TP-AP-DPA (A-2) and TP-AP-TPA (B-2) molecules.
Photoluminescence (PL) spectra of the synthesized compounds were acquired, and the corresponding data are shown in Fig. 3 and Table 1. TP-AP-TP showed a PL maximum (PLmax) value at 446 nm in the solution state.
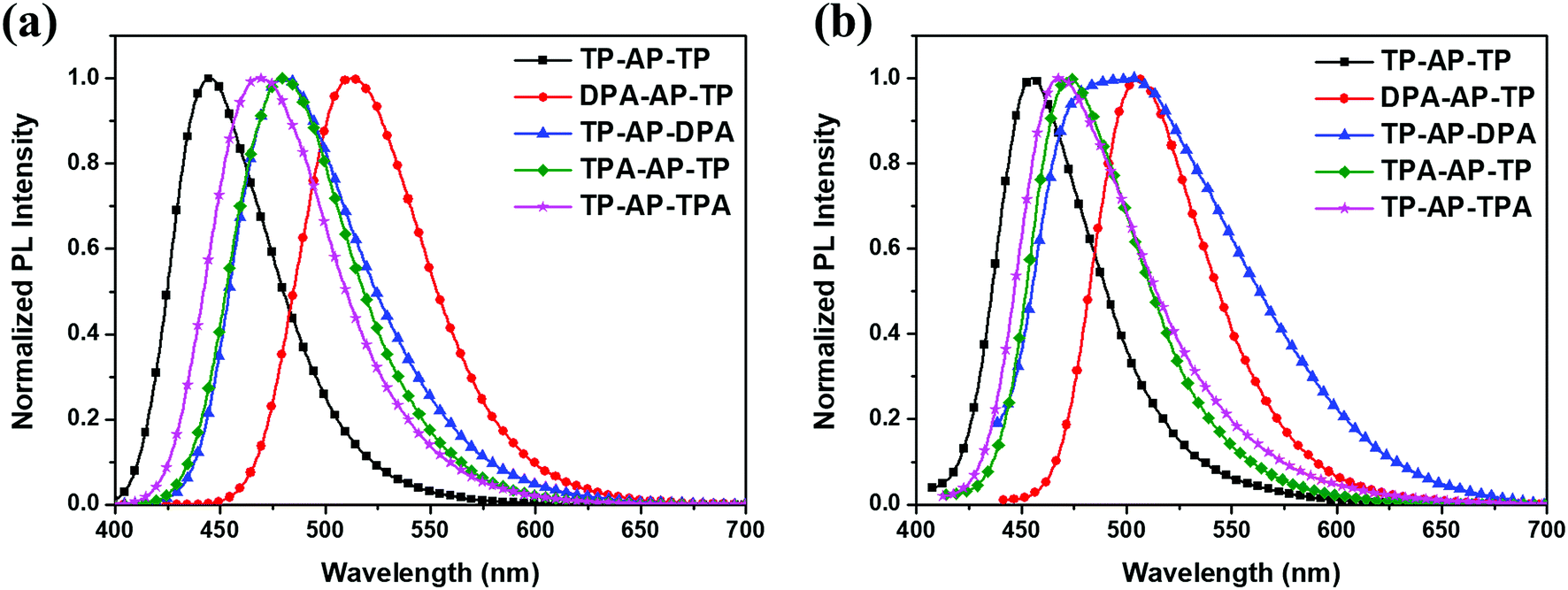 | ||
| Fig. 3 Normalized PL spectra of the synthesized materials: (a) solution state, (b) film state. | ||
Regarding the type (A) molecules, DPA-AP-TP (A-1) showed a PLmax at 513 nm, i.e., red shifted by 67 nm compared to that of TP-AP-TP, and TP-AP-DPA (A-2) showed a PLmax at 481 nm, i.e., red shifted by 35 nm. The large red shift of the PLmax value of DPA-AP-TP (A-1), in which a diphenylamine group was substituted into anthracene, was due to its greatly reduced band gap.10 On the other hand, type (B) molecules substituted with the triphenylamine group, which has a relatively small electron-donating effect, showed emissions in the shorter wavelength region compared to type (A) molecules: TPA-AP-TP (B-1) showed a PLmax at 479 nm, i.e., red shifted by 33 nm compared to that of TP-AP-TP; and TP-AP-TPA (B-2) showed a PLmax at 467 nm, i.e., red shifted by 21 nm.
The FWHM values of the PL spectra were also analyzed. In the solution state, TP-AP-TP showed a narrow value of 56 nm, whereas the type (A) and type (B) molecules, in which an aromatic amine group was substituted into the core, showed FWHM values increased to 63–67 nm. This increase can be interpreted as a diversification of the electronic transition channels due to a broadened distribution of intramolecular electron density following substitution of an electron donating side group into the core.14 Increases in the FWHM values of the type (A) and type (B) molecules were also shown in film state. An especially high FWHM value of 106 nm, with two PLmax values, at 487 nm and 502 nm, were observed for the film state of the TP-AP-DPA (A-2) molecule, in which diphenylamine was substituted into pyrene. This feature was probably caused by a solid-state-specific intermolecular excimer. To clarify whether the emissions at, say, 502 nm is excimer peak corresponding to PL emission from the molecule, excitation spectra were measured along with the UV-vis absorption spectrum (see Fig. S2†). The UV-vis absorption spectrum matched well with the excitation spectra. The excitation spectra monitored at 487 nm and 502 nm were observed to be very similar, indicating that they arose from the same excitation pathway. This finding is consistent with the value at 502 nm resulting from an excimer emission. Also, as shown in Fig. S3,† TP-AP-DPA (A-2) displays a dihedral angle between its pyrene and side group of only 68° and has a relatively small side group, and would hence appear to have a reduced ability to prevent intermolecular packing and excimer formation compared to DPA-AP-TP (A-1), which, while also having a relatively small side group, displays an increased dihedral angle between its anthracene and side group of 75°. Therefore, in addition to the size of the side group, the dihedral angle by itself may be a factor determining excimer formation. In case of TP-AP-TPA (B-2), it did not show excimer emission because of a relatively large side group.
2.2 Electroluminescence properties
Five different non-doped OLED devices (device configuration: ITO/4,4′,4′′-tris[2-naphthyl(phenyl)amino]triphenylamine [2-TNATA] 60 nm/N,N′-bis(naphthalen-1-yl)-N,N′-bis(phenyl)benzidine [NPB] 15 nm/synthesized emitting materials 35 nm/tris(8-hydroxyquinolinato)aluminum [Alq3] 20 nm/LiF 1 nm/Al 200 nm) were prepared using the five synthesized molecules as the respective emitting layers (EMLs), and the EL performances of the devices were measured. 2-TNATA was used as the hole-injection layer, NPB as the hole-transporting layer, and Alq3 as the electron-transporting layer. Emitting materials were applied as a single layer with a non-doped structure.For each of the five devices, the shape and maximum wavelength of the EL spectrum remained unchanged as the luminance was increased from 500 cd m−2 to 3000 cd m−2 (see Fig. S4†). All of the devices were confirmed to properly form excitons in the EML and operate stably. The OLED device properties are summarized in Fig. 4 and Table 3.
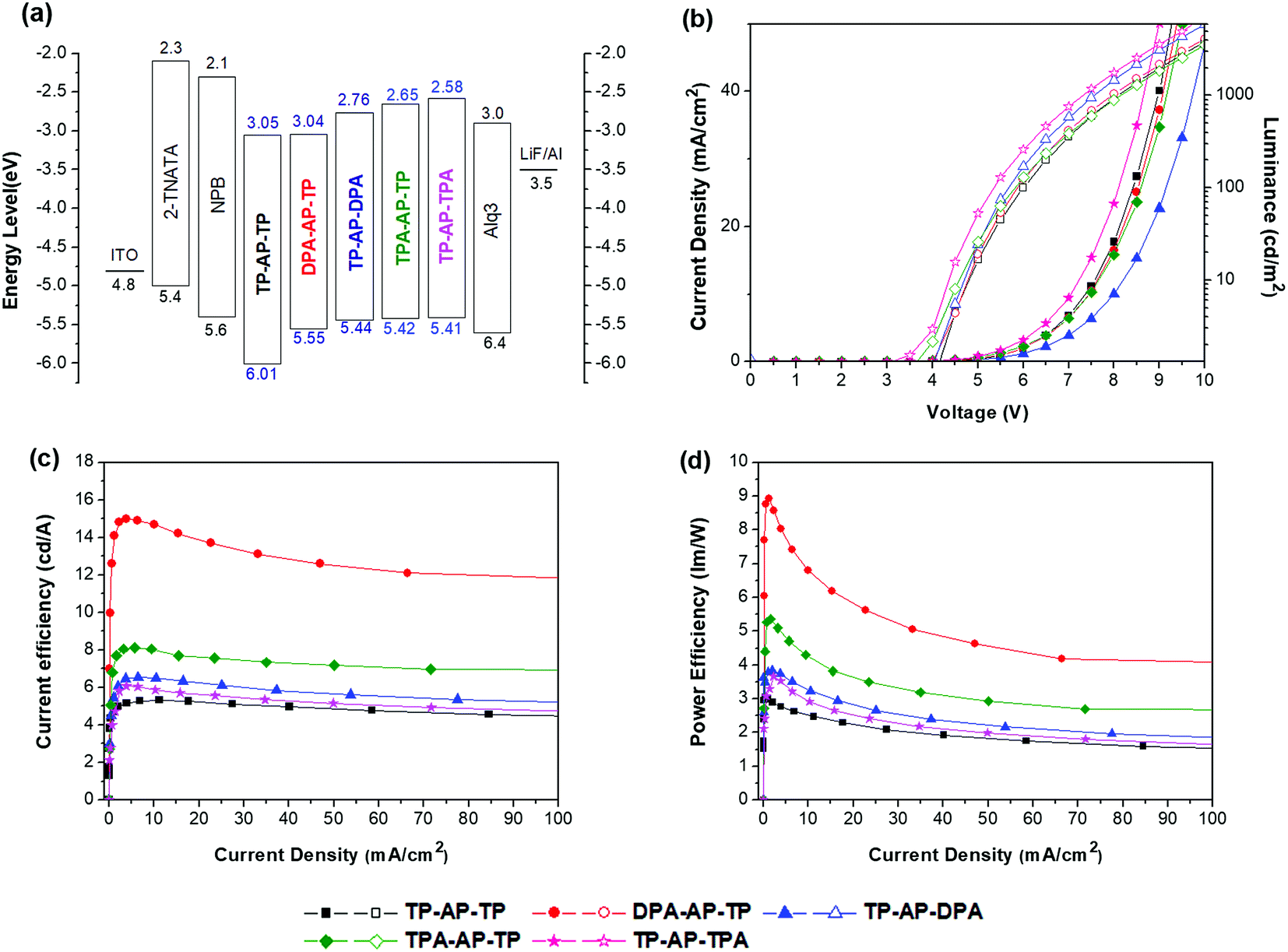 | ||
| Fig. 4 EL characteristics of devices using the synthesized compounds as EMLs: (a) energy levels of the organic materials, (b) I–V–L curves, (c) current efficiency (CE) versus current density, and (d) power efficiency (PE) versus current density. | ||
| Compound | Volt (v) | CE (cd A−1) | PE (lm W−1) | CIE (x,y) | EL (nm) | EL FWHM (nm) |
|---|---|---|---|---|---|---|
| TP-AP-TP | 7.19 | 5.34 | 2.58 | (0.15, 0.13) | 456 | 55 |
| DPA-AP-TP | 7.49 | 13.05 | 6.83 | (0.19, 0.56) | 501 | 57 |
| TP-AP-DPA | 6.93 | 6.50 | 3.27 | (0.24, 0.51) | 504 | 69 |
| TPA-AP-TP | 6.56 | 8.05 | 4.25 | (0.14, 0.24) | 473 | 53 |
| TP-AP-TPA | 6.96 | 5.89 | 2.96 | (0.16, 0.29) | 472 | 57 |
As was observed for its emission properties, TP-AP-TP of the reference material, which only consists of phenyl rings, yielded an EL maximum (ELmax) wavelength of 456 nm and blue color coordinates with Commission International de L'Eclairge (CIE) coordinates (x,y) of (0.15, 0.13). Also, it showed a CE of 5.34 cd A−1 and power efficiency (PE) of 2.58 lm W−1 under a current density of 10 mA cm−2. Compared to TP-AP-TP, devices that applied type (A) and type (B) molecules as the EML showed shifting of the emission wavelength to longer wavelengths, as found for PL, but both CE and PE were increased (see Fig. 5). These shifts were caused by the electron-donating aromatic amine moieties.10
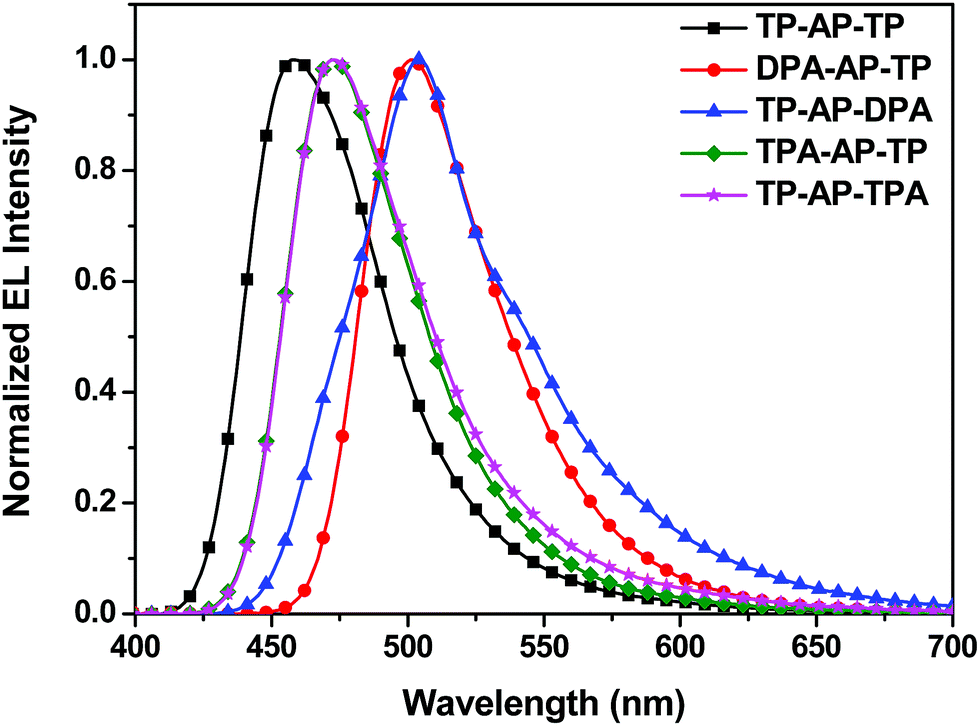 | ||
| Fig. 5 EL spectra of devices using the synthesized compounds. | ||
DPA-AP-TP (A-1) yielded the highest device efficiencies of 13.05 cd A−1and 6.83 lm W−1, but an emission with CIEx,y (0.19, 0.56) due to the squeezed band gap. TP-AP-DPA (A-2) showed EL efficiencies of 6.50 cd A−1and 3.27 lm W−1 and a CIEx,y (0.24, 0.51). Also, EL efficiencies of TPA-AP-TP (B-1) were 8.05 cd A−1 and 4.25 lm W−1, and EL efficiencies of TP-AP-TPA (B-2) were 5.89 cd A−1 and 2.96 lm W−1. They yielded color coordinate values of CIEx,y (0.14, 0.24) and CIEx,y (0.16, 0.29), respectively. MADN and DPVBi, which are well-known blue reference emitters, showed CE values of 2.86 and 3.92 cd A−1, PE values of 1.48 and 1.61 lm W−1, and EL maximum values of 454 and 465 nm, respectively [see Table S1†]. Comparing to the data of the reference emitters, TPA-AP-TP had over two times higher CE and PE values than those of MADN and DPVBi although EL maximum value was red-shifted 7–18 nm compared to MADN and DPVBi.
Comparing CE and CIEx,y values of type (A) and type (B) molecules, type (A) molecules substituted with the diphenylamine group showed higher CE values compared to type (B) molecules. However, the EL emission was shifted more to longer wavelengths, and CIEx,y values were also increased. On the other hand, type (B) molecules, with their triphenylamine group and its reduced electron-donating ability, displayed a relatively small shift of the EL wavelength and were found to have optimized CIEx,y values for the sky-blue color. The TPA-AP-TP (B-1) molecule, with its bulky triphenylamine side group substituted into the primary anthracene core, yielded especially excellent EL efficiencies of 8.05 cd A−1 and 4.25 lm W−1 as well as a narrow FWHM value of 53 nm. This study verified that in heterocore molecules such as the AP core, one of the two cores functions as the primary core and contributes more to the electronic transition than does the other, i.e., secondary, core—and that the largest changes to the optical and electronic properties of the final heterocore derivatives can be achieved by simply substituting an electron-donating side group into a position on the primary core. Such a methodology could be applied to develop excellent blue light-emitting materials applicable to OLED lighting.
3. Conclusions
This study examined hetero dual-core chromophores in which anthracene and pyrene are connected. The hetero dual-core chromophores included anthracene to be the primary core and pyrene to be the secondary core. We specifically investigated the effects of changing the side group on the performance properties of the final compounds. A nitrogen atom was introduced in order to increase electron donation to the dual core and hence increased device efficiency, and a bulky side group of triphenylamine was introduced to suppress intermolecular interactions. Especially, electron-donating side groups were substituted at different positions of TP-AP-TP, a molecule with a hetero dual-core structure, in order to determine whether one of its two cores, and if so which one, contributes more to changes in the optical and electronic properties of the resulting material.The electron density distribution within the molecule and the part of the molecule contributing to the electronic transition were confirmed to be affected by the substitution of the electronically optimized side group into the core. The TPA-AP-TP molecule, whose side group not only displays optimum electron donation to what was determined to be the main core, but also has a proper size, yielded a non-doped OLED device with high efficiency values of 8.05 cd A−1 and 4.25 lm W−1 as well as a narrow FWHM value of 53 nm. Comparing to the data of the reference emitters, TPA-AP-TP had two times higher CE and PE values than those of MADN and DPVBi.
This study verified that the largest changes to the optical and electronic properties of the final heterocore derivatives can be achieved by substituting an electron-donating side group into a position on the primary core.
This systematic methodology can be applied to develop new various other OLED emitters of blue light for OLED lighting based on many multi-core chromophores by introducing various electron-donating moieties to the main core.
4. Experimental section
4.1 General information
The requisite reagents were purchased from Sigma-Aldrich or TCI and were used without further purification, unless otherwise noted. The 1H-NMR spectra were recorded on Bruker Avance 300 spectrometers. The FAB+-mass and EI+-spectra were recorded on a JEOL, JMS-AX505WA, HP5890 series II. The optical absorption spectra were obtained by using a Lambda 1050 UV/vis/NIR spectrometer (PerkinElmer). A PerkinElmer luminescence spectrometer LS50 (Xenon flash tube) was used to perform photoluminescence spectroscopy. The glass-transition temperatures (Tg) of the compounds were determined with differential scanning calorimetry (DSC) under a nitrogen atmosphere by using a DSC4000 (PerkinElmer). Samples were heated to 500 °C at a rate of 10 °C min−1 and cooled at 10 °C min−1 then heated again under the same heating conditions as used in the initial heating process. Degradation temperatures (Td) were determined with thermo gravimetric analysis (TGA) by using a TGA4000 (PerkinElmer). Samples were heated to 700 °C at a rate of 10 °C min−1. The HOMO energy levels were determined with ultraviolet photoelectron yield spectroscopy (Riken Keiki AC-2). The LUMO energy levels were derived from the HOMO energy levels and the band gaps. All DFT and TD-DFT calculations were performed using the Gaussian09 (ref. 15) program. In each of the EL devices, 4,4′,4′′-tris[2-naphthyl(phenyl)amino]triphenylamine (2-TNATA) was used as the hole-injection layer, N,N′-bis(naphthalen-1-yl)-N,5N′-bis(phenyl)benzidine (NPB) was used for the hole transporting layer, one of the synthetic materials TP-AP-TP, DPA-AP-TP, TP-AP-DPA, TPA-AP-TP and TP-AP-TPA were used as the emitting layer (EML), emitting materials were applied as a single layer with a non-doped structure. Tris(8-hydroxyquinolinato)aluminum (Alq3) was used for the electron transporting (ETL), lithium fluoride (LiF) was used for the electron injection layer (EIL), and ITO was used as the anode and aluminum (Al) as the cathode. All organic layers were deposited under 10−6 Torr, with a rate of deposition of 1.0 Å s−1 to create an emitting area of 4 mm2. The LiF and aluminum layers were continuously deposited under the same vacuum conditions. The luminance efficiency data for the fabricated EL devices were obtained by using a Keithley 2400 electrometer. Light intensities were obtained with a Minolta CS-1000A. The operational stabilities of the devices were measured under encapsulation in a glove box.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:15) eluent to afford a white solid. (Yield 61%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.70 (s, 3H), 7.60 (d, 4H), 7.44 (t, 4H), 7.36 ppm (t, 2H).:n-hexane (1:7) as the eluent to afford a beige solid. (3.9 g, 32%). 1H-NMR (300 MHz, CDCl3, d): 8.65 (s, 1H), 8.55 (d, 1H), 8.41 (d, 1H), 8.32 (d, 1H), 8.24 (d, 1H), 8.16 (d, 2H), 8.06 (d, 1H), 7.93 (d, 1H) 7.72 (d, 1H), 7.47 (t, 2H), 7.33 (d, 1H), 7.30 (d, 2H), 7.22 (t, 2H).:n-hexane (1:7) as the eluent to afford a beige solid (TP-AP-TP) (1.0 g, 63%). 1H-NMR (500 MHz, CDCl3, d): 8.43 (d, 1H), 8.40 (d, 1H), 8.22 (d, 1H), 8.21 (d, 1H), 8.11 (d, 2H), 8.08 (s, 1H), 7.97 (m, 3H), 7.89 (m, 4H), 7.79 (m, 9H), 7.50 (m, 9H), 7.40 (m, 8H), 7.21 (t, 2H); 13C-NMR (500 MHz, CDCl3, d): 142.31, 141.99, 141.92, 141.04, 140.87, 140.12, 137.88, 137.39, 135.66, 134.40, 131.28, 131.02, 130.98, 130.72, 130.07, 129.71, 129.19, 129.00, 128.94, 128.92, 128.44, 127.91, 127.84, 127.82, 127.64, 127.42, 127.38, 127.22, 127.19, 125.94, 125.61, 125.39, 125.35, 125.20, 125.14, 125.10, 124.94, 124.74; HRMS (EI, m/z): [M]+ calcd for C56H42, 834.3287; found, 834.3289. Anal. calcd for C56H42: C 94.93, H5.07; found: C 94.23, H 5.06%.:hexane (1:5) eluent to afford a white solid. (Yield 61%) 1H NMR (300 MHz, DMSO, 25 °C, TMS): δ = 8.88 (s, 1H), 8.56 (d, 1H), 8.38 (t, 3H), 8.28 (t, 3H), 8.11 (m, 2H), 8.08 (d, 1H), 7.95 (m, 6H), 7.56 (t, 6H), 7.46 (t, 2H), 7.34 (t, 2H), 7.23 (m, 3H).:hexane (1:5) eluent to afford a white solid (yield 68%). 1H-NMR (300 MHz, [D8] THF 25 °C, TMS): 8.46 (d, 1H), 8.43 (d, 1H), 8.29 (m, 4H), 8.19 (d, 1H), 8.10 (d, 1H), 8.06 (m, 1H), 7.94 (m, 3H), 7.86 (d, 4H), 7.49 (t, 4H), 7.40 (m, 7H), 7.22 (m, 10H), 6.91 (m, 2H). EI+-mass: 773, HRMS (EI, m/z): [M+] calcd for C60H39N1, 773.3082; found, 773.3080. Anal. calcd for C60H39N1: C 93.11, H 5.08, N 1.81; found: C 92.62, H 5.07, N 1.80%.:hexane (1:4) eluent to afford a white solid. (Yield 76%) 1H NMR (300 MHz, [D8] THF 25 °C, TMS): δ = 8.46 (d, 1H), 8.42 (d, 1H), 8.29 (d, 2H), 8.18 (d, 1H), 8.07 (m, 2H), 7.86 (m, 9H), 7.49 (t, 5H), 7.37 (m, 18H), 7.20 (t, 2H), 7.08 (t, 2H). EI+-mass: 849, HRMS (EI, m/z): [M+] calcd for C66H43N1, 849.3395; found, 849.3393. Anal. calcd for C66H43N1: C 93.25, H 5.10, N 1.65; found: C 93.15, H 4.99, N 1.64%.:hexane (1:9) eluent to afford a white solid. (Yield 48%) 1H NMR (300 MHz, CDCl3 25 °C, TMS): δ = 8.46 (d, 1H), 8.64 (d, 2H), 8.03 (s, 1H), 7.82 (d, 2H), 7.72 (d,4H), 7.65 (m, 2H), 7.60 (m, 2H), 7.42 (m, 8H).:hexane (1:5) eluent to afford a white solid. (Yield 42%) 1H NMR (300 MHz, CDCl3 25 °C, TMS): δ = 8.60 (d, 1H), 8.48 (d, 1H), 8.38 (d, 1H), 8.26 (d, 1H), 8.15 (d, 1H), 8.07 (s, 1H), 7.95 (m, 3H), 7.88 (s, 1H), 7.83 (m, 6H), 7.52 (m, 5H), 7.43 (m, 6H), 7.22 (t, 2H).:hexane (1:4) eluent to afford a white solid (yield 58%). 1H-NMR (300 MHz, [D8] THF 25 °C, TMS): 8.42 (d, 1H), 8.30 (d, 1H), 8.19 (m, 3H), 8.09 (d, 1H), 7.90 (m, 10H), 7.49 (m, 4H), 7.37 (m, 7H), 7.24 (t, 6H), 7.11 (d, 4H), 6.97 (t, 2H). EI+-mass: 773, HRMS (EI, m/z): [M+] calcd for C60H39N1, 773.3082; found, 773.3092. Anal. calcd for C60H39N1: C 93.11, H 5.08, N 1.81; found: C 92.78, H 5.14, N 1.83%.:hexane (1:4) eluent to afford a white solid. (Yield 62%) 1H NMR (300 MHz, [D8] THF 25 °C, TMS): δ = 8.42 (d, 1H), 8.30 (d, 1H), 8.18 (m, 3H), 8.13 (d, 1H), 7.93 (m, 10H), 7.51 (m, 4H), 7.37 (m, 7H), 7.22 (t, 6H), 7.11 (d, 4H), 6.95 (t, 2H). EI+-mass: 849, HRMS (EI, m/z): [M+] calcd for C66H43N1, 849.3395; found, 849.3390. Anal. calcd for C66H43N1: C 93.25, H 5.10, N 1.65; found: C 92.90, H 4.97, N 1.70%.
:hexane (1:15) eluent to afford a white solid. (Yield 61%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.70 (s, 3H), 7.60 (d, 4H), 7.44 (t, 4H), 7.36 ppm (t, 2H).:n-hexane (1:7) as the eluent to afford a beige solid. (3.9 g, 32%). 1H-NMR (300 MHz, CDCl3, d): 8.65 (s, 1H), 8.55 (d, 1H), 8.41 (d, 1H), 8.32 (d, 1H), 8.24 (d, 1H), 8.16 (d, 2H), 8.06 (d, 1H), 7.93 (d, 1H) 7.72 (d, 1H), 7.47 (t, 2H), 7.33 (d, 1H), 7.30 (d, 2H), 7.22 (t, 2H).:n-hexane (1:7) as the eluent to afford a beige solid (TP-AP-TP) (1.0 g, 63%). 1H-NMR (500 MHz, CDCl3, d): 8.43 (d, 1H), 8.40 (d, 1H), 8.22 (d, 1H), 8.21 (d, 1H), 8.11 (d, 2H), 8.08 (s, 1H), 7.97 (m, 3H), 7.89 (m, 4H), 7.79 (m, 9H), 7.50 (m, 9H), 7.40 (m, 8H), 7.21 (t, 2H); 13C-NMR (500 MHz, CDCl3, d): 142.31, 141.99, 141.92, 141.04, 140.87, 140.12, 137.88, 137.39, 135.66, 134.40, 131.28, 131.02, 130.98, 130.72, 130.07, 129.71, 129.19, 129.00, 128.94, 128.92, 128.44, 127.91, 127.84, 127.82, 127.64, 127.42, 127.38, 127.22, 127.19, 125.94, 125.61, 125.39, 125.35, 125.20, 125.14, 125.10, 124.94, 124.74; HRMS (EI, m/z): [M]+ calcd for C56H42, 834.3287; found, 834.3289. Anal. calcd for C56H42: C 94.93, H5.07; found: C 94.23, H 5.06%.:hexane (1:5) eluent to afford a white solid. (Yield 61%) 1H NMR (300 MHz, DMSO, 25 °C, TMS): δ = 8.88 (s, 1H), 8.56 (d, 1H), 8.38 (t, 3H), 8.28 (t, 3H), 8.11 (m, 2H), 8.08 (d, 1H), 7.95 (m, 6H), 7.56 (t, 6H), 7.46 (t, 2H), 7.34 (t, 2H), 7.23 (m, 3H).:hexane (1:5) eluent to afford a white solid (yield 68%). 1H-NMR (300 MHz, [D8] THF 25 °C, TMS): 8.46 (d, 1H), 8.43 (d, 1H), 8.29 (m, 4H), 8.19 (d, 1H), 8.10 (d, 1H), 8.06 (m, 1H), 7.94 (m, 3H), 7.86 (d, 4H), 7.49 (t, 4H), 7.40 (m, 7H), 7.22 (m, 10H), 6.91 (m, 2H). EI+-mass: 773, HRMS (EI, m/z): [M+] calcd for C60H39N1, 773.3082; found, 773.3080. Anal. calcd for C60H39N1: C 93.11, H 5.08, N 1.81; found: C 92.62, H 5.07, N 1.80%.:hexane (1:4) eluent to afford a white solid. (Yield 76%) 1H NMR (300 MHz, [D8] THF 25 °C, TMS): δ = 8.46 (d, 1H), 8.42 (d, 1H), 8.29 (d, 2H), 8.18 (d, 1H), 8.07 (m, 2H), 7.86 (m, 9H), 7.49 (t, 5H), 7.37 (m, 18H), 7.20 (t, 2H), 7.08 (t, 2H). EI+-mass: 849, HRMS (EI, m/z): [M+] calcd for C66H43N1, 849.3395; found, 849.3393. Anal. calcd for C66H43N1: C 93.25, H 5.10, N 1.65; found: C 93.15, H 4.99, N 1.64%.:hexane (1:9) eluent to afford a white solid. (Yield 48%) 1H NMR (300 MHz, CDCl3 25 °C, TMS): δ = 8.46 (d, 1H), 8.64 (d, 2H), 8.03 (s, 1H), 7.82 (d, 2H), 7.72 (d,4H), 7.65 (m, 2H), 7.60 (m, 2H), 7.42 (m, 8H).:hexane (1:5) eluent to afford a white solid. (Yield 42%) 1H NMR (300 MHz, CDCl3 25 °C, TMS): δ = 8.60 (d, 1H), 8.48 (d, 1H), 8.38 (d, 1H), 8.26 (d, 1H), 8.15 (d, 1H), 8.07 (s, 1H), 7.95 (m, 3H), 7.88 (s, 1H), 7.83 (m, 6H), 7.52 (m, 5H), 7.43 (m, 6H), 7.22 (t, 2H).:hexane (1:4) eluent to afford a white solid (yield 58%). 1H-NMR (300 MHz, [D8] THF 25 °C, TMS): 8.42 (d, 1H), 8.30 (d, 1H), 8.19 (m, 3H), 8.09 (d, 1H), 7.90 (m, 10H), 7.49 (m, 4H), 7.37 (m, 7H), 7.24 (t, 6H), 7.11 (d, 4H), 6.97 (t, 2H). EI+-mass: 773, HRMS (EI, m/z): [M+] calcd for C60H39N1, 773.3082; found, 773.3092. Anal. calcd for C60H39N1: C 93.11, H 5.08, N 1.81; found: C 92.78, H 5.14, N 1.83%.:hexane (1:4) eluent to afford a white solid. (Yield 62%) 1H NMR (300 MHz, [D8] THF 25 °C, TMS): δ = 8.42 (d, 1H), 8.30 (d, 1H), 8.18 (m, 3H), 8.13 (d, 1H), 7.93 (m, 10H), 7.51 (m, 4H), 7.37 (m, 7H), 7.22 (t, 6H), 7.11 (d, 4H), 6.95 (t, 2H). EI+-mass: 849, HRMS (EI, m/z): [M+] calcd for C66H43N1, 849.3395; found, 849.3390. Anal. calcd for C66H43N1: C 93.25, H 5.10, N 1.65; found: C 92.90, H 4.97, N 1.70%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science & ICT (No. 2017M3A7B4041699). This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2017R1D1A1A09082138).Notes and references
- (a) C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef; (b) Y. R. Sun, N. Giebink, C. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908 CrossRef PubMed; (c) M. F. Wu, S. J. Yeh, C. T. Chen, H. Murayama, T. Tsuboi, W. S. Li, I. Chao, S. W. Liu and J. K. Wang, Adv. Funct. Mater., 2007, 17, 1887 CrossRef; (d) Z. Zhao, C. Deng, S. Chen, J. W. Y. Lam, W. Qin, P. Lu, Z. Wang, H. S. Kwok, Y. Ma, H. Qiu and B. Z. Tang, Chem. Commun., 2011, 47, 8847 RSC; (e) H. Wu, G. Zhou, J. Zou, C. L. Ho, W. Y. Wong, W. Yang, J. Peng and Y. Cao, Adv. Mater., 2009, 21, 4181 CrossRef; (f) M. C. Gather, A. Kohnen, A. Falcou, H. Becker and K. Meerholz, Adv. Funct. Mater., 2007, 17, 191 CrossRef; (g) J. Huang, X. Wang, A. J. deMello, J. C. deMello and D. D. C. Bradley, J. Mater. Chem., 2007, 17, 3551 RSC; (h) B. Chen, J. Ding, L. Wang, X. Jing and F. Wang, Chem. Commun., 2012, 48, 8970 RSC; (i) Z. Shen, P. E. Burrows, V. Bulović, S. R. Forrest and M. E. Thompson, Science, 1997, 276, 2009 CrossRef; (j) S. R. Forrest, Org. Electron., 2003, 4, 45 CrossRef; (k) A. R. Duggal, J. J. Shiang, C. M. Heller and D. F. Foust, Appl. Phys. Lett., 2002, 80, 3470 CrossRef; (l) B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef; (m) M. Yu, S. Wang, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem. C, 2015, 3, 861 RSC; (n) L. Zhao, S. Wang, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem. C, 2015, 3, 8859 Search PubMed; (o) S. Kim, B. Kim, J. Lee, H. Shin, Y.-I. Park and J. Park, Mater. Sci. Eng., R, 2016, 99, 1 CrossRef; (p) J. T. Smith, B. A. Katchman, D. E. Kullman, U. Obahiagbon, Y.-K. Lee, B. P. O'Brien, G. B. Raupp, K. S. Anderson and J. B. Christen, J. Disp. Technol., 2016, 12, 273 CrossRef CAS; (q) B. A. Katchman, J. T. Smith, U. Obahiagbon, S. Kesiraju, Y.-K. Lee, B. O'Brien, K. Kaftanoglu, J. B. Christen and K. S. Anderson, Sci. Rep., 2016, 6, 29057 CrossRef CAS PubMed; (r) J. Ynag, J. Huang, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC.
- (a) J. H. Jou, C. P. Wang, M. H. Wu, H. W. Lin, H. C. Pan and B. H. Liu, J. Mater. Chem., 2010, 20, 6626 RSC; (b) S. Krotkus, D. Kasemann, S. Lenk, K. Leo and S. Reineke, Light: Sci. Appl., 2016, 5, e16121 CrossRef CAS; (c) E. Angioni, M. Chapran, K. Ivaniuk, N. Kostiv, V. Cherpak, P. Stakhira, A. Lazauskas, S. Tamulevičius, D. Volyniuk, N. J. Findlay, T. Tuttle, J. V. Grazulevicius and P. J. Skabara, J. Mater. Chem. C, 2016, 4, 3851 RSC; (d) H.-J. Kim, M.-H. Shin, H.-G. Hong, B.-S. Song, S.-K. Kim, W.-H. Koo, J.-G. Yoon, S.-Y. Yoon and Y.-J. Kim, J. Disp. Technol., 2016, 12, 526 CrossRef; (e) Y. Chen, J. Wang, Z. Zhong, Z. Jiang, C. Song, Z. Hu, J. Peng, J. Wang and Y. Cao, Org. Electron., 2016, 37, 458 CrossRef CAS.
- (a) Y. L. Chang, Y. Song, Z. Wang, M. G. Helander, J. Qiu, L. Chai, Z. Liu, G. D. Scholes and Z. Lu, Adv. Funct. Mater., 2013, 23, 705 CrossRef CAS; (b) S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed; (c) M. Thomschke, S. Reineke, B. Lüssem and K. Leo, Nano Lett., 2012, 12, 424 CrossRef CAS PubMed; (d) L. Zhou, X. Li, X. Li, J. Feng, S. Song and H. Zhang, J. Phys. Chem. C, 2010, 114, 21723 CrossRef CAS; (e) H. P. Lin, F. Zhou, J. Li, X. W. Zhang, L. Zhang, X. Y. Jiang, Z. L. Zhang and J. H. Zhang, J. Phys. Chem. C, 2011, 115, 24341 CrossRef CAS; (f) S. A. Wang, W. Y. Hung, Y. H. Chen and K. T. Wong, Org. Electron., 2012, 13, 1576 CrossRef CAS; (g) Z. Wu, J. Luo, N. Sun, L. Zhu, H. Sun, L. Yu, D. Yang, X. Qiao, J. Chen, C. Yang and D. Ma, Adv. Funct. Mater., 2016, 26, 3306 CrossRef CAS.
- (a) X. Yang, H. Huang, B. Pan, M. P. Aldred, S. Zhuang, L. Wang, J. Chen and D. Ma, J. Phys. Chem. C, 2012, 116, 15041 CrossRef CAS; (b) L. Duan, D. Zhang, K. Wu, X. Huang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2011, 21, 3540 CrossRef CAS; (c) S. M. Shen, Y. C. Tsai and J. H. Jou, ACS Appl. Mater. Interfaces, 2011, 3, 3134 CrossRef PubMed; (d) X. Yang, Y. Zhao, X. Zhang, R. Li, J. Dang, Y. Li, G. Zhou, Z. Wu, D. Ma, W. Y. Wong, X. Zhao, A. Ren, L. Wang and X. Hou, J. Mater. Chem., 2012, 22, 7136 RSC; (e) R. Springer, B. Y. Kang, R. Lampande, D. H. Ahn, S. Lenk, S. Reineke and J. H. Kwon, Opt. Express, 2016, 24, 28131 CrossRef PubMed; (f) M. Du, Y. Feng, D. Zhu, T. Peng, Y. Liu, Y. Wang and M. R. Bryce, Adv. Mater., 2016, 28, 5963 CrossRef CAS PubMed.
- (a) N. C. Giebink, B. W. D'Andrade, M. S. Weaver, P. B. Mackenzie, J. J. Brown, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2008, 103, 44509 CrossRef; (b) I. R. de Moraes, S. Scholz, B. Lussem and K. Leo, Org. Electron., 2011, 12, 341 CrossRef; (c) H. Shin, H. Jung, B. Kim, J. Lee, J. Moon, J. Kim and J. Park, J. Mater. Chem. C, 2016, 4, 3833 RSC; (d) S. Lee, B. Kim, H. Jung, H. Shin, H. Lee, J. Lee and J. Park, Dyes Pigm., 2016, 136, 255 CrossRef.
- (a) S.-H. Lin, F.-I. Wu and R.-S. Liu, Chem. Commun., 2009, 6961 RSC; (b) K. Danel, T.-H. Huang, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Chem. Mater., 2002, 14, 3860 CrossRef CAS; (c) C.-H. Chien, C.-K. Chen, F.-M. Hsu, C.-F. Shu, P.-T. Chou and C.-H. Lai, Adv. Funct. Mater., 2009, 19, 560 CrossRef CAS; (d) C. J. Zheng, W. M. Zhao, Z. Q. Wang, D. Huang, J. Ye, X. M. Ou, X. H. Zhang, C. S. Lee and S. T. Lee, J. Mater. Chem., 2010, 20, 1560 RSC; (e) C. H. Wu, C. H. Chien, F. M. Hsu, P. I Shih and C. F. Shu, J. Mater. Chem., 2009, 19, 1464 RSC; (f) C. J. Kuo, T. Y. Li, C. C. Lien, C. H. Liu, F. I. Wu and M. J. Huang, J. Mater. Chem., 2009, 19, 1865 RSC; (g) S. K. Kim, B. Yang, Y. Ma, J. H. Lee and J. Park, J. Mater. Chem., 2008, 18, 3376 RSC; (h) H. Shin, H. Kang, B. Kim, Y. Park, Y. J. Yu and J. Park, Bull. Korean Chem. Soc., 2014, 35, 3041 CrossRef CAS; (i) Z. Li, Y. Sun, H. Li, J. Ren, C. Si, X. Lv, J. Yu, H. Wang, F. Shi and Y. Hao, Synth. Met., 2016, 217, 102 CrossRef CAS; (j) X. Wei, L. Bu, X. Li, H. Ågren and Y. Xie, Dyes Pigm., 2017, 136, 480 CrossRef CAS; (k) J. Y. Song, S. N. Park, S. J. Lee, Y. K. Kim and S. S. Yoon, Dyes Pigm., 2015, 114, 40 CrossRef CAS.
- (a) S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, M. F. Lo, S. T. Lee and C. S. Lee, J. Mater. Chem., 2011, 21, 1206 RSC; (b) T. M. Figueira-Duarte, P. G. D. Rosso, R. Trattnig, S. Sax, E. J. W. List and K. Mullen, Adv. Mater., 2010, 22, 990 CrossRef CAS PubMed; (c) K.-C. Wu, P.-J. Ku, C.-S. Lin, H.-T. Shih, F.-I. Wu, M.-J. Huang, J.-J. Lin, I.-C. Chen and C.-H. Cheng, Adv. Funct. Mater., 2008, 18, 67 CrossRef CAS; (d) M. Y. Lo, C. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808 CrossRef CAS PubMed; (e) K. R. Justin Thomas, N. Kapoor, M. N. K. Prasad Bolisetty, J.-H. Jou, Y.-L. Chen and Y.-C. Jou, J. Org. Chem., 2012, 77, 3921 CrossRef PubMed; (f) A. Islam, Q. Wang, L. Zhang, T. Lei, L. Hong, R. Yang, Z. Liu, R. Peng, L.-S. Liao and Z. Ge, Dyes Pigm., 2017, 142, 499 CrossRef CAS; (g) D. Karthik, K. R. J. Thomas, J.-H. Jou, S. Kumar, Y.-L. Chen and Y.-C. Jou, RSC Adv., 2015, 5, 8727 RSC.
- (a) E. J. W. List, R. Guentner, P. Scanducci de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374 CrossRef CAS; (b) M. S. Gong, H. S. Lee and Y. M. Jeon, J. Mater. Chem., 2010, 20, 10735 RSC; (c) Y. M. Jeon, J. Y. Lee, J. W. Kim, C. W. Lee and M. S. Gong, Org. Electron., 2010, 11, 1844 CrossRef CAS; (d) S. O. Jeon, Y. M. Jeon, J. W. Kim, C. W. Lee and M. S. Gong, Org. Electron., 2008, 9, 522 CrossRef CAS; (e) K.-T. Wong, R.-T. Chen, F.-C. Fang, C.-C. Wu and Y.-T. Lin, Org. Lett., 2005, 7, 1979 CrossRef CAS PubMed; (f) S. S. Reddy, V. G. Sree, K. Gunasekar, W. Cho, Y.-S. Gal, M. Song, J.-W. Kang and S.-H. Jin, Adv. Opt. Mater., 2016, 4, 1236 CrossRef CAS; (g) S. S. Reddy, V. G. Sree, W. Cho and S.-H. Jin, Chem.–Asian J., 2016, 11, 3275 CrossRef CAS PubMed.
- (a) L. Wang, Z. Y. Wu, W. Y. Wong, K. W. Cheah, H. Huang and C. H. Chen, Org. Electron., 2011, 12, 595 CrossRef CAS; (b) J. K. Park, K. H. Lee, S. Kang, J. Y. Lee, J. S. Park, J. H. Seo, Y. K. Kim and S. S. Yoon, Org. Electron., 2010, 11, 905 CrossRef CAS; (c) J. Y. Hu, Y. J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064 CrossRef CAS; (d) C. L. Wu, C. H. Chang, Y. T. Chang, C. T. Chen, C. T. Chen and C. J. Su, J. Mater. Chem. C, 2014, 2, 7188 RSC.
- (a) D. Bailey and V. E. Williams, Chem. Commun., 2005, 2569 RSC; (b) Y. Zhang, G. Cheng, Y. Zhao, J. Y. Hou, S. Y. Liu, S. Tang and Y. G. Ma, Appl. Phys. Lett., 2005, 87, 241112 CrossRef; (c) S. Tang, M. Liu, P. Lu, G. Cheng, M. Zeng, Z. Q. Xie, H. Xu, H. P. Wang, B. Yang, Y. G. Ma and D. H. Yan, Org. Electron., 2008, 9, 241 CrossRef CAS; (d) C.-C. Wu, Y.-T. Lin, K.-T. Wong, R.-T. Chen and Y.-Y. Chien, Adv. Mater., 2004, 16, 61 CrossRef CAS; (e) J. H. Jou, M. H. Wu, C. P. Wang, Y. S. Chiu, P. H. Chiang, H. C. Hu and R. Y. Wang, Org. Electron., 2007, 8, 735 CrossRef CAS; (f) S. Tang, W. J. Li, F. Z. Shen, D. D. Liu, B. Yang and Y. G. Ma, J. Mater. Chem., 2012, 22, 4401 RSC; (g) S. Tao, Y. l. Jiang, S. L. Lai, M. K. Fung, Y. C. Zhou, X. H. Zhang, W. M. Zhao and C. S. Lee, Org. Electron., 2011, 12, 358 CrossRef CAS.
- B. Kim, Y.-I. Park, J. Lee, D. Yokoyama, J. H. Lee, J. Kido and J. Park, J. Mater. Chem. C, 2013, 1, 432 RSC.
- (a) H. Lee, B. Kim, S. Kim, J. Kim, J. Lee, H. G. Shin, J. H. Lee and J. Park, J. Mater. Chem. C, 2014, 2, 4737 RSC; (b) K. Sumi, Y. Niko, K. Tokumaruz and G. Konishi, Chem. Commun., 2013, 49, 3893 RSC; (c) Z. Gao, Z. Wang, T. Shan, Y. Liu, F. Shen, Y. Pan, H. Zhang, X. He, P. Lu, B. Yang and Y. Ma, Org. Electron., 2014, 15, 2667 CrossRef CAS; (d) Q. Zhang, H. Kuwabara, W. J. Potscavage Jr, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070 CrossRef CAS PubMed.
- (a) Y. Park, B. Kim, C. Lee, A. Hyun, S. Jang, J. H. Lee, Y. S. Gal, T. H. Kim, K. S. Kim and J. Park, J. Phys. Chem. C, 2011, 115, 4843 CrossRef CAS; (b) S. Ye, J. Chen, C. Di, Y. Liu, K. Lu, W. Wu, C. Du, Y. Liu, Z. Shuai and G. Yu, J. Mater. Chem., 2010, 20, 3186 RSC.
- Y. Park, J. H. Lee, D. H. Jung, S. H. Liu, Y. H. Lin, L. Y. Chen, C. C. Wu and J. Park, J. Mater. Chem., 2010, 20, 5930 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11773f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |