
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sphingolipidomic study of davidiin-treated HepG2 human hepatocellular carcinoma cells using UHPLC-MS†
Li-Min Xie‡
a,
Lee-Fong Yau‡a,
Zhi-Hong Jiang ad,
Li-Yan Zhangb,
Yun Xia*c and
Jing-Rong Wang*a
ad,
Li-Yan Zhangb,
Yun Xia*c and
Jing-Rong Wang*a
aState Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Avenida Wai Long, Taipa, Macau, China. E-mail: jrwang@must.edu.mo; Fax: +853-28825886; Tel: +853-88972404
bCollege of Pharmacy, Guiyang University of Chinese Medicine, Guiyang 550025, China
cBaoshan Hospital of Integrated Chinese and Western Medicine, Shanghai 201999, China. E-mail: xuzhiyang1998@126.com
dInternational Institute for Translational Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
First published on 5th December 2017
Abstract
Davidiin, a natural product originating from Polygonum capitatum, has been proven to possess anti-hepatocellular carcinoma activity. To explore the mechanisms underlying the activity of davidiin, sphingolipids (SPLs) in HepG2 human hepatocellular carcinoma cells with or without the treatment of davidiin were comprehensively analyzed using an improved sphingolipidomic approach established in our lab. A total of 133 SPLs were identified and quantified by using the multiple reaction monitoring (MRM) technique. The results revealed an extensive elevation of dihydroceramide (DHCer) and sphinganine (Sa) induced by davidiin in HepG2 cells, which potentially mediate the cytotoxicity of davidiin. Based on the alteration of upstream and downstream products in the biosynthesis and metabolism network of SPLs, the elevated DHCer and Sa are proposed to result from the inhibition effect of davidiin on dihydroceramide desaturase (DES), which was further confirmed by in vitro assay. Given the emerging role of DES as a therapeutic target of cancer, our results not only provided evidence for the mechanisms underlying the cytotoxicity of davidiin, but also underscored the potential of ellagitannin as a new group of aromatic natural products acting on DES.
1. Introduction
Sphingolipids (SPLs) are a collection of complex and structurally diverse compounds that are biosynthesized from L-serine and palmitoyl CoA.1 SPLs have been found in eukaryotes, as well as in some prokaryotes and viruses,2 where they make up a small but vital fraction (2–20%) of the membrane lipids. It has been reported that SPLs are not only important structural constituents of the cellular membrane, but also essential messengers in intracellular signaling in diverse biological processes, including cell growth, differentiation, apoptosis, and immune responses.1,3–5Since SPL signaling operates upstream of several crucial apoptotic mediators, dysregulation of SPL metabolism, a common theme in human cancers, has been implicated as an important step in tumorigenesis, while alteration of SPL metabolic pathways has been suggested to be involved in various anti-cancer therapies.6 For instance, generation of ceramide (Cer), a proapoptotic species that can induce apoptosis, has been demonstrated to be a critical part of cancer therapy-induced cell death in response to several classes of cytotoxic chemotherapies, including vinca alkaloids (vincristine and vinblastine), anthracyclines (doxorubicin and daunorubicin), taxanes (paclitaxel), and topoisomerase inhibitors (irinotecan, etoposide).6–8 As another example, accumulation of Cer has been suggested as integral to outcomes of targeted anti-cancer agents, such as tyrosine kinase inhibitors imatinib and nilotinib.9,10 These studies implicated the extensive involvement of SPL manipulation in anti-cancer therapies, and underscore the importance of interdicting SPL metabolism as an anti-cancer strategy.
In biological systems, many pathways of SPL metabolism constitute an interconnected network. This network not only regulates the levels of individual biologically active SPLs, but also controls the interconversion of one lipid to another, as well as the balance among them. Therefore, sphingolipidomic analyses provided a powerful approach for viewing the overall interruption of SPL metabolism caused by anti-cancer agent.
Ultra-high performance liquid chromatography (UHPLC) provided adequate separation of isotopic/isomeric species.11 Quadrupole time-of-flight mass spectrometer (Q-TOF MS) offered high resolution and accurate mass measurements at both MS and MS/MS level, while triple quadrupole (QQQ) MS in multiple reaction monitoring (MRM) mode facilitated reliable and sensitive quantification. Therefore, LC-MS has become the most powerful tool for sphingolipidomic analysis owing to the high sensitivity, specificity and throughput capabilities.12 LC-MS strategy has been widely employed in sphingolipidomic studies for the discovery of biomarkers of various diseases, such as cancers,13–15 diabetes and atherosclerosis.16 The LC-MS based sphingolipidomic approach has also been extensively utilized in the evaluation of therapeutic effect of drug and exploration of drug target.17–19
Davidiin is an ellagitannin originating from Polygonum capitatum (P. capitatum). Previously, Wang et al. has reported that davidiin could downregulate EZH2 level via a proteasome-dependent mechanism, which provided important evidence for the antitumor effect of davidiin in hepatocellular carcinoma (HCC).20 Since a considerable number of cytotoxic agents, especially phenolic compounds such as N-(4-hydroxyphenyl) retinamide (4-HPR)18 and curcumin,21 have been shown to exert their activity via regulating SPL metabolism, we propose that davidiin may also alter the metabolism of SPL to exhibit its antitumor effect. Therefore, aiming at exploring the mechanisms underlying the anti-cancer activity of davidiin from the aspect of sphingolipids, we herein performed a comprehensive sphingolipidomic analyses in HepG2 human hepatocellular carcinoma cells with or without the treatment of davidiin by using a well-established UHPLC-MS method developed in our lab.11
2. Experimental
2.1. Chemicals and reagents
Davidiin was isolated from P. capitatum in our lab whose structure was identified by MS and nuclear magnetic resonance (NMR) (Bruker Ascend 600 NMR spectrometer), and its purity was >99%. C18 dihydroceramide (d18:0/18:0) and the internal standard cocktail (I.S.) composed of 25 μM each of 9 sphingolipid standards, including So (d17:1), Sa (d17:0), S1P (d17:1), Sa1P (d17:0), Cer (d18:1/12:0), Cer1P (d18:1/12:0), GalCer (d18:1/12:0), LacCer (d18:1/12:0), and SM (d18:1/12:0), was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Human Liver Microsomes (HLM) 50 donor pool, NADPH regenerating system solution A and solution B were purchased from BD Biosciences (Bedford, MA, USA). LC-MS grade methanol (MeOH) was purchased from J.T. Baker (Avantor Performance Materials, LLC. Center Valley, PA, USA). HPLC grade isopropanol (IPA) and chloroform (CHCl3) were purchased from RCI Labscan Limited (Bangkok, Thailand). Distilled water was prepared using a Milli-Q system (Millipore, Billerica, MA, USA). Bovine serum albumins (BSA), LC-MS grade acetic acid (CH3COOH), formic acid (HCOOH), ammonium acetate (NH4OAc, purity ≥ 98%), potassium dihydrogen phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4) and potassium hydroxide (KOH, purity ≥ 95%) were purchased from Sigma-Aldrich (St. Louis, MO, USA).2.2. Cell culture and treatment
HepG2 human hepatocellular carcinoma cell (ATCC, Rockville, MD) was maintained in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin (PS) (Gibco, New Zealand) at 37 °C in a 5% humidified CO2 incubator. For cell cytotoxicity assay, davidiin was dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 50 mM. Cytotoxicity was assessed by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma, St. Louis, MO, USA). HepG2 cells were seeded in 96-well plates at density of 5 × 103 cells per well. After overnight incubation, the cells were exposed to serial concentrations of davidiin (10–100 μM) for 72 h. Subsequently, 10 μL of MTT solution (5 mg mL−1) was added to each well and incubated at 37 °C for 4 h. Formazan product was dissolved in DMSO for optical density (OD) measurement at 570 nm. The assay was triplicated independently. For lipid extraction, HepG2 cells were seeded in 6-well plates under the same conditions as in the cell cytotoxicity assay. SPLs were extracted from the cells after 72 h treatment with davidiin.2.3. Extraction of SPLs
SPLs in cells were extracted by using a modified method based on our previously established method.11 In brief, cells in each well were washed by phosphate buffered solution (PBS) twice, and then harvested for SPL extraction. In the SPL extraction experiment, cells were transferred into borosilicate glass tubes, in which 10 μL of 2.5 μM I.S. and 0.75 mL extraction solvent MeOH/CHCl3 (2:1, v/v) were added. The mixture was incubated at 48 °C for 12 h after sonicated at room temperature for 30 s. Then the mixture was cooled at room temperature and 75 μL of KOH in MeOH (1 M) was added. The mixture was placed into a shaking incubator at 37 °C for 2 h, then 3 μL CH3COOH was used to neutralize the extracts. After that, a four-step extraction of the mixture was carried out to prepare the SPL extract for analysis. The QC sample was pooled with equal quantity of samples from different groups for the SPLs analysis.2.4. Sphingolipidomic analysis by using LC-MS
SPLs analysis was performed using our well-established UHPLC-MS method as previously descried.11 In brief, the chromatographic separation of SPLs was performed on an Agilent 1290 Infinity UHPLC system (Santa Clara, CA, USA) with an Agilent Eclipse Plus C18 column (100 × 2.1 mm, 1.8 μm) at 40 °C. The mobile phase consisted of (A) MeOH/H2O/HCOOH (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:0.2, v/v/v) and (B) MeOH/IPA/HCOOH (60:40:0.2, v/v/v), both containing 10 mM NH4OAc. The flow rate was 0.35 mL min−1, and the column temperature was maintained at 40 °C for each run.
:40:0.2, v/v/v) and (B) MeOH/IPA/HCOOH (60:40:0.2, v/v/v), both containing 10 mM NH4OAc. The flow rate was 0.35 mL min−1, and the column temperature was maintained at 40 °C for each run.
Qualitative analysis of SPLs was performed on an Agilent ultrahigh definition (UHD) 6550 Q-TOF MS. The injection volume was 10 μL and MS spectra and MS-MS spectra were acquired in positive mode with the mass range of m/z 110–1700 and m/z 40–1700, respectively. The acquisition and data analysis were processed with Agilent Mass Hunter Workstation Software (Agilent, USA).
Quantitative analysis of SPLs was carried out by using an Agilent 6460 QQQ MS (Santa Clara, CA, USA) in MRM mode. The injection volume was 5 μL. Data were processed with Agilent Mass Hunter Workstation Software. Further detail of the parameters, such as characteristic transitions, fragmentor and CE voltages optimized for each compound, and the methodology validations are similar as described before.
2.5. Data analysis
The screening and identification of SPLs were performed by searching with our personal database, which was built and updated based on the Agilent Mass Hunter Personal Compound Database and Library (PCDL) software and LIPID MAPS information (31643 SPLs until July 08 2015).The raw data obtained were processed with Agilent Mass Hunter Workstation Software. The processed SPLs quantitative data were firstly transferred into Microsoft Excel-type datasheet, then imported into SIMCA-P+ 14.0 (Umetrics, Umea, Sweden) for multivariate statistical analysis. PCA was used to visualize general clustering among different groups. PLS-DA was carried out to identify the differentially expressed SPLs between different groups based on VIP values (VIP > 1.0). Statistical analyses of the potential SPL biomarkers were performed by t-test.
2.6. In vitro dihydroceramide desaturase assay
Dihydroceramide desaturase (DES) activity was performed as described previously22 with some modifications. The solubilization of C18 dihydroceramide and davidiin was performed by preparing BSA-substrate complex. A stock solution containing 15 nmol of C18 dihydroceramide in CHCl3/MeOH (2:1, v/v) was dried under a stream of nitrogen and dissolved in 10 μL ethanol contained 0–400 μM davidiin. 30 nmol of BSA was dissolved in 90 μL of KPI buffer (0.5 M potassium phosphate buffer, pH 7.4). The ethanolic solution was added to the dissolved BSA and then vortexed vigorously for 30 s. This step was repeated twice after 1 min. Assay was conducted in a final reaction volume of 0.25 mL that contained BSA-substrate complex, 12.5 μL solution A, 2.5 μL solution B and KPI buffer. After preincubation at 37 °C for 5 min, the reaction was started by adding 25 μL HLM. After shaking gently at 37 °C for 120 min, the reaction was terminated by the addition of 200 μL CHCl3/MeOH (83:17, v/v) on ice. After centrifugation at 14000g for 5 min, the lower phase was collected and dried under a stream of nitrogen. The sample was then reconstituted in 500 μL MeOH and the amount of synthesized C18 ceramide was examined by LC-MS. The DES activity affected by the addition of davidiin was calculated by the following formula: (amount of synthesized C18 ceramide with different concentration of davidiin/amount of synthesized C18 ceramide without davidiin) × 100%.
3. Results and discussion
3.1. Cytotoxicity assay of davidiin in HepG2 cells
The cytotoxicity of davidiin in HepG2 cells was examined by incubating the cells with davidiin (10–100 μM) for 72 h. The cell viability of HepG2 cells under different concentration of davidiin was shown in Fig. 1. The mean IC50 value for davidiin from triplicate experiments was determined to be 59.0 ± 0.9 μM. This result is consistent with the IC50 value previously reported for davidiin of 60.9 ± 0.6 μM in the same cell line.20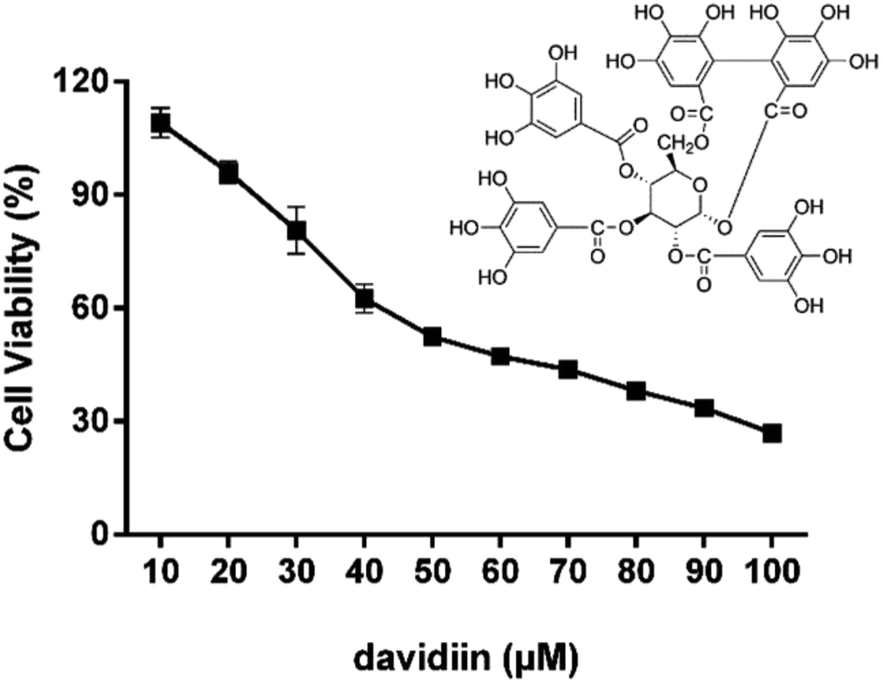 | ||
| Fig. 1 Cell viability of HepG2 cells treated with davidiin (10–100 μM) for 72 h (n = 3). | ||
3.2. Comprehensive profiling of SPLs in HepG2 cells
The SPLs in HepG2 cells were comprehensively profiled for the first time using the sphingolipidomic approach established in our lab.11 SPLs were separated using UHPLC and their high-resolution mass spectra were obtained by using Q-TOF MS. On the basis of high-resolution MS and MS/MS data, a total of 133 SPLs were identified from the pooled samples of HepG2 cells. The identified SPLs were then quantitatively analyzed by using QQQ MS in MRM mode. The identified SPLs and the MRM transitions used to monitor each SPL are listed in Table S1.† The identification and quantification results presented the structural diversity and distribution of SPLs in HepG2 cells, as described class-by-class below.Sphingomyelin (SM) is the most abundant class of SPLs in HepG2 cells, occupying over 80% of the overall SPLs. A total of 43 SMs were identified in HepG2 cells, including 9 dihydrosphingomyelins (DHSMs) and 34 sphingomyelins (SMs). Most SMs were found to possess a C18 sphingoid base backbone; d18:1 is the major type and SM (d18:1/16:0) is the most abundant species. The lengths of the N-acyl chains varied from 14 to 26 carbons, and the degree of unsaturation ranged from 0 to 5. Notably, some uncommon SMs were identified, including SM (d18:1/16:0 (OH)) which had a hydroxyl group on the N-acyl chain, and SM (d19:0/17:0) which contained an odd-numbered carbon chain in both sphingoid backbone and the N-acyl chain. Notably, the levels of individual SMs in the HepG2 cells varied greatly, for example some common species, such as SM (d18:1/16:0), were almost 1000 times more abundant than highly unsaturated species, e.g. SM (d18:1/23:3).
Cer constitutes approximately 10% of the overall SPLs, and bore the greatest structural diversity among the different classes of SPLs in HepG2 cells. A total of 52 Cers from 3 subclasses were identified, including 13 dihydroceramides (DHCers), 34 Cers, and 5 ceramide 1-phosphates (Cer1Ps). All DHCers possesses a C18 sphingoid base backbone with N-acyl chain length varied from 14 to 24 carbons. All Cers possessed C18 sphingoid base backbones except for 3 rare Cers, Cer (d16:1/22:0), Cer (d19:1/24:0), and Cer (d19:1/24:1). The N-acyl chain length of Cers varied from 14 to 25 carbons and the unsaturation degree ranged from 0 to 5. Almost all the identified Cer1Ps were highly unsaturated (unsaturation degree ≥4), including Cer1P (d18:1/24:4), Cer1P (d18:1/26:4), Cer1P (d18:1/28:4), and Cer1P (d18:1/29:4). Of note, a rare Cer1P with a very short N-acyl chain Cer1P (d18:0/3:0), was identified in HepG2 cells.
Glycosphingolipid accounted for about 2% of the overall SPLs in HepG2 cells. In total, 16 glycosphingolipids, including 3 hexosylceramides (HexCers) and 13 lactosylceramides (LacCers), were identified in HepG2 cells. All these HexCers possesses a d18:1 sphingoid backbone and highly-unsaturated N-acyl chain, e.g., HexCer (d18:1/20:3), HexCer (d18:1/26:3), and HexCer (d18:1/26:4). Most of identified LacCers also possesses a d18:1 sphingoid backbone, 4 of which possesses a hydroxyl group on the N-acyl chain, including LacCer (d18:1/16:0 (OH)), LacCer (d18:1/18:0 (OH)), LacCer (d18:1/20:0 (OH)), and LacCer (d18:1/24:0 (OH)). Compared with the high degree of unsaturation observed for the HexCers, the LacCers had relatively low unsaturation degree (1 or 2).
A total of 22 sphingoid bases which occupied about 2% of the overall SPLs in HepG2 cells were successfully characterized, including 13 sphinganines (Sas) and 9 sphingosines (Sos). Their carbon chain lengths varied from 14 to 24, the degree of unsaturation ranged from 0 to 2, and the number of hydroxyl groups ranged from 1 to 3.
3.3. Influence of davidiin on the SPL levels in HepG2 cells
As shown in Fig. 2, upon treatment with 50 μM davidiin for 72 h, significant alternations in the levels of several SPL class in HepG2 cells were observed, while the total SPL content showed only slight increase. Respectively, the levels of dihydrosphingolipids, including DHCer, DHSM, and Sa, showed remarkable elevations by 77.9%, 329.8% and 114.2% in davidiin-treated HepG2 cells compared with control. The levels of Cer1P showed less increase by 24.9% (P < 0.001) after treatment of davidiin.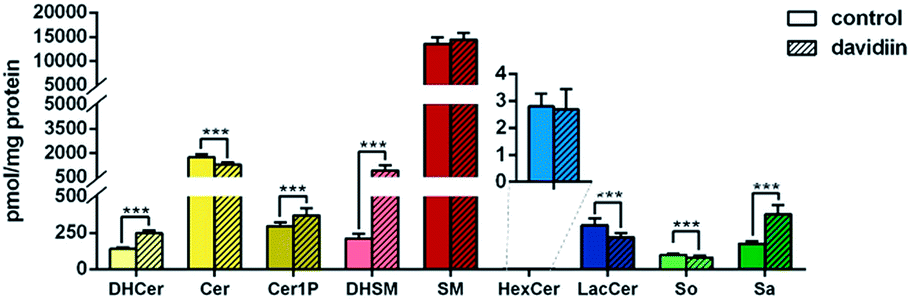 | ||
| Fig. 2 The total levels of the 9 sphingolipid subclasses in untreated (control) and davidiin-treated HepG2 cells (n = 20). Each column represents the mean ± SD (***P < 0.001). DHCer, dihydroceramide; Cer, ceramide; Cer1P, ceramide-1-phosphate; DHSM, dihydrosphingomyelin; SM, sphingomyelin; HexCer, hexosylceramide; LacCer, lactosylceramide; So, sphingosine; Sa, sphinganine. | ||
By contrast, the levels of Cer, LacCer, and So showed significant decreases (by 26.2%, 27.8%, and 19.7%, respectively) in davidiin-treated HepG2 cells (P < 0.001). No significant changes in the levels of SM and HexCer were observed.
3.4. Multivariate analyses of the sphingolipidomic data
Multivariate analysis was subsequently carried out to identify individual SPL markers of davidiin-treated HepG2 cells. As shown in Fig. 3a, the Principal Components Analysis (PCA) scores plot demonstrated a clear separation between control and davidiin-treated HepG2 cells (R2X = 0.857, Q2 = 0.811). The quality control (QC) samples were clustered into one group, indicating the good repeatability of the analysis. In addition, the changes in SPL levels were further analyzed by using Partial Least Squares Discriminant Analysis (PLS-DA) model for searching potential markers. As Fig. 3b showed, untreated and davidiin-treated HepG2 cells were well separated from each other with the value of R2X = 0.745, R2Y = 0.986 and Q2 = 0.984.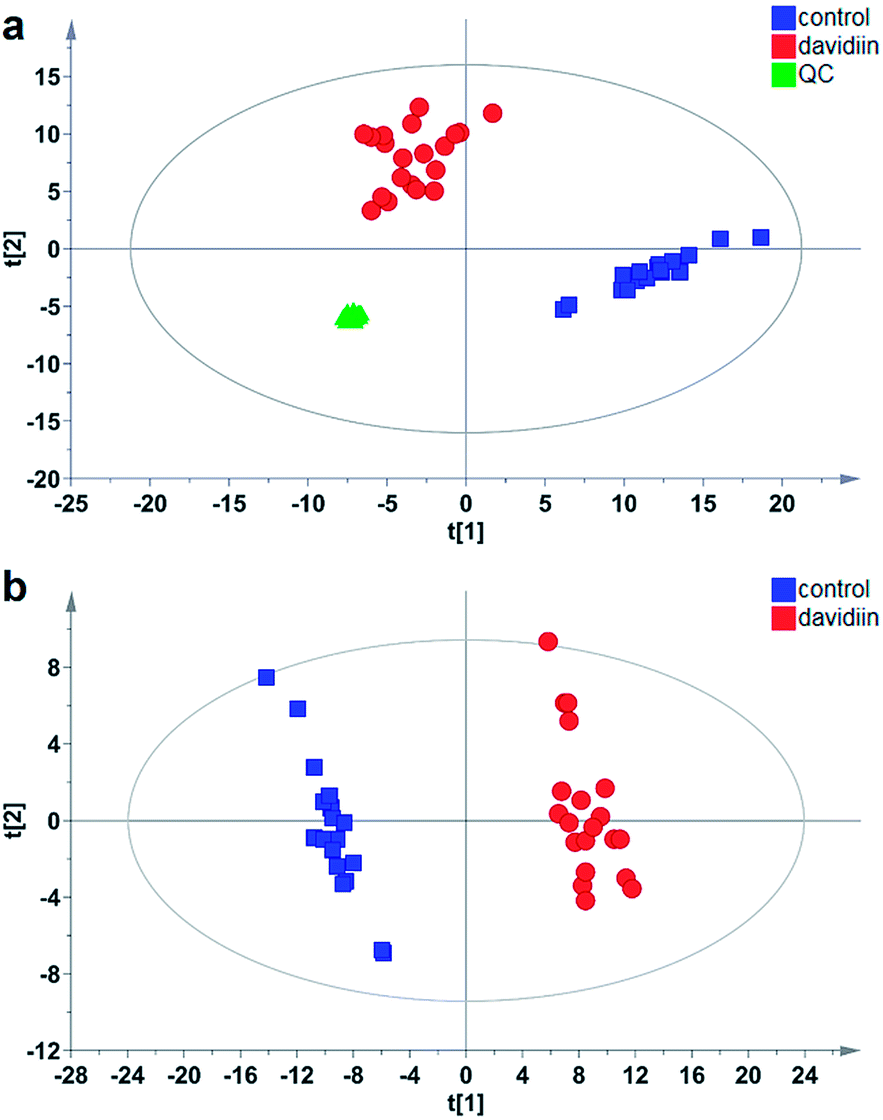 | ||
| Fig. 3 (a) The PCA scores plot for untreated (control) (blue, n = 20), davidiin-treated (red, n = 20) HepG2 cells, and QC (green, n = 20) (R2X = 0.857, Q2 = 0.811); (b) The PLS-DA scores plot for untreated (control) (blue, n = 20) and davidiin-treated (red, n = 20) HepG2 cells (R2X = 0.745, R2Y = 0.986 and Q2 = 0.984). | ||
SPLs with a Variable Importance Plot value (VIP) > 1 were usually considered as the significant variables contributing to the PLS-DA model, and were therefore regarded as potential markers. In our results, 80 out of 133 SPLs scored VIP > 1, including 36 Cers (12 DHCers and 24 Cers), 27 SMs (8 DHSMs and 19 SMs), 1 HexCer, 5 LacCers, 5 Sos, and 6 Sas. Their VIP value, contents and alterations in davidiin-treated HepG2 cells were shown in Table S2.† It can be seen that the level of potential SPL markers DHCer, DHSM and Sa showed consistent elevation in davidiin-treated HepG2 cells as compared to untreated HepG2 cells. Notably, all DHCer markers were increased by at least 1-fold. On the contrary, the level of Cer, HexCer and LacCer markers decreased generally. In the top 5 markers which demonstrated the greatest alteration, 4 were DHCers, in which Cer (d18:0/24:0), Cer (d18:0/14:0), Cer (d18:0/22:0) and Cer (d18:0/23:0) were increased by 11.6-fold, 10.2-fold, 9.7-fold and 7.1-fold respectively, implicating alternation in the level of DHCers is the major influence induced by the treatment of davidiin.
3.5. Time- and dose-dependence of the changes in SPL levels with davidiin treatment
The multivariate analyses revealed that DHCers showed extensive and significant changes in davidiin-treated HepG2 cells. Thus, we further examined the influence of incubation time and davidiin dosage on 6 DHCer potential markers, including Cer (d18:0/14:0), Cer (m18:0/16:0), Cer (d18:0/18:0), Cer (d18:0/22:0), Cer (d18:0/23:0) and Cer (d18:0/24:0).The levels of the 6 DHCer markers in HepG2 cells treated with 0, 25, 50, and 75 μM davidiin for 48 and 72 h were illustrated in the upper panel of Fig. 4a–f. It clearly showed dose-dependent increases in the levels of the 6 DHCer markers in HepG2 cells incubated for both 48 and 72 h. As DHCer can be converted into Cer by DES, we further examined the levels of corresponding Cer species of the 6 DHCer markers which were also scored VIP > 1. As shown in the lower panel of Fig. 4a–f, the levels of 6 Cers generally decreased along with the increase of davidiin dosages, which is just opposite to the change of the levels of DHCers. These phenomena indicated that the changes in the levels of DHCer and Cer induced by davidiin is reversely correlated.
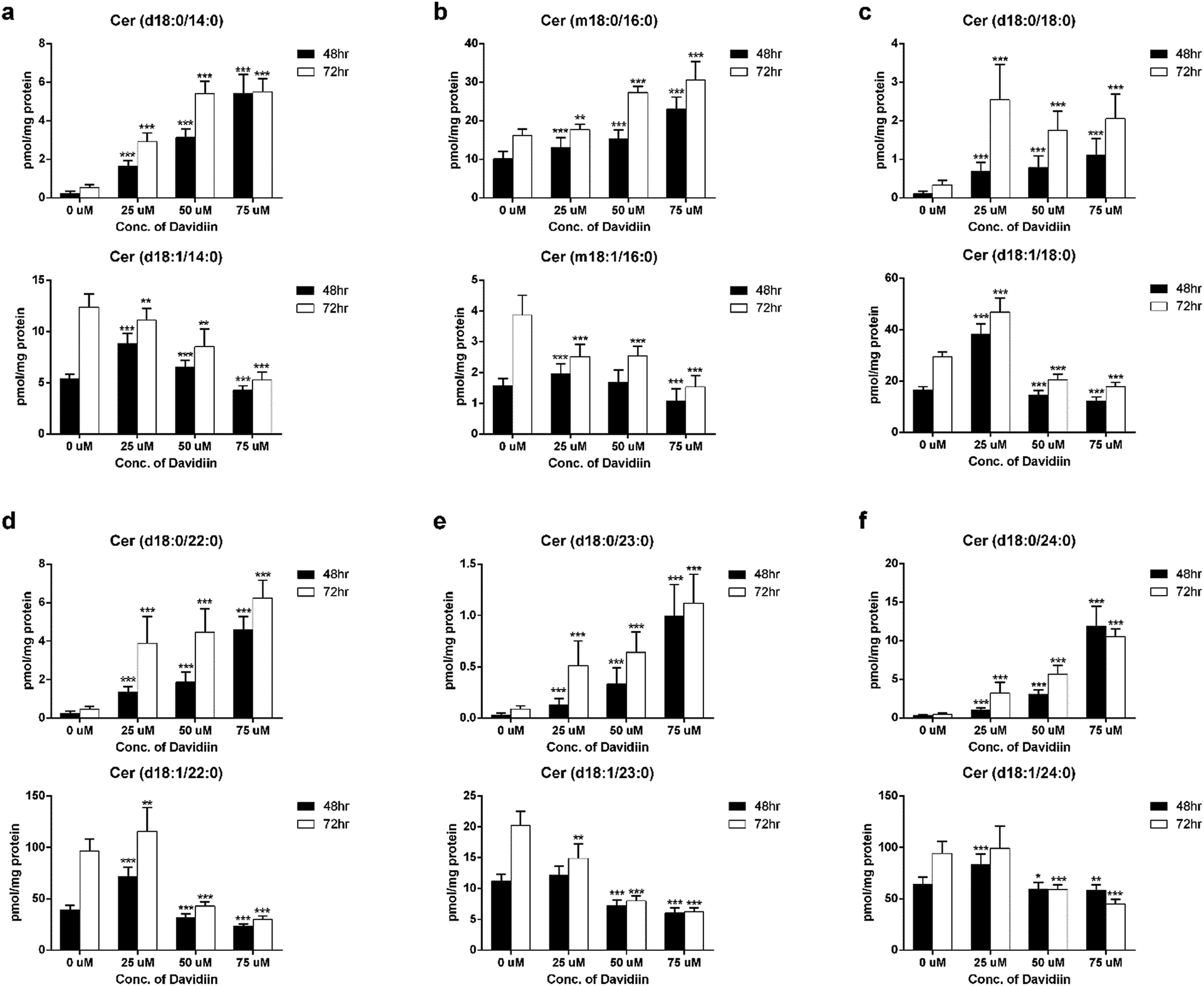 | ||
| Fig. 4 The levels of (a) Cer (d18:0/14:0) and Cer (d18:1/14:0), (b) Cer (m18:0/16:0) and Cer (m18:1/16:0), (c) Cer (d18:0/18:0) and Cer (d18:1/18:0), (d) Cer (d18:0/22:0) and Cer (d18:1/22:0), (e) Cer (d18:0/23:0) and Cer (d18:1/23:0), (f) Cer (d18:0/24:0) and Cer (d18:1/24:0) after treatment with 0, 25, 50, and 75 μM davidiin for 48 and 72 h (n = 10) (**P < 0.05, ***P < 0.001). | ||
The sphingolipidomic analysis revealed a significant and overall elevation of DHCer level in davidiin-treated HepG2 cells. Of note, 12 out of 13 DHCers were identified as potential markers and their levels showed significant and dose-dependent increase upon the treatment of davidiin. Although DHCers were previously regarded as ineffective or inactive lipids, an increasing amount of studies have suggested that DHCers are in fact biologically active in a series of processes such as autophagy, cell cycle arrest, etc.23 Notably, most of the evidence comes from studies on the action of anti-cancer drugs whose activity has been proven to involve the accumulation of DHCer. For example, N-(4-hydroxyphenyl) retinamide (4-HPR), also known as fenretinide, has been demonstrated to elevate DHCer and cell cycle arrest by direct inhibiting DES. Similarly, increase in the levels of DHCers resulting from the inhibition of DES has been suggested to be involved in the activities of tocopherol, resveratrol, curcumin, and celecoxib.17,21,23–25 A recent study further demonstrated that DHCer accumulation could mediate the cytotoxic autophagy of cancer cell.26 These studies collectively supported an important role of DHCer and/or their metabolites as mediators of anti-cancer agents' actions. On the basis of the above fundamentals, it can be speculated that the notable and dose dependent increase of DHCer level might mediate the cytotoxicity of davidiin towards HepG2 cells.
3.6. Inhibition of dihydroceramide desaturase by davidiin
DHCers are mediators in de novo Cer biosynthesis (Fig. 5). According to the biosynthesis and metabolism pathway of SPLs, the accumulation of DHCers might be resulted from stimulation of serine palmitoyl transferase (SPT), upregulation of ceramide synthases (CerS), activation of sphingomyelinase (SMase), and/or inhibition of dihydroceramide desaturase (DES). In our results, the elevation of DHCers was accompanied by simultaneous decrease in the levels of Cer counterparts, indicating a blockage in the desaturation step of Cer induced by davidiin, which was further supported by evidence from following two aspects. Firstly, Sas, the upstream species that can be converted into DHCers by CerS in the de novo biosynthesis of SPLs, as well as DHSMs, which can be produced from DHCer by sphingomyelin synthases, both elevated upon the treatment of davidiin, eliminating the possibilities that DHCer accumulation was resulted from the upregulation of CerS or activation of SMase. Secondly, the levels of Sos and LacCers, downstream metabolic products of Cers, were found to decrease along with Cers in response to the treatment of davidiin, implying down-regulation of Cer metabolism due to the decreased level of Cer. Thus, blocked desaturation of DHCer is the major step responsible for the accumulation of DHCer, and is the key event induced by davidiin. As DES is the key enzyme responsible for the conversion of DHCer into Cer, it can be deduced that accumulation of DHCer in davidiin-treated HepG2 cells is resulted from the inhibition of DES.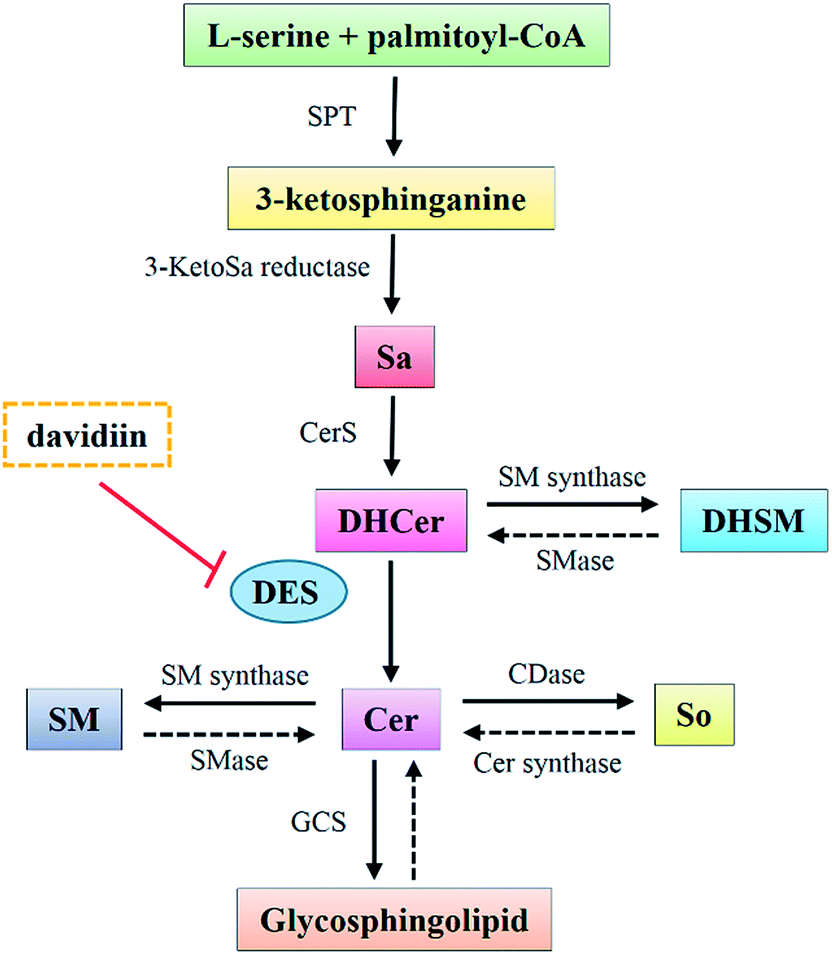 | ||
| Fig. 5 The de novo biosynthesis and conversion of ceramides. Sa, sphinganine; DHCer, dihydroceramide; DHSM, dihydrosphingomyelin; Cer, ceramide; SM, sphingomyelin; So, sphingosine; SPT, serine palmitoyl transferase; 3-KetoSa reductase, 3-ketosphinganine reductase; CerS, ceramide synthase; SM synthase, sphingomyelin synthase; SMases, sphingomyelinase; DES, dihydroceramide desaturase; CDase, ceramidase; Cer synthase, ceramide synthase; GCS, glucosylceramide synthase. | ||
In order to confirm the inhibition activity of davidiin on DES, an in vitro enzyme assay was carried out to incubate DES with 0, 50, 200 and 400 μM davidiin respectively. As a result, we demonstrated that davidiin could significantly inhibit the DES activity in dose-dependent (Fig. 6). The inhibition rate is 50.40 ± 11.31%, 47.98 ± 9.31% and 34.52 ± 10.88% upon treatment with 400 μM, 200 μM and 50 μM davidiin respectively.
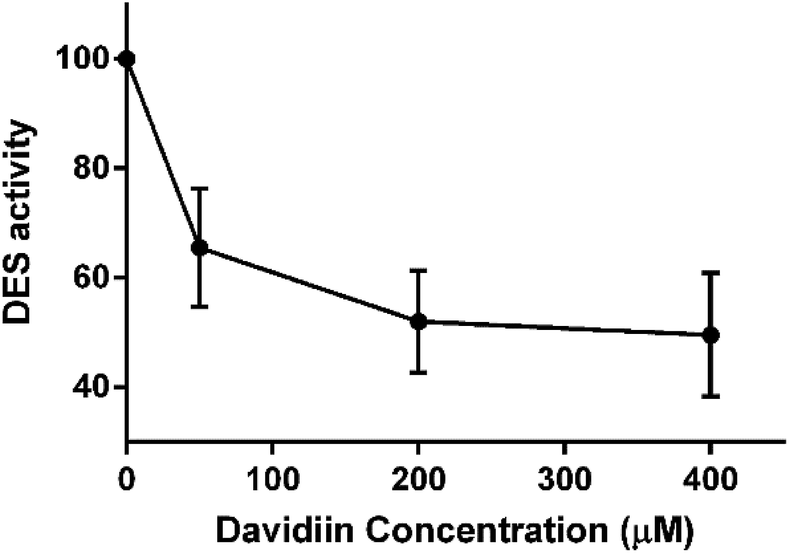 | ||
| Fig. 6 The DES activity under different concentration of davidiin (n = 3). DES, dihydroceramide desaturase. | ||
Previous studies have shown that several cytotoxic agents could induce accumulation of DHCer via similar pathways. Of note, most of these cytotoxic agents are aromatic compounds. For example, in 4-HPR-treated A2780 cells, the elevation of DHCer was caused by the activation of SPT and CerS, with concurrent inhibition of DES.18 Curcumin and celecoxib were also found to inhibit DES as well as leading to increase in DHCer levels.21 Resveratrol induced rise in DHCer level via inhibition of DES1 activity, which is responsible for autophagy promotion.17 Redox sensitive moiety in these aromatic drugs were supposed to contribute to their inhibition on DES.21 Davidiin sharing structure similarity with these aromatic drugs has been suggested to have potential effect on ROS level, therefore davidiin may inhibit DES via regulating ROS level. In addition, it has been demonstrated that inhibition of DES activity led to augmented amounts of dihydrosphingolipids.21 This is in accordance with the concomitant increase of DHCer, DHSM and Sas caused by davidiin, which provided evidence for the inhibition of DES by davidiin.
Apart from DHCers, Sas also elevated significantly in davidiin-treated cells. It has been reported that GT11, a DES inhibitor that mimics the effect of 4-HPR on DES, enhanced cytotoxicity of Sa.27 The concomitant elevations of Sas and DHCers induced by davidiin are quite similar to that observed for 4-HPR, an anti-cancer agent. Therefore, we speculate that the accumulation of Sas in davidiin-treated HepG2 cells was resulted from the inhibition on DES and such alteration also contributed to the cytotoxicity of davidiin.
4. Conclusions
In summary, the SPLs in HepG2 cells were comprehensively profiled for the first time using the sphingolipidomic approach established in our lab. We have identified and quantified 133 SPLs in HepG2 cells and explored 80 potential markers for davidiin-treated HepG2 cells. Our results revealed an extensive elevation of DHCers and Sas induced by davidiin in HepG2 cells, which potentially mediate the cytotoxicity of davidiin. The elevated DHCers and Sas are proved to result from the inhibition on DES by davidiin. This is the first study which report the action of davidiin on SPL metabolism. Given the emerging role of DES as therapeutic target of cancer, our results not only provided evidence for the mechanisms underlying the cytotoxicity of davidiin, but also underscored the potential of ellagitannin as a new group of aromatic natural products acting on DES.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 81260640) and Macao Science and Technology Development Fund (Project code: 023/2016/AFJ to W. J. R.).References
- S. Lahiri and A. H. Futerman, Cell. Mol. Life Sci., 2007, 64, 2270–2284 CrossRef CAS PubMed.
- J. T. Hannich, K. Umebayashi and H. Riezman, Cold Spring Harbor Perspect. Biol., 2011, 3, 725–738 Search PubMed.
- A. H. Merrill Jr, M. D. Wang, M. Park and M. C. Sullards, Trends Biochem. Sci., 2007, 32, 457–468 CrossRef PubMed.
- D. A. Brown and E. London, J. Biol. Chem., 2000, 275, 17221–17224 CrossRef CAS PubMed.
- R. Corriden, A. Hollands, J. Olson, J. Derieux, J. Lopez, J. T. Chang, D. J. Gonzalez and V. Nizet, Nat. Commun., 2015, 6, 8369 CrossRef CAS PubMed.
- T. H. Beckham, J. C. Cheng, S. T. Marrison, J. S. Norris and X. Liu, Adv. Cancer Res., 2013, 117, 1–36 CrossRef CAS PubMed.
- R. Bose, M. Verheij, A. Haimovitz-Friedman, K. Scotto, Z. Fuks and R. Kolesnick, Cell, 1995, 82, 405–414 CrossRef CAS PubMed.
- D. K. Perry, J. Carton, A. K. Shah, F. Meredith, D. J. Uhlinger and Y. A. Hannun, J. Biol. Chem., 2000, 275, 9078 CrossRef CAS PubMed.
- Y. Baran, A. Salas, C. E. Senkal, U. Gunduz, J. Bielawski, L. M. Obeid and B. Ogretmen, J. Biol. Chem., 2007, 282, 10922–10934 CrossRef CAS PubMed.
- W. C. Huang, C. C. Tsai, C. L. Chen, T. Y. Chen, Y. P. Chen, Y. S. Lin, P. J. Lu, C. M. Lin, S. H. Wang, C. W. Tsao, C. Y. Wang, Y. L. Cheng, C. Y. Hsieh, P. C. Tseng and C. F. Lin, FASEB J., 2011, 25, 3661–3673 CrossRef CAS PubMed.
- J. R. Wang, H. Zhang, L. F. Yau, J. N. Mi, S. Lee, K. C. Lee, P. Hu, L. Liu and Z. H. Jiang, Anal. Chem., 2014, 86, 5688–5696 CrossRef CAS PubMed.
- A. H. Merrill Jr, M. C. Sullards, J. C. Allegood, S. Kelly and E. Wang, Methods, 2005, 36, 207–224 CrossRef PubMed.
- A. H. Merrill Jr and Y. A. Hannun, Sphingolipid metabolism and cell signaling, Academic Press, 1999 Search PubMed.
- J. K. Nicholson, J. Connelly, J. C. Lindon and E. Holmes, Nat. Rev. Drug Discovery, 2002, 1, 153–161 CrossRef CAS PubMed.
- P. Knapp, L. Bodnar, A. Blachnio-Zabielska, M. Swiderska and A. Chabowski, Gynecol. Oncol., 2017, 147, 139–144 CrossRef CAS PubMed.
- M. Li, L. Yang, Y. Huang, Y. Bai and H. Liu, J. Test. Eval., 2017, 1, 245–254 Search PubMed.
- P. Signorelli, J. M. Munoz-Olaya, V. Gagliostro, J. Casas, R. Ghidoni and G. Fabrias, Cancer Lett., 2009, 282, 238–243 CrossRef CAS PubMed.
- M. Valsecchi, M. Aureli, L. Mauri, G. Illuzzi, V. Chigorno, A. Prinetti and S. Sonnino, J. Lipid Res., 2010, 51, 1832–1840 CrossRef CAS PubMed.
- S. A. Saddoughi, E. Garrett-Mayer, U. Chaudhary, P. E. O'Brien, L. B. Afrin, T. A. Day, M. B. Gillespie, A. K. Sharma, C. S. Wilhoit, R. Bostick, C. E. Senkal, Y. A. Hannun, J. Bielawski, G. R. Simon, K. Shirai and B. Ogretmen, Clin. Cancer Res., 2011, 17, 6097–6105 CrossRef CAS PubMed.
- Y. Wang, J. Ma, S. C. Chow, C. H. Li, Z. Xiao, R. Feng, J. Fu and Y. Chen, Tumor Biol., 2014, 35, 205–212 CrossRef PubMed.
- G. Fabrias, J. Munoz-Olaya, F. Cingolani, P. Signorelli, J. Casas, V. Gagliostro and R. Ghidoni, Prog. Lipid Res., 2012, 51, 82–94 CrossRef CAS PubMed.
- J. N. Abelson, M. I. Simon, A. H. Merrill Jr and Y. A. Hannun, Methods Enzymol., 1999, 311, 3–30 Search PubMed.
- M. M. Siddique, Y. Li, B. Chaurasia, V. A. Kaddai and S. A. Summers, J. Biol. Chem., 2015, 290, 15371–15379 CrossRef CAS PubMed.
- V. Gagliostro, J. Casas, A. Caretti, J. L. Abad, L. Tagliavacca, R. Ghidoni, G. Fabrias and P. Signorelli, Int. J. Biochem. Cell Biol., 2012, 44, 2135–2143 CrossRef CAS PubMed.
- Q. Jiang, X. Rao, C. Y. Kim, H. Freiser, Q. Zhang, Z. Jiang and G. Li, Int. J. Cancer, 2012, 130, 685–693 CrossRef CAS PubMed.
- S. Hernandez-Tiedra, G. Fabrias, D. Davila, I. J. Salanueva, J. Casas, L. R. Montes, Z. Anton, E. Garcia-Taboada, M. Salazar-Roa, M. Lorente, J. Nylandsted, J. Armstrong, I. Lopez-Valero, C. S. McKee, A. Serrano-Puebla, R. Garcia-Lopez, J. Gonzalez-Martinez, J. L. Abad, K. Hanada, P. Boya, F. Goni, M. Guzman, P. Lovat, M. Jaattela, A. Alonso and G. Velasco, Autophagy, 2016, 12, 2213–2229 CrossRef CAS PubMed.
- H. Wang, B. J. Maurer, Y. Y. Liu, E. Wang, J. C. Allegood, S. Kelly, H. Symolon, Y. Liu, A. H. Merrill Jr, V. Gouaze-Andersson, J. Y. Yu, A. E. Giuliano and M. C. Cabot, Mol. Cancer Ther., 2008, 7, 2967–2976 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1 and 2. See DOI: 10.1039/c7ra11266a |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |